

Summary
The G protein-coupled P2Y12 receptor (P2Y12R) is an important antithrombotic target and of great interest for pharmaceutical discovery. Its recently solved, highly divergent crystallographic structures in complex either with nucleotides (full or partial agonist) or with a nonnucleotide antagonist raise the question of which structure is more useful to understand ligand recognition. Therefore, we performed extensive molecular modeling studies based on these structures and mutagenesis, to predict the binding modes of major classes of P2Y12R ligands previously reported. Various nucleotide derivatives docked readily to the agonist-bound P2Y12R, but uncharged nucleotide-like antagonist ticagrelor required a hybrid receptor resembling the agonist-bound P2Y12R except for the top portion of TM6. Supervised molecular dynamics (SuMD) of ticagrelor binding indicated interactions with the extracellular regions of P2Y12R, defining possible meta-binding sites. Ureas, sulfonylureas, sulfonamides, anthraquinones and glutamic acid piperazines docked readily to the antagonist-bound P2Y12R. Docking dinucleotides at both agonist- and antagonist-bound structures suggested interactions with two P2Y12R pockets. Thus, our structure-based approach consistently rationalized the main structure-activity relationships within each ligand class, giving useful information for designing improved ligands.
Keywords: G protein-coupled receptors, purines, molecular modeling, nucleotides, structure-activity relationship, X-ray crystallographic structures
Graphical Abstract

Introduction
P2Y receptors (P2YRs) are a family of purinergic G protein-coupled receptors (GPCRs), activated by endogenous nucleotides such as ADP, ATP, UDP, UTP and UDP-glucose [1]. Based on phylogenetic proximity, sequence similarity and G-protein coupling, P2Y receptors can be divided into two subgroups: P2Y1-like (including P2Y1,2,4,6,11 subtypes) and P2Y12-like (including P2Y12,13,14 subtypes). Since purinergic signaling is an evolutionarily early molecular messenger system, these receptors are involved in numerous pathophysiological processes [2] and are widely distributed in both neuronal and non-neuronal tissues, making them interesting as potential drug targets.
The Gi-coupled P2Y12 receptor (P2Y12R), which responds to ADP as an endogenous agonist, was cloned in 2001 [3], but drugs targeting this receptor were developed before their actual site of action was known. P2Y12 receptor (P2Y12R) plays a major role in platelet aggregation, and antagonists of this receptor are useful as antithrombotic agents for the prevention of myocardial infarction and stroke [1, 4]. In particular, three P2Y12R antagonists are currently on the market, and one of these (clopidogrel) has been among the world’s best-selling drugs in the recent years [5, 6]. Another recently reported potential application for P2Y12R antagonists is for the treatment of inflammatory and neuropathic pain, through blockade of the receptor in the CNS [7].
Various P2YRs still lack pharmacological tool compounds, such as potent and selective synthetic agonists and antagonists. However, considerable progress has been made in exploring the structure-activity relationship (SAR) of antagonists for the P2Y12 subtype [1, 5]. P2Y12R agonists are essentially nucleotide derivatives [5, 8, 9] (compounds 1–8, Chart 1), while several different chemical classes have shown P2Y12 antagonist activity, including: ureas, sulfonylureas and sulfonamides [10–14]; anthraquinones [15]; glutamic acid piperazines [16–20]; nucleotide and nucleoside derivatives [21–28] and dinucleotides [29–31] (compounds 9–55, Chart 2, Table 1). The currently used antithrombotic drugs clopidogrel (56, Plavix) and prasugrel (57, Effient) (Chart 3) belong to the thienopyridine class of P2Y12 antagonists, which require metabolic activation in vivo prior to binding covalently to the receptor to induce an irreversible inhibition. Ticagrelor (47, Brilinta), developed through molecular optimization of nucleotide ligands, was the first reversible P2Y12 antagonist approved for clinical use [32]. The pharmacological action of some P2Y12R ligands, such as nucleoside 5′-triphosphates and dinucleotides, is still ambiguous, because they can behave as agonists, partial agonists or antagonists depending on structural features, the system under study and the receptor expression level [33–35]. ATP itself is a P2Y12R agonist [34], but its derivatives can have reduced efficacy.
Chart 1.

Structures of representative nucleotides as P2Y12R agonists and partial agonists (compounds 1–8).
Chart 2.

Structures of representative, reversible P2Y12R antagonists (nonnucleotides 9–41 and nucleotide-like 42–55) belonging to different chemical classes. Substituents, references and available pharmacological data at the hP2Y12R are reported in Table 1.
Table 1.
| Compound | Substitution | Pharmacological potency (nM) | |||
|---|---|---|---|---|---|
| R | R1 | R2 | R3 | IC50 | |
| 9 | Et | CF3 | CN | Ph | 320 |
| 10 | Et | H | Cl | Ph | 880 |
| 11 | Me | H | Cl | Ph | 9800 |
| 12 | i-Pr | H | Cl | Ph | 2400 |
| 13 | Et | CF3 | CN | 2-naphthyl | 190 |
| 14 | Et | H | Cl | Ph | 180 |
| 15 | Et | H | Cl | 5-Cl-2-thienyl | 90 |
| 16 | Me | H | Cl | 5-Cl-2-thienyl | 650 |
| 17 | Et | Me | CN | 5-Cl-2-thienyl | 6 |
| 18 | Et | Me | CN | Ph | 62 |
| 19 | - | - | - | benzyl | 11 |
| 20 | - | - | - | Ph | 120 |
| 21 | Me | H | Cl | 5-Cl-2-thienyl | 320 |
| 22 | Et | Me | CN | 5-Cl-2-thienyl | 97 |
| R | R1 | X | Y | IC50 | |
| 23 | Pr | Me | CH | CH | 27 |
| 24 | Et | Me | CH | CH | 336 |
| 25 | Pr | (CH2)2OH | CH | CH | 11 |
| 26 | Pr | Me | N | N | 17 |
| R | R1 | X | Ki | ||
| 27 | SO3Na | SO3Na | NH | 24 | |
| 28 | Me | SO3Na | NH | 540 | |
| 29 | SO3Na | H | NH | 1850 | |
| 30 | COOH | SO3Na | NH | 21 | |
| 31 | SO3Na | SO3Na | O | 63 | |
| R | R1 | X | Y | Ki | |
| 32 | Et | Ph | CH | CH | 15 |
| 33 | Et | H | CH | CH | 209 |
| 34 | Hept | H | CH | CH | 7 |
| 35 | Pent | piperidin-4-yloxy | CH | CH | 3.5 |
| 36 | Pent | O(CH2)2NEt2 | CH | N | 27 |
| 37 | Et | (CH2)2COOH | 7.3 | ||
| 38 | Bu | (CH2)2COOH | 0.8 | ||
| 39 | Et | (CH2)2OH | 23 | ||
| 40 | Et | Me | 111 | ||
| 41 | Bu | H | 7.7 | ||
| R | R1 | IC50 | |||
| 42 | SPr | H | 2.5 | ||
| 43 | S(CH2)2CF3 | (CH2)2SMe | 0.45 | ||
| R | R1 | Ki | |||
| 44 | COOH | butyl | 5.0 | ||
| 45 | COOH | (1R,2S)-2-phenylcyclo-propyl | 0.25 | ||
| 46 | CH2OH | (1R,2S)-2-phenylcyclo-propyl | 5.0 | ||
| 47 | O(CH2)2OH | (1R,2S)-2-(3,4-diF-phenyl)cyclo-propyl | 2.0 | ||
| R | R1 | IC50 | |||
| 48 | 1H-tetrazol-5-yl | (1R,2S)-2-phenylcyclo-propyl | 2 | ||
| X | Y | Z | IC50 | ||
| 49 | O | O | O | 7900 | |
| 50 | S | S | O | 2800 | |
| 51 | O | O | CHCl | 1300 | |
| 52 | S | O | CHCl | 660 | |
| 53 | S | S | CHCl | 120 | |
| 54 | O | O | CHF | 4000 | |
| 55 | O | O | CF2 | 6000 | |
Compounds 9–18, 21 and 22: radioligand binding assays with [125I]AZ11931285 and human platelet membranes [11–13]. Compounds 19 and 20: radioligand binding assays with [125I]AZ11931285 and hP2Y12R-transfected CHO-K1 cell membranes [12]. Compounds 23–26: radioligand binding assays with [33P]2MeSADP and P2Y12R-expressing CHO cells [43]. Compounds 27–31: radioligand binding assays with [3H]PSB-0413 and human platelet membranes [15]. Compounds 32–41: radioligand binding assays with [33P]2MeSADP and hP2Y12R-transfected CHO cell membranes [16, 18, 20]. Compounds 44–47: radioligand binding assays with [125I] antagonist of the P2Y12R and washed human platelets [23]. Compound 48: radioligand binding assays with [3H]2MeSADP and hP2Y12R-expressing cells [24]. Compounds 42, 43 and 49–55: inhibition of ADP-induced human platelet aggregation [21, 29, 30].
Chart 3.
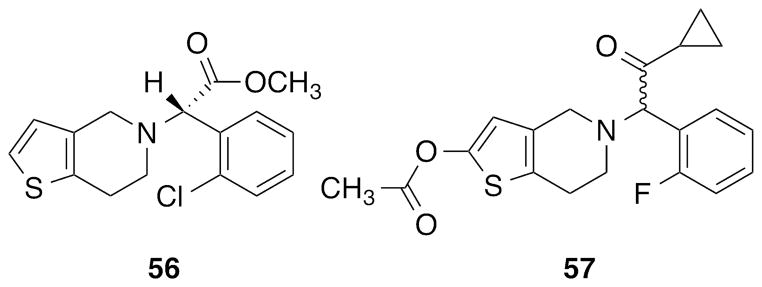
Structures of two irreversible P2Y12R antagonist prodrugs (thienopyridines 56 and 57).
Due to the great interest in the P2Y12R as a therapeutic target, previously only computational approaches could be applied to modeling its structure for use in SAR rationalization and rational drug design [36]. However, very recently the resolution of the crystallographic structures of the P2Y12R in complex with the full agonist 2-methylthio-adenosine-5′-diphosphate (2MeSADP, 3; PDB ID: 4PXZ) and partial agonist 2-methylthio-adenosine-5′-triphosphate (2MeSATP, 7; PDB ID: 4PY0) [37] and with the antagonist ethyl 6-(4-((benzylsulfonyl)carbamoyl)piperidin-1-yl)-5-cyano-2-methylnicotinate (AZD1283, 19; PDB ID: 4NTJ) [38] revealed atypical features of this receptor compared to other GPCR structures. These features, including the lack of a ‘conserved’ disulfide bridge in the antagonist-bound state, would have been impossible to predict by the previous modeling approaches. Therefore, there is now a need to review previous findings in light of this new structural information to gain a better understanding of recognition at this receptor. In this study, based on the newly solved P2Y12R crystallographic structures, we model the binding of most of the major classes of P2Y12R ligands reported. Due to the heterogeneity of binding and functional data found in the literature, our study is not intended to explain subtle differences in binding affinities among all the ligands but to identify a reasonable binding mode for each family of compounds. Thus, it is now possible to rationalize the main SAR findings previously reported for each ligand class, in order to facilitate the design of new and improved compounds. Insights obtained from molecular modeling analysis for one compound class, i.e. anthraquinones, are supported through mutagenesis studies.
Results
Comparison of P2Y12R Crystallographic Structures
We began by analyzing the available P2Y12R crystallographic structures, mainly focusing on the complexes with 3 (designated here as agonist-bound P2Y12R structure, resolution 2.50 Å) and with 19 (designated here as antagonist-bound P2Y12R structure, resolution 2.62 Å). The P2Y12R complex with nucleotide 7 was not included because it is very similar to the 3-P2Y12R complex, but is of lower resolution (3.10 Å). Crystallographic data revealed two very different conformations for the P2Y12R when bound to an agonist or an antagonist (Figures 4 and S1, Supporting Information), with the largest conformational change occurring in transmembrane helices (TMs) 6 and 7. In particular, the inward shift of TM6 and TM7 observed in the agonist and partial agonist complexes allows formation of an extensive ionic and polar interaction network with the phosphate groups of the nucleotides. On the other hand, the phenyl group of 19 is accommodated in a hydrophobic pocket between TMs 6 and 7 preventing the inward movement of these two helices and stabilizing an open conformation of the receptor. Another striking difference is that the disulfide bond connecting TM3 with the second extracellular loop (EL2), highly conserved among family A GPCRs, is clearly observed in the agonist-bound P2Y12R structure (Cys97(3.25)-Cys175(EL2)), while it is not resolved with the antagonist bound. Overall, the binding site is contracted in the agonist-bound structure as compared to the antagonist-bound structure; in the latter complex ELs are mainly unresolved, but they are closely associated with the nucleotide ligand in the agonist-bound structure. However, in both cases the binding cavity has a peculiar bifurcated shape that generates two pockets (Figure S1, Supporting Information). The first pocket, where both compounds 3 and 19 bind, is delimited by TMs 3, 4, 5, 6 and 7, while the second, empty pocket in both crystal structures, is delimited by TMs 1, 2, 3 and 7.
Figure 4.

Docking of anthraquinone and glutamic acid piperazine derivatives to the antagonist-bound P2Y12R crystal structure. (A) Side view of the docking pose of compound 27 (magenta carbon sticks) and crystal pose of compound 19 (green carbon sticks) at the antagonist-bound P2Y12R structure. (B) Side view of the docking poses of compounds 32 (magenta carbon sticks) and 41 (cyan carbon sticks) at the antagonist-bound P2Y12R crystal structure. Side chains of residues important for ligand recognition are shown in thin sticks (grey carbons), and red dashed lines indicate polar interactions formed by the docked compounds. Non-polar hydrogen atoms are not displayed.
Despite these large differences in receptor conformation between the two complexes, the binding poses of the two ligands partially overlay and show some conserved interactions (Figure 1). Both the nicotinate group of 19 and the adenine moiety of 3 are stabilized by a π-π stacking interaction with Tyr105 (3.33). The 2-thioether of the agonist and the ethyl ester of the antagonist inserts into the same side pocket delimited by Phe106 (3.34), Met152 (4.53), Leu155 (4.56), Ser156 (4.57) and Cys194 (5.43). The alkyl chains on these groups form hydrophobic interactions with the surrounding residues and help in anchoring the ligands because their fit with this side pocket gives greater complementarity with the binding site. Other polar interactions help stabilizing both ligands in this region of the binding site, such as two H-bonds between the adenine ring of 3 and Asn191 (5.40) and a H-bond between the ester carbonyl of 19 and Asn159 (4.60).
Figure 1.

Comparison between agonist- and antagonist-bound P2Y12R crystal structures. Side view of the crystallographic poses of (A) compound 3 (pink carbon sticks) at the agonist-bound P2Y12R structure and (B) compound 19 (green carbon sticks) at the antagonist-bound P2Y12R structure. Side chains of residues important for ligand recognition are shown in thin sticks (grey carbons) and polar interactions are indicated by red dashed lines. Non-polar hydrogen atoms are not displayed. 2D representation of the binding modes of (C) 3 and (D) 19 at the P2Y12R. Magenta arrows indicate H-bonds, red-purple lines indicate salt bridges and green lines indicate π-π stacking interactions. Residues are colored based on their features: positively charged residues in purple, polar residues in cyan and hydrophobic residues in green. (E) RMSD in Å of the residues located within 3 Å from the crystallographic poses of compounds 3 and 19 between the two P2Y12R crystal structures. Residues whose RMSD values are not reported are not resolved in the antagonist-bound P2Y12R structure.
The ribose moiety of compound 3 forms multiple H-bonding interactions that are not observed in the 19-P2Y12R complex; in fact, most of the residues interacting with the sugar, such as Cys97 (3.25), Asn159 (4.60), His187 (5.36) and Lys179 (EL2), are not solved or differently oriented in the antagonist-bound structure (see Figure 1). Interestingly, 3 binds with the ribose in a South conformation, in agreement with the previous observation that the North (N)-methanocarba derivative of 3 (MRS2365) is selective for the P2Y1R over the P2Y12 and P2Y13 subtypes [39].
The phosphate groups of agonist and partial agonist interact with numerous hydrophilic and positively charged residues, including Arg93 (3.21), Tyr105 (3.33), Cys175 (EL2), Arg256 (6.55), Tyr259 (6.58), Gln263 (6.62) and Lys280 (7.35). Among these amino acids, two cationic residues Arg256 (6.55) and Lys280 (7.35) and an aromatic residue Tyr259 (6.58), previously predicted by mutagenesis studies to be involved in receptor function and/or ligand binding and conserved in the P2Y12R-like subfamily [40, 41], interact also with compound 19. In particular, Arg256 (6.55) and Lys280 (7.35) interact with the acylsulfonamide moiety, and Tyr259 (6.58) helps stabilizing the phenyl group of the ligand.
Some water molecules in contact with the ligands have been solved in these crystal structures. There are two water molecules interacting with the sulfonamide moiety of compound 19 in the antagonist-bound P2Y12R structure and seven in proximity of compound 3 in the agonist-bound P2Y12R structure. In particular, four water molecules are in contact with the β-phosphate of compound 3 and mediate its interaction with some residues such as Arg19 (N-term). Two of these water molecules are in proximity of the N6 atom of the adenine ring, and one is closer to the ribose ring but not directly interacting with it. In both structures, the possible presence of other water molecules not observed in the crystals could mediate additional polar interactions with residues of the binding site.
Docking of P2Y12R ligands
We collected from the literature ~500 diverse small molecules that were experimentally tested at the P2Y12R [5, 8, 31] (representative derivatives shown in Figs. 1 and 2), and we docked them at both the antagonist- and agonist-bound P2Y12R structures to predict possible binding modes. Then, because of the significant conformational differences between the two crystals, for each family of ligands we selected the most appropriate structure for docking, based on fitting of the cavity, analysis of protein-ligand interactions and the ability to rationalize the reported SAR. As discussed below, in order to obtain a reasonable binding mode for some compounds it was necessary to build hybrid models of the P2Y12R that combined features of both the agonist- and antagonist-bound structures. The active metabolites of compounds 56 and 57 were not included in our docking study because of the covalent nature of their P2Y12R binding.
Figure 2.

Docking of P2Y12R agonists to the agonist-bound crystal structure: Side view of the docking poses of nucleoside 5′-diphosphates 1 (cyan carbon sticks) and 4 (magenta carbons sticks) and crystal pose of compound 3 (pink carbon sticks) at the agonist-bound P2Y12R structure. Non-polar hydrogen atoms are not displayed. The surface of the binding site is shown in grey. The view of TM5 is partially omitted.
Before performing docking of new ligands, the Glide42 docking algorithm was tested in its ability to reproduce the crystallographic poses of compounds 3 and 19 in the two structures. Self-docking gave good results in both cases, returning top scoring docking poses with root-mean-square deviation (RMSD) from the crystal pose of 0.2 Å and 0.9 Å for 3 and 19, respectively. However, cross-docking of 3 to the rigid antagonist-bound structure and of 19 to the rigid agonist-bound structure was not able to reproduce poses similar to the crystallographic ones due to the significant differences in receptor conformation between the two structures.
Docking of P2Y12R Agonists and Partial Agonists
Mononucleotide derivatives, e.g. 5′-diphosphates (compounds 1 – 4) and 5′-triphosphates (compounds 5 – 8) docked very easily to the agonist-bound structure with a conformation analogous to the one observed for the co-crystallized nucleoside 5′-diphosphate and 5′-triphosphate (Figure 2). All of the common interactions that stabilize the nucleotide molecules are conserved between the crystal structure and the docking poses. However, when docked to the antagonist-bound structure these compounds showed a different orientation in the binding cavity. In particular, the bicyclic ring of the base occupied the same region of the cavity in proximity to Tyr105 (3.33) but with a different orientation, and the ribose and phosphate groups are directed towards TM6 and TM7, interacting with Arg256 (6.55) and Lys280 (7.35) (Figure S2, Supporting Information). This is not surprising considering the outward position of these two helices in the antagonist-bound structure and the fact that the residues anchoring the ribose in the agonist complex are not in proximity to serve as recognition points in the antagonist-bound structure, as previously described.
Docking to the agonist-bound structure clearly shows the role of the substituent at the C2 position in modulating the receptor affinity and suggests the possibility of various substitutions (Figure 2). The insertion of a hydrophobic group in a small side pocket at the bottom of the binding site between TM3, TM4 and TM5 stabilizes the agonist binding mode and can explain the higher affinity of 3 compared to 1 (ADP). Moreover, more extended linear groups are tolerated at the C2 position as shown by the docking poses of 2-hexynyl-ADP (4) and 2-hexynyl-ATP (8) [9], which maintain all of the interactions observed in the crystal pose of 3. However, docking of 2-phenylethynyl-ADP and 2-phenylethynyl-ATP [9] did not give similar results; these compounds were not able to fit the binding site with a similar orientation because of the bulky C2 substituent. These findings are in agreement with the previously reported agonist activity of compounds 4 and 8 at the P2Y12R and the inactivity of the corresponding 2-phenylethynyl nucleotides [9].
Docking of P2Y12R Antagonists
Ureas, Sulfonylureas, Sulfonamides
Several urea, sulfonylurea and sulfonamide derivatives, analogues of compound 19, were reported to be P2Y12R antagonists (compounds 9–22), originating from a library of hit compounds identified by high throughput-screening that contained a piperazinyl-pyridine scaffold [11–14].
As expected, these derivatives when docked to the antagonist-bound structure showed a binding mode similar to 19, which was also in agreement with previous SAR findings (Figure 3A). Docking of such extended derivatives at the agonist-bound P2Y12R structure returned some lower-score docking poses. In these poses, the pyridine ring occupied the same region as in the 19-complex, and the rest of the molecule bent around TM3 with the distal aromatic group oriented towards TM2 (Figure S3, Supporting Information).
Figure 3.

Docking of urea, sulfonylurea, sulfonamide and amide derivatives to the antagonist-bound P2Y12R crystal structure. (A) Side view of the docking poses of compounds 13 (cyan carbon sticks), 18 (magenta carbons sticks) and 22 (pink carbons sticks) and crystal pose of compound 19 (green carbon sticks) at the antagonist-bound P2Y12R structure. (B) Side view of the docking pose of compound 26 (magenta carbons sticks) and crystal pose of compound 19 (green carbon sticks) at the antagonist-bound P2Y12R structure. Side chains of residues important for ligand recognition are shown in thin sticks (grey carbons), and polar interactions are indicated by red, dashed lines. Non-polar hydrogen atoms are not displayed.
Many features of the SAR of this class of compounds are in agreement with their binding mode observed at the antagonist-bound structure. SAR exploration of the aromatic substituent on the urea, sulfonylurea or sulfonamide group showed tolerance for phenyl, benzyl and naphthyl groups and preference for lipophilic substitutions on the ring [11]. This is in agreement with the docking observation that a hydrophobic pocket between TM6 and TM7, delimited by Phe252 (6.51), Ala 255 (6.54), Tyr259 (6.58), Leu276 (7.31), Val279 (7.34), Thr283 (7.38) and the alkyl portion of the side chains of Arg256 (6.55) and Lys280 (7.35), can accommodate such aromatic groups (Figure 3). A good fit of a ligand within this cavity could contribute significantly to its potency.
SAR investigations also revealed a key role for the 3-alkyl ester moiety [11, 12]. Docking results show that the carbonyl of the ester group forms a H-bond with Asn159 (4.60), and its alkyl moiety fits a small hydrophobic cavity between TM3, TM4 and TM5 (Figure 3A), as also observed for the crystal pose of compound 19. This binding mode suggests that both a good complementarity of the alkyl chain within the hydrophobic pocket and the presence of a good H-bond acceptor group are required for potency. This would explain why shortening the ethyl ester to methyl ester or inserting branches in the alkyl chain close to the carbonyl reduced the binding affinity, and replacing the ester with a carboxylic acid or a sulfonate group was not tolerated in P2Y12R binding. Moreover, replacement of the ethyl ester functionality with non-classical heterocyclic bioisosteres was tolerated only when a strong H-bond acceptor group and an alkyl group were present on the heterocycle at the right positions to match the ethyl ester spatially [13]. Interestingly, previously performed computational analysis based on ligand shape and electrostatic similarity proposed correctly positioned H-bond acceptor and alkyl groups as necessary structural motifs for binding. These hypotheses now can be further tested, knowing the structure of the receptor, as shown by the docking pose of compound 22 in Figure 4A.
Another compound bearing a sulfonylurea moiety but a different scaffold from the previously described compounds is Elinogrel (compound 58, Figure S4, Supporting information), a drug candidate that reached clinical trials but whose development was then terminated. Pharmacological data for derivatives of this compound have not been published; therefore, it is difficult to discuss the SAR of this compound. Nevertheless, when docked to the antagonist-bound P2Y12R structure compound 58 shows an orientation very similar to compound 19 with the thiophene ring inserted between TM6 and TM7, the sulfonylurea group interacting with Arg256 (6.55) and Lys280 (7.35), and the 2,4-dioxo-1,4-dihydroquinazoline ring interacting with Tyr105 (3.33).
Very recently, novel antagonists with micromolar binding activity at the human (h) P2Y12R were reported, starting from a 4-pyrazol-1-ylbenzamide hit obtained by high throughput screening [43]. Molecular optimization led to several derivatives with good affinity at the P2Y12R (compounds 23–26), among them the preclinical candidate N-[6-(4-butanoyl-5-methyl-1H-pyrazol-1-yl)pyridazin-3-yl]-5-chloro-1-[2-(4-methylpiperazin-1-yl)-2-oxoethyl]-1H-indole-3-carboxamide (26, SAR216471). Even though these compounds do not present a urea or sulfonamide group but rather an amide moiety, they showed a binding mode comparable to the one observed for the previously described derivatives (compounds 9–22). In fact, the docking pose of 26 at the antagonist-bound P2Y12R structure (Figure 3B) shows: the pyridazine ring forming an aromatic π-π stacking interaction with Tyr105 (3.33); the butanoyl chain located in proximity of TM3, TM4 and TM5 forming a H-bond with Asn159 (4.60); the amide group placed between Arg256 (6.55) and Lys280 (7.35); the indole moiety accommodated between TM6 and TM7 and the substituent on the indole pointing toward the extracellular environment in proximity of EL3 forming interactions with residues of the loop.
Anthraquinones
Following the observation of the antagonist activity at the P2Y12R of Reactive Blue 2, several derivatives belonging to the anthraquinone family have been reported as high-affinity P2Y12R antagonists (compounds 27–31) [15]. Docking of these derivatives to the P2Y12R agonist-bound crystal structure did not return any pose, because the compounds were too bulky to fit the contracted binding site. Therefore, to perform docking studies of anthraquinone derivatives we used the antagonist-bound crystal structure.
Figure 4A shows the superposition of the top-scoring docking pose of 1-amino-9,10-dihydro-9,10-dioxo-4-[[4-(phenylamino)-3-sulfophenyl]amino]-2-anthracenesulfonic acid (27, PSB-0739) with the crystal pose of 19. The anthraquinone core overlays with the pyridine ring of 19 and forms an aromatic π-π stacking interaction with Tyr105 (3.33). Furthermore, ring E of the anthraquinone derivative is located in the hydrophobic pocket between TM6 and TM7, as is the phenyl ring of 19. The stabilizing interactions formed by the insertion of an aromatic ring in this cavity, found to be important also for the urea and sulfonylurea series, explain the SAR finding that ring E is essential for P2Y12 affinity [15]. SAR studies also highlighted the importance for high affinity of acidic groups such as sulfonate or carboxylate at the 2 position of the anthraquinone scaffold and at the meta-position of ring D. This is in agreement with the observed interactions of these negatively charged groups with two positively charged residues of the binding site (Figure 4). Specifically, the sulfonic acid on ring D occupies the same region as the sulfonyl group of 19 and interacts with both Arg256 (6.55) and Lys280 (7.35), while the sulfonic acid on ring C interacts with Lys280 (7.35).
Available mutagenesis data confirm this binding mode. In fact, at the R256A-mutant P2Y12R the potency of 27 (bearing sulfonic acids on rings C and D) and 28 (bearing a sulfonic acid on ring D) was decreased, but the potency of 29 (lacking a sulfonic acid on ring D) was not affected [44]. The proximity of the sulfonic group on ring D to the side chain of Arg256 (6.55) in the docking poses of these derivatives can explain these mutagenesis results, while the sulfonic group on ring C is too far to strongly interact with that arginine residue.
To further prove this binding mode, we mutated Lys280 (7.35) and Cys194 (5.43) each to Ala and measured functional activity using a cAMP response element-driven luciferase reporter gene assay [45]. At the K280A-mutant receptor both compounds 28 and 29 showed lower apparent pKB-values (Figure 5). This is in agreement with the observation that Lys280 can interact with both sulfonic groups on rings C and D, therefore stabilizing the binding of both of these two derivatives. In addition, an increase in antagonist potency of compounds 28 and 29 was observed at the C194A-mutant receptor (Figure S5, Supporting Information), as also previously reported for 27 [45]. This can be explained by an increase in space and hydrophobicity of the region of binding of the anthraquinone core, resulting in a better accommodation of these compounds. In fact, Cys194 (5.43) is located at the bottom of the binding site in proximity of ring A.
Figure 5.

Effects of anthraquinones (A), (B) 28 and (C), (D) 29 on compound 3-mediated inhibition of forskolin-induced cAMP response element (CRE)-driven luciferase expression in CHO Flp-In cells expressing the hP2Y12R fused to ECFP (panels A and C) or K280A-mutant receptor fused to ECFP (panels B and D). Means ± S.E. of 4 to 24 experiments.
Interestingly, in the anthraquinone family substitutions on the A ring have not been reported; therefore, the possible beneficial effect on affinity of an interaction with Asn159 (4.60) and/or of the occupation of the small pocket between TM3, TM4 and TM5 cannot be probed. In fact, the carbonyl group on ring B and the amino group on ring C are too far to directly interact with Asn159 (4.60) or other polar residues of that region of the binding site. However, the possible presence of water molecules could mediate and/or possible side chain rearrangement could determine additional polar interactions with nearby residues, such as Gln98 (3.26), Ser101 (3.29), Asn159 (4.60) and His187 (5.36). We can speculate that insertion of a hydrophobic chain on the anthraquinone scaffold that fits the subpocket between TMs 3, 4 and 5 could possibly increase the potency of these compounds, as observed for other structural series.
Glutamic Acid Piperazines
Several groups have synthesized numerous glutamic acid piperazine derivatives having P2Y12R antagonist activity (compounds 32–41) [16–20]. Such compounds have been developed through the molecular optimization of quinoline derivatives (such as (S)-4-({[4-(1-carboxy-1-methylethoxy)-7-methylquinolin-2-yl]carbonyl}amino)-5-[4-(ethoxycarbonyl)piperazin-1-yl]-5-oxopentanoic acid, BX048, structure not shown) [57].
Docking studies showed that these derivatives are accommodated better at the antagonist-bound P2Y12R structure, as compared to the agonist-bound structure where the space is reduced. In particular, bulkier derivatives of the series could not fit the rigid binding site of the agonist-bound structure, while smaller derivatives could fit when bent around TM3 to occupy both pockets but only with poorer docking scores (Figure S6, Supporting Information). On the other hand, docking performed at the antagonist-bound structure returned poses with an orientation similar to 19 (Figure 4B).
As shown in Figure 4B, the piperazine ring occupies the same region as the pyridine ring of compound 19. In this case, even though Tyr105 (3.33) cannot form a stacking interaction with an aromatic ring of the ligand, as observed for compound 19, it helps in stabilizing the binding pose through hydrophobic interactions with the piperazine ring. The ester group forms an H-bond with Asn159 (4.60) and fits the pocket delimited by Phe106 (3.34), Met152 (4.53), Leu155 (4.56), Ser156 (4.57) and Cys194 (5.43), exactly as seen for the ester moiety of 19 in the crystal structure. The aromatic portion of these ligands inserts between TM6 and TM7, forming stabilizing hydrophobic interactions similar to the phenyl ring of 19. The glutamic acid moiety forms an ionic interaction with Lys280 (7.35), which even though contributing to the stability of the complex, does not seem to be essential as shown by the high affinity of compounds 39–41 for the P2Y12 R. These compounds present an analogous binding mode (Figure 4B), except that the ionic interaction is missing, which shows how optimization of the rest of the ligand structure can lead to compounds with high P2Y12 R affinity and not presenting the glutamic acid group. The different substitutions on the heteroaromatic ring are oriented toward the extracellular environment and make contacts with TM6, TM7 and EL3. Because this is a flexible region of the receptor, as highlighted by crystallographic data, it is more difficult to study the specific interactions formed by these substituents.
Nucleotide and Nucleoside Antagonists
Using agonist ATP as a chemical starting point, several nucleotide mimetics have been developed as P2Y12R antagonists, through optimization of pharmacodynamic and pharmacokinetic properties. Such optimization approaches led to the discovery of the first reversible P2Y12 antagonist approved for clinical use (compound 47) [32].
Docking of some nucleotide and nucleoside derivatives (compounds 42–48) to the antagonist-bound structure resulted in poses completely different from the one observed for nucleotides in the crystal structures and therefore did not seem very convincing. Instead, 43 and other nucleotide-mimetic antagonists with linear N6 substituents of up to four heavy atoms [21] when docked to the agonist-bound P2Y12R structure showed an orientation very similar to 3 (Figure 6A). This was true also for compounds with similar structure but not presenting phosphate groups (such as 44). Thus, as in the nucleotide crystal structures, such derivatives showed a π-π stacking interaction with Tyr105 (3.33), H-bonds with Cys97 (3.25), His187 (5.36), Lys179 (EL2) and Asn191 (5.40) and the C2 substituent accommodated in the side pocket delimited by TM3, TM4 and TM5. The N6 group was located at the bottom of the binding site directed towards TM6.
Figure 6.

Docking of nucleotide and nucleoside derivatives to the agonist-bound P2Y12R crystal structure and P2Y12R hybrid model. (A) Side view of the docking poses of compounds 43 (cyan carbon sticks) and 44 (green carbons sticks) and crystal pose of compound 3 (pink carbon sticks) at the agonist-bound P2Y12R structure. Non-polar hydrogen atoms are not displayed. The surface of the binding site is shown in grey. The view of TM5 is partially omitted. (B) Side view of the docking pose of compound 47 (green carbon sticks) at the P2Y12R hybrid model. Side chains of residues important for ligand recognition are shown in thin sticks (grey carbons) and polar interactions formed by compound 47 are indicated by red, dashed lines. Non-polar hydrogen atoms are not displayed. The crystal pose of compound 3 (pink carbon sticks) and the position of TM6 (white ribbon) in the agonist-bound P2Y12R structure are shown for comparison.
However, docking of compound 47 and other similar derivatives with a bulky N6 substituent (compounds 45–48) did not give satisfactory results with either the P2Y12R agonist- or antagonist-bound structure. In fact, at the agonist-bound structure the cavity was too small to fit 47 in an orientation similar to 3, because the bulky N6 substituent of 47 would coincide with TM6. Therefore, we decided to build a model of the P2Y12R to combine the features of both crystal structures. In particular, we built a hybrid model with an overall structure resembling the agonist-bound P2Y12R structure except for the upper part of TM6 (from Phe254 (6.53) to Ser262 (6.61)). That region of TM6 was modeled on the antagonist-bound P2Y12R structure. This region of the receptor shows major conformational changes between the two structures. In fact, when 3 binds, TM6 bends towards the binding site, and in particular the side chain of Arg256 (6.55) stretches to interact with the α-phosphate of the ligand. However, when 19 binds, TM6 is pushed outwards by the benzylsulfonyl-carbamoyl group of the antagonist, and the side chain of Arg256 (6.55) is directed toward TM5.
Using this hybrid model we were able to find a docking pose of uncharged carbocyclic compound 47 very similar to the one observed for 3 (Figure 6B). Some interactions formed by 3 are also observed in the docking pose of 47. The bicyclic core forms a π-π stacking interaction with Tyr105 (3.33) and a H-bond with Asn191 (5.40). The 2-thioether inserts into the hydrophobic side pocket between TM3, TM4 and TM5, and the 2′- and 3′-hydroxyl groups form H-bonds with Lys179 (EL2), His187 (5.36) and the backbone carbonyl of Cys97 (3.25). In addition, the N6 substituent of 47 is directed between TM6 and TM7, in a similar region as the phenyl ring of 19, and forms an aromatic interaction with Phe252 (6.51).
Considering this binding mode we were able to rationalize recently reported mutagenesis data for compound 47 [45]. R256A and K280A mutations did not affect the antagonist potency of 47, in agreement with the absence on the structure of 47 of an acidic group able to interact with these positively charged residues. Lys173 (EL2), Lys174 (EL2) and Cys248 (6.47) are not in direct contact with 47 in the proposed docking pose (more than 10 Å away) and therefore are not expected to be important in stabilizing its binding to the receptor, in agreement with mutagenesis results. Mutation of Ser101 (3.29) to Ala did not change the potency of 47; in fact, even though Ser101 (3.29) is in proximity of 47, it is not involved in any particular polar interaction with the ligand. C194A is a mutation that greatly affected the potency of 47. This residue is located in the lower part of the binding site in proximity of the exocyclic NH of 47. Cys194 (5.43) can have a role in stabilizing the binding of the ligand through polar interaction with the NH group and in shaping the cavity in a way favorable to this ligand orientation. Moreover, the residue is located in TM5 in proximity to the region where the conformational rearrangements occur, and therefore its mutation can affect this process.
Dinucleotides
Naturally occurring and synthetic dinucleotides (compounds 49–55) have shown partial agonist and/or antagonist activity towards the P2Y12R, depending on the system under study. The unusual bifurcation of the binding cleft in the P2Y12R crystal structures already suggested the possibility of the two nucleoside moieties of dinucleotide ligands reaching both pockets in the bound state [37, 46].
High-scoring docking poses were obtained for these derivatives at the antagonist-bound P2Y12R structure (Figure 7A). In particular, docking results show one nucleotide moiety accommodated in the pocket delimited by TMs 3, 4, 5, 6, and 7; while the second nucleotide moiety reaches the pocket delimited by TMs 1, 2, 3 and 7, with the phosphate groups interacting with positively charged residues on TMs 6 and 7. However, the orientation of the nucleotide moiety in the first pocket is different from the one observed for compound 3 in the crystal structure, since residues stabilizing the orientation of the ribose and of the base have different conformations in the two P2Y12R structures, i.e. Lys179 in EL2, Cys97 in TM3 because of the missing disulfide bond, His187 and Asn191 in TM5. On the other hand, docking of dinucleotides to the rigid agonist-bound P2Y12R crystal structure did not return any pose, probably because the binding site is contracted and EL2 is covering the pocket not leaving enough space for these extended ligands to bind. Therefore, modeling results seem to indicate that the receptor conformation that binds dinucleotides is intermediate between the two available crystal structures.
Figure 7.

Docking of dinucleotides to the antagonist- and agonist-bound P2Y12R crystal structures. (A) Top view of the docking pose of compound 53 (cyan carbon sticks) at the antagonist-bound P2Y12R structure. (B) Top view of the docking pose of compound 49 (magenta carbon sticks) at the agonist-bound P2Y12R structure obtained after Induced Fit Docking. The crystal pose of compound 3 (pink carbon sticks) is shown for comparison. Side chains of residues important for ligand recognition are shown in thin sticks (grey carbons), and red dashed lines indicate polar interactions formed by the docked compounds. Non-polar hydrogen atoms are not displayed.
To prove this hypothesis, we performed Induced Fit Docking [47] of dinucleotides at the agonist-bound structure. In fact, after optimization of the binding site residues, dinucleotides were able to fit the binding cavity. Docking poses showed one nucleotide moiety oriented and interacting similarly to 3 and the second nucleotide moiety accommodated less deeply in the binding site in proximity to TM2 and TM3, interacting also with residues in EL2 and the N-terminus (Figure 7B). The phosphate groups form H-bonding and ionic interactions with residues of TM6, TM7 and EL2, as observed in the agonist crystal pose.
Supervised Molecular Dynamics
In order to further elucidate the P2Y12R-ligand recognition process we used Supervised Molecular Dynamics (SuMD) to identify ligand-protein bound states that chronologically anticipate the orthosteric binding site recognition. In this framework, we selected compound 47 as a non-charged - at physiological pH - molecular probe to clarify the influence of: (a) interaction with TMs; (b) binding pocket topology and (c) hydration during the antagonist recognition process. Prior investigation of the ligand-recognition process, the apo-form of the P2Y12R hybrid model was equilibrated in a fully solvated membrane like environment and binding pocket hydration characterized using an approach previously reported [48]. Analysis of the Molecular Dynamics (MD) trajectories identified the stable formation of a salt bridge between Glu188 (5.37) and Arg256 (6.55) linking TM5 and TM6 (Figure 8A). The interaction between the two vicinal TMs is also stabilized by the formation of a hydrogen-bonding network between the polar side chains of Gln195 (5.44) and Thr260 (6.59) with water molecules and the glycerol 3-phosphate moiety of membrane lipids (POPC lipids). Such detail of interaction has not yet been observed in any ligand bound-P2Y12R crystal structures. In addition, the polar binding site of the P2Y12R revealed the presence of water clusters and other peculiar motifs (i.e. chloride ions) bridging charged residues present in TM1, TM2 and TM7. Time dependent organization of water clusters analysis on the produced MD trajectories [49] highlights the presence of structural water molecules bridging Lys80 (2.60), Asp84 (2.64), Arg93 (3.21), Glu281 (7.36) (Figure 8B) and Arg19 (N-term), Tyr21 (N-term), Lys174 (EL2), Glu273 (7.28) (Figure 9C). Chloride ions are also found to be coordination with Arg93 (3.21), as commonly found in X-ray crystal structures deposited in the Protein Data Bank [50].
Figure 8.

(A–C) Molecular details of key receptor-solvent/membrane interactions in P2Y12R prior to ligand recognition. Side chains of residues involved in key interaction networks are shown in sticks. Non-polar hydrogen atoms are not displayed. H-bonds and salt bridges are highlighted as dashed lines. Ions are depicted as spheres. (D) Overview of the apo-P2Y12R hybrid model in a membrane-like environment (POPC lipids).
Figure 9.

(A), (D) Overview of identified ligand-receptor bound states that chronologically anticipate the orthosteric binding site recognition of compound 47 at P2Y12R. Ligand is depicted as a colored arrow. Arrow coloring scheme enables distinction of the single independent SuMD simulations that generated depicted bound states (SuMD seeds). Receptor ribbon representation is viewed from the membrane side facing TM5 and TM6. (B), (C) Atomistic level detailed representation of characterized convergent metastable-states during the approaching of compound 47 (yellow carbon sticks) to the P2Y12R from the extracellular binding pocket vestibule. Side chains of residues within 4 Å from compound 47 are highlighted (white carbon sticks). H-bonds are highlighted as yellow dashed lines. Hydrogen atoms are not displayed.
We ran multiple independent SuMD simulations to fully sample the conformational landscape exploration of compound 47 during its P2Y12R recognition process and to characterize convergent metastable-states. Several discrete converging binding states were detected by clustering contacts and the orientation of the ligand along the binding pathway (Figure 9). EL2, EL3 and the N-terminus play an essential role in engaging interactions with the adenine core of the antagonist by providing favorable polar and hydrophobic contacts to anticipate the approach of the ligand from the extracellular binding pocket vestibule (Figure 9B). Meta-binding sites that are able to accommodate the ligand prior to recognition at the orthosteric binding site (Figure 9C) are composed of residues belonging to different TMs, such as TM5, TM6 and TM7 along with EL2. The 2-thioether of 47 is directed towards EL2 in proximity of Phe177 (EL2) and Ser178 (EL2); the 3,4-diF-phenyl-cyclopropyl group extends into a hydrophobic pocket delimited by TM6 and TM7 interacting with Tyr259 (6.58), Ala272 (7.27) and Leu276 (7.31). The 3 -hydroxyl group forms H-bonding interaction with Glu188 (5.37), which is involved in a strong salt bridge with Arg256 (6.55). Desolvation of non-bulk like water molecules and breaking of the salt bridge between Glu188 (5.37) and Arg256 (6.55) may represent one of the largest energetic barriers to overcome along the drug-receptor binding pathway in order to allow recognition of 47 at the orthosteric binding site. In support of this hypothesis, all major meta-binding sites characterized using independent SuMD simulations are located in the vestibule delimited by TM5, TM6, TM7 and EL2, above the orthosteric binding pocket (Figure 9D).
Discussion
The recent resolution of three crystallographic structures of the hP2Y12R provides new information to understand ligand recognition at this receptor and highlights the imprecision of previous modeling attempts. Moreover, the observation of two very different ligand-bound conformations of the P2Y12R provoked the question of which is the best structure to perform docking studies of specific ligands. To address these issues we performed extensive molecular docking studies of the major chemical classes previously reported as P2Y12R ligands.
As expected, the agonist-bound structure was found to be the best crystal form for docking of P2Y12R agonists in order to distinguish active and inactive compounds. The antagonist-bound structure returned agonist poses located in the same region of the binding site and interacting with a few common residues, but it was not suitable to determine the correct orientation of the ligands as it lacked several key interactions. On the other hand, the antagonist-bound structure was clearly the best target to dock bulky P2Y12R antagonists, such as urea, sulfonylurea, sulfonamide, anthraquinone and glutamic acid piperazine derivatives, while for other antagonists an optimal structure for docking was more difficult to identify. In particular, for compounds derived from nucleotide structures different results were obtained depending on the size of their N6 substituent. Derivatives with a linear N6 group up to four heavy atoms could fit the agonist-bound P2Y12R structure in an orientation similar to the one observed for the crystallized nucleotides, while derivatives with a bulkier N6 substituent needed a conformational rearrangement of the receptor to adopt an analogous conformation. This rearrangement was proposed based on the observation that TM6 assumed two very different conformations in the agonist- and antagonist-bound P2Y12R crystal structures. Also, in order to dock dinucleotides to the agonist-bound structure, an optimization of the binding site had to be performed to allow rearrangement of some side chains. Therefore, these docking studies are consistent with the great plasticity of the P2Y12R, already observed from crystallographic data. In fact, this receptor is able to adjust its conformation to accommodate ligands having very different size and chemical structure, mainly through the movement of TMs 6 and 7 and the rearrangement of residues in the ELs.
Despite the conformational differences associated with the binding of different ligands, our molecular modeling analysis also highlighted some common features. Figure 10 summarizes the superposition of the binding modes, crystallographic or predicted through modeling, of one representative compound for each different ligand class analyzed in this study (except dinucleotides), and Figure S7 (Supporting Information) shows a 2D schematic summary of the proposed binding modes for different class of compounds. The most common feature, both for agonists and antagonists, is the presence of an aromatic core located between TM3 and TM5 and interacting with Tyr105 (3.33). Only glutamic acid piperazines do not conserve this aromatic feature but rather place the piperazine ring in the same position, interacting with Tyr105 (3.33). Moreover, all ligand classes, except anthraquinones, have a linear alkyl chain, important to increase affinity for the receptor, inserted in a hydrophobic side pocket at the bottom of the binding site delimited by TM3, TM4 and TM5. Other features are common between some classes of ligands but not conserved in all of them. Nucleoside derivatives, anthraquinones and glutamic acid piperazines present a negatively charged group, such as phosphate, sulfonate or carboxylate, in place of the α-phosphate group in the crystal pose of compound 3, which interacts with Lys280 (7.35). The same region binds the hydroxyl-ethyloxy group of compound 47. Non-nucleotide antagonists share the presence of an H-bond acceptor group interacting with Asn159 (4.60) (except anthraquinones), a polar group, such as an amide, urea, sulfonamide or sulfonate, located between Arg256 (6.55) and Lys280 (7.35) and an aromatic/hydrophobic group inserted between TM6 and TM7 with the possibility of different, extended substitutents pointing toward EL3. The nucleotide-like derivative 47 also shows an aromatic ring located between TM6 and TM7, but in a slightly shifted position. All nucleotide-like ligands show peculiar H-bonding interactions formed by the ribose hydroxyl groups with Cys97 (3.25), His187 (5.36) and Lys179 (EL2).
Figure 10.

Comparison of the binding modes of different ligand classes at the P2Y12R. Superposition of the crystal poses of compounds 3 (pink carbon sticks) and 19 (green carbon sticks) and the docking poses of compounds 27 (magenta carbon sticks), 32 (blue carbon sticks), 43 (cyan carbon sticks) and 47 (dark green carbon sticks) at the P2Y12R. The antagonist-bound P2Y12R structure is shown in cyan ribbons. The view of TM5 is omitted. Similarities in the binding mode of different ligands are described.
SuMD simulations were performed to study the recognition process of compound 47 at the proposed hybrid model of the P2Y12R. Even though these simulations were not able to reach an orthosteric binding mode, they provided useful information on the approach of the ligand, identifying possible meta-binding sites. During its pathway towards the orthosteric binding site, compound 47 interacts with residues in the N-terminus, EL2, EL3 and the extracellular portions of TM5-TM7. The numerous polar and ionic interactions between residues in the extracellular region of the P2Y12R make it more difficult for the ligand to find its way to the final orthosteric pose. This is very different from what was observed previously for the A2AAR [48], where the entrance of the binding site is exposed and loops are not covering the orthosteric cavity. Therefore, these findings support the idea of high plasticity of the extracellular portion of the P2Y12R needed to accommodate different ligands. Moreover, analysis of the ligand recognition pathway could help in rationalizing some mutagenesis data highlighting the effect on ligand potency of specific residues that, even if not in direct contact with the ligand in its final orthosteric pose, can greatly affect its approach to the binding site.
Conclusions
We compared two highly divergent crystallographic structures of the P2Y12R to address the question of which structure is more applicable to understand recognition within each class of diverse, reversible P2Y12R ligands. Various mononucleotide derivatives docked readily to the agonist-bound P2Y12R, and dinucleotides could span two pockets of the binding site. Ureas, sulfonylureas, sulfonamides, anthraquinones and glutamic acid piperazines readily docked to the antagonist-bound P2Y12R. The uncharged nucleotide-like antagonist ticagrelor did not dock to the agonist-bound P2Y12R and required a hybrid model of the P2Y12R. Further examination of the interaction of ticagrelor with the P2Y12R used the recently reported method of SuMD to define possible meta-binding sites. Thus, our structure-based approach highlighted similarities and differences in the binding modes of different compounds and consistently rationalized the main SAR within each ligand class, giving useful information for the design of new and improved compounds and new insights in the P2Y12R recognition process.
Experimental Section
Molecular Modeling
P2Y12R structures
To perform molecular modeling studies we used two available P2Y12R crystallographic structures: the first in complex with compound 19 (antagonist-bound P2Y12R structure, PDB ID: 4NTJ) [38] and the second in complex with compound 3 (agonist-bound P2Y12R structure, PDB ID: 4PXZ) [37]. The residue numbering in parenthesis follows the arbitrary scheme by Ballesteros and Weinstein [51].
P2Y12R structures were prepared using the Protein Preparation Wizard tool implemented in the Schrödinger suite [52], adding all the hydrogen atoms and the missing side chains of residues whose backbone coordinates were observed in the structure. The orientation of polar hydrogens was optimized, the protein protonation states were adjusted and the overall structure was minimized with harmonic restraints on the heavy atoms, to remove strain. All the hetero groups and water molecules were deleted.
A hybrid model of the P2Y12R was built, to combine the features of both crystal structures, using the Prime homology modeling tool (energy-based method) of the Schrödinger suite [52]. To build this model, the agonist-bound P2Y12R crystal structure was used as a template for the entire P2Y12R structure except for the extracellular terminus of TM6 and EL3. The X-ray structure of the antagonist-bound P2Y12R, after superimposition with the agonist-bound P2Y12R structure, was used as template to build the extracellular terminus of TM6 (from Phe254 (6.53) to Ser262 (6.61)). No structural template was used for the modeling of EL3 (from Gln263 to Asp269).
Molecular docking
Structures of P2Y12R ligands, collected from the literature, were built and prepared for docking using the Builder and the LigPrep tools implemented in the Schrödinger suite [52]. In particular, possible ionization states at pH 7±1 were generated using Epik, tautomers were generated and geometries were optimized using the OPLS_2005 force field.
The SiteMap tool of the Schrödinger suite [52] was used to identify potential binding sites in the P2Y12R structures. A bifurcated cavity was identified on the extracellular side of the receptor and was selected as the docking site. Molecular docking of P2Y12R ligands to the P2Y12R crystal structures and hybrid model was performed by means of the Glide package from the Schrödinger suite. In particular, a Glide Grid was centered on the centroid of residues located within 5 Å from the previously identified cavity. The Glide Grid was built using an inner box (ligand diameter midpoint box) of 14 Å × 14 Å × 14 Å and an outer box (box within which all the ligand atoms must be contained) that extended 20 Å in each direction from the inner one. Docking of ligands was performed in the rigid binding site using both the standard precision (SP) and extra precision (XP) protocols. The top scoring docking conformations for each ligand were subjected to visual inspection and analysis of protein–ligand interactions to select the final binding conformations in agreement with the experimental data.
To obtain receptor conformations that more closely conform to the shape and binding mode of dinucleotide ligands we performed Induced Fit Docking (Standard Protocol) to the agonist-bound P2Y12R structure [47]. A docking box of 26 Å (with an inner box of 10 Å) was centered on the centroid of residues located within 5 Å from the SiteMap identified cavity. Prime refinement was performed on residues within 5 Å of ligand poses. Glide docking was performed using the SP protocol. Among the resulting complexes, the one in which the first half of the dinucleotide was closely resembling the orientation of compound 3 in the crystal was selected as optimized receptor to redock all the dinucleotide ligands. Redocking was performed to the new receptor structure by means of the Glide package. In particular, a Glide Grid was centered on the centroid of residues located within 5 Å from the bound dinucleotide. The Glide Grid was built using an inner box of 10 Å × 10 Å × 10 Å and an outer box that extended 26 Å in each direction from the inner one. Docking of ligands was performed in the rigid binding site using the XP protocol.
Supervised Molecular Dynamics
Molecular dynamics simulations have been performed with a 2 NVIDIA GTX 680 and 3 NVIDIA GTX 780 GPU cluster engineered by Acellera (http://www.acellera.com/). Trajectory analysis, figure and video generation have been performed using several functionalities implemented by Visual Molecular Dynamics [53], WORDOM [54], the PyMOL Molecular Graphics System (Version 1.5.0.4, Schrödinger, LLC, http://www.pymol.org/) and the Gnuplot graphic utility (http://www.gnuplot.info/). Ligand-P2Y12R interaction energies were calculated extrapolating the non-bonded energy interaction term of CHARMM27 Force Field using NAMD [55, 56]. Full methodological details have been previously reported [48].
Pharmacological studies
Mutagenesis studies and associated functional assays were performed as in Hoffmann et al [45].
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (Intramural Research Program of the NIDDK and R01HL077707).
Abbreviations used
- ADP
adenosine-5′-diphosphate
- ATP
adenosine-5′-triphosphate
- cAMP
cyclic adenosine monophosphate
- CHO
Chinese hamster ovary
- CNS
central nervous system
- CRE
cAMP response element
- ECFP
enhanced cyan fluorescent protein
- EL
extracellular loop
- GPCR
G protein-coupled receptor
- MD
Molecular Dynamics
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- RMSD
root-mean-square deviation
- SAR
structure-activity relationship
- SuMD
Supervised Molecular Dynamics
- TM
transmembrane helix
- UDP
uridine-5′-diphosphate
- UTP
uridine-5′-triphosphate
Footnotes
Supporting Information Available:
Additional figures; 3D coordinates of the modeled P2Y12R complexes with compounds 27, 32, 43 and 47; 3D coordinates of the docking poses of compounds 1, 3, 5, 7, 8, 42, 43 at the agonist-bound P2Y12R crystal structure, of compounds 18, 22, 27, 28, 29, 30, 31, 32, 41 at the antagonist-bound P2Y12R crystal structure and of compounds 42, 43, 47 at the hybrid P2Y12R model; 3D coordinates of the metastable-states of compound 47 at the P2Y12R obtained with SuMD simulations (corresponding to Figure 9 panels B and C); Pymol session file highlighting the interactions between the receptor and the membrane lipids shown in Figure 8; and a video showing the results from SuMD simulations. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burnstock G, Knight GE. Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol. 2004;240:31–304. doi: 10.1016/S0074-7696(04)40002-3. [DOI] [PubMed] [Google Scholar]
- 3.Hollopeter G, Jantzen HM, Vincent D, Li G, England L, Ramakrishnan V, Yang RB, Nurden P, Nurden A, Julius D, Conley PB. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature. 2001;409:202–207. doi: 10.1038/35051599. [DOI] [PubMed] [Google Scholar]
- 4.Hourani SM, Cusack NJ. Pharmacological receptors on blood platelets. Pharmacol Rev. 1991;43:243–98. [PubMed] [Google Scholar]
- 5.Jacobson KA, Deflorian F, Mishra S, Costanzi S. Pharmacochemistry of the platelet purinergic receptors. Purinergic Signal. 2011;7:305–324. doi: 10.1007/s11302-011-9216-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Debnath B, Al-Mawsawi LQ, Neamati N. Are we living in the end of the blockbuster drug era? Drug News Perspect. 2010;23:670–684. doi: 10.1358/dnp.2010.23.10.1506088. [DOI] [PubMed] [Google Scholar]
- 7.Horváth G, Gölöncsér F, Csölle C, Király K, Andó RD, Baranyi M, Koványi B, Máté Z, Hoffmann K, Algaier I, Baqi Y, Müller CE, Von Kügelgen I, Sperlágh B. Central P2Y12 receptor blockade alleviates inflammatory and neuropathic pain and cytokine production in rodents. Neurobiol Dis. 2014 doi: 10.1016/j.nbd.2014.06.011. pii: S0969–9961(14)00175–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herbert JM, Savi P. P2Y12, a new platelet ADP receptor, target of clopidogrel. Semin Vasc Med. 2003;3:113–122. doi: 10.1055/s-2003-40669. [DOI] [PubMed] [Google Scholar]
- 9.Cristalli G, Podda GM, Costanzi S, Lambertucci C, Lecchi A, Vittori S, Volpini R, Zighetti ML, Cattaneo M. Effects of 5′-phosphate derivatives of 2-hexynyl adenosine and 2-phenylethynyl adenosine on responses of human platelets mediated by P2Y receptors. J Med Chem. 2005;48:2763–2766. doi: 10.1021/jm0493562. [DOI] [PubMed] [Google Scholar]
- 10.Scarborough RM, Laibelman AM, Clizbe LA, Fretto LJ, Conley PB, Reynolds EE, Sedlock DM, Jantzen H. Novel tricyclic benzothiazolo[2,3-c]thiadiazine antagonists of the platelet ADP receptor (P2Y12) Bioorg Med Chem Lett. 2001;11:1805–1808. doi: 10.1016/s0960-894x(01)00313-4. [DOI] [PubMed] [Google Scholar]
- 11.Bach P, Boström J, Brickmann K, van Giezen JJ, Hovland R, Petersson AU, Ray A, Zetterberg FA. novel series of piperazinyl-pyridine ureas as antagonists of the purinergic P2Y12 receptor. Bioorg Med Chem Lett. 2011;21:2877–81. doi: 10.1016/j.bmcl.2011.03.088. [DOI] [PubMed] [Google Scholar]
- 12.Bach P, Boström J, Brickmann K, van Giezen JJ, Groneberg RD, Harvey DM, O’Sullivan M, Zetterberg F. Synthesis, structure-property relationships and pharmacokinetic evaluation of ethyl 6-aminonicotinate sulfonylureas as antagonists of the P2Y12 receptor. Eur J Med Chem. 2013;65:360–375. doi: 10.1016/j.ejmech.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 13.Bach P, Boström J, Brickmann K, Burgess LE, Clarke D, Groneberg RD, Harvey DM, Laird ER, O’Sullivan M, Zetterberg F. 5-alkyl-1,3-oxazole derivatives of 6-amino-nicotinic acids as alkyl ester bioisosteres are antagonists of the P2Y12 receptor. Future Med Chem. 2013;5:2037–56. doi: 10.4155/fmc.13.171. [DOI] [PubMed] [Google Scholar]
- 14.Bach P, Antonsson T, Bylund R, Björkman JA, Österlund K, Giordanetto F, van Giezen JJ, Andersen SM, Zachrisson H, Zetterberg F. Lead optimization of ethyl 6-aminonicotinate acyl sulfonamides as antagonists of the P2Y12 receptor, separation of the antithrombotic effect and bleeding for candidate drug AZD1283. J Med Chem. 2013;56:7015–24. doi: 10.1021/jm400820m. [DOI] [PubMed] [Google Scholar]
- 15.Baqi Y, Atzler K, Köse M, Glänzel M, Müller CE. High-affinity, non-nucleotide-derived competitive antagonists of platelet P2Y12 receptors. J Med Chem. 2009;52:3784–3793. doi: 10.1021/jm9003297. [DOI] [PubMed] [Google Scholar]
- 16.Parlow JJ, Burney MW, Case BL, Girard TJ, Hall KA, Hiebsch RR, Huff RM, Lachance RM, Mischke DA, Rapp SR, Woerndle RS, Ennis MD. Piperazinyl-glutamate-pyrimidines as potent P2Y12 antagonists for inhibition of platelet aggregation. Bioorg Med Chem Lett. 2009;19:6148–6156. doi: 10.1016/j.bmcl.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 17.Parlow JJ, Burney MW, Case BL, Girard TJ, Hall KA, Hiebsch RR, Huff RM, Lachance RM, Mischke DA, Rapp SR, Woerndle RS, Ennis MD. Piperazinyl-glutamate-pyridines as potent orally bioavailable P2Y12 antagonists for inhibition of platelet aggregation. Bioorg Med Chem Lett. 2009;19:4657–4663. doi: 10.1016/j.bmcl.2009.06.075. [DOI] [PubMed] [Google Scholar]
- 18.Parlow JJ, Burney MW, Case BL, Girard TJ, Hall KA, Harris PK, Hiebsch RR, Huff RM, Lachance RM, Mischke DA, Rapp SR, Woerndle RS, Ennis MD. Piperazinyl glutamate pyridines as potent orally bioavailable P2Y12 antagonists for inhibition of platelet aggregation. J Med Chem. 2010;53:2010–2037. doi: 10.1021/jm901518t. [DOI] [PubMed] [Google Scholar]
- 19.Parlow JJ, Burney MW, Case BL, Girard TJ, Hall KA, Harris PK, Hiebsch RR, Huff RM, Lachance RM, Mischke DA, Rapp SR, Woerndle RS, Ennis MD. Part II: piperazinyl-glutamate-pyridines as potent orally bioavailable P2Y12 antagonists for inhibition of platelet aggregation. Bioorg Med Chem Lett. 2010;20:1388–1394. doi: 10.1016/j.bmcl.2009.12.110. [DOI] [PubMed] [Google Scholar]
- 20.Zech G, Hessler G, Evers A, Weiss T, Florian P, Just M, Czech J, Czechtizky W, Görlitzer J, Ruf S, Kohlmann M, Nazaré M. Identification of high-affinity P2Y12 antagonists based on a phenylpyrazole glutamic acid piperazine backbone. J Med Chem. 2012;55:8615–8629. doi: 10.1021/jm300771j. [DOI] [PubMed] [Google Scholar]
- 21.Ingall AH, Dixon J, Bailey A, Coombs ME, Cox D, McInally JI, Hunt SF, Kindon ND, Teobald BJ, Willis PA, Humphries RG, Leff P, Clegg JA, Smith JA, Tomlinson W. Antagonists of the platelet P2T receptor: a novel approach to antithrombotic therapy. J Med Chem. 1999;42:213–220. doi: 10.1021/jm981072s. [DOI] [PubMed] [Google Scholar]
- 22.Xu B, Stephens A, Kirschenheuter G, Greslin AF, Cheng X, Sennelo J, Cattaneo M, Zighetti ML, Chen A, Kim SA, Kim HS, Bischofberger N, Cook G, Jacobson KA. Acyclic analogues of adenosine bisphosphates as P2Y receptor antagonists: phosphate substitution leads to multiple pathways of inhibition of platelet aggregation. J Med Chem. 2002;45:5694–5709. doi: 10.1021/jm020173u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Springthorpe B, Bailey A, Barton P, Birkinshaw TN, Bonnert RV, Brown RC, Chapman D, Dixon J, Guile SD, Humphries RG, Hunt SF, Ince F, Ingall AH, Kirk IP, Leeson PD, Leff P, Lewis RJ, Martin BP, McGinnity DF, Mortimore MP, Paine SW, Pairaudeau G, Patel A, Rigby AJ, Riley RJ, Teobald BJ, Tomlinson W, Webborn PJ, Willis PA. From ATP to AZD6140: the discovery of an orally active reversible P2Y12 receptor antagonist for the prevention of thrombosis. Bioorg Med Chem Lett. 2007;17:6013–6018. doi: 10.1016/j.bmcl.2007.07.057. [DOI] [PubMed] [Google Scholar]
- 24.Ye H, Chen C, Zhang HC, Haertlein B, Parry TJ, Damiano BP, Maryanoff BE. Carba-nucleosides as potent antagonists of the adenosine 5′-diphosphate (ADP) purinergic receptor (P2Y12 on human platelets. Chem Med Chem. 2008;3:732–736. doi: 10.1002/cmdc.200700310. [DOI] [PubMed] [Google Scholar]
- 25.Douglass JG, Patel RI, Yerxa BR, Shaver SR, Watson PS, Bednarski K, Plourde R, Redick CC, Brubaker K, Jones AC, Boyer JL. Lipophilic modifications to dinucleoside polyphosphates and nucleotides that confer antagonist properties at the platelet P2Y12 receptor. J Med Chem. 2008;51:1007–1025. doi: 10.1021/jm701348d. [DOI] [PubMed] [Google Scholar]
- 26.Douglass JG, deCamp JB, Fulcher EH, Jones W, Mahanty S, Morgan A, Smirnov D, Boyer JL, Watson PS. Adenosine analogues as inhibitors of P2Y12 receptor mediated platelet aggregation. Bioorg Med Chem Lett. 2008;18:2167–2171. doi: 10.1016/j.bmcl.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 27.Kortum SW, Lachance RM, Schweitzer BA, Yalamanchili G, Rahman H, Ennis MD, Huff RM, TenBrink RE. Thienopyrimidine-based P2Y12 platelet aggregation inhibitors. Bioorg Med Chem Lett. 2009;19:5919–5923. doi: 10.1016/j.bmcl.2009.08.059. [DOI] [PubMed] [Google Scholar]
- 28.Tu W, Fan J, Zhang H, Xu G, Liu Z, Qu J, Yang F, Zhang L, Luan T, Yuan J, Gong A, Feng J, Sun P, Dong Q. Synthesis and biological evaluation of cyclopentyl-triazolol-pyrimidine (CPTP) based P2Y12 antagonists. Bioorg Med Chem Lett. 2014;24:141–146. doi: 10.1016/j.bmcl.2013.11.055. [DOI] [PubMed] [Google Scholar]
- 29.Zamecnik PC, Kim B, Gao MJ, Taylor G, Blackburn GM. Analogues of diadenosine 5′,5‴-P1,P4-tetraphosphate (Ap4A) as potential anti-platelet-aggregation agents. Proc Natl Acad Sci U S A. 1992;89:2370–2373. doi: 10.1073/pnas.89.6.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang H, Yanachkov IB, Dix EJ, Li YF, Barnard MR, Wright GE, Michelson AD, Frelinger AL., 3rd Modified diadenosine tetraphosphates with dual specificity for P2Y1 and P2Y12 are potent antagonists of ADP-induced platelet activation. J Thromb Haemost. 2012;10:2573–2580. doi: 10.1111/jth.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang H, Yanachkov IB, Dix EJ, Yanachkova M, Li Y, Barnard MR, Wright GE, Michelson AD, Frelinger AL., 3rd Antiplatelet activity, P2Y1 and P2Y12 inhibition, and metabolism in plasma of stereoisomers of diadenosine 5′,5‴-P1,P4-dithio-P2,P3-chloromethylenetetraphosphate. PLoS One. 2014;9:e94780. doi: 10.1371/journal.pone.0094780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Husted S, van Giezen JJ. Ticagrelor: the first reversibly binding oral P2Y12 receptor antagonist. Cardiovasc Ther. 2009;27:259–274. doi: 10.1111/j.1755-5922.2009.00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kauffenstein G, Hechler B, Cazenave JP, Gachet C. Adenine triphosphate nucleotides are antagonists at the P2Y receptor. J Thromb Haemost. 2004;2:1980–1988. doi: 10.1111/j.1538-7836.2004.00926.x. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt P, Ritscher L, Dong EN, Hermsdorf T, Cöster M, Wittkopf D, Meiler J, Schöneberg T. Identification of determinants required for agonistic and inverse agonistic ligand properties at the ADP receptor P2Y12. Mol Pharmacol. 2013;83:256–266. doi: 10.1124/mol.112.082198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang H, Yanachkov IB, Michelson AD, Li Y, Barnard MR, Wright GE, Frelinger AL., 3rd Agonist and antagonist effects of diadenosine tetraphosphate, a platelet dense granule constituent, on platelet P2Y1, P2Y12 and P2X1 receptors. Thromb Res. 2010;125:159–165. doi: 10.1016/j.thromres.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deflorian F, Jacobson KA. Comparison of three GPCR structural templates for modeling of the P2Y12 nucleotide receptor. J Comput Aided Mol Des. 2011;25:329–338. doi: 10.1007/s10822-011-9423-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, Zhang K, Gao ZG, Paoletta S, Zhang D, Han GW, Li T, Ma L, Zhang W, Müller CE, Yang H, Jiang H, Cherezov V, Katritch V, Jacobson KA, Stevens RC, Wu B, Zhao Q. Agonist-bound structure of the human P2Y12 receptor. Nature. 2014;509:119–122. doi: 10.1038/nature13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang K, Zhang J, Gao ZG, Zhang D, Zhu L, Han GW, Moss SM, Paoletta S, Kiselev E, Lu W, Fenalti G, Zhang W, Müller CE, Yang H, Jiang H, Cherezov V, Katritch V, Jacobson KA, Stevens RC, Wu B, Zhao Q. Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature. 2014;509:115–118. doi: 10.1038/nature13083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chhatriwala M, Ravi RG, Patel RI, Boyer JL, Jacobson KA, Harden TK. Induction of novel agonist selectivity for the ADP-activated P2Y1 receptor versus the ADP-activated P2Y12 and P2Y13 receptors by conformational constraint of an ADP analog. J Pharmacol Exp Ther. 2004;311:1038–1043. doi: 10.1124/jpet.104.068650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoffmann K, Sixel U, Di Pasquale F, von Kügelgen I. Involvement of basic amino acid residues in transmembrane regions 6 and 7 in agonist and antagonist recognition of the human platelet P2Y12-receptor. Biochem Pharmacol. 2008;76:1201–1213. doi: 10.1016/j.bcp.2008.08.029. [DOI] [PubMed] [Google Scholar]
- 41.Ignatovica V, Megnis K, Lapins M, Schioth HB, Klovins J. Identification and analysis of functionally important amino acids in human purinergic 12 receptor using a Saccharomyces cerevisiae expression system. FEBS J. 2012;279:180–191. doi: 10.1111/j.1742-4658.2011.08410.x. [DOI] [PubMed] [Google Scholar]
- 42.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shaw DE, Shelley M, Perry JK, Francis P, Shenkin PS. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 43.Boldron C, Besse A, Bordes MF, Tissandié S, Yvon X, Gau B, Badorc A, Rousseaux T, Barré G, Meneyrol J, Zech G, Nazare M, Fossey V, Pflieger AM, Bonnet-Lignon S, Millet L, Briot C, Dol F, Hérault JP, Savi P, Lassalle G, Delesque N, Herbert JM, Bono F. N-[6-(4-Butanoyl-5-methyl-1H-pyrazol-1-yl)pyridazin-3-yl]-5-chloro-1-[2-(4-methylpiperazin-1-yl)-2-oxoethyl]-1H-indole-3-carboxamide (SAR216471), a Novel Intravenous and Oral, Reversible, and Directly Acting P2Y12 Antagonist. J Med Chem. 2014;57:7293–7316. doi: 10.1021/jm500588w. [DOI] [PubMed] [Google Scholar]
- 44.Hoffmann K, Baqi Y, Morena MS, Glänzel M, Müller CE, von Kügelgen I. Interaction of new, very potent non-nucleotide antagonists with Arg256 of the human platelet P2Y12 receptor. J Pharmacol Exp Ther. 2009;331:648–655. doi: 10.1124/jpet.109.156687. [DOI] [PubMed] [Google Scholar]
- 45.Hoffmann K, Lutz DA, Straßburger J, Baqi Y, Müller CE, von Kügelgen I. Competitive mode and site of interaction of ticagrelor at the human platelet P2Y12-receptor. J Thromb Haemost. 2014;12:1898–1905. doi: 10.1111/jth.12719. [DOI] [PubMed] [Google Scholar]
- 46.Jacobson KA, Paoletta S, Katritch V, Wu B, Zhao Q, Stevens RC, Kiselev E. Nucleotides Acting at P2Y Receptors: Connecting Structure and Function. Mol Pharmacol. 2015;88:220–230. doi: 10.1124/mol.114.095711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 48.Sabbadin D, Moro S. Supervised molecular dynamics (SuMD) as a helpful tool to depict GPCR-ligand recognition pathway in a nanosecond time scale. J Comp Inf Mod. 2014;54:372–376. doi: 10.1021/ci400766b. [DOI] [PubMed] [Google Scholar]
- 49.Sabbadin D, Ciancetta A, Moro S. Perturbation of fluid dynamics properties of water molecules during G protein-coupled receptor-ligand recognition: the human A2A adenosine receptor as a key study. J Chem Inf Model. 2014;54:2846–2855. doi: 10.1021/ci500397y. [DOI] [PubMed] [Google Scholar]
- 50.Carugo O. Buried chloride stereochemistry in the Protein Data Bank. BMC Struct Biol. 2014;14:19. doi: 10.1186/s12900-014-0019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ballesteros JA, Weinstein H. Integrated methods for the construction of three dimensional models and computational probing of structure function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 52.Schrödinger Suite 2014. Schrödinger, LLC; New York, NY: [Google Scholar]
- 53.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–28. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 54.Seeber M, Felline A, Raimondi F, Muff S, Friedman R, Rao F, Caflisch A, Fanelli F. Wordom: a user-friendly program for the analysis of molecular structures, trajectories, and free energy surfaces. J Comput Chem. 2011;32:1183–1194. doi: 10.1002/jcc.21688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.MacKerell AD, Jr, Banavali N, Foloppe N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 2000;56:257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 56.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Post JM, Alexander S, Wang YX, Vincelette J, Vergona R, Kent L, Bryant J, Sullivan ME, Dole WP, Morser J, Subramanyam B. Novel P2Y12 adenosine diphosphate receptor antagonists for inhibition of platelet aggregation (II): pharmacodynamic and pharmacokinetic characterization. Thromb Res. 2008;122:533–540. doi: 10.1016/j.thromres.2008.04.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.