

Summary
We report a new cellular interaction between the infecting transposable phage Mu and the host E. coli replication machinery during repair of Mu insertions, which involves filling-in of short target gaps on either side of the insertion, concomitant with degradation of extraneous long flanking DNA (FD) linked to Mu. Using the FD as a marker to follow repair, we find that after transposition into the chromosome, the unrepaired Mu is indefinitely stable until the replication fork arrives at the insertion site, whereupon the FD is rapidly degraded. When the fork runs into a Mu target gap, a double strand end (DSE) will result; we demonstrate fork-dependent DSEs proximal to Mu. These findings suggest that Pol III stalled at the transpososome is exploited for coordinated repair of both target gaps flanking Mu without replicating the intervening 37 kb of Mu, disassembling the stable transpososome in the process. This work is relevant to all transposable elements, including retroviral elements like HIV-1, which share with Mu the common problem of repair of their flanking target gaps.
Introduction
Genomes of virtually all organisms harbor transposable elements (TEs) whose past as well as present activity continues to shape genome structure, function and evolution (Huang et al., 2012). Active human TEs have been estimated to generate about one new insertion per 10–100 human births (Kazazian, 1999). In single individuals, a significant number of de novo insertions influence a range of phenotypes, both life enhancing (creating somatic heterogeneity in the brain; (Singer et al., 2010, Perrat et al., 2013)), and life threatening (primarily cancer-causing; (Kazazian, 2004, Mills et al., 2007, Chenais, 2013)). Understanding the mechanism and regulation of these events is important for controlling their incidence.
The cutting and joining reactions of transposition that link the transposon to the target are well studied (Craig, 2002). The majority of DNA transposons, including retroviruses and retroviral-like transposons, transpose by a non-replicative mechanism i.e. without duplicating themselves in the process. These reactions leave short gaps in the target DNA on either side of the transposon. Transposition is not complete until these gaps are repaired. Yet this essential step is not as yet deciphered. Because a majority of these transposons are not duplicated, it is assumed that gap-filling polymerases fill the gaps, but conclusive evidence for a specific polymerase is lacking (Sasakawa et al., 1981, Syvanen et al., 1982, Yoder & Bushman, 2000). In this study we have used transposable phage Mu to investigate gap repair in vivo, because of unique features of Mu that make the analysis possible.
As a temperate transposable phage, Mu uses transposition to integrate into the E. coli host chromosome to generate a prophage during the lysogenic phase, and to amplify its genome over a hundred-fold during the lytic phase (Symonds et al., 1987). During both phases, the chemical steps of transposition are the same: single-stranded DNA cleavages at Mu ends followed by strand transfer (ST) of the cleaved ends to phosphodiester bonds spaced 5 bp apart on the target DNA (Mizuuchi, 1992, Chaconas & Harshey, 2002). However, the structure of the Mu donor is different during the two phases, and has a bearing on how the ST intermediate is resolved (Fig. 1A and Fig. S1) (Harshey, 2015). During the lytic phase, Mu transposes from one site to another on the E. coli chromosome while remaining part of the covalently closed chromosome (Fig. 1B, 60 min). Here, the ST intermediate is resolved by target-primed replication by the Restart primosome, which fills the flanking target gaps while replicating across Mu (Fig. S1) (Nakai et al., 2001). During the lysogenic phase, the infecting Mu genome is linear, is linked to several hundred base pairs of non-Mu flanking DNA (FD), and is non-covalently closed by the phage N protein (Fig. 1A) (Harshey & Bukhari, 1983, Puspurs et al., 1983, Gloor & Chaconas, 1986). Here, the ST intermediate is resolved without replication (Akroyd & Symonds, 1983, Harshey, 1984, Liebart et al., 1982, Chaconas et al., 1983), during which the FD is degraded concomitant with gap repair (Au et al., 2006). Using a convenient assay for monitoring FD degradation in vivo, we have learned that the first event in repair is removal of the FD by the RecBCD exonuclease (Choi et al., 2014a), whose entry past the N-protein block is controlled by the transpososome and facilitated by ClpX (Choi & Harshey, 2010, Choi et al., 2014b). In vitro experiments reveal that RecBCD action is required for stimulating endonucleolytic cleavage within the transpososome-protected DNA, leaving 4-nt flanks outside both Mu ends (Fig. 1A). This structure is likely the substrate for gap repair by host enzymes. The infection phase of non-replicative Mu transposition is an ideal system to investigate the gap repair process not only because of its high efficiency, where every infecting Mu genome integrates into the E. coli chromosome within 10 minutes, but also because we can track repair events using the FD degradation assay. We know that when integration of infecting Mu is blocked, the unintegrated N-linked Mu genome is indefinitely stable (Harshey & Bukhari, 1983, Puspurs et al., 1983, Gloor & Chaconas, 1986). Thus, degradation of the FD is timed to coincide with some event that follows integration. We demonstrate in this study that this event is arrival of the E. coli replication fork.
Fig. 1. Repair of Mu insertions is prevented if chromosome replication is prevented.

(A) Known steps in the non-replicative (repair) pathway of Mu transposition. This pathway is used during integration of infecting Mu. The infecting genome is linear, and attached at both ends to long flanking DNA (FD) protected by Mu N protein. This DNA is variable in length (60 – 150 bp at the L end and 0.5 – 3 Kbp at the R end). MuN circularizes the DNA non-covalently, and protects it from nucleases. MuA catalyzes cleavage and strand transfer (integration) of Mu into the E. coli genome, assisted by MuB protein and host HU protein. The N protein is removed only after integration by an unknown mechanism assisted by the transpososome (purple ball), and the FD is degraded by RecBCD. Degradation is slowed in the absence of ClpX. In vitro, the final product of RecBCD degradation leaves 4 nt of the FD. This strand transfer intermediate with short flanks is likely the substrate for the final steps in repair, where the 5 bp target gaps are filled to generate a simple insertion. (B) Mu life cycle. After infection, Mu integrates into the E. coli genome, the FD is degraded, the Mu insertion is repaired, and Mu enters the lytic cycle. The approximate time (0–60 min) of these events is indicated. (C) Schematic of known mechanisms for replication initiation at oriC and during Restart of Mu replication in E. coli. (D) Preparation for the FD detection assay. At various times after infection, genomic DNA was subjected to pulse-field agarose gel electrophoresis (PFGE) to separate integrated Mu from free Mu, and the gDNA was excised for analysis by PCR. The (−) lane is an uninfected control where Mu DNA was added to the gDNA prior to electrophoresis, to assess contamination of the excised gDNA band with free Mu. Mu-length DNA at 60 min reflects packaged virions. (E) PCR assay for FD detection. Strains were infected with Mu at 37°C for the indicated times, and the isolated gDNA was tested by PCR to detect Mu or FD (lacZ) sequences using appropriate primers; lacZ sequences linked to infecting Mu are not found in the host (Au et al., 2006). + is Mu virion DNA and − is gDNA from the uninfected control lane in panel D. WT (BW25113); RecB− (JW2788); ClpX− (JW0428). (F) Monitoring inactivation of replication in DnaAts (SS1424) and DnaCts (SS1021) mutants by measuring chromosome equivalents. The mutants were held at 42°C for indicated times, without shaking, and fixed in ethanol before staining with the fluorescent DNA stain SYTOX Green. Stained cells were analyzed by flow cytometry as described under Experimental Procedures. By 60 min, there are no new rounds of replication in either mutant, as judged by the shift in the initial DNA content of ~2–4 chromosome equivalents to 1 chromosome equivalent. (G) FD removal depends on chromosome replication. DnaAts and DnaCts mutants were infected with Mu at both 30°C and 42°C. The latter infections were carried out after replication arrest for 90 min. In the WT (MG1655) infection at 42°C, FD is removed faster than at the lower temperatures. The PriA mutant (SS1448) and its WT parent (SS996) were infected at 30°C. Strains are listed and described in Table S1.
Results
Repair of Mu insertions depends on the replication fork
The cutting and joining reactions of Mu transposition do not generate broken DNA ends (Mizuuchi, 1992), yet E. coli proteins that repair double strand breaks (DSBs) are required to recover Mu insertions after infection (Jang et al., 2012). This suggests that one of the Mu target gaps might be converted to a break, perhaps by a replication fork. We will use the terminology DSE (double strand end) for the single DNA break created when a fork runs over a gap, and DSB for DNA breaks generated spontaneously or introduced by restriction enzymes. We had envisioned two alternative scenarios for generating a DSE after Mu integration - either arrival of the native fork at the Mu insertion site, or start of replication on the FD-linked Mu insertion (Jang et al., 2012). In E. coli, chromosome replication initiates at a unique origin oriC (Kornberg & Baker, 1992), where the initiator protein DnaA first recruits the DnaB helicase from the DnaB-DnaC complex, followed by loading of the Pol III holoenzyme (Fig. 1C, top). Bidirectional replication forks proceed from oriC to the terminus ter, but can collapse if they encounter nicks, gaps or other forms of DNA damage (Michel et al., 2007). Collapsed forks are reassembled by several Restart pathways (Gabbai & Marians, 2010), the two main pathways requiring PriA and DnaT. The function of Restart proteins is similar to DnaA in recruiting DnaB for Pol III assembly (Fig. 1C, bottom). Mu uses the PriA Restart primosome to initiate replication specifically on Mu DNA during lytic growth (Fig. S1) (Nakai et al., 2001).
To determine the role of the replication enzymes in the repair of Mu-linked FD, we selectively prevented replication forks initiating at oriC by using temperature sensitive (ts) mutants of DnaA and DnaC, or prevented Restart forks by using a PriA mutant, and monitored FD repair. At various times after Mu infection, E. coli genomic DNA (gDNA) was subjected to pulse-field agarose gel electrophoresis to separate integrated Mu from free Mu (Fig. 1D). The gDNA band was excised from the gel, and a PCR assay was used to detect Mu (primers 1, 2) and FD sequences (primers 3, 4) (Fig. 1E). In a wild type host infection, both Mu and FD are found integrated in the gDNA by 15 min, but the FD disappears soon after (Fig. 1E; WT). The FD is not degraded in a RecB mutant (Fig. 1E; RecB−), and degradation is delayed in a ClpX mutant, as reported earlier and (Fig. 1E; ClpX−) (Au et al., 2006, Choi & Harshey, 2010, Choi et al., 2014b). A different integration assay showed additionally that the FD was processed to a short length in vivo (Fig. S2), similar to that seen in vitro with RecBCD (Fig. 1A) (Choi et al., 2014a).
When oriC replication is prevented in DnaAts and DnaCts mutants at 42°C, the ts mutants finish ongoing rounds of replication but do not re-initiate new ones (Wechsler & Gross, 1971), an observation we reconfirmed using FACS analysis for measuring chromosome ploidy, which shifts from two chromosome equivalents to one within ~ 60 min (Fig. 1F). Ninety minutes after replication inactivation at 42°C, the cells were infected with Mu. Both mutants supported Mu integration at 42°C (Fig. 1G; see Mu panels in the Dnats mutants), consistent with earlier data showing that Mu integration is independent of chromosome replication (Nakai & Taylor, 1985). However, in the absence of replication, the FD was not removed (Fig. 1G, compare FD panels at 30°C vs 42° in the ts mutants). The FD was degraded normally in the absence of PriA. These results were confirmed using an alternate assay, where the short product of FD degradation was not detected in non-replicating cells (Fig. S3). We conclude that chromosome replication is required for repair of Mu insertions, and that PriA-dependent replication initiating from Mu is not involved.
The entire Pol III replisome machinery is necessary for FD removal: gap-filling polymerases are not required
To determine if a specific component of the replisome is required for FD removal, ts mutants in all available components – helicase DnaB, β-clamp DnaN, β-clamp loader DnaX, and Pol III α subunit DnaE – were tested (Fig. 2A). Replication ceases immediately at 42°C in these replication elongation mutants (McMacken et al., 1987). Mu integration was apparently normal when replication was arrested (Fig. 2B, 42°C and Fig. S4); however, FD degradation was blocked in all the strains only at 42°C.
Fig. 2. The entire Pol III replisome machinery is necessary for FD removal: gap-filling polymerases are not required.

(A) Replisome components tested in this experiment. Newly synthesized DNA is depicted by dotted lines. (B) Prior to Mu infection, replication was inactivated in the ts mutants by incubating cells at 42°C for 30 min. Mu and FD sequences were monitored as described in Fig. 1D and E. WT (SS996); DnaBts (SS6699); DnaEts (SS6239); DnaNts (SS6700); DnaXts (SS6698). (C) The polA (Pol I) mutant (SS2357) and its WT parent were infected with Mu at 30°C, while the triple umuC dinB polB (Pol II, IV, V) mutant (SS7346) and its WT parent were infected at 37°C, prior to analysis of Mu and FD. (D) SOS induction with or without Mu infection was monitored by expression of GFP under the control of the sulA promoter (PsulA-gfp) as described under Experimental Procedures (McCool et al., 2004). Mu infections were carried out in lexA3 (SS4294) and lexA71 (SS4610) derivatives as well as in their WT parent, all carrying a chromosomal sulA-gfp fusion. lexA3 is defective for SOS induction, while lexA71 is constitutively induced.
E. coli has four other polymerases that assist in gap repair either during normal replication (Pol I), or during repair of DNA damage (Pol II, IV, V) (Fijalkowska et al., 2012). Both Mu integration and FD degradation were similar to wild type in the single Pol I mutant as well as in the triple damage-inducible polymerase mutants (Fig. 2C). Thus, none of the gap-filling polymerases were required for Mu repair. These data do not address whether Mu integration might trigger an SOS response, the damage-inducible polymerases serving as a backup repair mechanism. To test this, we measured SOS induction by monitoring GFP fluorescence expressed from the promoter of the SOS-induced gene sulA with or without Mu infection in a wild type strain, or its lexA3 and lexA71 derivatives (Fig. 2D) (McCool et al., 2004); lexA3 is defective for SOS induction, and serves as negative control, while lexA71 is constitutively induced. The data show that Mu infection does not induce an SOS response (Fig. 2D). We conclude that the Pol III holoenzyme, and not gap-filling polymerases, carries out Mu repair.
Permitting blocked forks to resume replication, resumes Mu repair
We interpret the above data to mean that interaction of the E. coli replication fork with the Mu transpososome is required to allow entry of RecBCD into linear FD ends that are normally protected by the phage N protein (Fig. 1A). To test this interpretation, Mu was first allowed to integrate in replication-blocked cells, followed by restart of replication either at the oriC site (DnaCts mutant) or at any elongation site (DnaEts mutant) on the chromosome, by a timed shift-down to 30°C. The DnaAts mutant was not used in these experiments because Mu can enter the lytic mode of growth in this mutant (Toussaint & Faelen, 1974, McBeth & Taylor, 1982), during which the Restart fork would replicate Mu, bypassing FD repair, as also seen in RecBCD and other mutants which permit Mu lytic growth without FD repair ((Choi & Harshey, 2010, Choi et al., 2014a); see also Fig. S3).
In the absence of chromosome replication, the FD remained stable at 42°C when monitored across a 2 hr window in both DnaC and DnaE mutants (Fig. 3A, top panel; 120 min time-point not shown). When replication-blocked cells harboring integrated but unrepaired Mu in their genomes were shifted down to 30°C after 15, 30 and 60 min at the non-permissive temperature, the FD was degraded in both mutants within 15–30 min after the shift-down (Fig. 3A, bottom 3 panels).
Fig. 3. Permitting blocked forks to resume replication, resumes Mu repair.

(A) The indicated ts mutants were infected with Mu after inactivation of replication at 42°C for 30 min (DnaEts) or 90 min (DnaCts). Following infection (0 min), cells held at 42°C (top row) were shifted down to 30°C (15 – 60 min, rows 2–4) to release the replication block, and monitored for presence of the FD. (B) Schematic depicting the time taken by the bidirectional fork originating at oriC to reach ter. Mu integrates all over the chromosome. On average, the fork would encounter Mu in ~20 min.
For E. coli growing in a rich medium, it takes ~40 min for the bi-directional fork to travel from oriC to ter (Fig. 3B) (Cooper & Helmstetter, 1968). Since Mu integration events occur all over the E. coli chromosome (Manna et al., 2004, Ge et al., 2011), on average, the fork would encounter a Mu insertion in approximately half that time, consistent with the average observed time for FD degradation. We conclude that repair of Mu insertions does not begin until the replication fork arrives at the Mu site.
FD is degraded by replication forks initiating at oriC-independent sites
Does a fork have to initiate at oriC to promote events that result in FD repair? To address this question we used an RNase HI mutant (rnhA), where oriC-independent initiation can occur from RNA-DNA hybrids (R-loops) at multiple ectopic oriK sites using the PriA pathway (the R-loops are normally removed by RNase HI; (Kogoma, 1997)). The mutant strain grows poorly because of fork collisions associated with unregulated replication, and because of an SOS-constitutive phenotype brought on by DNA breaks (Fig. 4A, B) (Maduike et al., 2014). The DnaAts mutation was moved into this strain to additionally prevent oriC replication; the double mutant grows even more slowly (Fig. 4A). Nonetheless, both mutants still supported Mu integration at 30°C as well as at 42°C, and although slightly delayed, the FD was clearly degraded at both temperatures (Fig. 4C). We conclude that the FD degradation takes place in the presence of replication forks even when replication is not initiated at oriC.
Figure 4. oriC-independent forks also promote FD repair.

(A) Growth of the rnhA (N4704), dnaA46 (ts; AU1054), and rnhA dnaA46 double mutant (AU1066) strains on LB agar plates at 30°C and 42°C. The double mutant grows poorly even at 30°C. (B) Schematic depiction of replication originating at multiple ectopic locations in the RnhA mutant. (C) Mu integration and FD degradation in rnhA and rnhA dnaA46 strains at 30°C and 42°C. The latter infections were carried out after replication arrest for 90 min. The WT parent for these strains is MG1655, shown in Fig. 1G.
Release of the replication block generates Mu-proximal DSBs
When the replication fork runs into the Mu transpososome, it is expected to stall because the Mu transpososome is extremely stable (Surette et al., 1987, Choi et al., 2014a). If the fork travers the Mu gap on one strand, a DSE would be generated on the other strand. To test for the presence of the expected DSE at the junction of Mu and the replication fork, we marked both Mu and the potential DSE with fluorescent markers. Mu was fluorescently labeled by first incorporating a TetO array into the prophage genome (Fig. 5A), such that phage viability was not affected; similar phage titers were obtained upon induction of both wild type and TetO-engineered prophages (Fig. 5B, C). When the Mu::TetO phage were used to infect cells expressing TetR-mCherry at an MOI of 1, > 80% of the cells had single mCherry foci (Fig. 5D, E).
Figure 5.

Visualizing integrated Mu with TetR-mCherry.
(A) The TetO array was substituted for the SE region of the prophage, which is dispensable for phage growth. A compensatory deletion in another non-essential region near the R end restored the original Mu DNA length, ensuring that head-full packaging would yield viable phage progeny (Symonds et al., 1987). See Experimental Procedures for construction details. (B) Lysis profiles of Mu::TetO (SJ012) and Mu wild-type (HM8305) prophage strains. Similar phage titers were obtained from both strains (see C). Error bars indicate standard deviation from the mean of triplicate data sets obtained from three independent colonies of the same strain. (C) Plaque morphologies of wild-type and Mu::TetO, titered on BW25113. Mutations in the SE region are known to affect plaque size (inset) (Symonds et al., 1987). (D) Snapshot of Mu::TetO infection (MOI = 1) into WT strain BW25113 expressing TetR-mCherry (pDB317), photographed before (−Mu) and after ~15–20 min of infection (+Mu). (E) Quantitation of uninfected and infected cells in D by counting mCherry foci. > 80% of the cells had mCherry foci, and a majority of these had single foci.
DSEs were fluorescently labeled using two different proteins, Mu Gam and RecA. Mu Gam binds directly to a DSE (d’Adda di Fagagna et al., 2003), while RecA binds to the single-strand region of a processed DSE (Cox, 2001, Dillingham & Kowalczykowski, 2008). Fluorescent fusions to these proteins bind to DSEs in vivo (Shee et al., 2013, Renzette et al., 2005). Since overexpression of Gam interferes with repair of DSEs and cell viability (see Fig. S5), its expression was controlled by the tightly regulated rhamnose-inducible promoter in pGam-GFP, repressing it with 0.2% glucose until needed; rhamnose was added only 30 min before Mu infection. RecA-GFP is expressed constitutively from its normal chromosomal location.
When cells expressing TetR-mCherry and Gam-GFP were infected with Mu::TetO at an MOI of 1 where the majority of cells had an mCherry focus (Fig. 5), < 0.1% of the cells had visible GFP foci before infection; however, their numbers increased at least 30-fold after Mu infection (Fig. 6A). The green foci were binned into different categories with respect to their proximity to the red foci. Of the foci that appeared after Mu infection, ~ 40% either co-localized with or were proximal to Mu (Fig. 6A, right). The overall low numbers and low intensity of Gam-GFP foci may be due to limiting its expression to only 30 min prior to Mu infection, which was necessitated because prolonged expression had a negative impact on cell viability (Fig. S5).
Fig. 6. Mu insertions generate replication-dependent DSBs in their vicinity.

Mu::TetO is detected by TetR-mCherry (labeled Mu-mCherry) and DSBs by either Gam-GFP or RecA-GFP in WT (BW25113) or its DnaEts derivative (SJ005). (A) Left, snapshot of Mu-mCherry foci relative to Gam-GFP foci upon Mu infection of WT. Right, quantitation of the position of the green and red foci. GFP foci were scored as ‘at Mu’ when they completely overlapped with mCherry foci, ‘near Mu’ when at least their edges touched, and ‘random’ when they did not touch. Background fluorescence with no detectable foci was scored ‘diffuse’. (B) As in A, but with a RecA-GFP expressing strain. Foci with large or aberrant morphology are classified ‘other’. Using microbeTracker software (Sliusarenko et al., 2011) the average area of a cell and of a fluorescent focus were calculated to be 1.78 μm2 and 0.0675 μm2 respectively, yielding a total of 26 independent sites that any focus can occupy in the cell. Two foci that touch occupy 4 times the area of one focus. The probability that any two foci will be stochastically near each other is then modeled by 4/262 or slightly less than 0.6%, well below the experimentally observed 50%, which include foci that completely overlap. (C) Left, replication-arrested cells were infected with Mu and monitored for Mu-mCherry and Gam-GFP foci (top panel). The same frame of cells was photographed after releasing the replication block by shifting cells to 25°C (bottom panel). White arrow points to GFP focus that appeared after the temperature shift. Right, quantitation of foci after fork release, as in A. See also Fig. S6. See Experimental Procedures for details.
When RecA-GFP was used to monitor DSEs, the GFP foci were bright and 24% of uninfected cells had a single focus, consistent with the frequency of RecA foci detected in cells growing in rich media (Fig. 6B) (Renzette et al., 2005). Infection with Mu increased their numbers by 13%. Here, >50% of the total number of the foci seen after infection were associated with Mu, suggesting that the majority of the Mu-induced RecA foci are near Mu. That only a fraction of the cells with Mu foci had Gam or RecA foci, could be due to fast repair of the DSE. This inference is supported by the observation that Mu infection does not induce the SOS response (Fig. 2D); induction of an SOS response requires that the DSE persist (Kuzminov, 1999, Lusetti & Cox, 2002). GFP foci not associated with Mu might be accounted for recession of the DSE by RecBCD to large distances from the site of the break (Shee et al., 2013).
To test if appearance of the GFP foci was dependent on replication, replication-arrested cells (DnaEts) were infected with Mu in cells expressing Gam-GFP. New GFP foci appeared only when the replication block was removed (Fig. 6C and S6); 50% of these foci were associated with Mu. (This experiment could not be conducted using RecA-GFP because of a high background of GFP foci in the DnaEts strain at 42°C (Massoni & Sandler, 2013, Renzette et al., 2005)). The independent probability of localization of the GFP foci next to Mu-mCherry foci was estimated to be less than 0.6%, a number much lower than the observed 50% co-localization (see Fig. 6 legend).
We conclude that Mu insertions generate DSEs in their vicinity that are dependent on chromosomal replication.
Discussion
This work opens a new window into the repair of transposon insertions, the majority of which share with Mu the problem of repair of flanking target gaps. Progress in this area of transposon research has languished likely because of the difficulty of studying the repair of such short gaps. Mu has provided the first insight into this problem primarily because of the unusual feature of long FD sequences linked to the infecting Mu genome, which are repaired concomitant with gap repair (Fig. 1A). The development of a convenient in vivo assay for monitoring FD repair (Au et al., 2006), has allowed us to make large strides into understanding the repair process, both in vivo and in vitro. In vivo, the RecBCD exonuclease was found to be responsible for removal of the FD only after Mu integration. Thus, some post-integration event(s) must allow RecBCD access by releasing the N protein protecting the FD (Choi et al., 2014a). In vitro, a strand transfer substrate with FD lengths similar to the in vivo substrate but lacking N, showed that RecBCD degrades the FD until it encounters the transpososome, leaving 19 bp of FD, which is shorted to 4 nt with added cell extracts (Choi et al., 2014a). The present study confirms the generation of the short FD product in vivo (Figs. S2 and S3). The major finding of this study is that the event that triggers post-integration FD removal is arrival of the oriC replication fork at the Mu insertion site (Figs. 1–4). Thus, proximity of the replisome to the transpososome is required to initiate N removal. A requirement for the native fork explains why the DSB repair machinery is required to recover Mu insertions (Jang et al., 2012). We show that fork-dependent DSEs appear proximal to Mu (Fig. 6), and establish that gap-filling polymerases are not required for Mu repair (Fig. 2).
Model for Pol III-mediated coordinated repair of flanking target gaps without replicating the intervening Mu DNA
Why does Mu depend on the Pol III replisome and not gap-filling polymerases, and why does it employ a repair scheme that generates additional DNA damage for repair? We suggest that the Mu transpososome partners with the replisome for multiple tasks. First, the transpososome uses the incoming Pol III to signal just-in-time FD degradation, protecting the flanking gaps until the polymerase is on site to fill these gaps. Next, Pol III stalled at the transpososome is exploited for coordinated repair of both target gaps flanking Mu without replicating the intervening 37 kb of Mu. Finally, the replisome is used for transpososome disassembly concomitant with gap repair, as discussed below.
According to the data in Figs. 2–6, replisome interaction with the transpososome is somehow sensed at the distant N protein, destabilizing/removing N to allow RecBCD entry (Fig. 7A). At the same time, Pol III is in position for its leading strand subunit to fill the proximal Mu gap. However, the fork stalls because the transpososome is blocking its path. The Pol III subunit on the lagging strand gets dislodged while remaining attached to the leading strand subunit via the clamp loader (Fig. 7B; the DSE may not be released as yet, but is shown as such to facilitate demonstration of the next step). The dislodged subunit is now available to reload on the target DNA flanking the distal Mu gap, which although 37 kb downstream, is actually proximal to Pol III because of a hairpin bend (140°) in the target DNA held within the transpososome (Montano et al., 2012). As the polymerase moves forward to fill both gaps, the transpososome must be removed because the gaps are protected within it (Lavoie et al., 1991, Mizuuchi et al., 1991). Such a replisome-transpososome partnership ensures that the gaps are not exposed prior to transpososome disassembly, that gap repair is synchronized at both ends, and that gaps are filled without having to replicate the entire length of the intervening Mu DNA (non-replicative transposition). The remaining 4 nt of FD must be trimmed by a nuclease prior to sealing the DNA with ligase. The lagging strand DSE is subsequently repaired by homologous recombination, and the stalled replication fork reinstated by the Restart machinery (Fig. 7B). DSE repair is obligatory for cell viability, since absence of the recombination-restart proteins was seen to lower recovery of Mu insertions by two orders of magnitude (Jang et al., 2012).
Fig. 7. Model depicting how the Mu transpososome first exploits incoming Pol III for just-in-time FD degradation, and next exploits Pol III stalled at the DSB for coordinated repair of both target gaps flanking Mu.
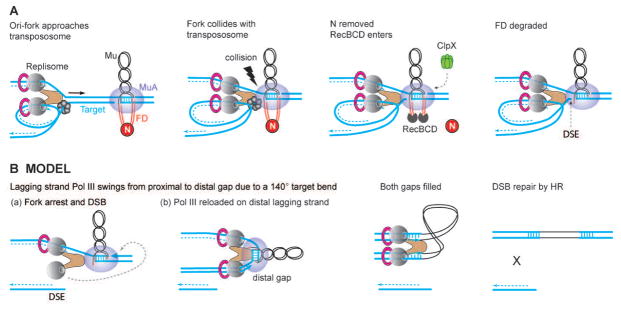
(A) Sequence of repair events as deduced in this, and in prior work (Choi & Harshey, 2010, Choi et al., 2014a). The Mu transpososome does not begin FD repair until the replication fork arrives. Interaction between the two complexes generates a ‘signal’ for N removal and RecBCD entry. The fork stalls because the transpososome is blocking its progress. (B) Scheme for how the DSB and the bent target is exploited for repair of both gaps during non-replicative Mu transposition. E. coli replisomes contain three polymerase molecules (Reyes-Lamothe et al., 2010), but only two are shown for clarity. When the polymerase stalls at the transpososome, the Pol III subunit on the lagging strand (or the extra third subunit) reloads on the distal gap brought into proximity by the hairpin target bend within the transpososome (Montano et al., 2012). As the polymerase moves forward, the transpososome is dislodged and both gaps are filled simultaneously. The DSE may not form until this step, but is shown earlier only to accommodate the target bend. A nuclease must trim the 4 nt flanks, and a ligase seal the remaining nicks. The DSE is repaired by homologous recombination (HR).
The co-ordinate gap repair model we propose, in which Mu is not replicated, is consistent with the observation that Mu is not replicated immediately after integration (Harshey, 1984, Liebart et al., 1982). We note that when the DSE generated at the Mu gap is repaired, and the stalled fork eventually resumes its travel, Mu will be replicated as a normal segment of the chromosome and inherited by both daughter cells. An alternative model where the gaps are not co-ordinately repaired will have to invoke two DSEs at each end: repair-restart at the first DSE, replicate through Mu, repair-restart at the second DSE. The model we propose is parsimonious, reasonable and testable.
Why has Mu evolved an elaborate mechanism for removal and repair of the FD given that it can bypass FD repair to enter lytic growth in DnaA, RecBCD and some MuA mutants (this work and (Choi & Harshey, 2010, Choi et al., 2014a)). We believe that a mechanism limiting Mu replication to short gaps at the Mu ends gives the phage a chance to enter the prophage state.
Repair of other transposon insertions
The short gaps generated in the target are a universal feature of transposition. The 4 nt FD overhangs in Mu are similar to 3 nt or 2 nt overhangs on the 5′ flanking DNA of transposition intermediates of Tn7 and retroviral-like transposons, respectively (Bainton et al., 1991, Craigie, 2002, Sandmeyer et al., 2002). Although generated by different mechanisms in each element, the overhangs will encounter the common fate of being removed during repair of the gaps. Besides a common structure shared by the strand transfer intermediates of Mu and retroviral elements, the transpososomes of these elements also share many structural features, including their extraordinary stability and a bent target conformation (Montano et al., 2012, Maertens et al., 2010). The bend positions a short target DNA segment within two transposase active sites, each of which holds one cleaved 3′OH end of the transposon; nucleophilic attack of the two DNA 3′OHs on target phosphodiester bonds spaced 5 bp apart completes the joining reaction of transposition. This chemistry is shared by all transposons, so it is likely that the target bend is also a shared by all transpososomes (Harshey, 2015). While Mu has an elaborate mechanism to disassemble the transpososome during transition to replication (Fig. S1), the results in this paper suggest that the replisome might participate in transpososome disassembly during non-replicative transposition, which is the most prevalent transposition mechanism. In vitro attempts to recapitulate the gap-filling reaction after retroviral transposition has identified several polymerases, ligase and FEN-1 nuclease, but the in vivo chromatin substrate is substantially different, and the participating enzymes are likely to be different (Daniel, 2006). Interestingly, like with Mu, DSB repair proteins have been implicated in repair of retroviral integration events (Daniel, 2006, Yang et al., 2010). All these shared features hint at a common pathway of gap repair in all transposons.
Perspective
Mu has played a central role in the development of the mobile DNA element field (Harshey, 2012). Pioneering in vitro Mu experiments led to the unraveling of the phosphoryl transfer chemistry for all transposable elements (Mizuuchi & Baker, 2002). High-throughput integration assays modeled after Mu (Craigie et al., 1991), led to the development and marketing of the HIV integrase inhibitor Raltegravir (Summa et al., 2008). We expect our in vivo study on the repair of Mu insertions to provide equally important insights into the long-standing problem of how DNA transposons, retrotransposons, and retroviruses disassemble their highly stable transpososome intermediates and repair their insertions without duplicating themselves in the process, presenting perhaps a new target for drug development.
Beyond the repair of transposition events, our work has implications for understanding how polymerases stalled at DNA damage sites could signal removal of proteins that block repair of DNA lesions such as inter-strand crosslinks (Stingele et al., 2014), or bypass the lesion entirely by re-initiating replication downstream of the damage (Ho & Scharer, 2010, McVey, 2010, Pathania et al., 2011).
Experimental Procedures
Strains and growth conditions
All strains used in this work were derivatives of E. coli K-12 and listed in Table S1. They were propagated in LB media, except for priA and polA mutants, which were grown in 56/2 minimal medium: 0.06 M Na2HPO4, 0.04 M KH2PO4, 0.02% MgSO4·7H20, 0.2% (NH4)2SO4, 0.001% Ca(NO3)2, and 0.00005% FeSO4·7H20, 0.2% glucose, with casamino acids at 50 μg/ml (Willetts et al., 1969). P1 transduction was used to move mutations between strains (Miller, 1992).
Blocking replication in Dnats mutants
For all Dnats mutants, cells grown at 30°C were shifted to 42°C for variable times, depending on the mutation. DnaAts and DnaCts mutants were incubated for 90 min at 42°C to allow ongoing replication to terminate (Wechsler & Gross, 1971), while all other ts mutants were incubated for 30 min at 42°C, because these other mutants stop replication immediately at the non-permissive temperature (McMacken et al., 1987).
Construction of pGam-GFP
Plasmids are listed in Table S2 and oligonucleotide primers in Table S3. The Gam-GFP fusion was amplified from the genome of strain SMR14334 using primers P1 and P2. The ~ 1.2 kb Gam-GFP product was digested by Sal I and Xba I and ligated into the same enzyme sites of the vector pRHA-113, where it was placed under the control of the rhamnose-inducible promoter. Similarly, a GFP-only control was amplified from same strain SMR14334 using primers P2 and P3. The resulting plasmids pGam-GFP and pGFP were verified by sequencing.
Construction of prophage Mu::TetO
The Mu lysogen strain HM8305 was used to construct prophage Mu::TetO (SJ012) as follows. First, kan was introduced next to the TetO array in pRS306X112TetO at the SalI-SpeI sites (using primers P4/P5) to give pTetO(kan). pMuHF was then constructed to introduce ~500 bp homology corresponding to either side of the SE (semi-essential) region (primers P6/P7 and P8/P9) in the prophage into which TetOkan was to be substituted. Next, TetOkan was cloned within the two arms of the SE region in pMuHF to give pTetO(kan)-MuHF. This plasmid was digested with SalI and recombined into the Mu prophage to produce a deletion of the SE (~3.6 kb; 4319 bp to 7953 bp on the Mu genome) concomitant with a substitution (~6kb) with TetOkan, using λ-Red recombination (Datsenko & Wanner, 2000). The kan cassette was removed using Flp recombinase from pCP20. Finally, a non-essential region spanning ~2.4 kb at the right end of Mu (33,883 to 36,300 bp) was deleted by substitution with a kan cassette (primers P10 and P11), followed by removal of kan as before. The latter deletion was required to maintain a genome length that could be packaged into viable phage.
Phage preparation
Wild-type Mu prophage inserted into lacZ gene in strain HM8305 carries a temperature sensitive (ts) allele of the lysogenic repressor and is induced by repressor inactivation at a high temperature as described (Au et al., 2006). Phage from a 1 liter culture were precipitated with PEG 8000 and concentrated on a CsCl density gradient. Final concentration of purified phage was ~1011 pfu/ml, estimated by plating on BW25113. Phage from the Mu::TetO lysogen were prepared in a smaller 50 ml culture and not concentrated on CsCl gradient. Instead, when cells reached an OD600 of 0.4, they were concentrated by pelleting and resuspensing in 10 ml of pre-warmed LB and induced as described above. Phage titers were ~109 pfu/ml.
Phage infection and purification of chromosomal DNA
200 μl of overnight cultures were transferred into 20 ml LB media (5 mM CaCl2 and 5 mM MgSO4) and grown till an OD600 of ~ 0.3–0.4. All infections with wild type Mu phage were at MOI = 5 at either 30°C, 37°C or 42°C. Mu infections in the Dnats strains were carried out at 42°C. At various times after infection, total DNA was extracted, subjected to pulse field gel electrophoresis, and the gDNA band isolated as described (Au et al., 2006).
Detecting integrated Mu and its flanking DNA in the E. coli genome by PCR
Mu and flanking DNA (FD) sequences inserted into the E. coli genome were amplified by standard PCR as described, and the products visualized on 1% agarose gels after staining with ethidium bromide (Choi & Harshey, 2010). Mu integration was detected by PCR using primers within the MuB gene, and FD DNA by primers that amplified the junction between right end of Mu and lacZ (Table S3). PCR was performed with 50–100 ng of template DNA, 10 pmol of primers, 10 μmol of deoxynucleoside triphosphates, 2.5 units of Taq polymerase (Qiagen), 1x PCR buffer, and 1x Q solution in 25 μl. The PCR conditions were 94°C for 2 min; 30–40 cycles of 94°C for 30 s, 62°C for 30 s, and 72°C for 30 s; and a final extension at 72°C for 7 min as described. SS996 derived host strains include a partial lacZ gene, but this did not interfere with the assay because amplification was based on a second primer annealing inside Mu. Real-time or qPCR assays, were programmed in the ViiA7 sequence detector (Applied Biosystems) and the level of integrated Mu DNA was normalized to a chromosomal locus of 16S rRNA.
Iterative primer extension (IPE) assay
This assay has been described (Pato, 2004). One-directional PCR was performed with 1μg of total genomic DNA as a template, 5 pmol of a single primer which was 5′end-labeled with γ-32P, Taq 2X master mix solution (Promega). The primer was extended as follows: 94°C for 2 min; 60 cycles of 94°C for 30 s, 55°C for 30 s, and 68°C for 20 s; and a final extension at 72 for 7 min. PCR products were separated on a 10% polyacrylamide sequencing gel and quantified with a phosphorimager, Typhoon 9500 (GE Life Science).
Flow cytometry
Flow cytometry was used to determine the number of chromosomal origins per cell. 1 ml culture samples were fixed by adding 9 ml of 95% ethanol and maintained at 4°C. These cultures were centrifuged at 6000g for 10 min in 4°C and the cell pellet was washed with and resuspended in TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0). Cells were diluted to OD600 of 0.1 and stained by addition of 5 μM SYTOX Green (Invitrogen) by incubating in the dark for 15 min. A total of ~20,000 viable cells were sorted in a BD Accuri™ flow cytometer, and the data were analyzed by FlowJo software.
Measuring the SOS response using GFP
This assay measures GFP expression from the promoter of the SOS-induced gene sulA (PsulA-gfp; (McCool et al., 2004). Mu infections were carried out either in WT (SS996), or its lexA3 (SS4294) and lexA71 (SS4610) derivatives. lexA3 is defective for SOS induction, while lexA71 is constitutively induced. 30 min after Mu infection, cells were harvested and diluted 1:100 into TE buffer. Cells were sorted in the BD Accuri™ flow cytometer and analyzed as described above.
Visualizing Mu and DSBs in vivo
Mu::TetO location was visualized by expressing TetR-mCherry from pDB317 using 100 μM sodium salicylate. Double strand breaks (DSBs) were assayed by appearance of either Gam-GFP foci (Shee et al., 2013) or RecA-GFP foci (Renzette et al., 2005). Gam-GFP expression protocol was modified slightly from that of Shee et al. who expressed it for 4 hr prior to visualization (Shee et al., 2013). Prolonged expression of Gam-GFP affects cell viability (Fig. S5). Therefore, expression was controlled by the tightly regulated rhamnose-inducible promoter in pGam-GFP by repressing it with 0.2% glucose until needed. 500 μM rhamnose was added only 30 min before Mu infection. RecA-GFP is expressed constitutively from its normal chromosomal location in the recA-gfp strain (SJ006).
Mu::TetO infection was carried out at an MOI of 1 in host strains already expressing TetR-mCherry. 15 min after infection, cells were placed onto 1% agarose pads at room temperature as described(Skinner et al., 2013), and visualized within 5–10 min with an Olympus BX53 fluorescence microscope. Images were captured using cellSens standard software (version 1.6) from Olympus.
Western blot
After Mu infection for 30 min, 5 × 108 cells were harvested and lysed in lysis buffer (50 mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol, 1% β-mercaptoethanol, 12.5 mM EDTA and 0.02 % bromophenol blue). After boiling at 95°C for 5 min, samples were applied to 10% SDS-PAGE gel, transferred to a PVDF membrane (Bio-Rad) after electrophoresis, and the blot was probed with a polyclonal anti-MuB antibody (Parsons & Harshey, 1988), followed by HRP-conjugated goat anti-rabbit IgG (Bio-Rad), and detected using ECL western blotting analysis reagents (GE Healthcare) (Ausubel & al, 2003).
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health grant GM33247, and in part by the Robert Welch Foundation grant F-1351. We thank Susan Rosenberg, Steven Sandler and James Walker for strains, and Makkuni Jayaram, Benedicte Michel and David Walker for discussions.
References
- Akroyd JE, Symonds N. Evidence for a conservative pathway of transposition of bacteriophage Mu. Nature. 1983;303:84–86. doi: 10.1038/303084a0. [DOI] [PubMed] [Google Scholar]
- Au TK, Agrawal P, Harshey RM. Chromosomal integration mechanism of infecting Mu virion DNA. J Bacteriol. 2006;188:1829–1834. doi: 10.1128/JB.188.5.1829-1834.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel FM, et al. Current Protocols in Molecular Biology. Jon Wiley & Sons Inc; New York, NY: 2003. [Google Scholar]
- Bainton R, Gamas P, Craig NL. Tn7 transposition in vitro proceeds through an excised transposon intermediate generated by staggered breaks in DNA. Cell. 1991;65:805–816. doi: 10.1016/0092-8674(91)90388-f. [DOI] [PubMed] [Google Scholar]
- Chaconas G, Harshey RM. Transposition of phage Mu DNA. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. Washington DC: ASM Press; 2002. pp. 384–402. [Google Scholar]
- Chaconas G, Kennedy DL, Evans D. Predominant integration end products of infecting bacteriophage Mu DNA are simple insertions with no preference for integration of either Mu DNA strand. Virology. 1983;128:48–59. doi: 10.1016/0042-6822(83)90317-3. [DOI] [PubMed] [Google Scholar]
- Chenais B. Transposable elements and human cancer: a causal relationship? Biochim Biophys Acta. 2013;1835:28–35. doi: 10.1016/j.bbcan.2012.09.001. [DOI] [PubMed] [Google Scholar]
- Choi W, Harshey M. DNA repair by the cryptic endonuclease activity of Mu transposase. Proc Natl Acad Sci U S A. 2010;107:10014–10019. doi: 10.1073/pnas.0912615107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W, Jang S, Harshey RM. Mu transpososome and RecBCD nuclease collaborate in the repair of simple Mu insertions. Proc Natl Acad Sci U S A. 2014a;111:14112–14117. doi: 10.1073/pnas.1407562111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W, Saha RP, Jang S, Harshey RM. Controlling DNA degradation from a distance: a new role for the Mu transposition enhancer. Mol Microbiol. 2014b;94:595–608. doi: 10.1111/mmi.12781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S, Helmstetter CE. Chromosome replication and the division cycle of Escherichia coli B/r. J Mol Biol. 1968;31:519–540. doi: 10.1016/0022-2836(68)90425-7. [DOI] [PubMed] [Google Scholar]
- Cox MM. Recombinational DNA repair of damaged replication forks in Escherichia coli: questions. Annu Rev Genet. 2001;35:53–82. doi: 10.1146/annurev.genet.35.102401.090016. [DOI] [PubMed] [Google Scholar]
- Craig NL, Craigie R, Gellert M, Lambowitz A. Mobile DNA II. ASM Press; Washington, D.C: 2002. [Google Scholar]
- Craigie R. Retroviral DNA integration. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. Washington, DC: ASM Press; 2002. pp. 613–630. [Google Scholar]
- Craigie R, Mizuuchi K, Bushman FD, Engelman A. A rapid in vitro assay for HIV DNA integration. Nucleic Acids Res. 1991;19:2729–2734. doi: 10.1093/nar/19.10.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Adda di Fagagna F, Weller GR, Doherty AJ, Jackson SP. The Gam protein of bacteriophage Mu is an orthologue of eukaryotic Ku. EMBO Rep. 2003;4:47–52. doi: 10.1038/sj.embor.embor709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel R. DNA repair in HIV-1 infection: a case for inhibitors of cellular co-factors? Current HIV research. 2006;4:411–421. doi: 10.2174/157016206778560027. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillingham MS, Kowalczykowski SC. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol Mol Biol Rev. 2008;72:642–671. doi: 10.1128/MMBR.00020-08. Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijalkowska IJ, Schaaper RM, Jonczyk P. DNA replication fidelity in Escherichia coli: a multi-DNA polymerase affair. FEMS Microbiol Rev. 2012;36:1105–1121. doi: 10.1111/j.1574-6976.2012.00338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbai CB, Marians KJ. Recruitment to stalled replication forks of the PriA DNA helicase and replisome-loading activities is essential for survival. DNA repair. 2010;9:202–209. doi: 10.1016/j.dnarep.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Lou Z, Cui H, Shang L, Harshey RM. Analysis of phage Mu DNA transposition by whole-genome Escherichia coli tiling arrays reveals a complex relationship to distribution of target selection protein B, transcription and chromosome architectural elements. J Biosci. 2011;36:587–601. doi: 10.1007/s12038-011-9108-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloor G, Chaconas G. The bacteriophage Mu N gene encodes the 64-kDa virion protein which is injected with, and circularizes, infecting Mu DNA. J Biol Chem. 1986;261:16682–16688. [PubMed] [Google Scholar]
- Harshey RM. Transposition without duplication of infecting bacteriophage Mu DNA. Nature. 1984;311:580–581. doi: 10.1038/311580a0. [DOI] [PubMed] [Google Scholar]
- Harshey RM. The Mu story: how a maverick phage moved the field forward. Mob DNA. 2012;3:21. doi: 10.1186/1759-8753-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harshey RM. Transposable phage Mu. In: Chandler M, Craig NL, editors. Mobile DNA III and microbiolspec. 5. Vol. 2. Washington, D.C: ASM press; 2015. doi:10.1128/microbiolspec MDNA3-0007-2014. pp. In press. [Google Scholar]
- Harshey RM, Bukhari AI. Infecting bacteriophage Mu DNA forms a circular DNA-protein complex. J Mol Biol. 1983;167:427–441. doi: 10.1016/s0022-2836(83)80343-x. [DOI] [PubMed] [Google Scholar]
- Ho TV, Scharer OD. Translesion DNA synthesis polymerases in DNA interstrand crosslink repair. Environmental and molecular mutagenesis. 2010;51:552–566. doi: 10.1002/em.20573. [DOI] [PubMed] [Google Scholar]
- Huang CR, Burns KH, Boeke JD. Active transposition in genomes. Annu Rev Genet. 2012;46:651–675. doi: 10.1146/annurev-genet-110711-155616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S, Sandler SJ, Harshey RM. Mu insertions are repaired by the double-strand break repair pathway of Escherichia coli. PLoS Genet. 2012;8:e1002642. doi: 10.1371/journal.pgen.1002642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH., Jr An estimated frequency of endogenous insertional mutations in humans. Nat Genet. 1999;22:130. doi: 10.1038/9638. [DOI] [PubMed] [Google Scholar]
- Kazazian HH., Jr Mobile elements: drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- Kogoma T. Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol Mol Biol Rev. 1997;61:212–238. doi: 10.1128/mmbr.61.2.212-238.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg A, Baker TA. DNA Replication. W. H. Freeman & Co; 1992. [Google Scholar]
- Kuzminov A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev. 1999;63:751–813. doi: 10.1128/mmbr.63.4.751-813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie BD, Chan BS, Allison RG, Chaconas G. Structural aspects of a higher order nucleoprotein complex: induction of an altered DNA structure at the Mu-host junction of the Mu type 1 transpososome. EMBO J. 1991;10:3051–3059. doi: 10.1002/j.1460-2075.1991.tb07856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebart JC, Ghelardini P, Paolozzi L. Conservative integration of bacteriophage Mu DNA into pBR322 plasmid. Proc Natl Acad Sci U S A. 1982;79:4362–4366. doi: 10.1073/pnas.79.14.4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusetti SL, Cox MM. The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annu Rev Biochem. 2002;71:71–100. doi: 10.1146/annurev.biochem.71.083101.133940. [DOI] [PubMed] [Google Scholar]
- Maduike NZ, Tehranchi AK, Wang JD, Kreuzer KN. Replication of the Escherichia coli chromosome in RNase HI-deficient cells: multiple initiation regions and fork dynamics. Mol Microbiol. 2014;91:39–56. doi: 10.1111/mmi.12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens GN, Hare S, Cherepanov P. The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature. 2010;468:326–329. doi: 10.1038/nature09517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna D, Breier AM, Higgins NP. Microarray analysis of transposition targets in Escherichia coli: the impact of transcription. Proc Natl Acad Sci U S A. 2004;101:9780–9785. doi: 10.1073/pnas.0400745101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massoni SC, Sandler SJ. Specificity in suppression of SOS expression by recA4162 and uvrD303. DNA repair. 2013;12:1072–1080. doi: 10.1016/j.dnarep.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBeth DL, Taylor AL. Growth of bacteriophage Mu in Escherichia coli dnaA mutants. J Virol. 1982;44:555–564. doi: 10.1128/jvi.44.2.555-564.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCool JD, Long E, Petrosino JF, Sandler HA, Rosenberg SM, Sandler SJ. Measurement of SOS expression in individual Escherichia coli K-12 cells using fluorescence microscopy. Mol Microbiol. 2004;53:1343–1357. doi: 10.1111/j.1365-2958.2004.04225.x. [DOI] [PubMed] [Google Scholar]
- McMacken R, Silver L, Georgopoulos C. DNA replication. In: Escherichia coli and Salmonella typhimurium. In: Neidhardt FC, editor. Cellular and Molecular Biology. Washington D.C: ASM Press; 1987. pp. 564–612. [Google Scholar]
- McVey M. Strategies for DNA interstrand crosslink repair: insights from worms, flies, frogs, and slime molds. Environmental and molecular mutagenesis. 2010;51:646–658. doi: 10.1002/em.20551. [DOI] [PubMed] [Google Scholar]
- Michel B, Boubakri H, Baharoglu Z, LeMasson M, Lestini R. Recombination proteins and rescue of arrested replication forks. DNA repair. 2007;6:967–980. doi: 10.1016/j.dnarep.2007.02.016. [DOI] [PubMed] [Google Scholar]
- Miller JH. A short course in bacterial genetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1992. [Google Scholar]
- Mills RE, Bennett EA, Iskow RC, Devine SE. Which transposable elements are active in the human genome? Trends Genet. 2007;23:183–191. doi: 10.1016/j.tig.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Mizuuchi K. Transpositional recombination: mechanistic insights from studies of Mu and other elements. Annu Rev Biochem. 1992;61:1011–1051. doi: 10.1146/annurev.bi.61.070192.005051. [DOI] [PubMed] [Google Scholar]
- Mizuuchi K, Baker TA. Chemical mechanisms for mobilizing DNA. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. Washington DC: ASM Press; 2002. pp. 12–23. [Google Scholar]
- Mizuuchi M, Baker TA, Mizuuchi K. DNase protection analysis of the stable synaptic complexes involved in Mu transposition. Proc Natl Acad Sci U S A. 1991;88:9031–9035. doi: 10.1073/pnas.88.20.9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montano SP, Pigli YZ, Rice PA. The Mu transpososome structure sheds light on DDE recombinase evolution. Nature. 2012;491:413–417. doi: 10.1038/nature11602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai H, Doseeva V, Jones JM. Handoff from recombinase to replisome: insights from transposition. Proc Natl Acad Sci U S A. 2001;98:8247–8254. doi: 10.1073/pnas.111007898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai H, Taylor AL. Host DNA replication forks are not preferred targets for bacteriophage Mu transposition. J Bacteriol. 1985;163:282–290. doi: 10.1128/jb.163.1.282-290.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons RL, Harshey R. Autoregulation of phage Mu transposase at the level of translation. Nuc Acids Res. 1988;16:11285–11301. doi: 10.1093/nar/16.23.11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania S, Nguyen J, Hill SJ, Scully R, Adelmant GO, Marto JA, Feunteun J, Livingston DM. BRCA1 is required for postreplication repair after UV-induced DNA damage. Mol Cell. 2011;44:235–251. doi: 10.1016/j.molcel.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pato ML. Replication of Mu prophages lacking the central strong gyrase site. Res Microbiol. 2004;155:553–558. doi: 10.1016/j.resmic.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Perrat PN, DasGupta S, Wang J, Theurkauf W, Weng Z, Rosbash M, Waddell S. Transposition-driven genomic heterogeneity in the Drosophila brain. Science. 2013;340:91–95. doi: 10.1126/science.1231965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puspurs AH, Trun NJ, Reeve JN. Bacteriophage Mu DNA circularizes following infection of Escherichia coli. EMBO J. 1983;2:345–352. doi: 10.1002/j.1460-2075.1983.tb01429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzette N, Gumlaw N, Nordman JT, Krieger M, Yeh SP, Long E, Centore R, Boonsombat R, Sandler SJ. Localization of RecA in Escherichia coli K-12 using RecA-GFP. Mol Microbiol. 2005;57:1074–1085. doi: 10.1111/j.1365-2958.2005.04755.x. [DOI] [PubMed] [Google Scholar]
- Reyes-Lamothe R, Sherratt DJ, Leake MC. Stoichiometry and architecture of active DNA replication machinery in Escherichia coli. Science. 2010;328:498–501. doi: 10.1126/science.1185757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandmeyer SB, Aye M, Menees T. Ty3, a position-specific, gypsy-like element in Saccharomyces cerevisiae. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. Washington, DC: ASM Press; 2002. pp. 663–683. [Google Scholar]
- Sasakawa C, Uno Y, Yoshikawa M. The requirement for both DNA polymerase and 5′ to 3′ exonuclease activities of DNA polymerase I during Tn5 transposition. Mol Gen Genet. 1981;182:19–24. doi: 10.1007/BF00422761. [DOI] [PubMed] [Google Scholar]
- Shee C, Cox BD, Gu F, Luengas EM, Joshi MC, Chiu LY, Magnan D, Halliday JA, Frisch RL, Gibson JL, Nehring RB, Do HG, Hernandez M, Li L, Herman C, Hastings P, Bates D, Harris RS, Miller KM, Rosenberg SM. Engineered proteins detect spontaneous DNA breakage in human and bacterial cells. eLife. 2013;2:e01222. doi: 10.7554/eLife.01222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer T, McConnell MJ, Marchetto MC, Coufal NG, Gage FH. LINE-1 retrotransposons: mediators of somatic variation in neuronal genomes? Trends in neurosciences. 2010;33:345–354. doi: 10.1016/j.tins.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner SO, Sepulveda LA, Xu H, Golding I. Measuring mRNA copy number in individual Escherichia coli cells using single-molecule fluorescent in situ hybridization. Nat Protoc. 2013;8:1100–1113. doi: 10.1038/nprot.2013.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliusarenko O, Heinritz J, Emonet T, Jacobs-Wagner C. High-throughput, subpixel precision analysis of bacterial morphogenesis and intracellular spatio-temporal dynamics. Mol Microbiol. 2011;80:612–627. doi: 10.1111/j.1365-2958.2011.07579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J, Schwarz MS, Bloemeke N, Wolf PG, Jentsch S. A DNA-dependent protease involved in DNA-protein crosslink repair. Cell. 2014;158:327–338. doi: 10.1016/j.cell.2014.04.053. [DOI] [PubMed] [Google Scholar]
- Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Gonzalez Paz O, Hazuda DJ, Jones P, Kinzel O, Laufer R, Monteagudo E, Muraglia E, Nizi E, Orvieto F, Pace P, Pescatore G, Scarpelli R, Stillmock K, Witmer MV, Rowley M. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. Journal of medicinal chemistry. 2008;51:5843–5855. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- Surette MG, Buch SJ, Chaconas G. Transpososomes: stable protein-DNA complexes involved in the in vitro transposition of bacteriophage Mu DNA. Cell. 1987;49:253–262. doi: 10.1016/0092-8674(87)90566-6. [DOI] [PubMed] [Google Scholar]
- Symonds N, Toussaint A, Van de Putte P, Howe MM. Phage Mu. Cold Spring Harbor Laboratory; Cold Spring Harbor, New York: 1987. [Google Scholar]
- Syvanen M, Hopkins JD, Clements M. A new class of mutants in DNA polymerase I that affects gene transposition. J Mol Biol. 1982;158:203–212. doi: 10.1016/0022-2836(82)90429-6. [DOI] [PubMed] [Google Scholar]
- Toussaint A, Faelen M. The dependence of temperate phage Mu-1 upon replication functions of E. coli K12. Mol Gen Genet. 1974;131:209–214. doi: 10.1007/BF00267960. [DOI] [PubMed] [Google Scholar]
- Wechsler JA, Gross JD. Escherichia coli mutants temperature-sensitive for DNA synthesis. Mol Gen Genet. 1971;113:273–284. doi: 10.1007/BF00339547. [DOI] [PubMed] [Google Scholar]
- Willetts NS, Clark AJ, Low B. Genetic location of certain mutations conferring recombination deficiency in Escherichia coli. J Bacteriol. 1969;97:244–249. doi: 10.1128/jb.97.1.244-249.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YX, Guen V, Richard J, Cohen EA, Berthoux L. Cell context-dependent involvement of ATR in early stages of retroviral replication. Virology. 2010;396:272–279. doi: 10.1016/j.virol.2009.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder KE, Bushman FD. Repair of gaps in retroviral DNA integration intermediates. J Virol. 2000;74:11191–11200. doi: 10.1128/jvi.74.23.11191-11200.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.