

Abstract
Visceral leishmaniasis is a neglected parasitic disease that has a high fatality rate in the absence of treatment. New drugs that are inexpensive, orally active, and effective could be useful tools in the fight against this disease. We previously showed that N2,N4-disubstituted quinazoline-2,4-diamines displayed low- to sub-micromolar potency against intracellular Leishmania, and lead compound N4-(furan-2-ylmethyl)-N2-isopropyl-7-methylquinazoline-2,4-diamine (4) exhibited modest efficacy in an acute murine model of visceral leishmaniasis. In the present work, thirty-one N2,N4-disubstituted quinazoline-2,4-diamines that had not previously been examined for their antileishmanial activity were evaluated for their potency and selectivity against Leishmania donovani, the causative parasite of visceral leishmaniasis. Quinazoline-2,4-diamines with aromatic substituents at both N2 and N4 exhibited potent in vitro antileishmanial activity but relatively low selectivity, while compounds substituted with small alkyl groups at either N2 or N4 generally showed lower antileishmanial potency but were less toxic to a murine macrophage cell line. Based on their in vitro antileishmanial potency, N4-benzyl-N2-(4-chlorobenzyl)quinazoline-2,4-diamine (15) and N2-benzyl-N4-isopropylquinazoline-2,4-diamine (40) were selected for in vivo evaluation of their pharmacokinetic and antileishmanial properties. While 15 displayed a longer plasma half-life and a greater area under the curve than 40, both compounds showed low efficacy in an acute murine visceral leishmaniasis model. Although the present study did not identify new quinazoline-2,4-diamines with promising in vivo efficacy, the reduced in vitro toxicity of derivatives bearing small alkyl groups at either N2 or N4 may provide clues for the design of safe and effective antileishmanial quinazolines.
Keywords: Leishmania, Neglected disease, Parasitic disease
1. Introduction
Visceral leishmaniasis (VL) is a protozoan parasitic disease affecting mainly poor populations in developing tropical and subtropical areas of the world. Of the approximately 300,000 new cases of VL that occur annually, over 90% are in India, Bangladesh, Ethiopia, Sudan, South Sudan, and Brazil. In the absence of chemotherapy, the mortality rate for those affected by symptomatic VL can approach 100%,1 fatalities most often arise from secondary infections or hemorrhage. Since a VL vaccine is not available and drugs are critically important for the treatment of symptomatic disease, access to effective drugs is crucial for VL patients. Unfortunately, VL is a neglected disease affecting impoverished populations in the developing world that does not receive adequate attention regarding the development of more effective treatment and prevention.
Antimonials have been used as first line VL drugs for over a century. The relatively recent development of resistance to antimonials on the Indian subcontinent has led to the use of other drugs in this region, although sodium stibogluconate is still considered to be a first line treatment for visceral leishmaniasis in the Americas2,3 and in Ethiopia.4 In addition to problems with drug resistance, the side effects of antimonials are well known, and in some cases these side effects may worsen the clinical outcome of VL.2 Paromomycin, liposomal amphotericin B, and miltefosine are also used for VL treatment. A combination of paromomycin and sodium stibogluconate recently replaced sodium stibogluconate monotherapy as the recommended treatment for VL in South Sudan,5 and paromomycin monotherapy has been approved for use against VL in India.6 Liposomal amphotericin B is highly effective against VL on the Indian subcontinent, even when given in a single dose,7
and various combinations of amphotericin B, paromomycin, and miltefosine evaluated in this region displayed cure rates >97%.8 Improved access to VL drugs superior to antimonials, such as liposomal amphotericin B and miltefosine, has aided in the development of VL elimination plans in India9 and in Bangladesh,10 and a correlation between reductions in VL mortality in India and the use of effective drugs has been noted.11 Paromomycin, liposomal amphotericin B, and miltefosine display shortcomings, however, which include expense (liposomal amphotericin B and miltefosine), route of administration (liposomal amphotericin B and paromomycin), potential side effects (miltefosine), and decreased effectiveness (miltefosine).12 To overcome the limitations of existing VL drugs, new oral agents that are inexpensive, safe, and effective are needed.13 Such agents could be particularly useful as components of VL combination therapy.14
Although strategies have been devised to develop new oral VL drugs, progress toward this goal has been slow.15 Only one compound from a new class, the nitroheterocycle fexinidazole,16 is currently in clinical trials for VL, and much of the preclinical drug development for VL carried out by the Drugs for Neglected Diseases initiative also focuses on nitroheterocycles.17 As part of our continuing efforts to discover new classes of compounds as VL candidates, we identified and developed 2,4-substituted quinazoline diamines 1–4 that possess activity against intracellular Leishmania donovani.18 In almost all cases, submicromolar EC50 values were obtained against a β-lactamase expressing L. donovani strain when at least one of the amino groups was substituted with a benzyl group. 2,4-Quinazoline diamines bearing an N4-furan-2-ylmethyl group were generally less potent, although they typically displayed single digit micromolar EC50 values against the β-lactamase expressing L. donovani strain. N4-(Furan-2-ylmethyl)-N2-isopropyl-7-methylquinazoline-2,4-diamine (4) also displayed a submicromolar EC50 value against the β-lactamase expressing L. donovani strain and moderate efficacy in a murine model of visceral leishmaniasis, while two 2,4-quinazoline diamines bearing N2-alkyl substituents paired with an N4-benzyl group were less active in vivo. In the current paper, we further explore the structural requirements for antileishmanial activity in this series of 2,4-quinazoline diamines and seek to further probe the potential of this class of compounds as new VL candidates (Fig. 1).
Figure 1.
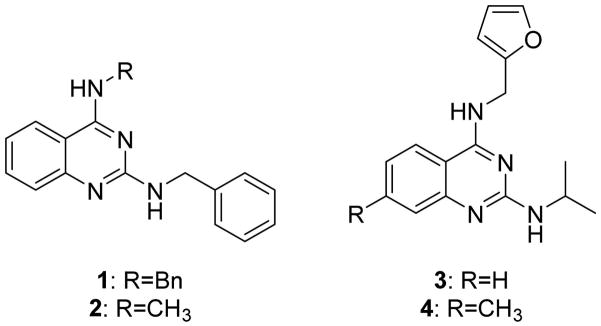
N2,N4-Disubstituted quinazoline diamines 1–4 with potent antileishmanial activity.
2. Results and discussion
2.1. Chemistry
The synthesis of the target compounds is shown in Scheme 1. N2,N4-Disubstituted quinazoline-2,4-diamines were prepared as previously reported starting from 2,4-dichloro-quinazoline 5 and corresponding amines via sequential substitution reactions.18,19 Similarly, treatment of 5 with benzyl alcohol afforded 4-(benzyloxy)-2-chloroquinazoline 6, which was then further reacted with benzylamine to give 18. 2-Aryl- or 4-aryl-substituted quinazolines were prepared following previous reports.20,21 Treatment of 2,4-dichloro-quinazoline 5 with sodium hydroxide followed by benzylamine provided 2-(benzylamino)quinazolin-4(3H)-one 7. Intermediate 7 was chlorinated with phosphorus oxychloride yielding 4-chloro-substituted quinazoline 8, which was then arylated via palladium catalysis to give 19 and 20. 2-Aryl-quinazoline 21 was prepared similarly starting from N-benzyl-2-chloroquinazolin-4-amine 9.
Scheme 1.

Synthetic routes for the preparation of antileishmanial quinazolines.
2.2. Biological evaluation
Our earlier study showed the in vitro potency of N2,N4-dibenzylquinazoline-2,4-diamine 1 against intracellular L. donovani.18 The antileishmanial potency and selectivity of analogs of this compound are given in Table 1. All but two of these compounds displayed EC50 values against intracellular L. donovani ranging from 0.39 to 1.1 μM. No clear trend for in vitro antileishmanial activity is observed for compounds substituted with methyl, methoxy, and chloro substituents at the para position of the benzyl groups at N2 and N4, although a methoxy group appears to decrease antileishmanial activity if it is placed at the para position of the N4 benzyl group as in 11 (EC50 = 2.5 μM) and 17 (EC50 = 6.0 μM). Replacing a nitrogen atom with an oxygen atom at position 4 does not appear to affect antileishmanial activity (compare 1 with 18). The antileishmanial selectivity indexes (SI) of these compounds was modest; seven displayed SI values of 5.1–8.2 for intracellular L. donovani compared to murine J774.A1 macrophages while three of these molecules exhibited SI values <3. The effect of an aryl substitution at position 4 when paired with a benzyl substitution at the N2 atom was also examined, as was an aryl substitution at position 2 when paired with a benzyl substitution at the N4 atom (Table 2). These compounds (19–21) were not active against L. donovani up to a concentration of 10 μM.
Table 1.
Probing the para-position of benzyl substituents of N2,N4-disubstituted quinazoline-2,4-diamines
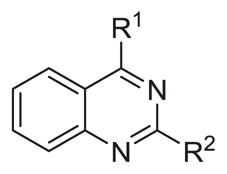
| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 |
L. donovani EC50a (μM) |
J774A.1 EC50b (μM) | SI |
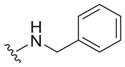
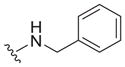
| 1 |
|
|
0.67 ± 0.27c | 5.5 ± 1.4c | 8.2 |
| 10 |
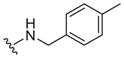
|
|
0.39 ± 0.16 | 2.1 ± 0.2 | 5.4 |
| 11 |
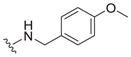
|
|
2.5 ± 0.2 | 5.2 ± 2.0 | 2.1 |
| 12 |
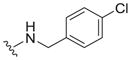
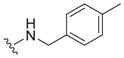
|
|
0.82 ± 0.23 | 4.6 ± 2.4 | 5.6 |
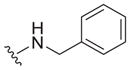
| 13 |
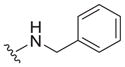
|
|
0.89 ± 0.10 | 4.5 ± 2.1 | 5.1 |
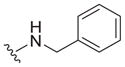
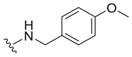
| 14 |
|
|
0.72 ± 0.10 | 4.1 ± 2.5 | 5.7 |
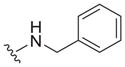
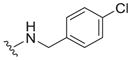
| 15 |
|
|
0.61 ± 0.13 | 3.2 ± 0.1 | 5.2 |
| 16 |
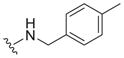
|
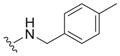
|
1.1 ± 0.5 | 2.9 ± 1.1 | 2.6 |
| 17 |
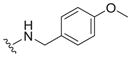
|
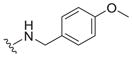
|
6.0 ± 2.9 | 2.2 ± 1.4 | 0.37 |
| 18 |
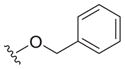
|
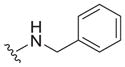
|
0.65 ± 0.13 | 3.3 ± 0.1 | 5.1 |
EC50 value is the mean ± standard deviation of at least three independent experiments. The control drug for the in vitro intracellular antileishmanial assay is amphotericin B, which displays an EC50 = 41 ± 8 nM against L. donovani (n = 20).
EC50 value is the mean ± standard deviation of at least three independent experiments. Podophyllotoxin is the control compound for the in vitro cytotoxicity assay, exhibiting an EC50 = 21 ± 5 nM against the J774.A1 macrophages (n = 11).
From Van Horn et al.18
Table 2.
Probing 2-phenyl or 4-phenyl-substituted quinazolines
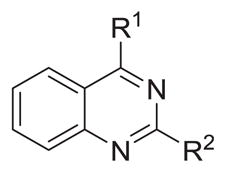
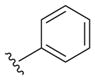
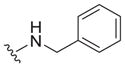
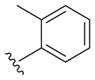
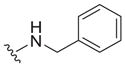
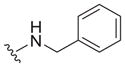
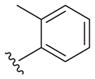
The reported value is based on two determinations. The control drug for the in vitro intracellular antileishmanial assay is amphotericin B, which displays an EC50 = 41 ± 8 nM against L. donovani (n = 20).
Not determined.
The most potent quinazoline-2,4-diamine against intracellular L. donovani in vitro from our previous study18 was N2-benzyl-N4-methylquinazoline-2,4-diamine 2, which displayed an EC50 of 0.15 μM against intracellular L. donovani and a selectivity index of 100. Table 3 shows a series of N4-methylated analogs of this compound. A dramatic decrease in activity is observed as smaller substituents were placed at N2. Aside from the reduction in activity compared to 2, there is no clear SAR trend in this series, as antileishmanial potency fluctuated when going from the N2-cyclopentyl derivative 23 (EC50 = 4.4 μM) to the N2-isopropyl and N2-ethyl derivatives 24 and 25 (EC50 values >25 μM) to the N2-methyl derivative 26 (EC50 = 7.6 μM). Those N4-methyl derivatives that were tested displayed less toxicity to J774 macrophages compared to the dibenzylated quinazolines shown in Table 1, however.
Table 3.
Probing N2-substituents of N4-methyl-quinazoline-2,4-diamines
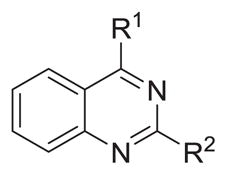
| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 |
L. donovani EC50a (μM) |
J774A.1 EC50b (μM) |
SI |
| 2 |
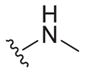
|
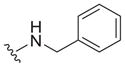
|
0.15 ± 0.02c | 15 ± 1c | 100 |
| 22 |
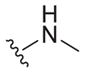
|
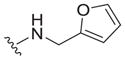
|
20 ± 5 | ND | ND |
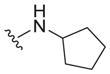
| 23 |
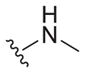
|
|
4.4 ± 1.2 | 39 ± 10 | 8.9 |
| 24 |
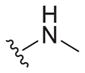
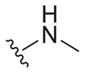
|
|
>25 | ND | ND |
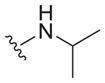
| 25 |
|
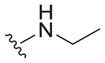
|
>25 | ND | ND |
| 26 |
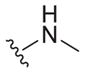
|
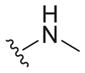
|
7.6 ± 1.3 | >133 | >17.5 |
| 27 |
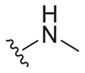
|
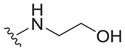
|
>25 | ND | ND |
EC50 value is the mean ± standard deviation of at least three independent experiments. When a mean value could not be ascertained, the reported value is based on at least two determinations. The control drug for the in vitro intracellular antileishmanial assay is amphotericin B, which displays an EC50 = 41 ± 8 nM against L. donovani (n = 20).
EC50 value is the mean ± standard deviation of at least three independent experiments. When a mean value could not be ascertained, the reported value is based on two determinations. Podophyllotoxin is the control compound for the in vitro cytotoxicity assay, exhibiting an EC50 = 21 ± 5 nM against the J774.A1 macrophages (n = 11).
From Van Horn et al.18
Our earlier work showed that N4-(furan-2-ylmethyl)-N2-isopropyl-7-methylquinazoline-2,4-diamine 4 exhibited promising activity in L. donovani-infected mice when given at a dose of 15 mg/kg/day for five days by the ip route.18 We thus synthesized analogs where either the furan-2-ylmethyl substituent at N4 or the isopropyl substituent at N2 were held constant and substituents at the other exocyclic nitrogen atom were varied (Tables 4 and 5) in an effort to identify compounds with improved in vitro activity to take forward to in vivo evaluation. Of the compounds possessing an N4-(furan-2-ylmethyl) substituent (Table 4), only the derivative 31 bearing a cyclohexyl group at N2 displayed in vitro potency similar to reference compound 3, but 31 showed negligible selectivity for intracellular L. donovani compared to J774 macrophages. When the N2-isopropyl group was held constant (Table 5, compounds 32–37), promising in vitro antileishmanial activity was observed with the N4-benzyl (32, EC50 = 2.0 μM), N4-phenyl (33, EC50 = 1.9 μM), and N4-isopropyl (35, EC50 = 2.3 μM) substituted analogs. Of these three compounds, the antileishmanial selectivity is best with the diisopropyl derivative 35 (SI >13). In the series of N4-isopropyl derivatives (Table 5, compounds 38–40), the N2-benzyl derivative 40 displayed outstanding in vitro potency against L. donovani and good selectivity (SI = 19).
Table 4.
Probing N4-substituents of N2-furfuryl-quinazolines
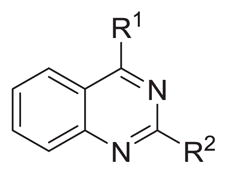
| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 |
L. donovani EC50a (μM) |
J774A.1 EC50b (μM) |
SI |
| 3 |
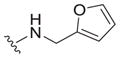
|
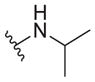
|
2.5 ± 0.4c | 17 ± 6c | 6.8 |
| 28 |
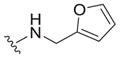
|
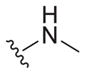
|
>25 | ND | ND |
| 29 |
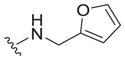
|
|
>25 | ND | ND |
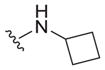
| 30 |
|
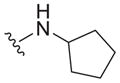
|
>25 | ND | ND |
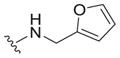
| 31 |
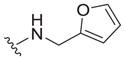
|
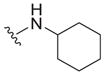
|
4.0 ± 2.5 | 5.3 ± 0.8 | 1.3 |
EC50 value is the mean ± standard deviation of at least three independent experiments. When a mean value could not be ascertained, the reported value is based on at least two determinations. The control drug for the in vitro intracellular antileishmanial assay is amphotericin B, which displays an EC50 = 41 ± 8 nM against L. donovani (n = 20).
EC50 value is the mean ± standard deviation of at least three independent experiments. Podophyllotoxin is the control compound for the in vitro cytotoxicity assay, exhibiting an EC50 = 21 ± 5 nM against the J774.A1 macrophages (n = 11).
From Van Horn et al.18
Table 5.
Probing 4- or 2-substitutions of N2-iso-propyl or N4-iso-propyl mono-substituted quinazoline-2,4-diamines
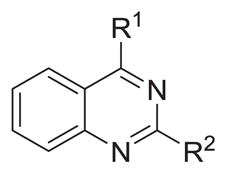
| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 |
L. donovani EC50a (μM) |
J774A.1 EC50b (μM) |
SI |
| 32 |
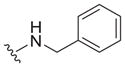
|
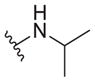
|
2.0 ± 0.1 | 6.7 ± 1.2 | 3.4 |
| 33 |
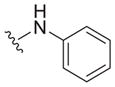
|
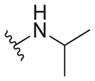
|
1.9 ± 0.2 | 12 ± 0 | 6.3 |
| 34 |
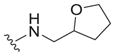
|
|
8.9 ± 0.1 | 46 ± 4 | 5.2 |
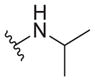
| 35 |
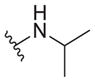
|
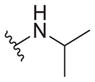
|
2.3 ± 0.5 | >30 | >13 |
| 36 |
|
|
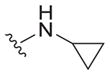
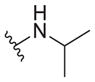
6.1 ± 2.8 | 31 ± 5 | 5.1 |
| 37 |
|
|
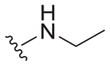
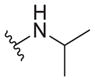
6.3 ± 1.9 | >30 | >4.8 |
| 38 |
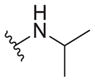
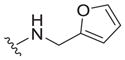
|
|
8.0 ± 2.0 | >40 | >5 |
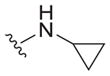
| 39 |
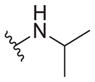
|
|
11 ± 3 | 8.3 ± 5.8 | 0.75 |
| 40 |
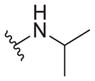
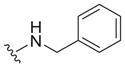
|
|
0.38 ± 0.09 | 7.2 ± 0.2 | 19 |
EC50 value is the mean ± standard deviation of at least three independent experiments. The control drug for the in vitro intracellular antileishmanial assay is amphotericin B, which displays an EC50 = 41 ± 8 nM against L. donovani (n = 20).
EC50 value is the mean ± standard deviation of at least three independent experiments. When a mean value could not be ascertained, the reported value is based on three determinations. Podophyllotoxin is the control compound for the in vitro cytotoxicity assay, exhibiting an EC50 = 21 ± 5 nM against the J774.A1 macrophages (n = 11).
Regarding general SAR trends, quinazoline-2,4-diamines bearing benzyl substituents at both N2 and N4 displayed potent antileishmanial activity in vitro, with EC50 values typically ranging from 0.4 to 1.1 μM, but the SI values for such compounds are less than 10. A phenyl substituent at either position 2 or position 4 caused a loss of activity when the substituent at the other such position was benzylamine, but an N4-phenyl group was permitted when the N2 substituent was isopropyl. Compounds bearing small alkyl substitutions at either N2 or N4 were typically less active in vitro, but had the advantage of being less toxic to the macrophage cell line. Compounds were identified that showed both reasonable potency against intracellular L. donovani and relatively low toxicity against the macrophage cell line, such as 26 and 35. These compounds offer a starting point for the design of new antileishmanial quinazolines aimed at maximizing antileishmanial potency while minimizing host toxicity.
Compounds 15 and 40 were selected for the in vivo evaluation of PK properties and for efficacy in a murine model of visceral leishmaniasis on the basis of their sub-micromolar in vitro antileishmanial potency (EC50 values of 0.61 and 0.38 μM, respectively) and fair to good selectivity (SI values of 5.2 and 19, respectively). The mean plasma and tissue concentration–time profiles of 15 and 40 after ip administration (10 mg/kg) are shown in Figure 2 and the relevant pharmacokinetic parameters are listed in Table 6. Compared with 40, 15 showed a much slower decrease of plasma concentration with a longer terminal t1/2 (9.3 h vs 2.1 h). Both compounds accumulated in the target tissues (Table 6) with tissue-to-plasma partition coefficients ranging from 3.2 to 16 for 40 and 20 to 21 for 15. After adjusting for dose, 15 exhibited 2.8-fold greater plasma AUC than 40. Longer plasma half-life and greater AUC of 15 are consistent with 15 being more metabolically stable (Table 6) and exhibiting greater tissue partitioning. Both 15 and 40 were then selected for in vivo antileishmanial evaluation. First, these compounds were examined for their toxicity to BALB/c mice. While 15 and 40 were toxic to the mice at a dose of 30 mg/kg/day × 5 by the ip route, the compounds had no apparent adverse effects on the mice when given by this route at 10 mg/kg/day × 5. This dose was thus chosen for evaluation in our murine model of visceral leishmaniasis. 22 Quinazoline 40 reduced liver parasitemia by only 23.8 ± 7.5% and compound 15 decreased liver parasitemia by 12.0 ± 3.2%, both given ip, compared to infected control groups. The same dose of the control drug miltefosine exhibited 93.6 ± 1.4% inhibition of liver parasitemia when given ip, consistent with our previously reported results.18,23
Figure 2.

Mean plasma (○), liver (■), and spleen (▲) concentration–time profiles of compound 15 (A) and compound 40 (B) after ip administration at a dose of 10 mg/ kg to mice. Symbols and error bars represent the mean and standard error of triplicate determinations.
Table 6.
Pharmacokinetics of 15 and 40 after ip administration in mice
| Parametera | Compound 15
|
Compound 40
|
||||
|---|---|---|---|---|---|---|
| Plasma | Liver | Spleen | Plasma | Liver | Spleen | |
| AUC (μM·h) | 15.7 | 7.14 | ||||
| t1/2 (h) | 9.3 | 2.1 | ||||
| Cmax (μM)b | 1.4 | 25.3 (8.51) | 26.6 (11.4) | 2.15 | 20.2 (0.54) | 16.5 (0.18) |
| Tmax (h) | 0.5 | 0.25 | ||||
| Vz/F (L/kg) | 22.3 | 1.15 | ||||
| Mic t1/2 (min)c | 88 | 42 | ||||
AUC, the area under the concentration–time curve from time zero to time infinity. Cmax and Tmax, maximum concentration and time to reach Cmax. t1/2, terminal half-life. Vz/F, apparent volume of distribution.
Values represent compound concentrations in liver and spleen at 4 h (at 12 h) post dose.
In vitro mouse liver microsomal half-life.
3. Conclusion
As illustrated by the in vivo studies reported here with 15 and 40, the toxicity of the 2,4-quinazoline diamines in mice continues to limit the dose of these compounds that can be administered, restricting the ability to conduct efficacy studies in the murine model of visceral leishmaniasis. The synthesis and evaluation of the 2,4-quinazoline diamines reported here provides further information regarding the antileishmanial SAR of these molecules, however, and lends clues for the synthesis of new analogs with lower toxicity. Future efforts will involve the preparation and testing of 2,4-quinazoline diamines containing isopropyl or related small alkyl substitutions at the exocyclic nitrogen atoms to maximize efficacy and exposure and minimize toxicity in vivo.
4. Experimental
4.1. General methods
All commercially available chemical reagents, except for the boronic acids, and anhydrous solvents were purchased from either Sigma Aldrich, Oakwood Products, Inc. or TCI America and used without any further purification. Boronic acids used were purchased through Frontier Scientific. NMR spectra were recorded at ambient temperature on a 500 MHz Varian NMR spectrometer in the solvent indicated. All 1H NMR experiments are reported in δ units, parts per million (ppm) downfield of TMS and were measured relative to the signals for chloroform (7.26 ppm), methanol (3.31 ppm) and dimethylsulfoxide (2.50 ppm). All 13C NMR spectra were reported in ppm relative to the signals for chloroform (77 ppm), methanol (49 ppm) and dimethylsulfoxide (39.5 ppm) with 1H decoupled observation. Data for 1H NMR are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, sext = sextet, sept = septet, oct = octet, m = multiplet), integration and coupling constant (Hz), whereas 13C NMR analyses were reported in terms of chemical shift. NMR data were analyzed by using MestReNova Software ver. 5.3.2–4936. The purity of the final compounds was determined to be ≥95% by LC/MS using an Agilent 1100 LC/MSD-VL with electrospray ionization. High resolution mass spectra (HRMS) were performed on an Agilent LC/MSD TOF system G3250AA. Analytical thin layer chromatography (TLC) was performed on silica gel 60 F254 pre-coated plates (0.25 mm) from EMD Chemical Inc. and components were visualized by ultraviolet light (254 nm). EMD silica gel 60 (particle size 40–63 μm) 230–400 mesh was used for all flash column chromatography.
4.2. Quinazolines
4.2.1. N2,N4-Disubstituted quinazoline-2,4-diamines
Compounds 1–5, 9–18, as well as 22–40 have been reported previously.18,19
4.2.2. N-Benzyl-4-(benzyloxy)quinazolin-2-amine (18)
Benzyl alcohol (59.8 mg; 0.55 mmol), 2,4-dichloroquinazoline 5 (100 mg; 0.5 mmol) and sodium hydride (13.3 mg; 0.55 mmol) were combined in dry tetrahydrofuran (2.0 mL). The reaction mixture was first cooled in an ice bath until addition of all material was complete and allowed to come to room temperature. The reaction was monitored by TLC and LCMS until no starting material was observed. The reaction mixture was then treated with water (10 mL) and diluted with dichloromethane (10 mL). The organic layer was separated, washed with equal volumes of water three times, and subsequently dried over sodium sulfate. Purification of the crude material was completed by column chromatography (hexanes/ethylacetate = 6:1) to afford the product 4-(benzyloxy)-2-chloroquinazoline (6) (106 mg; 0.39 mmol) as a white solid in 78% yield. 1H NMR (500 MHz, methanol-d4) δ 8.18 (ddd, J = 8.2, 1.5, 0.7 Hz, 1H), 7.96–7.91 (m, 1H), 7.83–7.79 (m, 1H), 7.64 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.59–7.54 (m, 2H), 7.46–7.34 (m, 3H), 5.66 (d, J = 1.7 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 167.58, 154.96, 151.63, 135.51, 135.44, 128.70, 128.61, 128.54, 128.46, 128.13, 126.68, 123.85, 114.51, 69.51. Compound 6 (100 mg; 0.37 mmol) and benzylamine (79.2 mg; 0.74 mmol) were combined in ethanol (1.2 mL). The reaction was heated in a sealed tube and monitored by TLC and LCMS until no starting material was observed. The reaction was quenched with water (5.0 mL) and diluted with dichloromethane (5.0 mL). The organic layer was separated and washed with the equal volume of water three times, then dried over sodium sulfate Purification of the crude material was completed by column chromatography (dichloromethane/methanol = 10:1) to afford product 18 (98.4 mg; 0.78 mmol) as a white solid in 78% yield. 1H NMR (500 MHz, methanol-d4) δ 7.89 (ddd, J = 8.2, 1.5, 0.6 Hz, 1H), 7.53 (ddd, J = 8.4, 7.0, 1.4 Hz, 1H), 7.37–7.16 (m, 10H), 7.11 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 4.75 (s, 2H), 4.61 (s, 2H). 13C NMR (126 MHz, methanol-d4) δ 162.49, 161.54, 152.82, 142.37, 141.35, 134.37, 129.86, 129.82, 128.99, 128.74, 128.35, 128.18, 125.08, 123.92, 122.75, 112.87, 46.39, 45.79. HRMS: m/z calculated for C22H19N3O [M+H]+ 342.142; found 342.1439.
4.2.3. 2-(Benzylamino)quinazolin-4(3H)-one (7)
2-(Benzylamino)quinazolin-4(3H)-one (7) was prepared over a reaction sequence of two steps. First, 2,4-dichloroquinazoline 5 (1.38 g; 6.9 mmol) was stirred in 1 N sodium hydroxide solution (3.0 mL) and was monitored by TLC and LCMS until no starting material was observed. The solution was then diluted with water (5.0 mL) and filtered. The filtrate was then neutralized with 6 M acetic acid, which afforded a white precipitate. The precipitate was then filtered, dried and used without further purification to afford the intermediate 2-chloroquinazolin-4(3H)-one as a white solid in 95% yield. 1H NMR (500 MHz, methanol-d4) δ 8.18 (dd, J = 8.0, 1.5 Hz, 1H), 7.82 (ddd, J = 8.2, 7.2, 1.6 Hz, 1H), 7.61 (d, J = 8.2 Hz, 1H), 7.54 (ddd, J = 8.1, 7.2, 1.1 Hz, 1H). 13C NMR (126 MHz, methanol-d4) δ 156.79, 133.20, 128.14, 127.00, 118.60, 118.26, 114.62, 111.44, 107.02. Subsequently, the crude material of intermediate 2-chloroquinazolin-4(1H)-one was then reacted with benzylamine (0.07 g; 6.6 mmol) in ethanol (1 mL) in a sealed tube at 150 °C. The progression of the reaction was monitored by TLC and LCMS until no starting material was observed. Once the reaction was cooled and quenched with water (5.0 mL), a precipitate was formed and filtered off. The resulting crystalline product was washed with cold ethanol (twice with 3.0 mL) to afford the titled compound 7 (80 mg; 0.32 mmol) in 96% yield without further purification. 1H NMR (500 MHz, methanol-d4) δ 8.01 (dd, J = 8.0, 1.3 Hz, 1H), 7.61 (ddd, J = 8.6, 7.2, 1.6 Hz, 1H), 7.41–7.37 (m, 2H), 7.37–7.30 (m, 3H), 7.29–7.24 (m, 1H), 7.20 (s, 1H), 4.64 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 162.08, 151.10, 150.64, 139.42, 134.38, 128.55, 127.56, 127.13, 126.07, 124.84, 121.90, 117.70, 43.80.
4.2.4. N-Benzyl-4-chloroquinazolin-2-amine (8)
2-(Benzylamino)quinazolin-4(3H)-one (7) (0.70 g; 2.9 mmol) was added to phosphorus oxychloride (3.5 mL). This solution was refluxed at 110 °C until no starting material was observed by TLC and LCMS. The reaction mixture was then poured over ice and diluted with ethyl acetate (25 mL). The organic layer was separated and washed with equal volume of water three times, then dried over sodium sulfate. Purification of the crude material was completed by column chromatography (hexane/ethylacetate = 10:1) affording product 8 (0.563 g; 2.1 mmol) as a white crystalline solid in 75% yield. 1H NMR (500 MHz, chloroform-d3) δ 8.01 (ddd, J = 8.3, 1.5, 0.7 Hz, 1H), 7.71 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 7.61 (d, J = 8.5 Hz, 1H), 7.42–7.38 (m, 2H), 7.37–7.32 (m, 2H), 7.30–7.27 (m, 2H), 4.75 (d, J = 5.9 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 159.41, 150.15, 136.07, 135.48, 128.48, 127.58, 127.35, 126.63, 124.75, 117.98, 115.77, 44.68.
4.2.5. General procedure for the arylation of chloro-substituted quinazolines to yield aryl-substituted quinazolines 19–21
Mono-chloro-substituted quinazoline (0.37 mmol), boronic acid (0.37 mmol), and 5 mol % of tetrakis-palladium catalyst, were mixed with toluene (1.25 mL) and 1 M solution of disodium carbonate (0.25 mL) in a round bottom flask and kept at room temperature under argon. The progression of the reaction was monitored by TLC and LCMS until no starting material was observed. The reaction mixture was then diluted with dichloromethane (2.0 mL) and the organic layer was separated and washed with an equal volume of water three times and subsequently dried over sodium sulfate. Purification of the final product was completed by column chromatography (hexane/ethylacetate = 5:1).
4.2.5.1. N-Benzyl-4-phenylquinazolin-2-amine (19)
Compound 19 was isolated as a white crystalline solid (90.5 mg; 0.79 mmol) in 79% yield. 1H NMR (500 MHz, Methanol-d4) δ 7.79 (d, J = 7.7 Hz, 1H), 7.73–7.67 (m, 3H), 7.62 (d, J = 8.4 Hz, 1H), 7.59–7.53 (m, 3H), 7.44–7.40 (m, 2H), 7.34–7.28 (m, 2H), 7.25–7.18 (m, 2H), 4.75 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 169.01, 158.76, 152.68, 140.19, 136.80, 133.58, 129.48, 129.21, 128.19, 127.98, 127.12, 126.81, 126.35, 125.54, 121.95, 117.46, 43.89. HRMS: m/z calculated for C21H17N3 [M+H]+ 312.1495; found 312.1495.
4.2.5.2. N-Benzyl-4-(o-tolyl)quinazolin-2-amine (20)
Compound 20 was isolated as a yellow crystalline solid (96.3 mg; 0.29 mmol) in 80% yield. 1H NMR (500 MHz, methanol-d4) δ 7.69 (ddd, J = 8.4, 6.8, 1.5 Hz, 1H), 7.63 (d, J = 8.5 Hz, 1H), 7.46–7.42 (m, 2H), 7.42–7.36 (m, 2H), 7.35–7.27 (m, 5H), 7.25–7.21 (m, 1H), 7.15 (ddd, J = 8.1, 6.8, 1.2 Hz, 1H), 4.74 (d, J = 13.9 Hz, 2H), 2.09 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 179.84, 168.44, 161.78, 149.72, 145.99, 144.69, 143.40, 139.72, 138.39, 138.24, 137.66, 136.74, 136.26, 136.02, 135.04, 131.62, 127.94, 53.52, 28.72. HRMS: m/z calculated for C22H19N3 [M+H]+ 326.1652; found 326.1653.
4.2.5.3. N-Benzyl-2-(o-tolyl)quinazolin-4-amine (21)
Compound 21 was isolated as a beige crystalline solid (108.4 mg; 0.33 mmol) in 90% yield. 1H NMR (500 MHz, methanol-d4) δ 8.19 (ddd, J = 8.3, 1.3, 0.7 Hz, 1H), 7.82–7.75 (m, 2H), 7.56–7.51 (m, 2H), 7.38 (ddt, J = 7.6, 1.4, 0.7 Hz, 2H), 7.33–7.20 (m, 6H), 4.89 (s, 2H), 2.29 (s, 3H). 13C NMR (101 MHz, methanol-d4) δ 164.30, 149.20, 139.81, 139.18, 136.36, 132.67, 130.14, 129.22, 128.35, 127.99, 126.98, 126.54, 126.35, 125.69, 125.05, 121.87, 113.17, 44.02, 19.16. HRMS: m/z calculated for C22H19N3 [M+H]+ 326.1652; found 326.1653.
4.3. In vitro assays
4.3.1. Intracellular L. donovani assay
The activity of compounds against intracellular Leishmania donovani strain MHOM/ET/67/L82 β-lactamase expressing parasites was determined as described previously.23
4.3.2. Macrophage toxicity assay
A cytotoxicity counterscreen was performed using the J774A.1 murine macrophage cell line by the general method described by Salem and Werbovetz24 with modifications. Briefly, J774A.1 macrophages were maintained in RPMI + GlutaMAX™ medium (Gibco) supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum at 37 °C in a humidified 5% CO2 atmosphere. Macrophages were plated at a density of 5 × 103 cells/well in the presence or absence of serial dilutions of test compounds or podophyllotoxin standard in a final volume of 100 μL. After 72 h incubation under the same conditions described above, 25 μL of a 5 mg/mL solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) in water was added to each well, and the plate was returned to the incubator for 2 h. A solution of 10% SDS (w/v) in 50% aqueous dimethylformamide was then added to each well, then the plate was incubated for an additional 3–5 h at 37 °C. Optical densities for each well were then read at 570 nm using a SpectraMax Plus microplate reader (Amersham Biosciences, Piscataway, NJ). The program SoftMax Pro (Amersham Biosciences, Piscataway, NJ) was used to determine IC50 values according to the dose-response equation , where x is the concentration of test compound, y is the absorbance at 570 nm, a is the upper asymptote, b is the slope, c is the EC50 value, and d is the lower asymptote.
4.3.3. Metabolic stability of compounds 15 and 40
Metabolic stability of 15 and 40 was evaluated using pooled mouse liver microsomes supplemented with NADPH as described previously.18 Plasma (2 μL) or tissue homogenates (10 μL) were mixed with 200 μL of 7:1 (v/v) methanol-water containing 10 nM compound 4 (internal standard). After vortex-mixed for 30 s, mixtures were centrifuged to pellet proteins at 4400 rpm for 10 min. The supernatant was transferred to a new tube and dried using a 96-well microplate evaporator (Model SPE Dry 96; Biotage, LLC, Charlotte, NC, USA) under nitrogen gas at 50 °C, followed by reconstitution with 200 μL of 15% methanol. The reconstituted samples and microsomal incubation mixtures were analyzed for drug concentration using a Waters Xevo TQ-S triple quadrupole mass spectrometer (Foster City, CA) coupled with aWaters Acquity UPLC I-Class system. Compounds were separated on aWaters UPLC BEH C18 column (2.1 × 50 mm; 1.7 μm) coupled with an ACQUITY column in-line filter (0.2 μm; Waters Corporation). The UPLC mobile phases consisted of (A) water containing 0.1% (v/v) formic acid and (B) methanol containing 0.1% (v/v) formic acid. The initial gradient began with 30% B and increased linearly to 70% over 2.5 min. The column was washed with 95% B for 0.7 min and the system re-equilibrated with 30% B for 1 min prior to the next injection (total analysis time of 4 min). A 0.4 mL/min flow rate was used throughout the UPLC gradient. The autosampler temperature was set at 6 °C and the UPLC column was heated at 50 °C. The sample injection volume was 1 μL. The characteristic single reaction monitoring (SRM) transitions for 15 and 40 were m/z 375.2→106.1 and 293.2→251.1 under positive ion mode. The calibration curves for 15 and 40 ranged from 0.025 to 25 μM for plasma and tissue samples. The intraday accuracy (deviation from true concentrations) and precision (coefficient of variation) of the method were within 15%.
Microsomal half-life (Mic t1/2) values were obtained by fitting the one-phase exponential decay equation to the percentage of substrate remaining versus time curves (GraphPad Prism, version 5.04, San Diego, CA). Noncompartmental pharmacokinetic analysis of plasma concentration versus time curves was carried out to obtain the area under the concentration–time curve (AUC), maximum concentration (Cmax), time to reach Cmax (Tmax), and terminal half-life (t1/2) (WinNonlin Version 5.3, Pharsight, Mountain View, CA).
4.4. In vivo studies
4.4.1. Animals
Protocols for animal studies were approved by the Institutional Animal Care and Use Committees (IACUC) of the University of Kansas (in vivo PK studies) or The Ohio State University (procurement of murine peritoneal macrophages and murine visceral leishmaniasis studies). Male Swiss Webster mice (weighing 20–25 g) were purchased from the Charles River Laboratories (O’Fallon, MO). Female CD-1 mice and female BALB/c mice (both 6–8 weeks of age) were obtained from Harlan Laboratories (Indianapolis, IN).
4.4.2. Pharmacokinetic analysis of compounds 15 and 40
Compounds 15 and 40 were dissolved in a mixture of DMSO/PEG400/ethanol/water (0.5:5:1:3.5, v/v), the same vehicle used for the efficacy study. Minor reversible overt toxicity (e.g., hypoactivity) was observed with the vehicle treated Swiss Webster mice. The compounds were administered intraperitoneally (ip) to mice at a dose of 10 mg/kg body weight (or approximately 34 and 26 μmol/kg, respectively; dose volume = 5 mL/kg), the same dose used in the efficacy study. Blood sampling time schedules were 0.25, 0.5, 1, 2, 4, 8, and 12 h (n = 3 at each blood sampling time point) after dosing. Blood was collected via the submandibular vein (~40 μL per bleed) or heart (terminal bleed) into lithium heparincoated Microvet®tubes (Sarstedt Inc., Newton, NC). Terminal blood and tissue samples were collected at 4 and 12 h post ip dose. Liver and spleen of each mouse were excised, rinsed with distilled water, blotted dry with tissue paper, and frozen on dry ice. A piece of frozen tissue was weighed and homogenized in two volumes (v/w) of distilled water (assuming 1 g of wet tissue is equal to 1 mL in volume) for liver (3-fold dilution) or three volumes (v/w) for spleen (4-fold dilution) using a sonic dismembrator (Model 100, Fisher Scientific, Waltham, MA). Plasma and tissue homogenates were stored at −70 °C until UPLC-MS/MS analysis as described below.
4.4.3. Murine visceral leishmaniasis models
The efficacy of compounds in the acute murine visceral leishmaniasis model was determined by the general method described previously.22 In these experiments, compounds 15 and 40 were dissolved in DMSO as a 20 mg/ml stock solution and then diluted 1:20 with vehicle (50% PEG 400, 10% ethanol, 35% water resulting in a clear solution after vortexing. The infected control group also received this vehicle, while miltefosine was dissolved in PBS.22
Acknowledgments
This work was funded by NIH Grant R21 AI101529. The funding agency had no involvement in designing the study, in collecting, analyzing, or interpreting the data, in writing the manuscript, or in deciding to publish the results. The authors would also like to thank Dr. Dennis E. Kyle for participating in helpful discussions regarding this manuscript.
Footnotes
Dedicated to the memory of Martin John Rogers for his sincere efforts to facilitate neglected disease research, particularly in the area of drug discovery and development
References and notes
- 1. [accessed December 26, 2014]; http://www.who.int/topics/leishmaniasis/en/
- 2.Belo V, Struchiner C, Barbosa D, Nascimento B, Horta M, da Silva E, Werneck G. PLoS Negl Trop Dis. 2014;8:e2982. doi: 10.1371/journal.pntd.0002982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martins-Melo F, Lima M, Ramos A, Jr, Alencar C, Heukelbach J. PLoS ONE. 2014;9:e93770. doi: 10.1371/journal.pone.0093770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leta S, Dao T, Mesele F, Alemayehu G. PLoS Negl Trop Dis. 2014;8:e3131. doi: 10.1371/journal.pntd.0003131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abubakar A, Ruiz-Postigo J, Pita J, Lado M, Ben-Ismail R, Argaw D, Alvar J. PLoS Negl Trop Dis. 2014;8:e2720. doi: 10.1371/journal.pntd.0002720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sundar S, Jha T, Thakur C, Sinha P, Bhattacharya S. N Engl J Med. 2007;356:2571. doi: 10.1056/NEJMoa066536. [DOI] [PubMed] [Google Scholar]
- 7.Sundar S, Chakravarty J, Agarwal D, Rai M, Murray H. N Engl J Med. 2010;362:504. doi: 10.1056/NEJMoa0903627. [DOI] [PubMed] [Google Scholar]
- 8.Sundar S, Sinha P, Rai M, Verma D, Nawin K, Alam S, Chakravarty J, Vaillant M, Verma N, Pandey K, Kumari P, Lal C, Arora R, Sharma B, Ellis S, Strub-Wourgaft N, Balasegaram M, Olliaro P, Das P, Modabber F. Lancet. 2011;377:477. doi: 10.1016/S0140-6736(10)62050-8. [DOI] [PubMed] [Google Scholar]
- 9.Thakur C, Kumar A, Thakur M, Thakur S. Indian J Med Res. 2009;129:102. [PubMed] [Google Scholar]
- 10.Chowdhury R, Mondal D, Chowdhury V, Faria S, Alvar J, Nabi S, Boelaert M, Dash A. PLoS Negl Trop Dis. 2014;8:e3020. doi: 10.1371/journal.pntd.0003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muniaraj M. Trop Parasitol. 2014;4:10. doi: 10.4103/2229-5070.129143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sundar S, Singh A, Rai M, Prajapati V, Singh A, Ostyn B, Boelaert M, Dujardin J, Chakravarty J. Clin Infect Dis. 2012;55:543. doi: 10.1093/cid/cis474. [DOI] [PubMed] [Google Scholar]
- 13.Sundar S, Chakravarty J. Expert Opin Investig Drugs. 2015;24:43. doi: 10.1517/13543784.2014.954035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matlashewski G, Arana B, Kroeger A, Be-Nazir A, Mondal D, Nabi S, Banjara M, Das M, Marasini B, Das P, Medley G, Satoskar A, Nakhasi H, Argaw D, Reeder J, Olliaro P. Lancet Global Health. 2014;2:e683. doi: 10.1016/S2214-109X(14)70318-3. [DOI] [PubMed] [Google Scholar]
- 15.Hayden E. Nature. 2014;505:142. doi: 10.1038/505142a. [DOI] [PubMed] [Google Scholar]
- 16.Wyllie S, Patterson S, Stojanovski L, Simeons F, Norval S, Kime R, Read K, Fairlamb A. Sci Transl Med. 2012;4:119re1. doi: 10.1126/scitranslmed.3003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. [accessed December 26, 2014]; http://www.dndi.org/diseases-projects/portfolio.html.
- 18.Van Horn K, Zhu X, Pandharkar T, Yang S, Vesely B, Vanaerschot M, Dujardin JC, Rijal S, Kyle D, Wang M, Werbovetz K, Manetsch R. J Med Chem. 2014;57:5141. doi: 10.1021/jm5000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Horn K, Burda W, Fleeman R, Shaw L, Manetsch R. J Med Chem. 2014;57:3075. doi: 10.1021/jm500039e. [DOI] [PubMed] [Google Scholar]
- 20.Samrin F, Sharma A, Ali Khan I, Puri S. J Heterocycl Chem. 2012;49:1391. [Google Scholar]
- 21.Miyaura N, Suzuki A. Chem Rev. 1995;95:2457. [Google Scholar]
- 22.Delfín DA, Morgan RE, Zhu X, Werbovetz KA. Bioorg Med Chem. 2009;17:820. doi: 10.1016/j.bmc.2008.11.031. [DOI] [PubMed] [Google Scholar]
- 23.Zhu X, Pandharkar T, Werbovetz K. Antimicrob Agents Chemother. 2012;56:1182. doi: 10.1128/AAC.05412-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salem MM, Werbovetz KA. J Nat Prod. 2005;68:108. doi: 10.1021/np049682k. [DOI] [PubMed] [Google Scholar]