

Abstract
Background
Reactive oxygen species (ROS) contributes to adult tumorigenesis; however, their roles in pediatric solid tumors are unknown. Here, we sought to define the steady-state ROS levels in neuroblastoma and examine whether aggressive cellular behavior, which may predict treatment failure, is regulated by ROS.
Methods
Neuroblastoma sections were assessed for 4-hydroxynonenal (4HNE), which represents intracellular lipid peroxidation, a byproduct of elevated ROS. Human neuroblastoma cell lines, MYCN-amplified BE(2)-C and MYCN-non-amplified SK-N-SH, were examined for our study. Superoxide and hydroperoxide oxidation products were detected by staining for DHE and CDCFH2, using the oxidation-insensitive analog CDCF as a negative control. Cells were treated with N-acetylcysteine (NAC; 10 mM) daily for 5 d and analyzed.
Results
Higher expression of 4HNE was observed in undifferentiated tumor sections as compared to benign differentiated tumors. Interestingly, increased levels of ROS were detected in MYCN-amplified BE(2)-C cells. Moreover, GRP-R-induced ROS production stimulated upregulation of the HIF-1α/VEGF pathway and an increase in cell growth. Antioxidant NAC decreased HIF-1α/VEGF expression, inhibited BE(2)-C cell growth.
Conclusion
. We report a novel observation that shifting the redox balance toward higher ROS results in more aggressive neuroblastoma phenotype. Our data suggest that ROS plays a critical role in refractory neuroblastoma.
Keywords: ROS, Neuroblastoma, GRP-R, HIF-1α, VEGF
INTRODUCTION
Neuroblastoma is the most common extracranial solid cancer in infants and children, arising from the neural crest elements of the sympathetic nervous system. Children with high-risk metastatic disease have poor overall outcomes.1 In particular, metastatic neuroblastoma in children older than 18 months of age at diagnosis is lethal for most patients despite aggressive multimodality therapy. Therefore, new therapeutic strategies are necessary to improve the survival and cure rates of patients with high-risk neuroblastoma.2, 3
It has been increasingly recognized that reactive oxygen species (ROS) (i.e., superoxide, hydrogen peroxide, hydroxyl radical) induce metabolic oxidative stress and may play an important role in cytotoxicity, genotoxicity and carcinogenesis.4–6 Recent studies suggest that malignant cells have higher steady-state levels of ROS than normal tissue cells.4, 7 The excess ROS can react with a broad range of biomolecules including lipids, protein and DNA to form other radicals or cytotoxic byproducts, which could further contribute to carcinogenesis. Another mechanism by which increased ROS production affects cell proliferation and carcinogenesis is by acting as second messengers, affecting redox regulated signaling and gene expression.6, 8, 9 Recently, ROS has also been implicated in enhancing the aggressiveness of solid tumors like breast cancer.10 Though much is known about ROS in adult solid tumors, little information is known with about the role of ROS in pediatric tumors.
Our laboratory has demonstrated the significance of gastrin-releasing peptide receptor (GRP-R), a G-protein coupled receptor, in neuroblastoma tumorigenesis.11 Expression of GRP-R correlates to aggressiveness of the disease12 and targeting GRP-R using short-hairpin RNA (shRNA) inhibited liver metastasis in an in vivo spleen-liver metastasis model.11 Interestingly, targeting GRP-R using a specific pharmacologic inhibitor, RC-3095, decreased ROS-induced oxidative damage in an animal model of gastritis.13 This study demonstrates that GRP-R utilizes ROS to mediate its downstream effects, and therefore, GRP-R may elicit its protumorigenic effects in neuroblastomas by altering ROS levels.
In this study we sought to determine whether ROS might function as a downstream effector of GRP-R signaling in neuroblastoma. We utilized two human neuroblastoma cell lines, SK-N-SH and BE(2)C, that have been previously reported to differ in their basal GRP-R expression levels.11 Both cell lines are derived from bone marrow aspirates taken after initial treatment, at the time of disease relapse, from separate patients with disease in the high-risk category. Interestingly, these cell lines differ markedly in their functional behavior. The high-GRP-R-expressing BE(2)-C cell line exhibits a shorter doubling time, increased migration and anchorage-independent growth and a propensity to form more metastatic liver foci in a shorter time after splenic injection in a murine metastatic model compared to the low-GRP-R expressing SK-N-SH cell line.11
Here we report that the steady-state levels of ROS production (i.e., superoxide, hydrogen peroxide) affected neuroblastoma cell growth and proliferation, and may do so by regulating HIF-1α/VEGF expression. ROS production was higher in the BE(2)-C cells, which also express higher GRP-R and is more aggressive, compared to SK-N-SH. Tumor specimens from undifferentiated neuroblastoma had higher 4-hydroxynonenal protein adduction, an indicator of lipid peroxidation and ROS level within the tissue. Silencing GRP-R in BE(2)-C cells decreased ROS production. Conversely, SK-N-SH cells overexpressing GRP-R had higher steady-state levels of ROS. Antioxidant treatment with N-acetylcysteine (NAC) inhibited the BE(2)-C cell growth and colony formation, and also attenuated the GRP-R-induced increase in growth and colony formation in GRP-R overexpressing SK-N-SH cells. Finally, we report that GRP-R-mediated ROS production may affect neuroblastoma cell growth and proliferation via upregulation of hypoxia inducible factor (HIF)-1α and its downstream target vascular endothelial growth factor (VEGF). Overall, these results demonstrate that GRP-R regulates ROS generation in neuroblastoma cells and increased ROS mediates the pro-growth effects of GRP-R signaling.
MATERIALS AND METHODS
Cell culture, plasmids and transfections
Human neuroblastoma cell lines, BE(2)-C and SK-N-SH, were cultured in RPMI 1640 medium (Cellgro) with 10% fetal bovine serum (FBS) (Sigma) and 1% penicillin-streptomycin. Cells were maintained and experiments were performed at 37°C in a humidified 5% CO2 incubator. For GRP-R overexpression in SK-N-SH cells and GRP-R silencing in BE(2)-C, pEGFP-GRP-R and pENTR/H1/TO (Invitrogen) were used, respectively. The sequence targeting GRP-R (NM_005314) is underlined in the following shRNA sequence: 5′-CACCGTAACGTGTGCTCCAGTGGACGAATCCAC TGGAGCACA CGTTA-3′, the nonspecific control shCON was: 5′-CACCGGGCGCGCTTTGT AGGATTCGC CGAAGCGAATCCTACAAAGCGCGCC-3′.11 Transfection was accomplished by Lipofectamine 2000 system and cells were used for experiments 24 h after transfection.
Reagents
NAC was purchased from Sigma (St. Louis, MO) and prepared as a 1 M stock solution in PBS with sodium bicarbonate. In all experiments, NAC was used at a concentration of 10 mM. Bicarbonate in PBS without NAC was applied to cells as a vehicle control. Antibody against GRP-R was from Abcam (Cambridge, MA); VEGF antibody was purchased from Cell Signaling Technology (Beverly, MA); antibody against HIF-1α was from Novus (Littleton, CO); antibody against β-actin was from Sigma.
Measurement of intracellular ROS production
Steady-state levels of specific ROS molecules were estimated using fluorescent oxidation probes and detection by flow cytometry (FACScan Flow Cytometer, Becton Dickinson Immunocytometry System, Inc., Mountain View, CA). For all experiments, 100,000 cells/dish were plated in a 60-mm tissue culture dish, grown at 37°C for 48 h then trypsinized into a single cell suspension and washed with 5 mM pyruvate- containing PBS once and labeled with the respective probes for total cellular superoxide (O2 ·−; dihydroethidium, DHE, Molecular Probes, Eugene, OR, excitation/emission: 488/585 nm, 10 μM in 0.1% DMSO, 40 min), mitochondrial superoxide (MitoSOX™, Molecular Probes, excitation/emission: 488/585 nm, 2 μM in 0.1% DMSO, 20 min) and hydroperoxides (5, 6-carboxy-2′, 7′-dichlorodihydrofluorescein diacetate, CDCFH2-DA, Molecular Probes, excitation/emission: 488/530 nm, 10 μg/mL in 0.1% DMSO, 15 min). The CDCFH2-DA probe crosses the cell membrane to enter the cytoplasm, where the diacetate moiety is cleaved by intracellular esterases to trap the CDCFH2 portion of the probe intracellularly. Cytoplasmic hydroperoxides then oxidize the probe to remove the hydrogen atoms and a fluorescent signal is produced. An oxidation insensitive fluorescent probe (5, 6-carboxy-2′, 7′-dichlorofluorescein diacetate CDCF, Molecular Probes, excitation/emission: 488/530 nm, 10 μg/mL in 0.1% DMSO, 15 min) was utilized as a negative control to detect cell line differences in probe uptake, esterase cleavage, probe efflux and nonspecific probe activation. After labeling, cells were kept on ice. For each measurement, the mean fluorescent intensity (MFI) was assessed for 10,000 cells and corrected for background autofluorescence from unlabeled cells. MFI data was then normalized to control for each group at each respective time point, as indicated.5, 14
Western blot analysis
Protein (40 μg) was resolved on 12% SDS-PAGE and electroblotted into nitrocellulose membranes (Bio Rad, Hercules, CA, USA). The membrane was incubated with monoclonal antibody diluted at 1:1,000 in 5% non-fat dry milk for 1 h at room temperature. After washing, the membrane was incubated with a secondary antibody at a 1:10,000 dilution for 1 h at room temperature (RT). After washing, signals were detected on an X-ray film using an enhanced ECL detection system (Perkin Elmer, Alameda, CA)
Tissue immunohistochemistry
Neuroblastoma tumor samples from eight patients were obtained from the discarded tissue portion of the pathologic samples (IRB#: 091331). Tumor samples were fixed in formalin for 3 d and embedded in paraffin wax. Paraffin-embedded sections (5 μm) were deparaffinized in three xylene washes followed by a graded alcohol series, antigen retrieval performed with 10 mM sodium citrate buffer, peroxidase treatment and then blocked with serum-free blocking solution for 1 h at RT. Sections were incubated with primary antibody against 4HNE overnight at 4°C. They were washed with PBS and incubated with secondary antibody for 30 min at RT. Sections were developed with DAB reagent. All sections were counterstained with hematoxylin, and then dehydrated with ethanol and xylene. Coverslips were mounted on slides and observed by light microscopy.
Cell proliferation and soft agar colony formation assay
Growth curve were constructed by plating 5×104 cells/dish in 60 mm tissue culture dishes. After 24 h, the cells were treated with NAC daily for 5 d. Media were changed daily and fresh NAC (10 mM) was added. The cell numbers were counted from days 1 through 5. Cell proliferation was assessed using CCK-8 kit daily. The values, corresponding to the number of viable cells, were read at OD450 with the FlexStation 3 Microplate Reader (Molecular Devices, Sunnyvale, CA). For soft agar colony formation assay, cells were trypsinized and resuspended in RPMI 1640 medium containing 0.4% agarose and 7.5% FBS. BE(2)-C cells with or without NAC treatment were overlaid onto a bottom layer of solidified 0.8% agarose in RPMI 1640 medium containing 5% FBS, at cell concentrations of 2.5×103 cells per well of a 6-well plate, and incubated for 3 weeks. Colonies were stained with 0.05% crystal violet, photographed, and quantified.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism version 4.00 for Windows (GraphPad Software, San Diego, CA). Data were expressed as means ± SEM unless otherwise specified. One-way ANOVA analysis with Tukey’s post analysis was used to study the differences among three or more means. Significance was determined at p < 0.05.
RESULTS
Steady-state levels of ROS correlated with GRP-R expression and aggressive undifferentiated phenotype
It is unknown whether ROS plays a critical role in neuroblastoma. To test whether ROS production by neuroblastoma cells promote cell proliferation and metastatic behavior, we first measured the steady-state levels of superoxide and hydrogen peroxide in two human neuroblastoma cell lines, BE(2)-C and SK-N-SH. These cell lines demonstrate quite contrasting cellular behavior, as well as GRP-R expression. BE(2)-C is highly proliferative with short doubling time, along with high propensity to metastasize in vivo when compared to the less aggressive SK-N-SH cells. BE(2)-C also expresses a higher level of GRP-R than SK-N-SH. We used fluorescent oxidation probe detection by flow cytometry to measure superoxide (O2·−)(dihydroethidium, DHE) and hydrogen peroxide (5, 6-carboxy-2′, 7′-dichlorodihydrofluorescein, CDCFH2) in the cell populations of interest. Steady-state levels of intracellular O2·− and hydrogen peroxide were approximately 2-fold higher in BE(2)-C compared to SK-N-SH cells (Fig. 1A, B). To serve as a negative control, we tested each cell line with an oxidation-insensitive probe (5, 6-carboxy-2′, 7′-dichlorofluorescein, CDCF). Differences in CDCF fluorescence between cell lines would indicate differences in probe uptake, probe cleavage by esterases and probe efflux from the cell, none of which are dependent on oxidation of the probe by the respective ROS molecules. There was no significant difference in CDCF fluorescence between BE(2)-C and SK-N-SH cell lines (Fig. 1C). These data demonstrate that ROS production is different between neuroblastoma cell lines; differential ROS levels correlated with aggressive cellular phenotype, as well as GRP-R expression.
Figure 1. Steady-state ROS levels correlated with GRP-R expression.

(A, B) MYCN-amplified BE(2)-C cells demonstrated increased DHE and CDCFH2 oxidation when compared to SK-N-SH cells. (C) An oxidation-insensitive probe (CDCF) demonstrated no significant differences in fluorescence between cell lines, as a negative control (mean ± SEM; three to nine treatment dishes performed in three separate experiments; *=p < 0.05 vs. control). Normalized MFI is the mean fluorescent index (MFI)/10,000 cells by flow cytometry, expressed as a ratio for each group relative to BE(2)-C cells. (D) Anti-4-HNE antibody showed increased staining (brown) by immunohistochemistry in undifferentiated neuroblastoma (left panel) compared to differentiated ganglioneuroma (right panel) in sections from paraffin-embedded patient tumor samples. Representative images are shown.
We further sought to test whether a similar relationship of ROS production existed in patient tumors that varied in their degree of differentiation. To test our hypothesis that ROS production correlates with undifferentiation of neuroblastoma, we selected paraffin-embedded discarded tumor sections from patient with different histopathologic neuroblastoma subtypes, and performed antibody staining against 4-hydroxynonenal (4-HNE), a lipid peroxidation product that is stable after ex vivo tissue processing and storage. The 4-HNE levels appeared higher in undifferentiated neuroblastoma (highest risk) compared to differentiated neuroblastoma (low risk) (Fig. 1D, representative image). Two patient tissue sections were analyzed for each of four categories of pathologic differentiation status: ganglioneuroma, ganglioneuroblastoma, differentiating neuroblastoma and undifferentiated neuroblastoma. Three individuals scored each section for staining intensity and the results for each sample were averaged. There was a trend toward higher staining density in the undifferentiated sections (mean score 7.00) compared to ganglioneuroma (mean score 3.33), suggesting a correlation between lipid peroxidation status and differentiation state. We did not find a statistically significant difference between the four groups due to the small sample size available for analysis. A similar difference in GRP-R expression is seen in specimens with similar degrees of differentiation, as we have previously reported.12 Taken together these data suggest that ROS production and GRP-R expression correlate in neuroblastoma and ROS production might correlate with undifferentiated phenotype, which have much higher risk of treatment resistance, relapse and metastasis than more differentiated subtypes of disease.
GRP-R-induced steady-state level of ROS in neuroblastoma cells
We next sought to examine whether GRP-R signaling plays a direct role in the regulation of ROS production. BE(2)-C cells were transfected with shRNA against GRP-R (shGRP-R) or control vector (shCON) and the intracellular steady-state level of hydroperoxide was determined by measuring CDCFH2 oxidation. BE(2)-C/shGRP-R cells exhibited lower levels of CDCFH2 oxidation (~ 1.5 fold) and MitoSOX™ when compared to BE(2)-C/shCON cells (Fig. 2A, B). These findings suggest that ROS production is at least partially under the control of GRP-R signaling. To further test this observation, we utilize the SK-N-SH cell line, which naturally expresses a low level of GRP-R, and overexpress the receptor by plasmid transfection, offering a system to study changes specific to the overexpression of the receptor and overactivation of its signaling pathway. CDCFH2 and MitoSOX™ probe oxidation was higher in the SK-N-SH cell lines after transfection with GRP-R compared to transfection with control plasmid (Fig. 2C, D).
Figure 2. GRP-R-induced ROS production in neuroblastoma cells.

(A, B) Targeted silencing of GRP-R by shRNA was performed by lentiviral transfection in BE(2)-C cells, as measured by CDCFH2 (A) and MitoSOX (B) oxidation. (C, D) GRP-R overexpression by plasmid transfection in SK-N-SH cells resulted in increases in CDCFH2 (C) and MitoSOX (D) oxidation. (E, F) GRP-R overexpressing SK-N-SH cells were treated with RC-3095, a GRP-R antagonist and CDCFH2 (E) and MitoSOX (F) oxidations were measured. RC-3095 reversed the increase observed after GPR-R overexpression. Probe oxidation in A–D is expressed as Mean MFI/10,000 cells. For E and F, MFI values are normalized and expressed relative to control cells (mean ± SEM; *=p < 0.05 vs. control).
RC-3095 is a specific small molecule antagonist of GRP-R that we have employed previously to define the role of autocrine GRP-R signaling in neuroblastoma.15 Here, we tested whether RC-3095 would reverse the increased ROS production seen after GRP-R overexpression, to provide further evidence that increased ROS production is due to increased receptor levels and not an off target effect of transfection or other unrelated mechanism. Indeed, RC-3095 (1 μM) abolished the GRP-R induced increase in oxidation of each of the two ROS probes (Fig. 2E, F).
Taken together, these data suggest that GRP-R signaling directly affects ROS production. As we have previously shown the critical role of GRP-R signaling in neuroblastoma growth and metastatic behavior, these data additionally support the conclusion that ROS production may contribute to the observed differences in behavior of different neuroblastoma cell lines.
NAC inhibited neuroblastoma cell tumorigenicity
Overproduction of ROS may drive tumor cell proliferation by activating pro-survival oncogenes.6–8 To determine whether inhibition of ROS could decrease the growth potential of neuroblastoma, BE(2)-C cells were treated with NAC (10 mM), a non-specific thiol antioxidant. Daily NAC treatment significantly inhibited the proliferative rate of BE(2)-C cells after 3 days, and this effect persisted through 5 days of treatment (Fig. 3A; left panel). Doubling times (Td) of BE(2)-C cells were calculated using the equation: Td=0.693t/ln (Nt/N0), where Nt and N0 represent cell number at time t and time zero respectively. The doubling time of untreated control BE(2)-C cells is 22 h, while NAC treatment prolongs this time to 37 h. NAC treatment decreased BE(2)-C cell viability after 3 d and this decrease persisted to day 5 (Fig. 3A; right panel). In addition, NAC inhibited the ability of BE(2)-C cells to undergo anchorage-independent growth assessed by soft agar colony formation assay, an assay commonly used to measure in vitro an approximation of metastatic potential. NAC treated cells showed more than 80% reduction in colony formation after 3 days as compared to the control group (Fig. 3B; left panel). By day 5 of NAC treatment, colony formation was completely attenuated compared to control (Fig. 3B; right panel). These data clearly indicate anti-tumorigenic actions of antioxidant therapy in neuroblastoma cells.
Figure 3. NAC inhibited neuroblastoma cell proliferation and soft agar colony formation.

(A) Cell culture media for BE(2)-C cells was changed daily and supplemented with NAC (10 mM). Monolayer cultures were trypsinized and cells counted daily from t=1 to t=5 d (left panel). BE(2)-C cell viability was assessed after NAC treatment (right panel). Value at each time point represents mean of n=6 samples from two separate experiments (mean ± SEM; *=p < 0.05 vs. control). (B) BE(2)-C cells were plated in 0.4% agarose gel suspension to measure anchorage-independent colony formation and treated with NAC (10 mM) or vehicle (control). NAC treatment decreased the number of colonies compared to vehicle-treated controls (left bar graph; mean ± SEM; *=p < 0.05 vs. control). A representative image of control and NAC-treated soft agar plates are shown (right). Data at each time point represents n=4 samples performed in triplicate in two separate experiments (mean ± SEM; *=p < 0.05 vs. control).
NAC suppressed GRP-R-induced increase in neuroblastoma tumorigenicity
The use of ROS scavengers is an emerging therapeutic strategy in combating adult malignancies.6, 16 We have previously shown that interfering with GRP-R signaling can abrogate the growth and metastatic potential of neuroblastoma both in vitro and in vivo.11 We utilized the same GRP-R overexpression system as in Figure 2. We observed a 70% increase in cell proliferation by CCK-8 assay (Fig. 4A, filled circles) and a 20% increase in the number of colonies in 0.4% soft agar anchorage-independent culture conditions (Fig. 4B, single hatch) with GRP-R overexpression when compared to SK-N-SH cells transfected with control plasmid. NAC treatment completely reversed the observed increases in proliferation produced by GRP-R in both culture conditions, despite continued GRP-R overexpression (Fig. 4A and B, filled squares and solid bars, respectively). These data suggest that antioxidant therapy can reverse GRP-R-induced changes in cellular behavior.
Figure 4. NAC inhibited GRP-R-induced increase in neuroblastoma cell growth.
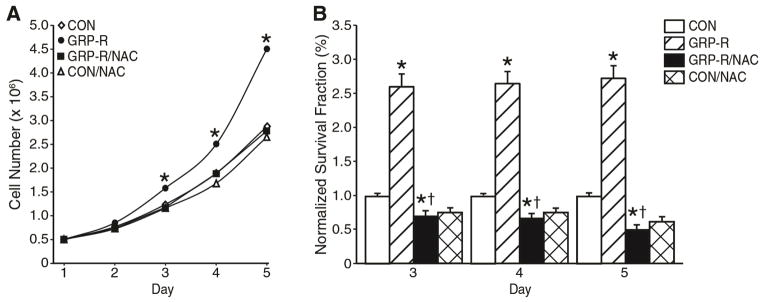
GRP-R was overexpressed in SK-N-SH cells as in Fig. 3. GRP-R and control-plasmid transfected cells were treated with NAC (10 mM) daily for 5 d. NAC inhibited proliferative rate of both control and GRP-R overexpressing cells by CCK-8 assay (A) and decreased anchorage-independent colony formation by soft agar assay (B). Value at each time point represents n=6 samples performed in triplicate in two separate experiments for (A) and n=4 samples in triplicate in two separate experiments for (B) (mean ± SEM; *=p < 0.05 vs. GRP-R/NAC and GRP-R groups compared to vehicle-transfected SK-N-SH control. †= p < 0.05 for GRP-R/NAC compared to GRP-R overexpression group).
GRP-R-induced ROS production increased HIF-1α/VEGF expression
ROS production is known to regulate multiple signal transduction pathways, including the HIF-1α/VEGF signaling pathway.17 To ascertain whether GRP-R-induced ROS production can stimulate HIF-1α and its downstream target VEGF, we first compared basal levels of these signaling mediators in BE(2)-C and SK-N-SH cells (Fig. 5A). BE(2)-C cells with higher GRP-R expression also demonstrated a higher basal expression of HIF-1α and VEGF, suggesting that GRP-R signaling may stimulate activation of HIF-1α/VEGF signaling pathways. To further confirm the role of GRP-R signaling on HIF-1α/VEGF pathways, both HIF-1α and VEGF protein levels were measured in BE(2)-C cells after GRP-R silencing and in GRP-R overexpressing SK-N-SH cells. Expression of HIF-1α and its downstream target, VEGF, was clearly decreased in BE(2)-C/shGRP-R cells compared to BE(2)-C/shCON cells (Fig. 5B). In contrast, SK-N-SH cells with induced overexpression of GRP-R had higher expression of both HIF-1α and VEGF compared to vector-transfected control (Fig. 5C). Moreover, NAC treatment also decreased expression of both HIF-1α and VEGF protein levels (Fig. 5D). These data suggest that ROS overproduction may mediate the GRP-R pro-angiogenic effects15 and that NAC may represent a way to diminish the GRP-R-mediated contribution to neuroblastoma growth and maintenance.
Figure 5. GRP-R-induced ROS production increased expression of HIF-1α/VEGF.

(A) Basal expression of HIF-1α and VEGF in BE(2)-C and SK-N-SH cells assessed by Western blot analysis. (B) HIF-1α and VEGF protein levels measured by Western blotting after GRP-R silencing by shRNA and compared to non-targeted shRNA-transfected control cells. (C) GRP-R was overexpressed by plasmid transfection in SK-N-SH cells and HIF-1α and VEGF protein levels were determined by Western blotting. (D) NAC (10 mM) decreased HIF-1α and VEGF protein levels in BE(2)-C cells when compared to control. Experiments were repeated on two separate occasions and β-actin was used as protein loading control.
DISCUSSION
Over 60% of patients with neuroblastoma present with metastasis to bone marrow at the time of diagnosis.1, 18 Relatively hypoxic microenvironment of bone marrow is a sanctuary for metastatic tumor-initiating cells, and therefore, ROS may be a critical driver of metastatic neuroblastoma foci. ROS production contributes to carcinogenesis and tumor cell proliferation.6–8 Moreover, recent studies demonstrate that tumor cells exhibit higher steady-state levels of ROS compared to their normal cellular counterparts.4, 6 However, these findings are derived from studies in adult cancers and little is known about the role of ROS in neuroblastoma.
Although excessive ROS production in normal cells appears cytotoxic, the elevated steady-state level of ROS appears to be preferred by tumor cells. The increased ROS production from aberrant metabolism in tumor cells contributes to many aspects of tumor cell survival and proliferation.8 Increased ROS production can further increase the genomic instability and enhance the metastatic potential of tumor cells.16, 19 These findings are all based on adult solid tumors and it is therefore critical to investigate if ROS has similar roles in pediatric cancers. Understanding the role of ROS in neuroblastoma and how it can potentially contribute to neuroblastoma cell proliferation may provide a different view to current neuroblastoma therapy.
Here, we report that NAC treatment significantly inhibited neuroblastoma cell proliferation and anchorage-independent colony growth, indicating that suppression of ROS production could be an effective means of decreasing neuroblastoma tumorigenicity. Interestingly, ROS production correlated with the relative expression of GRP-R in neuroblastoma cell lines and in human neuroblastoma patient samples. This is a novel observation and suggests that ROS production is directly involved in neuroblastoma pathology and positively correlated to the degree of clinical risk-group cateogorization. Earlier studies have reported that blocking GRP-R reduces ROS production and inflammatory responses,13, 20 thus highlighting that there may be significant therapeutic potential to target GRP-R/ROS in neuroblastoma.
Our previous studies have highlighted the critical nature of GRP-R signaling in neuroblastoma. GRP-R expression is higher in undifferentiated neuroblastomas in comparison to differentiated ganglioneuromas and targeting GRP-R inhibited tumor metastasis in vivo,11, 12 making it an ideal candidate target for novel therapeutic strategies in treating aggressive neuroblastomas. In line with that, the data from the present study demonstrates that one of the biological effects of targeting GRP-R is decreased ROS production. Steady-state levels of superoxide and H2O2 may serve as a sensing mechanism for controlling key cellular processes through regulation of the redox signaling,14, 21, 22 and reduced ROS production could decrease activation of signaling pathways involved in cancer cell survival, proliferation and motility. Moreover, the antioxidant NAC decreased the oncogenic properties of GRP-R in vitro, confirming the functional significance of targeting GRP-R/ROS signaling in neuroblastoma.
Previous studies in adult solid tumors have demonstrated that increased ROS production by cancer cells enhances HIF-1α/VEGF expression and eventually tumor growth.17 VEGF is a signaling protein produced by cells that stimulates angiogenesis and is thought to play a critical role in neuroblastoma tumor metastasis.23 VEGF is a redox-regulated protein that can be activated by increased HIF-1α expression in circumstances of excessive ROS production. Interestingly, we have previously reported that gastrin-releasing peptide, GRP, the specific activating ligand for GRP-R, stimulates neuroblastoma angiogenesis in vitro and in vivo.15 Here we demonstrate that the angiogenic properties of GRP-R could be suppressed by antioxidant therapy, and may occur by inhibiting activation of the HIF-1α/VEGF signaling system. Taken together, our data suggests that G-protein coupled receptor-mediated ROS production is tumorigenic in neuroblastoma and tumor behavior can be modulated by a widely available and safe medication. To the best of our knowledge, this is the first report describing a causal relationship between GRP-R and ROS production in neuroblastoma. Future studies will be needed to determine the sources of cellular ROS production in response to growth factor activation, the specific ROS isoforms responsible for the tumorigenic behavior, and the exact mechanism of action of NAC in this context. It will also be critical to determine which signaling protein intermediates, whether HIF-1α, VEGF, or some other factor is responsible for transducing the pro-oncogenic ROS signal to alter gene expression patterns toward a resistant, metastatic phenotype.
Acknowledgments
This work was supported by a grant R01 DK61470 from the National Institutes of Health and the Rally Foundation for Cancer Research.
Footnotes
Presented at the 10th Annual Academic Surgical Congress in Las Vegas, NV, February 3–5, 2015
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362:2202–11. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishola TA, Chung DH. Neuroblastoma. Surg Oncol. 2007;16:149–56. doi: 10.1016/j.suronc.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Park JR, Eggert A, Caron H. Neuroblastoma: biology, prognosis, and treatment. Hematol Oncol Clin North Am. 2010;24:65–86. doi: 10.1016/j.hoc.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 4.Aykin-Burns N, Ahmad IM, Zhu Y, Oberley LW, Spitz DR. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem J. 2009;418:29–37. doi: 10.1042/BJ20081258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu Y, Kalen AL, Li L, Lehmler HJ, Robertson LW, Goswami PC, et al. Polychlorinated-biphenyl-induced oxidative stress and cytotoxicity can be mitigated by antioxidants after exposure. Free Radic Biol Med. 2009;47:1762–71. doi: 10.1016/j.freeradbiomed.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010;44:479–96. doi: 10.3109/10715761003667554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–91. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 8.Gius D, Spitz DR. Redox signaling in cancer biology. Antioxid Redox Signal. 2006;8:1249–52. doi: 10.1089/ars.2006.8.1249. [DOI] [PubMed] [Google Scholar]
- 9.Mattson D, Bradbury CM, Bisht KS, Curry HA, Spitz DR, Gius D. Heat shock and the activation of AP-1 and inhibition of NF-kappa B DNA-binding activity: possible role of intracellular redox status. Int J Hyperthermia. 2004;20:224–33. doi: 10.1080/02656730310001619956. [DOI] [PubMed] [Google Scholar]
- 10.Kim H, Choi JA, Park GS, Kim JH. BLT2 up-regulates interleukin-8 production and promotes the invasiveness of breast cancer cells. PLoS one. 2012;7:e49186. doi: 10.1371/journal.pone.0049186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiao J, Kang J, Ishola TA, Rychahou PG, Evers BM, Chung DH. Gastrin-releasing peptide receptor silencing suppresses the tumorigenesis and metastatic potential of neuroblastoma. Proc Natl Acad Sci U S A. 2008;105:12891–6. doi: 10.1073/pnas.0711861105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim S, Hu W, Kelly DR, Hellmich MR, Evers BM, Chung DH. Gastrin-releasing peptide is a growth factor for human neuroblastomas. Ann Surg. 2002;235:621–9. doi: 10.1097/00000658-200205000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petronilho F, Araujo JH, Steckert AV, Rezin GT, Ferreira GK, Roesler R, et al. Effect of a gastrin-releasing peptide receptor antagonist and a proton pump inhibitor association in an animal model of gastritis. Peptides. 2009;30:1460–5. doi: 10.1016/j.peptides.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 14.Zhu Y, Park SH, Ozden O, Kim HS, Jiang H, Vassilopoulos A, et al. Exploring the electrostatic repulsion model in the role of Sirt3 in directing MnSOD acetylation status and enzymatic activity. Free Radic Biol Med. 2012;53:828–33. doi: 10.1016/j.freeradbiomed.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang J, Ishola TA, Baregamian N, Mourot JM, Rychahou PG, Evers BM, et al. Bombesin induces angiogenesis and neuroblastoma growth. Cancer Lett. 2007;253:273–81. doi: 10.1016/j.canlet.2007.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tochhawng L, Deng S, Pervaiz S, Yap CT. Redox regulation of cancer cell migration and invasion. Mitochondrion. 2013;13:246–53. doi: 10.1016/j.mito.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 17.Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang XR, Jiang BH. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007;67:10823–30. doi: 10.1158/0008-5472.CAN-07-0783. [DOI] [PubMed] [Google Scholar]
- 18.Smith MA, Seibel NL, Altekruse SF, Ries LA, Melbert DL, O’Leary M, et al. Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol. 2010;28:2625–34. doi: 10.1200/JCO.2009.27.0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhandary B, Marahatta A, Kim HR, Chae HJ. Mitochondria in relation to cancer metastasis. J Bioenerg Biomembr. 2012;44:673–7. doi: 10.1007/s10863-012-9464-x. [DOI] [PubMed] [Google Scholar]
- 20.Pereira DV, Steckert AV, Mina F, Petronilho F, Roesler R, Schwartsmann G, et al. Effects of an antagonist of the gastrin-releasing peptide receptor in an animal model of uveitis. Invest Ophthalmol Vis Sci. 2009;50:5300–3. doi: 10.1167/iovs.09-3525. [DOI] [PubMed] [Google Scholar]
- 21.Lee YJ, Galoforo SS, Berns CM, Chen JC, Davis BH, Sim JE, et al. Glucose deprivation-induced cytotoxicity and alterations in mitogen-activated protein kinase activation are mediated by oxidative stress in multidrug-resistant human breast carcinoma cells. J Biol Chem. 1998;273:5294–9. doi: 10.1074/jbc.273.9.5294. [DOI] [PubMed] [Google Scholar]
- 22.Spitz DR, Sim JE, Ridnour LA, Galoforo SS, Lee YJ. Glucose deprivation-induced oxidative stress in human tumor cells. A fundamental defect in metabolism? Ann N Y Acad Sci. 2000;899:349–62. doi: 10.1111/j.1749-6632.2000.tb06199.x. [DOI] [PubMed] [Google Scholar]
- 23.Kang J, Rychahou PG, Ishola TA, Mourot JM, Evers BM, Chung DH. N-myc is a novel regulator of PI3K-mediated VEGF expression in neuroblastoma. Oncogene. 2008;27:3999–4007. doi: 10.1038/onc.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]