

Abstract
Mitsugumin 29 (MG29) is related to the fatigue and aging processes of skeletal muscle. To examine the roles of MG29 in conjunction with its binding protein, the canonical-type transient receptor potential cation channel 3 (TRPC3), in skeletal muscle, the binding region of MG29 to TRPC3 was studied along with the functional relevance of the binding in mouse primary skeletal myotubes using co-immunoprecipitation assays and Ca2+ imaging experiments. The N-terminus and the I–II loop of MG29 constitute the binding region for TRPC3. The myotubes that expressed the MG29 mutant missing the entire TRPC3-binding region showed a disrupted binding between endogenous MG29 and TRPC3 and a reduction in Ca2+ transients in response to membrane depolarization without affecting ryanodine receptor 1 activity, the resting cytosolic Ca2+ level, and the amount of releasable Ca2+ from the sarcoplasmic reticulum. Among the proteins mediating Ca2+ movements in skeletal muscle, TRPC4 expression was significantly decreased by the MG29 mutant. Therefore, MG29 could be a new factor for regulating Ca2+ transients during skeletal muscle contraction possibly via a correlation with TRPC3 and TRPC4.
Keywords: mitsugumin 29, skeletal muscle, TRPC3, TRPC4
1. INTRODUCTION
The main task of skeletal muscle is contraction by excitation-contraction (EC) coupling. During skeletal EC coupling, dihydropyridine receptor (DHPR, a Ca2+ channel in the transverse (t)-tubule membrane) senses the depolarization of the t-tubule membrane induced by α-motor neurons, and the depolarization-sensed DHPR physically interacts with and activates the ryanodine receptor 1 (RyR1, a Ca2+ channel on sarcoplasmic reticulum (SR) membrane) [1,2,3]. The activated RyR1 allows the release of Ca2+ ions from the SR to the cytoplasm, and the Ca2+ ions activate contractile proteins for skeletal muscle contraction. RyR1 is also activated by sub-micromolar Ca2+, via a Ca2+-induced Ca2+ release (CICR) mechanism, and CICR is required for maximizing Ca2+ transients for skeletal EC coupling [1,2,3]. The canonical-type transient receptor potential cation channel 3 (TRPC3) assists in the maximization [4]. The junctional membrane complex (JMC), where the t-tubule and the SR membranes are juxtaposed, provides a structural context for the proper arrangement of the proteins mentioned above [2,3,5]. Junctophilins (JPs) contribute to the formation of the JMC [3,6,7,8].
Mitsugumin 29 (MG29), one of the synaptophysin proteins, is a small 29 kDa protein [9]. Unlike most other proteins in skeletal and cardiac muscle, MG29 is exclusively expressed in skeletal muscle (in both t-tubule and SR membranes) [9,10,11]. Skeletal muscle from MG29-deficient mice show swollen and irregular t-tubules, incomplete SR structures, and a partial malformation of JMC [12,13]. Along with these morphological changes, MG29-deficient mice exhibit functional abnormalities in skeletal muscle: low twitch force and impaired store-operated Ca2+ entry (SOCE) [13,14,15]. In addition, MG29-deficient mice are more susceptible to fatigue [14,16,17], as shown in animal models with chronic degenerative skeletal muscle diseases or disorders such as atrophy and age-related sarcopenia [18,19]. In accordance with this, in aged mouse skeletal muscle, MG29 expression is significantly decreased [20].
The TRPCs are non-selective cation channels on plasma/t-tubule membranes that allow Ca2+ entry into various cells [2,3,21]. TRPC1, TRPC3, TRPC4, and TRPC6 are mainly expressed in skeletal muscle (TRPC2 with extremely lower expression level than others) [2,3,22]. Among them, the roles of TRPC3 in skeletal muscle have been well examined. TRPC3-deficient mice show an impaired walking behavior due to abnormal skeletal muscle coordination [23]. TRPC3-transgenic mice induce a phenotype similar to the mouse model of Duchenne muscular dystrophy by showing an increased SOCE and similar changes in gene expressions [24]. The expression levels of TRPC3 are tightly regulated during the differentiation of skeletal myoblasts to myotubes [4]. TRPC3 interacts with six skeletal proteins that are expressed in JMC, including MG29 [25].
Considering that both MG29 and TRPC3 have relevance to skeletal muscle functions and that MG29 is a TRPC3-binding protein, it is possible that the interaction between MG29 and TRPC3 plays a certain role in skeletal muscle functions. Therefore, in the present study, the binding region of MG29 to TRPC3 was examined, and MG29 mutants missing the TRPC3-binding region, either in part or entirely, were expressed in mouse primary skeletal myotubes and Ca2+ transients in the myotubes were examined.
2. MATERIALS AND METHODS
2.1 cDNA constructions and the expression of MG29 portions
Using mouse MG29 cDNA (GenBank accession No. AB010140.1) as a template, various portions or deletion mutants of MG29 were synthesized via PCR using the primers presented in Supplementary Material 1. Each PCR product was inserted into a pGEX-4T-1 vector (at the EcoRI and SalI sites for GST-fused portions) or into a pGFP mammalian expression vector (at the BamHI and EcoRV sites for Δ33-MG29 and Δ116-MG29). Each GST-fused portion was expressed in E. coli (DH5α), was solubilized in a lysis buffer, and was pulled down using GST beads (Amersham Biosciences, Pittsburgh, PA, USA) followed by SDS-PAGE and Coomassie Blue staining, as previously described [26,27,28].
2.2 Preparation of the triad sample, co-immunoprecipitation, and immunoblot assay
The triad vesicles containing TRPC3 were prepared from rabbit fast-twitch skeletal muscle and were solubilized, as previously described [4,6,25,29]. All surgical interventions and methods of animal care were conducted under the guidelines, as previously described [6,27,30]. The solubilized triad sample or the lysate of myotubes was subjected to co-immunoprecipitation assay using anti-TRPC3 antibody [28,30]. For immunoblot assays, various antibodies were used: anti-RyR1, anti-DHPR, anti-SERCA1a, anti-MG29, anti-JP1, anti-JP2, and anti-GST antibodies (1:1,000) from Thermo Scientific Inc. (Rockford, IL, USA), anti-TRPC3 and anti-TRPC4 antibodies (1:800) from Alomone Laboratories (Jerusalem 9104201, Israel), and anti-Orai1, anti-STIM1, and anti-α-actin antibodies (1:1,000) from Abcam (Cambridge, MA, USA).
2.3 Cell culture and cDNA transfection
Primary skeletal myoblasts derived from mouse neonates were allowed to proliferate or differentiate to myotubes, and were transfected with each cDNA to express wild-type MG29, Δ33-MG29, or Δ116-MG29, as previously described [6,27,29,30,31].
2.4 Ca2+ imaging experiments
Mouse primary skeletal myotubes on 96-well plates were loaded with either 5 μM of fura-2 (for resting cytosolic Ca2+ levels), fluo-5N (which is suitable for detecting high levels of Ca2+ ranging from micro to mill molar, for the amount of releasable Ca2+ from the SR), or with fluo-4 (for other measurements) in an imaging buffer, as previously described [6,27,29,30,31]. Ca2+ transients in the myotubes were measured using an inverted stage microscope (Nikon Eclipse TS100, Melville, NY, USA) equipped with a Nikon 40X oil-immersion objective (NA 1.30), a high-speed monochromator with a 75W xenon lamp (FSM150Xe, Bentham Instruments, Verona, VA, USA), a 12-bit charge-coupled device camera (DVC-340M-OO-CL, Digital Video Camera Company, Austin, TX 78744, USA), and an auto-perfusion system (AutoMate Scientific, Berkeley, CA, USA) [6,27,29,30,31]. The data were analyzed using image acquisition and analysis software (High-Speed InCyt Im1 or Im2, v5.29, Intracellular Imaging Inc., Cincinnati, OH, USA).
2.5 Statistical analysis
The results are presented as the means ± S.E. for the number of experiments presented in the figure legends. The significant differences were analyzed using a paired t-test (GraphPad InStat, v2.04, GraphPad Software, La Jolla, CA, USA). The differences were considered significant at p < 0.05. The graphs were prepared using Origin v7 software (OriginLab, Northampton, MA, USA).
3. RESULTS AND DISCUSSION
3.1. The N-terminus and the I–II loop of MG29 bind to TRPC3
To examine the binding region of MG29 to TRPC3, MG29 portions, with the exception of the transmembrane domains, were constructed as GST-fused proteins (Fig. 1A). Each portion was expressed in E. coli, and was pulled down with GST beads followed by SDS-PAGE and Coomassie Blue staining (Fig. 1B, left). Each MG29 portion was successfully expressed. Co-immunoprecipitation of TRPC3 with each MG29 portion was conducted using a solubilized triad sample containing TRPC3, the lysate of E. coli expressing each GST-fused MG29 portion, and anti-TRPC3 antibody (Fig. 1B, right). Among them, the N-terminus and the I–II loop was bound to TRPC3. In the case of the III–IV loop, two different sizes were expressed, and the upper band is for the expected size according to its number of amino acids. However, neither was bound to TRPC3. The N-terminus was sub-divided into smaller portions in order to narrow the region (Supplementary Material 2). None of the smaller portions were bound to TRPC3, suggesting that the intact N-terminus of MG29 is required for the binding of MG29 to TRPC3. This is reasonable because MG29 is a small protein (29 kDa). Overall, the region from 1 to 116 amino acids of MG29 covering the N-terminus and the I–II loop could constitute the TRPC3-binding region.
Figure 1. Co-immunoprecipitation of TRPC3 with each MG29 portion.
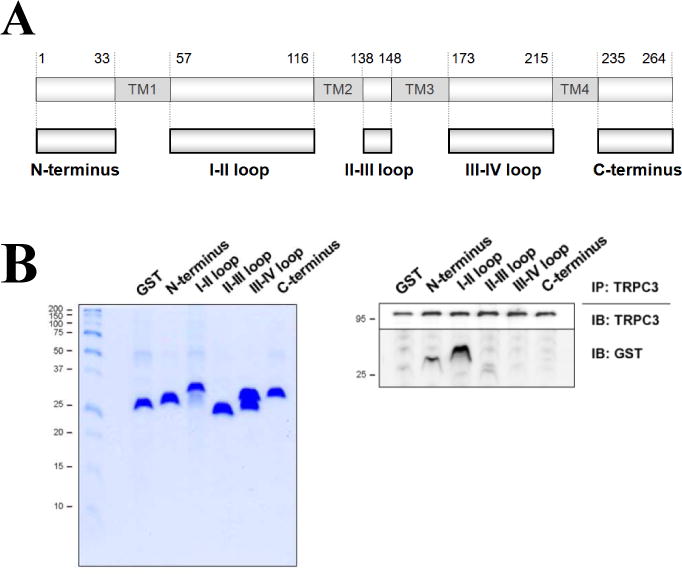
(A) Schematic diagrams of full-length MG29 and various MG29 portions. Numbers indicate the sequence of amino acids. TM indicates transmembrane domain. (B) Immobilized GST-fused MG29 portions on GST beads were separated on SDS-PAGE gel (12%) followed by Coomassie Blue staining (left). Each portion was successfully expressed in E. coli. Co-immunoprecipitation of TRPC3 with each GST-fused MG29 portion was conducted using anti-TRPC3 antibody, followed by immunoblot assay with anti-TRPC3 or anti-GST antibodies (right). GST was used as a negative control. IP or IB translates to immunoprecipitation or immunoblot. Three independent experiments were conducted and a representative result is presented. The N-terminus and the I–II loop of MG29 were bound to TRPC3.
Based on the three-dimensional (3D) structure of the TRPC3-binding region of MG29 predicted by the RaptorX program [32] (Supplementary Material 3A and 3B), the binding of MG29 to TRPC3 could be mediated on both sides of the plasma/t-tubule membrane: an un-structured random coil and a short α-helix in the N-terminus in the cytoplasm, and 3 tandem β-strands in the I–II loop in the extracellular space. The unstructured random coil in the N-terminus was predicted to exist in an intrinsically disordered state [33] (Supplementary Material 3C), which means that it could adopt a fixed 3D structure after binding to its partners, such as TRPC3. Phosphorylation sites exist predominately in intrinsically disordered proteins [34], and, indeed, 4 residues in the un-structured random coil (20%) were predicted to be phosphorylation sites (Supplementary Material 3D).
3.2 In skeletal myotubes, the MG29 mutant missing the entire TRPC3-binding region results in a reduction in Ca2+ transients for skeletal EC coupling
To examine the role of the binding between MG29 and TRPC3 in the context of full-length MG29 and in skeletal muscle, two deletion mutants of MG29 were constructed (Fig. 2A): one was Δ33-MG29 missing a portion of the TRPC3-binding region (N-terminus only), and the other was Δ116-MG29 missing the entire TRPC3-binding region. Each mutant was expressed in mouse primary skeletal myotubes, and their successful expressions were confirmed by the presence of the GFP signal (Fig. 3B). As expected from the fact that MG29 is not responsible for the differentiation of myoblasts to myotubes [13,14], the expressions of neither mutant interfered with the differentiation (i.e., myotube formations).
Figure 2. A reduction in Ca2+ transients in response to membrane depolarization, and the disruption of the binding between endogenous MG29 and TRPC3 in mouse primary skeletal myotubes expressing Δ116-MG29.
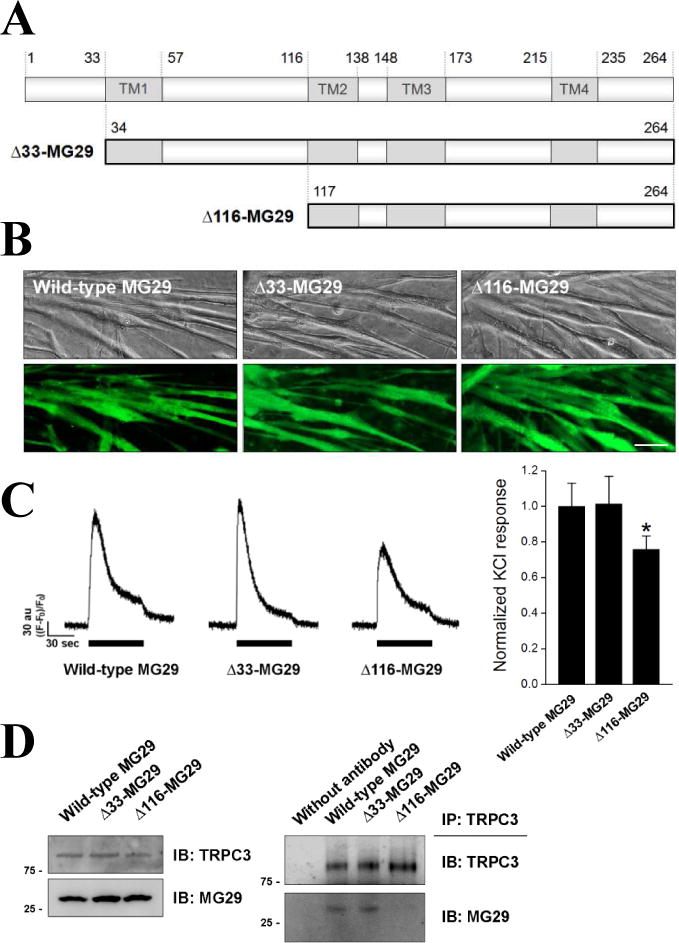
(A) Schematic diagram of two MG29 deletion mutants: Δ33-MG29 and Δ116-MG29. (B) Successful expressions of each mutant in mouse primary skeletal myotubes were confirmed by the presence of the GFP signal. Bar represents 50 μm. (C) KCl inducing membrane depolarization was applied to the myotubes expressing each mutant. Histograms are shown for normalized peak amplitude to the mean value of those from wild-type MG29 (92 wild-type MG29, 90 Δ33-MG29, or 95 Δ116-MG29 myotubes). *, significant difference compared with wild-type MG29 (p < 0.05). A significant reduction in Ca2+ transients by Δ116-MG29 was found. (D) The lysate of myotubes expressing each mutant was subjected to immunoblot assay with anti-TRPC3 or anti-MG29 antibodies (left), or to co-immunoprecipitation assay using anti-TRPC3 antibodies followed by immunoblot assay with anti-TRPC3 or anti-MG29 antibodies (right). Three independent experiments were conducted and a representative result is presented. There was no significant change in the expression level of TRPC3 or MG29, however, the binding between endogenous MG29 and TRPC3 was disrupted by Δ116-MG29.
Figure 3. A significant decrease in the TRPC4 expression in mouse primary skeletal myotubes expressing Δ116-MG29.
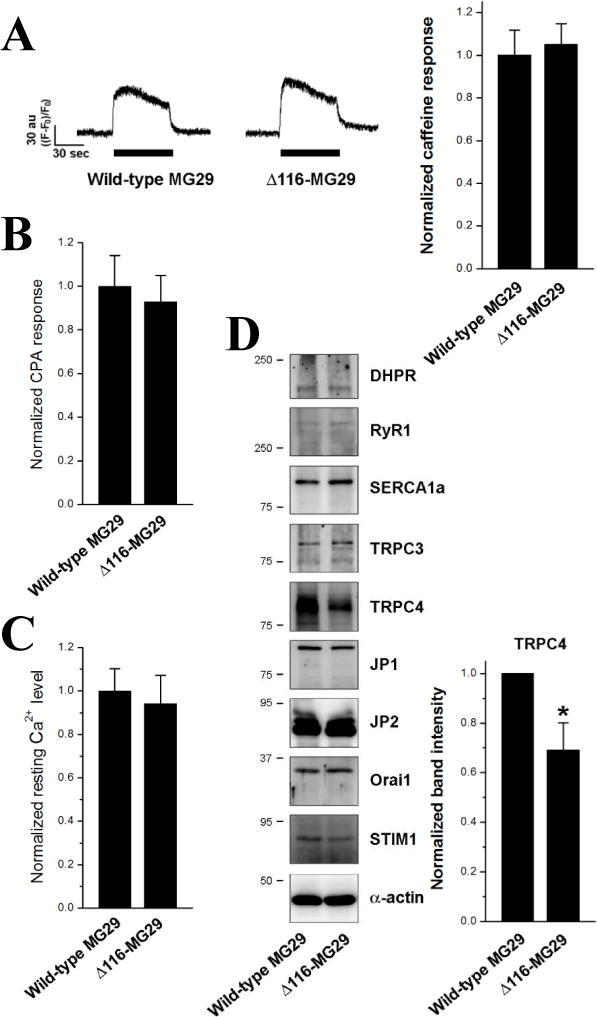
(A) Caffeine was applied to the myotubes expressing Δ116-MG29. There was no significant change in Ca2+ transients in response to caffeine by Δ116-MG29 compared with wild-type MG29 control. The amount of releasable Ca2+ from the SR to the cytoplasm in response to CPA (10 μM) (B) or resting cytosolic Ca2+ level (C) was examined in the myotubes (40 myotubes per each for CPA response, and 84 wild-type MG29 or 85 Δ116-MG29 myotubes for resting cytosolic Ca2+ level). The results are summarized as histograms. Neither the amount of releasable Ca2+ from the SR nor the resting cytosolic Ca2+ level was significantly changed by Δ116-MG29. (D) The lysate of the myotubes expressing Δ116-MG29 was subjected to immunoblot assays with various antibodies. α-actin was used as a loading control. Three independent experiments were conducted for each protein and a representative result is presented. *, significant difference compared with wild-type MG29 (p < 0.05). TRPC4 expression was significantly decreased by Δ116-MG29 compared with wild-type MG29 control (summarized as histograms on the right).
Ca2+ transients from the SR to the cytoplasm in response to KCl (a membrane depolarizer) were measured in the myotubes expressing either of the MG29 mutants (Fig. 2C). KCl depolarizes t-tubule membranes, activates DHPR, and induces Ca2+ transients through RyR1 for skeletal muscle contraction (i.e., KCl induces Ca2+ transients for skeletal EC coupling) [4,6,29,30,31]. Unlike myotubes expressing Δ33-MG29, myotubes expressing Δ116-MG29 showed a significantly reduced response to KCl compared with wild-type MG29 control (Fig. 2C, histogram, 0.76 ± 0.08 vs. 1.00 ± 0.13), suggesting that the absence of the entire TRPC3-binding region in MG29 could induce a reduction in Ca2+ transients for skeletal EC coupling. It is unlikely that the reduction in the Ca2+ transients by Δ116-MG29 results from a change in the channel activity of DHPR, because the expression level of DHPR was not changed by Δ116-MG29 (Fig. 3D), and the major function of DHPR in skeletal muscle is to activate RyR1 by sensing the depolarization of the t-tubule membrane instead of serving as a Ca2+ channel [1,2,3].
3.3 In skeletal myotubes, the binding between MG29 to TRPC3 could participate in regulating Ca2+ transients for EC coupling
The expression levels of endogenous MG29 or TRPC3 in the myotubes expressing either of the MG29 mutants were examined (Fig. 2D, left), and there was no significant change in the expression level of MG29 or TRPC3 by either mutant. Considering that both MG29 mutants are missing the TRPC3-binding region, either in part or entirely, the binding ability of endogenous MG29 to TRPC3 in the myotubes expressing either of the MG29 mutants was examined by co-immunoprecipitation assay using anti-TRPC3 antibody (Fig. 2D, right). Myotubes expressing Δ116-MG29 showed an almost disrupted binding between endogenous MG29 and TRPC3, suggesting that the existence of Δ116-MG29 in the myotubes induces a disruption of the binding between endogenous MG29 and TRPC3. MG29 homo-dimerizes (Figure 4C in [35]), which is a possible explanation for how the disrupted binding that the dimerization of endogenous MG29 with TRPC3-unbinding Δ116-MG29, but not with Δ33-MG29, disrupts the binding between endogenous MG29 and TRPC3. Also, the I–II loop might be a region for the homo-dimerization of MG29 as well as for a part of TRPC3-binding.
To examine the possibility that the reduction in the Ca2+ transients for skeletal EC coupling by Δ116-MG29 resulted from a change in RyR1 activity [30,31], RyR1 activity in the myotubes was assessed by applying caffeine, which is a direct agonist of RyR1 (Fig. 3A). There was no significant change in Ca2+ transients in response to caffeine by Δ116-MG29 compared with wild-type MG29 control, suggesting that RyR1 activity is not changed by Δ116-MG29. To measure the amount of releasable Ca2+ from the SR to the cytoplasm, cyclopiazonic acid (CPA) inducing Ca2+ depletion from the SR was applied to the myotubes in the absence of extracellular Ca2+ (Fig. 3B), because RyR1 activity is affected by the luminal Ca2+ level of the SR via CICR mechanism [1,3,36]. There was also no significant change in the amount of releasable Ca2+ from the SR by Δ116-MG29. The involvement of cytosolic Ca2+ levels via the CICR mechanism was also ruled out, because there was no significant change in the resting cytosolic Ca2+ level (Fig. 3C). Therefore, these results suggest again that the reduced Ca2+ transients for skeletal EC coupling in myotubes expressing Δ116-MG29 could be due to the disrupted interaction between endogenous MG29 and TRPC3 by Δ116-MG29. TRPC3-knockdown skeletal myotubes show a reduction in Ca2+ transients for skeletal EC coupling without a change in the amount of releasable Ca2+ from the SR and in the expression levels of DHPR and RyR1 [4], which is similar to those found in the myotubes expressing TRPC3-unbinding Δ116-MG29 in the present study (Figs. 2 and 3). Therefore, MG29 could participate in regulating Ca2+ transients for skeletal EC coupling via binding to TRPC3 and regulating TRPC3 activity.
Muscle fibers from MG29-deficient mice show an increase in susceptibility to muscle fatigue due to a reduction in the amount of releasable Ca2+ from the SR and an increase in CICR through RyR1 [14,16,17]. Considering no change in the resting cytosolic Ca2+ level, the amount of releasable Ca2+ from the SR, and RyR1 activity in the myotubes expressing Δ116-MG29 (Fig. 3), the TRPC3-binding region of MG29 could be unrelated to the fatigability of skeletal muscle. These facts suggest possibilities that the TRPC3-binding region of MG29 is involved in short-term Ca2+ transients (i.e., during EC coupling, as shown in Fig. 2C), and that the other region of MG29 is mainly related to long-term Ca2+ homeostasis such as muscle fatigue. In addition, it seems that the TRPC3-binding region of MG29 is also unrelated to the aging of skeletal muscle, because gross changes in JMC, which is easily found in aged skeletal muscle fibers [15,20], were not induced by the expression of Δ116-MG29 (Supplementary Material 4).
3.4 In skeletal muscle, MG29, TRPC3 and TRPC4 could be correlated
To find additional protein(s) that participates in the reduced Ca2+ transients for skeletal EC coupling by Δ116-MG29, the expression levels of major proteins mediating Ca2+ movements in skeletal muscle were examined by immunoblot assays with the lysate of myotubes expressing Δ116-MG29 (Fig. 3D). There was no significant change in the expression levels of 3 major proteins that mediate Ca2+ movements between the SR and the cytoplasm during skeletal muscle contraction and relaxation: DHPR, RyR1 and SERCA1a. There was also no significant change in the expression levels of proteins that are responsible for SOCE: Orai1 and STIM1, suggesting a low possibility that a change in SOCE via Orai1 is a cause of the reduced Ca2+ transients by Δ116-MG29. There was also no change both in the expression levels of proteins that mediate the formation of JMC: neither in JP1 and JP2, nor in overall JMC formations (Supplementary Material 4).
It is interesting that TRPC4 expression was significantly decreased in the myotubes expressing TRPC3-unbinding Δ116-MG29 (Fig. 4D, right, more than 30% reduction compared with wild-type MG29 control), suggesting that the decrease in TRPC4 expression could assist in the reduced Ca2+ transients for skeletal EC coupling by Δ116-MG29. TRPC3-knockdown in mouse muscular dysgenic skeletal myotubes also dramatically decreases TRPC4 expression [37]. Therefore, MG29 could play a role in skeletal muscle functions in conjunction with TRPC4 as well as TRPC3. This suggests that, along with TRPC3 [4], TRPC4 could not only serve as a protein mediating SOCE [2,3,21], but may also take part in regulating the Ca2+ transients for skeletal EC coupling. This is the first clue that TRPC4 could be functionally related to TRPC3 in skeletal muscle, although, among TRPCs, TRPC4 differs a great deal from TRPC3 [21].
Supplementary Material
Acknowledgments
This work was supported by the Basic Science Research Program (No. NRF-2010-0022731 to E.H.L.) and by the Mid-career Researcher Program (No. NRF-2014R1A2A1A11050963 to E.H.L.) through the National Research Foundation of Korea grants funded by the Korea government (Ministry of Education (MOE), and Ministry of Science, ICT and Future Planning (MSIP), respectively). This work was also supported by a NIH grant (RO1-AG028614 to J.M.). We acknowledge the help of Hong Lim Kim (Laboratory of Electron Microscope, Integrative Research Support Center at The Catholic University of Korea) for great expertise in transmission electron microscopy observations.
Abbreviations
- MG29
mitsugumin 29
- TRPC
canonical-type transient receptor potential cation channel
- EC
excitation-contraction
- SR
sarcoplasmic reticulum
- SERCA1a
sarcoplasmic/endoplasmic reticulum Ca2+-ATPase 1a
- RyR
ryanodine receptor
- DHPR
dihydropyridine receptor
- STIM1
stromal interaction molecule 1
- t-tubule
transverse-tubule
- JMC
junctional membrane complex
- JP
junctophilin
- SOCE
store-operated Ca + entry
- CICR
Ca2+-induced Ca2+ release
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare that they have no conflicts of interest.
References
- 1.Zucchi R, Ronca-Testoni S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol Rev. 1997;49:1–51. [PubMed] [Google Scholar]
- 2.Lee EH, Kim do H, Allen PD. Interplay between intra- and extracellular calcium ions. Mol Cells. 2006;21:315–329. [PubMed] [Google Scholar]
- 3.Lee EH. Ca2+ channels and skeletal muscle diseases. Prog Biophys Mol Biol. 2010;103:35–43. doi: 10.1016/j.pbiomolbio.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 4.Lee EH, Cherednichenko G, Pessah IN, Allen PD. Functional coupling between TRPC3 and RyR1 regulates the expressions of key triadic proteins. J Biol Chem. 2006;281:10042–10048. doi: 10.1074/jbc.M600981200. [DOI] [PubMed] [Google Scholar]
- 5.Franzini-Armstrong C. STUDIES OF THE TRIAD : I. Structure of the Junction in Frog Twitch Fibers. J Cell Biol. 1970;47:488–499. doi: 10.1083/jcb.47.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woo JS, Cho CH, Lee KJ, Kim do H, Ma J, Lee EH. Hypertrophy in skeletal myotubes induced by junctophilin-2 mutant, Y141H, involves an increase in store-operated Ca2+ entry via Orai1. J Biol Chem. 2012;287:14336–14348. doi: 10.1074/jbc.M111.304808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito K, Komazaki S, Sasamoto K, Yoshida M, Nishi M, Kitamura K, Takeshima H. Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J Cell Biol. 2001;154:1059–1067. doi: 10.1083/jcb.200105040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 9.Takeshima H, Shimuta M, Komazaki S, Ohmi K, Nishi M, Iino M, Miyata A, Kangawa K. Mitsugumin29, a novel synaptophysin family member from the triad junction in skeletal muscle. Biochem J. 1998;331(Pt 1):317–322. doi: 10.1042/bj3310317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimuta M, Komazaki S, Nishi M, Iino M, Nakagawara K, Takeshima H. Structure and expression of mitsugumin29 gene. FEBS Lett. 1998;431:263–267. doi: 10.1016/s0014-5793(98)00770-4. [DOI] [PubMed] [Google Scholar]
- 11.Komazaki S, Nishi M, Kangawa K, Takeshima H. Immunolocalization of mitsugumin29 in developing skeletal muscle and effects of the protein expressed in amphibian embryonic cells. Dev Dyn. 1999;215:87–95. doi: 10.1002/(SICI)1097-0177(199906)215:2<87::AID-DVDY1>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 12.Komazaki S, Nishi M, Takeshima H, Nakamura H. Abnormal formation of sarcoplasmic reticulum networks and triads during early development of skeletal muscle cells in mitsugumin29-deficient mice. Dev Growth Differ. 2001;43:717–723. doi: 10.1046/j.1440-169x.2001.00609.x. [DOI] [PubMed] [Google Scholar]
- 13.Nishi M, Komazaki S, Kurebayashi N, Ogawa Y, Noda T, Iino M, Takeshima H. Abnormal features in skeletal muscle from mice lacking mitsugumin29. J Cell Biol. 1999;147:1473–1480. doi: 10.1083/jcb.147.7.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan Z, Yang D, Nagaraj RY, Nosek TA, Nishi M, Takeshima H, Cheng H, Ma J. Dysfunction of store-operated calcium channel in muscle cells lacking mg29. Nat Cell Biol. 2002;4:379–383. doi: 10.1038/ncb788. [DOI] [PubMed] [Google Scholar]
- 15.Weisleder N, Ma J. Altered Ca2+ sparks in aging skeletal and cardiac muscle. Ageing Res Rev. 2008;7:177–188. doi: 10.1016/j.arr.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagaraj RY, Nosek CM, Brotto MA, Nishi M, Takeshima H, Nosek TM, Ma J. Increased susceptibility to fatigue of slow- and fast-twitch muscles from mice lacking the MG29 gene. Physiol Genomics. 2000;4:43–49. doi: 10.1152/physiolgenomics.2000.4.1.43. [DOI] [PubMed] [Google Scholar]
- 17.Brotto MA, Nagaraj RY, Brotto LS, Takeshima H, Ma JJ, Nosek TM. Defective maintenance of intracellular Ca2+ homeostasis is linked to increased muscle fatigability in the MG29 null mice. Cell Res. 2004;14:373–378. doi: 10.1038/sj.cr.7290237. [DOI] [PubMed] [Google Scholar]
- 18.Romanick M, Thompson LV, Brown-Borg HM. Murine models of atrophy, cachexia, and sarcopenia in skeletal muscle. Biochim Biophys Acta. 2013;1832:1410–1420. doi: 10.1016/j.bbadis.2013.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faulkner JA, Brooks SV, Zerba E. Muscle atrophy and weakness with aging: contraction-induced injury as an underlying mechanism. J Gerontol A Biol Sci Med Sci. 1995;50:124–129. doi: 10.1093/gerona/50a.special_issue.124. Spec No. [DOI] [PubMed] [Google Scholar]
- 20.Weisleder N, Brotto M, Komazaki S, Pan Z, Zhao X, Nosek T, Parness J, Takeshima H, Ma J. Muscle aging is associated with compromised Ca2+ spark signaling and segregated intracellular Ca2+ release. J Cell Biol. 2006;174:639–645. doi: 10.1083/jcb.200604166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 22.Vandebrouck C, Martin D, Colson-Van Schoor M, Debaix H, Gailly P. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J Cell Biol. 2002;158:1089–1096. doi: 10.1083/jcb.200203091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hartmann J, Dragicevic E, Adelsberger H, Henning HA, Sumser M, Abramowitz J, Blum R, Dietrich A, Freichel M, Flockerzi V, Birnbaumer L, Konnerth A. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron. 2008;59:392–398. doi: 10.1016/j.neuron.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Millay DP, Goonasekera SA, Sargent MA, Maillet M, Aronow BJ, Molkentin JD. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc Natl Acad Sci U S A. 2009;106:19023–19028. doi: 10.1073/pnas.0906591106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woo JS, Kim do H, Allen PD, Lee EH. TRPC3-interacting triadic proteins in skeletal muscle. Biochem J. 2008;411:399–405. doi: 10.1042/bj20071504. [DOI] [PubMed] [Google Scholar]
- 26.Lee KJ, Park CS, Woo JS, Kim do H, Ma J, Lee EH. Mitsugumin 53 attenuates the activity of sarcoplasmic reticulum Ca(2+)-ATPase 1a (SERCA1a) in skeletal muscle. Biochem Biophys Res Commun. 2012;428:383–388. doi: 10.1016/j.bbrc.2012.10.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woo JS, Lee KJ, Huang M, Cho CH, Lee EH. Heteromeric TRPC3 with TRPC1 formed via its ankyrin repeats regulates the resting cytosolic Ca2+ levels in skeletal muscle. Biochem Biophys Res Commun. 2014;446:454–459. doi: 10.1016/j.bbrc.2014.02.127. [DOI] [PubMed] [Google Scholar]
- 28.Lee EH, Rho SH, Kwon SJ, Eom SH, Allen PD, Kim do H. N-terminal region of FKBP12 is essential for binding to the skeletal ryanodine receptor. J Biol Chem. 2004;279:26481–26488. doi: 10.1074/jbc.M309574200. [DOI] [PubMed] [Google Scholar]
- 29.Lee KJ, Hyun C, Woo JS, Park CS, Kim do H, Lee EH. Stromal interaction molecule 1 (STIM1) regulates sarcoplasmic/endoplasmic reticulum Ca(2)(+)-ATPase 1a (SERCA1a) in skeletal muscle. Pflugers Arch. 2014;466:987–1001. doi: 10.1007/s00424-013-1361-6. [DOI] [PubMed] [Google Scholar]
- 30.Woo JS, Hwang JH, Ko JK, Weisleder N, Kim do H, Ma J, Lee EH. S165F mutation of junctophilin 2 affects Ca2+ signalling in skeletal muscle. Biochem J. 2010;427:125–134. doi: 10.1042/BJ20091225. [DOI] [PubMed] [Google Scholar]
- 31.Lee KJ, Woo JS, Hwang JH, Hyun C, Cho CH, Kim do H, Lee EH. STIM1 negatively regulates Ca(2)(+) release from the sarcoplasmic reticulum in skeletal myotubes. Biochem J. 2013;453:187–200. doi: 10.1042/BJ20130178. [DOI] [PubMed] [Google Scholar]
- 32.Kallberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7:1511–1522. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gunasekaran K, Tsai CJ, Kumar S, Zanuy D, Nussinov R. Extended disordered proteins: targeting function with less scaffold. Trends Biochem Sci. 2003;28:81–85. doi: 10.1016/S0968-0004(03)00003-3. [DOI] [PubMed] [Google Scholar]
- 34.Collins MO, Yu L, Campuzano I, Grant SG, Choudhary JS. Phosphoproteomic analysis of the mouse brain cytosol reveals a predominance of protein phosphorylation in regions of intrinsic sequence disorder. Mol Cell Proteomics. 2008;7:1331–1348. doi: 10.1074/mcp.M700564-MCP200. [DOI] [PubMed] [Google Scholar]
- 35.Zhao X, Weisleder N, Thornton A, Oppong Y, Campbell R, Ma J, Brotto M. Compromised store-operated Ca2+ entry in aged skeletal muscle. Aging Cell. 2008;7:561–568. doi: 10.1111/j.1474-9726.2008.00408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ikemoto N, Ronjat M, Meszaros LG, Koshita M. Postulated role of calsequestrin in the regulation of calcium release from sarcoplasmic reticulum. Biochemistry. 1989;28:6764–6771. doi: 10.1021/bi00442a033. [DOI] [PubMed] [Google Scholar]
- 37.Woo JS, Cho CH, Kim do H, Lee EH. TRPC3 cation channel plays an important role in proliferation and differentiation of skeletal muscle myoblasts. Exp Mol Med. 2010;42:614–627. doi: 10.3858/emm.2010.42.9.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.