

Lee et al. show that elevating miR-431 improved the myogenic capacity of old myoblasts, while inhibiting endogenous miR-431 lowered myogenesis. In an in vivo model of muscle regeneration following cardiotoxin injury, ectopic miR-431 injection greatly improved muscle regeneration and reduced SMAD4 levels.
Keywords: miR-431, SMAD4, muscle aging, myoblast, differentiation, regeneration
Abstract
The myogenic capacity of myoblasts decreases in skeletal muscle with age. In addition to environmental factors, intrinsic factors are important for maintaining the regenerative potential of muscle progenitor cells, but their identities are largely unknown. Here, comparative analysis of microRNA (miRNA) expression profiles in young and old myoblasts uncovered miR-431 as a novel miRNA showing markedly reduced abundance in aged myoblasts. Importantly, elevating miR-431 improved the myogenic capacity of old myoblasts, while inhibiting endogenous miR-431 lowered myogenesis. Bioinformatic and biochemical analyses revealed that miR-431 directly interacted with the 3′ untranslated region (UTR) of Smad4 mRNA, which encodes one of the downstream effectors of TGF-β signaling. In keeping with the low levels of miR-431 in old myoblasts, SMAD4 levels increased in this myoblast population. Interestingly, in an in vivo model of muscle regeneration following cardiotoxin injury, ectopic miR-431 injection greatly improved muscle regeneration and reduced SMAD4 levels. Consistent with the finding that the mouse miR-431 seed sequence in the Smad4 3′ UTR is conserved in the human SMAD4 3′ UTR, inhibition of miR-431 also repressed the myogenic capacity of human skeletal myoblasts. Taken together, our results suggest that the age-associated miR-431 plays a key role in maintaining the myogenic ability of skeletal muscle with age.
Skeletal muscle exhibits a degenerative age-associated decline in mass and function, termed sarcopenia (Narici and Maffulli 2010). Sarcopenia can be triggered by a variety of conditions, including hormonal imbalances, motor neuron degeneration, inflammation, and oxidative stress (Frontera et al. 1991; Hughes et al. 2001; Goodpaster et al. 2006). Recent studies revealed that muscle stem cells, also known as satellite cells, showed age-associated phenotypes that included loss of self-renewal capacity, reduced regenerative potential, and senescence (Bernet et al. 2014; Cosgrove et al. 2014; Sousa-Victor et al. 2014). The gradual reduction of satellite cells is a major underlying cause of sarcopenia with age (Brack et al. 2005; Shefer et al. 2006; Collins et al. 2007). However, after activation of satellite cells into myoblasts, the mechanisms responsible for the decreased myogenic capacity of aged myoblasts have remained unclear.
So far, most research on muscle differentiation and regeneration has primarily focused on the regulation of myogenesis by myogenic regulatory factors (MRFs) such as MYOD, MEF2, and MYOG (Sabourin and Rudnicki 2000; Sartorelli and Caretti 2005; Wang and Rudnicki 2012). In addition, several recent studies have shown that microRNAs (miRNAs) are also involved in the maintenance of muscle stem cell quiescence and in muscle diseases, including skeletal muscle hypertrophy and muscular disorders (Eisenberg et al. 2007; Thum et al. 2007; Cheung et al. 2012; Dey et al. 2012; Liu et al. 2012). However, the roles of miRNAs in age-associated muscular disorders are largely unknown. miRNAs are noncoding RNAs spanning ∼22 nucleotides that play an important role in gene expression at the post-transcriptional level via mRNA degradation and/or inhibition of translation (Ambros 2004; Bartel 2004). The finding that skeletal muscle-specific knockout of Dicer, a key enzyme for maturation of precursor miRNAs (pre-miRNA), causes a decrease in muscle mass and abnormal muscle formation in mice, causing perinatal death, suggests that miRNAs have a key role in normal muscle development (O'Rourke et al. 2007). In addition, we recently found that the miRNA–mRNA regulatory network in skeletal muscle changes with age, suggesting that miRNAs may be involved in the process of muscle aging (Kim et al. 2014).
Transforming growth factor β (TGF-β) signaling inhibits myogenesis by down-regulating the transcriptional activity of MRFs (Martin et al. 1992; Liu et al. 2001). Moreover, previous studies showed that activated TGF-β signaling delays muscle differentiation and inhibits aged muscle regeneration (Carlson et al. 2008, 2009; Gopinath and Rando 2008; Jang et al. 2011). Old muscle cells produce high levels of TGF-β, which in turn binds and activates TGF-β receptors in resident satellite cells. Activated TGF-β receptors phosphorylate SMAD2 and SMAD3, and phosphorylated SMAD2/3 form a complex with SMAD4 (the common mediator SMAD [Co-SMAD]). The SMAD complex translocates into the nucleus and activates a transcriptional program that interferes with the regenerative capacity of satellite cells. However, satellite cells isolated from old muscle tissues also exhibit less myogenic capacity, suggesting that one or several intrinsic cellular factors participate in age-associated muscle degeneration.
Here, analysis of miRNA expression profiles of myoblasts prepared from young and old mouse skeletal muscles uncovered a novel age-associated miRNA, miR-431. We describe that miR-431 regulates SMAD4 expression and hence modulates the skeletal myogenic program in myoblasts with age.
Results
Differential expression of miRNAs between young and old myoblasts
To find miRNAs differentially expressed in skeletal muscle with advancing age, we analyzed miRNA expression profiles in young and old mouse myoblasts using next-generation sequencing (NGS). We isolated primary myoblasts from hindlimb muscles of 3-mo-old (hereafter designated “young myoblasts”) and 27-mo-old (hereafter designated “old myoblasts”) mice. Two proteins, PAX7 and MYOD, were used as specific markers for primary myoblasts (Wang and Rudnicki 2012). By immunofluorescence analysis, ∼99% of isolated cells expressed both PAX7 and MYOD (Supplemental Fig. 1). Consistent with previous reports (Schultz and Lipton 1982; Fulle et al. 2005; Wagers and Conboy 2005), old myoblasts showed decreased myogenic capability, with decreased numbers of differentiated myotubes positive for myosin heavy chain (MyHC) (Supplemental Fig. 2A,B) and decreased levels of MyHC abundance (Supplemental Fig. 2C) relative to young myoblasts. The levels of Myog mRNA, encoding the myogenic transcription factor, as well as MyHC mRNA were also significantly down-regulated in old myoblasts (Supplemental Fig. 2D).
Among the 118 mature miRNAs that showed significant changes (greater than twofold) between young and old myoblasts (Fig. 1A), 47 miRNAs were significantly up-regulated, and 71 miRNAs were down-regulated in old myoblasts (Tables 1, 2). We recently reported that 57% of miRNAs down-regulated in old muscle tissues were located in the Dlk-Dio3 region of chromosome 12 (Kim et al. 2014). Interestingly, 63 of the 71 miRNAs (89%) down-regulated in old myoblasts were also located in the Dlk-Dio3 genomic region, suggesting that miRNAs expressed from this locus may be relevant to the process of muscle aging. We thus focused on the miRNAs located in this genomic region.
Figure 1.
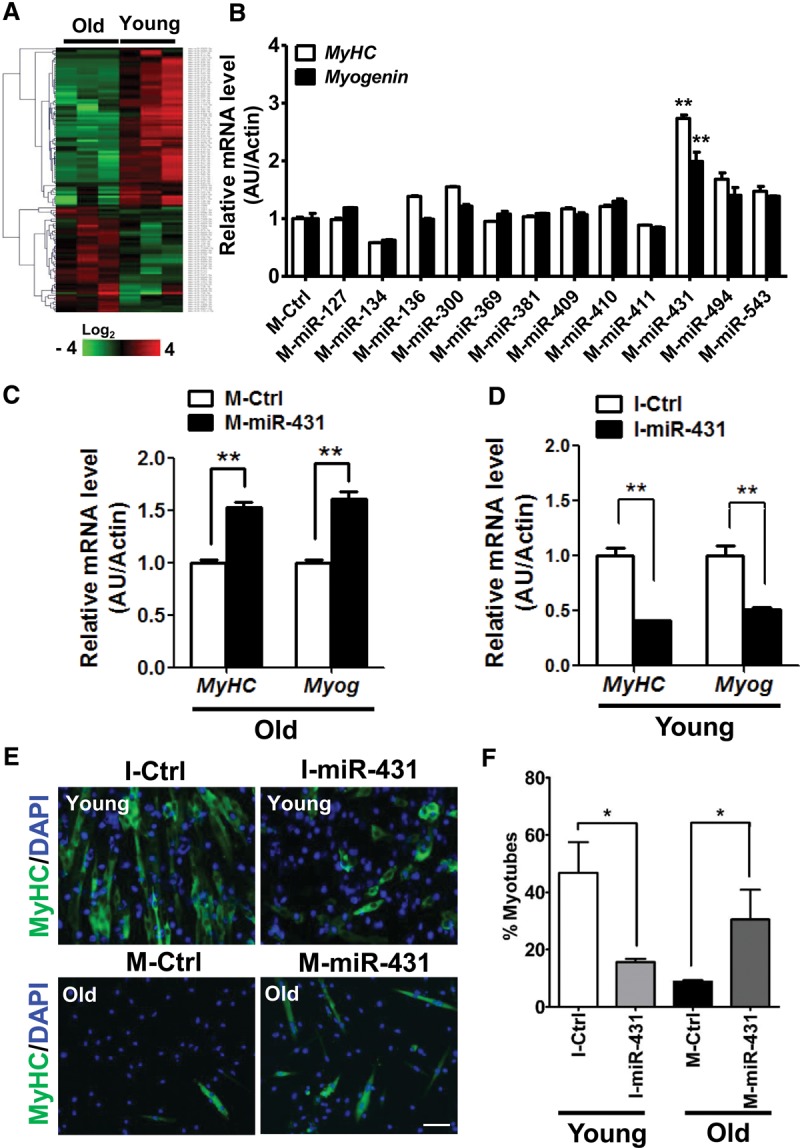
miR-431 promotes differentiation of old myoblasts. (A) Unsupervised hierarchical clustering of 118 differentially regulated miRNAs with aging; 47 miRNAs were up-regulated, and 71 miRNAs were down-regulated. Each column represents miRNA levels in young (n = 3) and old (n = 3) myoblasts isolated from 3-mo-old and 27-mo-old mice each. The intensity represents the magnitude of the difference. Red and green denote high and low expression, respectively. (B) After transfection with the miRNA mimics indicated, old myoblasts were induced to differentiate in differentiation medium. The relative expression of differentiation marker genes such as MyHC (white bar; n = 3 for each group) and Myog (black bar; n = 3 for each group) were quantified by RT-qPCR. The results were normalized to the amount of Actb (β-actin) mRNA. The data are presented as the means ± SD. (**) P < 0.01. (C) After transfection with Ctrl-mimic (M-Ctrl) or miR-431 mimic (M-miR-431), old myoblasts were induced to differentiate in differentiation medium and analyzed for the levels of differentiation markers such as MyHC and Myog mRNAs. (D) Young myoblasts transfected with the miR-431 inhibitor (I-miR-431) showed decreased levels of the differentiation markers tested (MyHC and Myog mRNAs). The results were normalized by the average of Actb mRNA. Data are presented as the means ± SD. (**) P < 0.01. (E) Representative images of differentiated myotubes in I-miR-431 transfected young myoblasts (top panels), M-miR-431 transfected old myoblasts (bottom panels), and the corresponding control transfections; immunofluorescence staining was used to detect MyHC (green) and DAPI (blue). Bar, 200 µm. (F) The fusion index was assessed by calculating the percentage of MyHC-positive cells that contained two or more nuclei versus total MyHC-positive cells. Six different views were randomly selected for measurement of the fusion index. (*) P < 0.05.
Table 1.
miRNAs up-regulated in old myoblasts

Table 2.
miRNAs down-regulated in old myoblasts

Exogenous miR-431 improves myogenic differentiation of old myoblasts
We reasoned that the myogenic capability of old myoblasts could be restored by supplementation of the miRNAs that showed reduced levels with age. We selected 12 such miRNAs from the Dlk-Dio3 region, transfected them into old myoblasts, and analyzed the levels of markers MyHC mRNA and Myog mRNA to monitor myogenesis. We found the highest induction of MyHC and Myog mRNAs in old myoblasts transfected with a mimic (M) of miR-431 (M-miR-431) (Fig. 1B,C). Moreover, both MyHC and Myog mRNA were reduced in young myoblasts transfected with an inhibitor (I) of miR-431 (the antagomiR I-miR-431) (Fig. 1D). These results strongly suggested that miR-431, one of the miRNAs showing reduced levels in old myoblasts, is an important regulatory miRNA of myogenesis with age. Notably, M-miR-431 did not elevate MyHC and Myog mRNAs in young myoblasts, likely because the levels of miR-431 were already high in young myoblasts. Likewise, I-miR-431 did not further decrease MyHC and Myog mRNAs in old myoblasts (Supplemental Fig. 3), suggesting that the levels of miR-431 might be saturated in young myoblasts but depleted in old myoblasts, consistent with our NGS results.
Next, we asked whether transfection of M-miR-431 might be able to restore differentiation of old myoblasts, as determined by assessing myotube morphology and the number of MyHC-positive myotubes. Interestingly, M-miR-431 induced myogenesis of old myoblasts, with the appearance of more spindle-like, elongated myotubes, and, conversely, I-miR-431 suppressed the myogenic capability of young myoblasts (Fig. 1E). The number of MyHC-positive cells that contained two or more nuclei relative to the total MyHC-positive cells was significantly increased in M-miR-431 transfected old myoblasts (Fig. 1F), further suggesting that miR-431 plays an important role in maintaining the age-dependent myogenic capacity of myoblasts.
miR-431 regulates SMAD4 expression through direct binding to the Smad4 3′ untranslated region (UTR)
In order to identify the target mRNAs regulated by miR-431, we searched for putative targets using TargetScan (http://www.targetscan.org) and miRanda (http://www.microRNA.org). One potential target of miR-431 was SMAD4, a protein of interest given that SMAD4 negatively regulates myogenic differentiation (Dey et al. 2012; Khanna et al. 2014). Together with phosphorylated SMAD2/3 (a modification elicited via TGF-β signaling), the SMAD complex delays muscle regeneration in old mice. We thus asked whether miR-431 regulates SMAD4 expression.
Among the 71 miRNAs down-regulated in old myoblasts (Table 2), putative target sites for four miRNAs—miR-411, miR-434, miR-673, and miR-431—were identified on the 3′ UTR of mouse Smad4 mRNA (Fig. 2A). Reporter analysis using a construct that expressed the luciferase-Smad4 3′ UTR and miRNA mimics indicated that among the four miRNAs, only miR-431 reduced luciferase activity (Fig. 2B). This inhibition was specific, as deletion of the miR-431 site (Mut 3′ UTR) on the Smad4 3′ UTR abolished this repression (Fig. 2C).
Figure 2.

miR-431 regulates SMAD4 expression by directly interacting with the Smad4 3′ UTR. (A) Schematic depicting the sites of putative interaction of down-regulated miRNAs within the 3′ UTR of the Smad4 mRNA. (B) The effect of M-miR-431 on the activity of a luciferase-Smad4 3′ UTR reporter construct. (**) P < 0.01. (C) Effect of miR-431 on the activity of luciferase reporters bearing either a wild-type Smad4 3′ UTR or a mutant (MT) Smad4 3′ UTR with a deletion of the miR-431 site. (D) Lysates (300 µg) from young or old muscle tissues were incubated with biotinylated (Bi)-miR431-5p or Bi-cel-miR-67 (Ctrl miR) at 4°C with rotation. Four hours later, the RNA was isolated from pull-down material using streptavidin beads and was analyzed by RT-qPCR for Smad4 mRNA enrichment; normalization was carried out using Gapdh mRNA for young or old muscle tissue. The data represent the means ± SEM of three independent experiments. (*) P < 0.05. (E) Lysates from young myoblasts were incubated with antisense oligomers (ASOs) complementary to Smad4 mRNA or a negative control RNA (Gm14635 lincRNA). The RNA isolated after pull-down using streptavidin beads and Smad4 mRNA (normalized to Gapdh mRNA) (left) or for miR431-5p (normalized to RNU6B) (right) were measured by RT-qPCR analysis. (F) Levels of SMAD4 in I-miR-431 transfected young myoblasts (left) and in M-miR-431 transfected old myoblasts (right), each relative to control cells.
Direct interaction between miR-431 and endogenous Smad4 mRNA was confirmed by pull-down experiments using biotinylated (Bi)-miR-431 or Smad4 antisense oligomers (ASOs) as baits. C2C12 cells were first transfected with Bi-miR-431 or (control) Bi-cel-miR-67, and RNA was isolated from cell lysates by pull-down using streptavidin-coupled beads. Smad4 mRNA was found enriched in the RNA isolated by pull-down from lysates of Bi-miR-431 transfected cells, as detected by RT-qPCR analysis (Supplemental Fig. 4A). Smad4 mRNA was also enriched after pull-down using Bi-miR-431 and muscle tissue lysates (Fig. 2D). Conversely, miR-431 was significantly enriched in Bi-Smad4 ASO pull-down using lysates from either primary myoblasts or C2C12 cells (Fig. 2E; Supplemental Fig. 4B). Together, these results support the notion that endogenous Smad4 mRNA and endogenous miR-431 interact physically.
As anticipated, SMAD4 protein levels were lower in old myoblasts transfected with the miR-431 mimic and were higher in young myoblasts transfected with the miR-431 antagomir (I-miR-431) than in control transfected myoblasts (Fig. 2F). Similar results were found in the mouse myoblast cell line C2C12 following treatment with M-miR-431 or I-miR-431 (Supplemental Fig. 4C). Taken together, these results indicate that miR-431 repressed the expression of SMAD4 by direct binding to Smad4 mRNA.
Another putative miR-431 target predicted by TargetScan was SMURF1, an E3 ubiquitin ligase that regulates SMAD4 expression (Moren et al. 2005). We thus studied whether SMURF1 might also be regulated by miR-431. We found that SMURF1 levels were not elevated in old myoblasts, where miR-431 levels were low; additionally, SMURF1 levels remained unchanged after M-miR-431 transfection in old myoblasts (Supplemental Fig. 5). These results indicated that miR-431 is unlikely to target SMURF1, at least in muscle cells, thus ruling out the possibility that miR-431 regulates SMAD4 levels indirectly via SMURF1-regulated proteolysis.
SMAD4 knockdown promotes myogenesis in old myoblasts
Given the facts that miR-431 can bind to the Smad4 mRNA, in turn reducing SMAD4 expression levels, and that miR-431 abundance decreased in old myoblasts, we investigated the expression levels of SMAD4 as well as other downstream effectors of TGF-β signaling (SMAD2 and SMAD3) in young and old myoblasts. Interestingly, SMAD3 and SMAD4 were robustly up-regulated in old myoblasts, while SMAD2 levels remained unchanged (Fig. 3A). To determine whether the increase in SMAD3 and SMAD4 was associated with decreased myogenic capability in old myoblasts, we knocked down SMAD3 or SMAD4 in old myoblasts using siRNA and examined myogenesis relative to control siRNA transfected old myoblasts. Knockdown of SMAD4 significantly (and selectively) up-regulated the expression levels of myogenic markers (MyHC and Myog mRNAs), while knockdown of SMAD3 did not (Fig. 3B). Consistently, the number of differentiated myotubes was significantly higher in siSmad4 transfected than in control siRNA transfected old myoblasts (Fig. 3C), suggesting that SMAD4 is a major regulatory SMAD protein responsible for the age-associated decline in myogenic capacity.
Figure 3.

Knockdown of SMAD4 promotes myogenesis of old myoblasts. (A, top) Lysates of four independently isolated young and old myoblasts were subjected to immunoblot analysis with antibodies to detect the indicated proteins, and ACTB was used as a loading control. (Bottom) Quantification of Western blotting signals is presented as the means ± SD. (*) P < 0.05. (B, top) Twenty-four hours after old myoblasts were transfected with siCtrl and siSmad3 or siSmad4, myogenesis was induced using differentiation medium. Differentiation markers MyHC mRNA and Myog mRNA were identified by RT-qPCR analysis. (Bottom) The effect of SMAD4 knockdown by assessing the same parameters. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. (C) Immunofluorescence staining for MyHC (green) and DAPI (blue) to detect differentiated myotubes in old myoblasts expressing normal or silenced levels of SMAD4. Bar, 200 µm. (Right) The fusion index was measured by the percentage of MyHC-positive cells that contained two or more nuclei versus total MyHC-positive cells. Six different views were randomly selected for measurement of the fusion index. (*) P < 0.05. (D) Expression levels of the indicated proteins, as determined by Western blot analysis in young and old myoblasts cultured in differentiation medium for up to 3 d (DM3). ACTB was used as a loading control. (E) Relative expression of miR-431 in the cells described in D. The data were normalized to the amount of U6 snoRNA and were expressed relative to the normalized value for young myoblasts in growth medium. (GM) Growth medium; (DM) differentiation medium. (F) The effect of miR-431 on the repression of myogenesis due to TGF-β treatment. After transfection with M-Ctrl or M-miR-431, old myoblasts were incubated in differentiation medium with 5 ng/mL TGF-β1 for 2 d. The levels of MyHC mRNA and Myog mRNA were measured by RT-qPCR analysis. The results were normalized to Actb mRNA levels in each sample, as measured by RT-qPCR analysis. Data are presented as the means ± SD. (**) P < 0.01. (G) The effect of miR-431 on the SMAD signaling induced by the treatment with TGF-β1. C2C12 cells were transfected with M-miR-431 or I-miR-431 together with the 4xSBE-luc reporter construct. Cells were treated with 5 ng/mL TGF-β1 for 16 h before luciferase analysis. The values represent fold induction relative to the basal activity of 4xSBE-luc in a dual-luciferase assay. (*) P < 0.05.
We next analyzed the expression levels of SMAD4 and the Smad4 mRNA regulator miR-431 during myogenesis of young and old myoblasts. SMAD4 levels gradually decreased as old myoblasts differentiated, while miR-431 levels increased (Fig. 3D,E). In young myoblasts, basal miR-431 levels were high and increased further with myogenesis. Accordingly, SMAD4 was lowest during differentiation of young myoblasts. Together, these results indicated that SMAD4 might be down-regulated by miR-431 in order for myoblasts to differentiate into mature myotubes and that these functions are impaired in old myoblasts.
Since TGF-β signaling inhibits muscle differentiation and delays regeneration in aged muscle (Carlson et al. 2008, 2009; Gopinath and Rando 2008; Jang et al. 2011), we asked whether miR-431 could control TGF-β signaling by regulating SMAD4 levels. Exogenous transfection of miR-431 ameliorated the repression of myogenesis seen in myoblasts treated with recombinant TGF-β1 (Fig. 3F). Moreover, miR-431 was capable of suppressing the increased luciferase activity of the 4xSBE-luc reporter, which contains four SMAD-binding elements in its promoter region (Zawel et al. 1998), in C2C12 cells treated with TGF-β1 (Fig. 3G). Myostatin (MSTN) and Activin A (ActA) are two members of a TGF-β superfamily known to regulate muscle homeostasis (McPherron et al. 1997; Lee et al. 2010; Massague 2012). Consistent with the effects of TGF-β1 treatment, MSTN- and ActA-induced luciferase activity of 4xSBE-luc was reduced by miR-431 (Supplemental Fig. 6). However, miR-431 did not affect the rise in SMAD2/3 phosphorylation following treatment with TGF-β1 (Supplemental Fig. 7). Our findings suggest that miR-431 affects SMAD signaling by controlling SMAD4 protein levels but not by controlling SMAD2/3 phosphorylation status.
Increased SMAD4 abundance during muscle regeneration in aged mice
To investigate the function of SMAD4 and miR-431 in muscular tissue regeneration in vivo, we employed a mouse model of muscle injury (Charge and Rudnicki 2004; Liu et al. 2012). To activate satellite cells into myoblasts, we induced skeletal muscle injury and regeneration by injecting cardiotoxin (CTX) into tibialis anterior (TA) muscle of young and old mice. Consistent with previous reports (Conboy and Rando 2012), old TA muscles showed impaired regeneration after CTX injection, whereas young TA muscles regenerated well (Supplemental Fig. 8A). During muscle regeneration for the following 14 d, we compared the expression levels of SMAD4 and miR-431. Similar to the expression pattern during myoblast differentiation, SMAD4 levels were higher in old muscle than in young muscle. Interestingly, SMAD4 levels increased dramatically after CTX injection and declined to the basal level as young muscle regenerated. However, in old TA muscles, SMAD4 levels were increased further by CTX injury and remained elevated for 14 d (Fig. 4A; Supplemental Fig. 8B). In contrast, expression of miR-431 remained lower in old TA muscles than in young TA muscles (Fig. 4A).
Figure 4.

Levels of SMAD4 and miR-431 during regeneration of injured muscles. (A) SMAD4 expression patterns were determined using Western blot analysis in CTX-injured young and old TA muscle tissues for up to 14 d post-injury (DPI). (Top) GAPDH was included as a loading control. RT-qPCR analysis of miR-431 in CTX-injured young and old TA muscle tissues for up to 14 d. (Bottom) Expression levels were measured in triplicate, and the data were normalized to the amount of U6 snoRNA and expressed relative to the normalized value for young TA muscle tissues at day 0. (B) At day 14 after CTX injury, young and old TA muscle tissue sections were stained with antibodies that recognized PAX7 (green) and SMAD4 (red), revealing the colocalization of both antigens in myoblasts localized in old muscle tissues. Bar, 20 µm. (White dotted lines) Muscle fibers. (***) P < 0.001.
To investigate where SMAD4 was expressed in regenerating muscle tissues, we performed immunohistochemical analysis of tissue sections at day 14 after CTX injection. Interestingly, SMAD4 was intensely present in PAX7-positive myoblasts (Fig. 4B), not in mature muscle fibers. Old muscle contained more (80%) SMAD4-positive myoblasts than young muscle (∼20%), suggesting the possibility that the elevated SMAD4 in aged myoblasts contributes to impaired regeneration of old muscle tissues.
miR-431 restores regeneration in old mice
To further investigate the therapeutic potential of miR-431 on aged muscle regeneration, we examined whether miR-431 or siSmad4 could improve the impaired muscle regeneration seen in old mice in vivo. We injected miR-431 or siSmad4 into old TA muscles on days 2 and 5 after CTX injury (Fig. 5A) and confirmed that SMAD4 levels were down-regulated in siSmad4-treated or M-miR-431-treated muscle tissues at day 6 after CTX injury (Supplemental Fig. 9). Two weeks later, we investigated the degree of muscle regeneration by performing immunohistochemical analysis of DAPI and laminin in tissue sections. As shown in Figure 5B, the newly formed myofibers with central nuclei had significantly larger diameters in SMAD4 knockdown TA muscle tissues than in control muscle tissues. Furthermore, miR-431 enhanced muscle regeneration in old mice, as muscles injected with miR-431 displayed significantly larger regenerating fibers than control muscles (Fig. 5C). These results support the idea that miR-431 improves muscle regeneration in old mice by targeting SMAD4, underscoring its therapeutic potential for healthy muscle aging.
Figure 5.
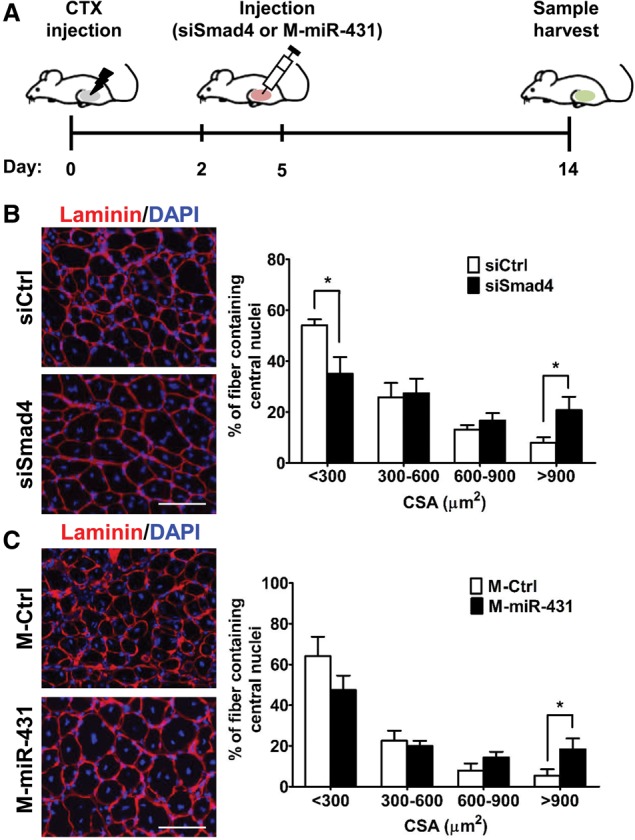
siSmad4-treated or miR-431-treated old muscles showed enhanced regenerative capacity. (A) Scheme of the experimental procedures. CTX (20 µM) was injected into TA muscle tissues of old mice at day 0. Two days and 5 d later, injured muscle tissues were injected with siSmad4 or miR-431 mimic, and contralateral muscle tissues were injected with control siRNA or miRNA mimic. At 14 d after injury, muscle tissues were analyzed by immunohistochemistry. (B,C) Representative immunohistochemistry images (left panels) of laminin (red) and DAPI (blue) staining of siSmad4-treated (B) and miR-431 mimic-treated (C) regenerating fibers (n = 4) on day 14 after injury. Each cross-section area (CSA) was measured by ImageJ software, and six different views were randomly selected for measurement of CSA; (*) P < 0.05. Bar, 100 µm.
miR-431 regulates differentiation of human skeletal muscle myoblasts (HSMMs)
Since the miR-431 seed sequence is conserved between the human SMAD4 3′ UTR and the mouse Smad4 3′ UTR (Supplemental Fig. 10), we anticipated that miR-431 might regulate human SMAD4 expression levels as well. To test this possibility, we analyzed SMAD4 protein levels and differentiation indices in HSMMs transfected with M-miR-431 or I-miR-431. Consistent with the results obtained in mouse myoblasts, M-miR-431 transfection down-regulated SMAD4 protein levels in HSMMs, whereas I-miR-431 increased SMAD4 levels (Fig. 6A). While M-miR-431 did not promote myogenic differentiation significantly in HSMMs (Supplemental Fig. 11), I-miR-431 repressed myogenic differentiation of HSMMs robustly (Fig. 6B,C). Perhaps the lack of response of HSMMs to M-miR-431 is related to the presence of high levels of miR-431, as HSMM cells originated from a young (17-yr-old) donor (Lonza). Together with the results in mouse myoblasts, our data collectively indicate that down-regulation of miR-431 in myoblasts is an important intrinsic cue to delay myogenesis, a major phenotype of aged myoblasts.
Figure 6.
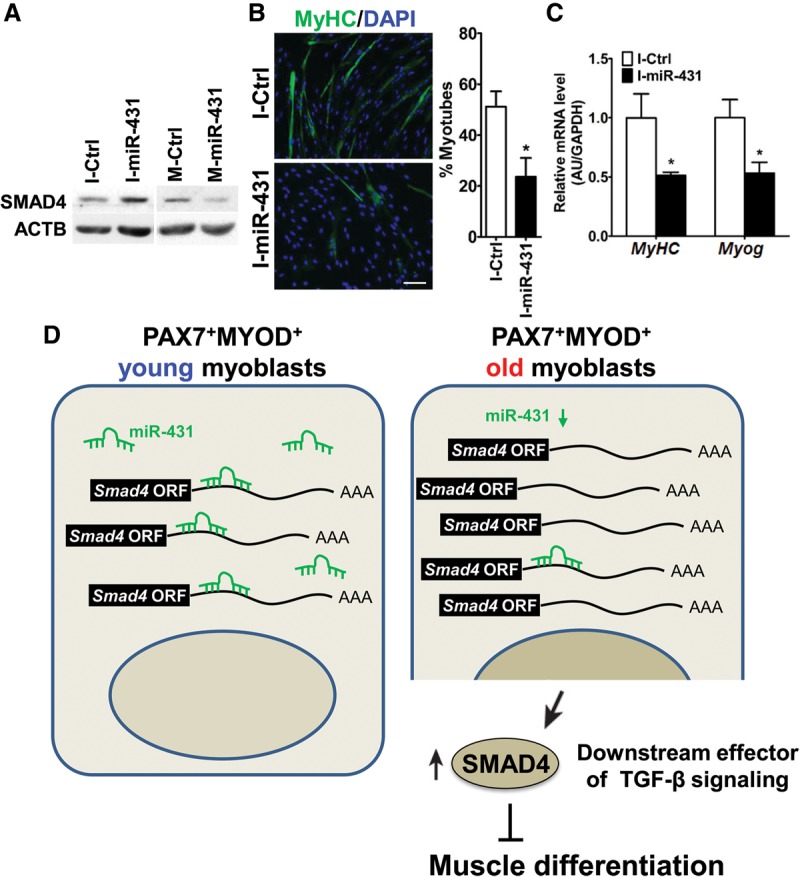
The effect of miR-431 inhibition on the differentiation of HSMMs. (A) Levels of SMAD4 protein after transfection of HSMMs in growth medium (GM) with I-miR-431 or M-miR-431. Protein levels were detected by Western blot analysis, and β-actin (ACTB) was used as a loading control. (B) After transfection with I-Ctrl or I-miR-431, HSMMs were placed in differentiation medium (DM), and the numbers of differentiated myotubes were compared with those in control cells after immunofluorescence staining for MyHC (green) and DAPI (blue). Bar, 200 µm. (C) Levels of differentiation markers (MyHC and Myog mRNAs) in I-miR-431 transfected HSMMs. Data represent the means ± SD. (*) P < 0.05. (D) Proposed model for the regulation of myoblast differentiation by miR-431 in different ages. Briefly, decreased miR-431 in old PAX7+MYOD+ myoblasts results in increased levels of SMAD4, a downstream effector of signaling by the myogenesis repressor TGF-β, thereby interrupting normal muscle differentiation.
Discussion
Our study provides evidence supporting a key role for miR-431 in the maintenance of myogenic capability of myoblasts with age. We identified 118 age-related miRNAs in myoblasts from different ages using NGS. Among them, we focused on miR-431, which we found to regulate SMAD4 expression by directly targeting the Smad4 3′ UTR. Interestingly, in myoblasts of aged mice, SMAD4 protein levels increased dramatically, while miR-431 levels declined. We found that ectopic elevation of miR-431 improved the differentiation of old myoblasts in culture and restored the regeneration of muscle in old mice in vivo. Conversely, antagonization of miR-431 delayed the differentiation of human myoblasts and elevated SMAD4 expression, in agreement with the fact that the miR-431 site on the human SMAD4 3′ UTR is conserved. Taken together, our findings suggest that miR-431 is required for maintenance of the myogenic capability of PAX7+MYOD+ myoblasts (Fig. 6D), underscoring its potential usefulness as a therapeutic target to slow down muscle aging.
Muscle-specific miRNAs (myomiRs), including miR-1, miR-133, and miR-206, are highly expressed in cardiac and skeletal muscles. Several studies revealed that myomiRs are highly relevant to muscle development and diseases, including muscle hypertrophy and atrophy (Callis et al. 2007; McCarthy and Esser 2007; McCarthy et al. 2009; Townley-Tilson et al. 2010; McCarthy 2011). miR-1 and miR-206 promoted myogenic differentiation, miR-133 repressed myogenesis by increasing myoblast proliferation, and miR-1 and miR-133 levels declined in hypertrophic muscle tissues. Recent studies in miR-206 knockout and mdx mice showed that miR-206 enhanced skeletal muscle regeneration and delayed progression of Duchenne muscular dystrophy (Liu et al. 2012). However, none of these myomiRs showed different abundance as a function of age in our screen of young and old myoblasts, suggesting that they may not be involved in the age-related impairment of myogenesis. Instead, we identified miR-431 as the first candidate age-related myomiR. The TGF-β/Smad signaling pathway is well known to inhibit muscle development, myogenesis, and age-related muscle regeneration (Massague et al. 1986; Liu et al. 2001, 2004). The aged muscle produces excessive TGF-β, which in turn induces high levels of phospho-SMAD3 in muscle stem cells, thus inhibiting their myogenic capability (Carlson et al. 2008). In addition to this age-related increase in extracellular signals that block muscle regeneration, we propose that a decrease in an intrinsic cellular factor, miR-431, delays myogenesis and impairs muscle regeneration with advancing age.
Overexpression of miR-26a promoted muscle differentiation and regeneration through down-regulation of SMAD1 and SMAD4 (Dey et al. 2012), two factors that were down-regulated by miR-26a in vascular smooth muscle cells (Leeper et al. 2011). Since miR-26a expression levels did not change between young and old myoblasts, miR-431 may be the first age-related miRNA responsible for SMAD4-dependent impairment of muscle differentiation and regeneration in old mice. Our findings further suggest that SMAD4 may be a promising new target in therapeutic efforts (possibly involving a rise in miR-431 function) to prevent age-related muscle loss. It will be interesting to see whether miR-431 and other age-related miRNAs are also involved in dysfunctions such as impaired self-renewal and differentiation observed in aged PAX7+MYOD− satellite cells. Since differentiation into myotubes requires activation of satellite cells (PAX7+ MYOD+ myoblasts), which in turn differentiate into myotubes or proliferate themselves (Wang and Rudnicki 2012), the mechanisms underlying this myoblast-to-myotube transition point to key regulators with implications for therapy in muscle recovery from injury, a most debilitating problem that afflicts the aging population.
Our recent report demonstrated that 57% of down-regulated miRNA in old muscle tissues was located in the Dlk-Dio3 region on chromosome 12 (Kim et al. 2014), and here we also found that 89% of miRNAs down-regulated in old myoblasts were also located in this genomic region. Therefore, we hypothesized that miRNAs located in the Dlk-Dio3 region could have an important role in the aging process for myoblasts, such as delayed myogenesis. We thus primarily focused on this genomic region and discovered that miR-431 was a novel miRNA influencing the myogenic capability of old myoblasts. It remains to be seen whether the miRNAs within the Dlk-Dio3 region are involved in the aging processes in not only muscle but also other organs. Future investigations into the relevance of other Dlk-Dio3 miRNAs for maintenance of muscle homeostasis may provide further important insight into our understanding of muscle aging.
In conclusion, we found that the conserved miR-431 regulates SMAD4 expression in muscle cells from both humans and mice, promotes regeneration in old mouse skeletal muscle, and enhances differentiation of old myoblasts. Taking our findings together, we propose that miR-431 is a valuable candidate for the development of therapies aimed at maintaining muscle homeostasis during aging.
Materials and methods
Mice
Young (3-mo-old) and old (27-mo-old) C57BL/6 mice were purchased from the Laboratory Animal Resource Center (Korea Research Institute of Bioscience and Biotechnology [KRIBB]). All animal experiments were performed according to protocols approved by the Animal Care and Use Committee of KRIBB.
Isolation of primary myoblasts and cell culture
Primary myoblasts were isolated from the hindlimb muscles of 3-mo-old or 27-mo-old mice as previously described (Rando and Blau 1994). Briefly, the muscles were minced using scissors and incubated with dissociation buffer for 20 min at 37°C. The dissociation buffer included 2.4 U/mL dispase II (Roche), 1% collagenase D (Roche), and 2.5 µM CaCl2. The slurry was triturated using a serological pipette and subsequently passed through a 70-µm nylon mesh (BD Bioscience) to remove debris. The cells were collected by centrifugation at 1000 rpm for 3 min and resuspended in growth medium, which consisted of Ham's F-10 (HyClone) supplemented with 20% FBS, antibiotics, and 5 ng/mL bFGF. To eliminate fibroblasts, the cells were plated on noncoated plates for 1 h, and the floating cells (the myoblasts) were transferred to collagen-coated culture dishes. To maintain primary myoblasts, the growth medium was changed every 2 d. Myoblasts from young and old mouse donors that had been cultured for the same number of passages were used for all experiments. The differentiation of primary myoblasts was induced by culturing the cells in differentiation medium that consisted of DMEM (HyClone) supplemented with antibiotics and 5% horse serum for 5 d.
HSMMs (Lonza Walkersville, Inc.) isolated from a 17-yr-old donor were cultured in growth medium consisting of skeletal muscle basal medium 2 (Lonza Walkersville, Inc.) supplemented with antibiotics, human epidermal growth factor (hEGF), dexamethasone, L-glutamine, and 10% fetal bovine serum. Differentiation was initiated 24–48 h after seeding by changing to the differentiation medium containing DMEM/F12 (Gibco), antibiotics, and 2% horse serum.
C2C12 cells were purchased from American Type Culture Collection and cultured in growth medium consisting of DMEM (Gibco) with antibiotics and 10% fetal bovine serum. Differentiation was initiated 24–48 h after seeding by changing to the differentiation medium containing DMEM (Gibco), antibiotics, and 5% horse serum.
miRNA sequencing and analysis
Small RNA was enriched from total myoblast RNA (n = 12) using the mirVana miRNA isolation kit (Ambion) following the manufacturer's protocol, and miRNA libraries were prepared following the Illumina library preparation protocol (Illumina, Inc.). Each library was indexed with the Illumina adaptor (6-base barcode). The small RNA library was size-fractionated on a 6% TBE urea polyacrylamide gel, and the 140- to 160-base-pair (bp) fraction was excised from the gel. The purified miRNA library was quantified on the Agilent DNA 1000 chip. All of the indexed samples were pooled and sequenced using an Illumina GAIIx instrument (50-bp reads). Before mapping the raw small RNA sequencing reads on the reference genome, the quality of the reads was evaluated by FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc), and attached adaptor sequences at the 3′ ends were removed by in-house PERL script. Adapter-trimmed small RNA reads were then mapped on the reference genome mm10 downloaded from the University of California at Santa Cruz genome browser (http://genome.ucsc.edu) by Burrows-Wheeler Aligner (http://bio-bwa.sourceforge.net) software with default options. For known miRNAs (miRBase version 20), we calculated the FPKM (fragments per kilobase per million mapped reads) for each precursor and mature miRNA from the mapped reads by custom Python script. miRBase was used to filter known miRNAs, and Rattus norvegicus was used as a related species. The data were deposited to Gene Expression Omnibus (GEO) under accession number GSE66267.
qPCR and miRNA expression analysis
RNA preparation and cDNA synthesis were performed according to standard protocols. qRT–PCR analysis was performed using StepOnePlus (Applied Biosystems) with a 20-µL reaction volume containing cDNA, primers, and SYBR Master Mix (Applied Biosystems). The data were normalized to the abundance of Actb mRNA in each reaction. The primer sequences are listed in Supplemental Table 1.
For analysis of mature miRNA expression, TaqMan miRNA assays were performed according to the manufacturer's recommended protocols (Applied Biosystems). Briefly, each reverse transcriptase reaction contained 10 ng of total RNA isolated using the mirVana miRNA isolation kit, 50 nM stem–loop reverse transcription primer, 0.25 mM each dNTP, 3.33 U/μL MultiScribe reverse transcriptase, 0.25 U/μL RNase inhibitor, and 1× reverse transcriptase buffer (all purchased from Applied Biosystems). The RT-qPCR reactions were carried out in 96-well plates using an Applied Biosystems instrument. U6 or RNU6B snRNA served as an endogenous normalization control.
Transfection and luciferase assays
siRNAs, miRNA mimics, and miRNA inhibitors (30 nM each) were transfected into primary myoblasts, C2C12 myoblasts, or HSMMs using RNAiMAX (Invitrogen) according to the manufacturer's protocol. miRNA mimics and miRNA inhibitors were purchased from mirVana (Invitrogen) or AccuTarget (Bioneer) (Supplemental Tables 2–4). siRNAs (Supplemental Table 5) were purchased from Dhamacon (Thermo Scientific). For luciferase assays, the 3′ UTR fragment (nucleotide positions 0–1265) of Smad4 mRNA containing a miR-431-binding site was cloned into the pcDNA3-luc vector in which the luc-coding sequence exists between HindIII and BamHI in the multiple cloning site (Kim et al. 2010). The resulting plasmid construct expressed a luciferase chimeric RNA driven by a constitutive promoter, allowing for functional analysis of miRNA translational control of mRNA. The mutant Smad4 3′ UTR with a deletion of the miR-431-binding region (position 1067–1073 bp) was also cloned into the same pcDNA3-luc vector for luciferase assays. 293T cells were transfected with miRNAs and luciferase plasmids. As an internal control, pGL3 vector (Promega) was cotransfected. For analysis of luciferase activities using 4xSBE-luc (Zawel et al. 1998), 500 ng of 4xSBE-luc vector was transfected into C2C12 cells with miRNA mimics or inhibitors (30 nM each) using Lipofectamine 2000 reagent (Invitrogen). A control reporter vector (pRL-TK) was cotransfected for normalization. Twenty-four hours after transfection, cells were serum-starved for 6 h, whereupon they were either left untreated or treated for 16 h with 5 ng/mL TGF-β (R&D Systems), 100 ng/mL MSTN (R&D Systems), or 100 ng/mL ActA (R&D Systems). Forty-eight hours after transfection, luciferase assay measurements were performed using a dual-luciferase reporter assay (Promega) and a Victor X3 instrument (PerkinElmer).
Pull-down assay using biotinylated miR431-5p or ASOs as a bait
C2C12 cells were transfected with biotinylated oligomers (60 nM each; IDT): Bi-miR431-5p (5′-UGUCUUGCAGGCCGUCAUGCA-3′ biotin) and control Bi-cel-miR-67 (5′-UCACAACCUCCUAGAAAGAGUAGA-3′ biotin) using Lipofectamine 2000 (Invitrogen). Twenty-four hours after transfection, cells were rinsed with PBS and lysed in 1 mL of lysis buffer (20 mM Tris at pH 7.5, 100 mM KCl, 5 mM MgCl2, 0.3% NP-40, 100 U of RNase inhibitor [Thermo Fisher], protease inhibitor [Roche]) and incubated for 10 min on ice as described earlier (Lal et al. 2011). The cytoplasmic lysate was cleared by centrifugation at 12,000g for 10 min. Dynabeads (100 μL) were added and further incubated for 4 h at 4°C with rotation. Beads were washed four times with 1 mL of ice-cold lysis buffer, RNA was isolated using TRIzol, and the enrichment of Smad4 mRNA was measured by RT-qPCR and normalized to Gapdh mRNA levels. For young and old muscle tissues, 300 µg lysates was incubated with 100 pmol of Bi-miR431-5p or control Bi-cel-miR-67 for 4 h at 4°C with rotation in 1 mL of lysis buffer before pull-down of the biotinylated miR-431 using the above method. For antisense oligo pull-down of Smad4 mRNA, young myoblasts or C2C12 cells were lysed in a buffer (20 mM Tris-HCl at pH 7.5, 100 mM KCl, 5 mM MgCl2, 0.5% NP-40) containing protease inhibitors (Roche) and RNase inhibitor (Thermo Fisher). Lysates were incubated with 100 pmol of biotin-labeled oligomers complementary to Smad4 mRNA (CCAAACGTCACCTTCACCTT, GGCAGCAAACACATCTCTCA, GCTGGCTGAGCAGTAAATCC, GCCCTGAAGCTATCTGCAAC, TCAATCGCTTCTGTCCTGTG, CAGACAGACTGATGGCTGGA, GTGGAAGCCACAGGAATGTT, GCAGGACTTCATCCAAGAGC, TTCACTTTCCCCACCTTGTC, GCAGAACAGTGAAGCAGCAG, CCTGAAGTCGTCCATCCAAT, GGGGAAGAAACCCTGAAGTC, CAAGCAAAAGCGATCTCCTC, and CTCCACAGACGGGCATAGAT) or long noncoding RNA Gm14635 (CGTTCCATCATTGCAAGCCC, ACCGTCATCTTCCAGGGAGA, and TTCGGGGATGTACGAAAGCA) in 1× TENT buffer (10 mM Tris-HCl at pH 8.0, 1 mM EDTA at pH 8.0, 250 mM NaCl, 0.5% [v/v] Triton X-100) and protease and RNase inhibitors for 1 h at room temperature with rotation as described (Yoon et al. 2012). Dynabeads (50 μL) were added and further incubated for 30 min at room temperature with rotation. Beads were washed three times with ice-cold PBS buffer, RNA was isolated using TRIzol, and the enrichment of Smad4 mRNA was analyzed by RT-qPCR and normalized to Gapdh mRNA. The same RNA was used to analyze the abundance of miR-431-5p in Smad4 mRNA-enriched samples using QuantiMir kit (System Biosciences, Inc.).
Immunoblot analysis
Muscle tissues and isolated myoblasts were homogenized in Pro-Prep lysis buffer (iNtRON Biotechnology, Inc.) containing protease and phosphatase inhibitors. The lysates were centrifuged at 15,000g for 20 min at 4°C, and the resulting supernatants were subjected to SDS-PAGE followed by immunoblot analysis. Antibodies for immunoblotting recognized MYHC (Santa Cruz Biotechnology), MYOG (Santa Cruz Biotechnology), ACTB (Santa Cruz Biotechnology), SMURF1 (Thermo Scientific), phospho-SMAD2/3 (Cell Signaling Technology), SMAD2/3 (Cell Signaling Technology), SMAD4 (Santa Cruz Biotechnology), and GAPDH (Santa Cruz Biotechnology).
Muscle regeneration following CTX injection
We injected CTX (Sigma) into the TA muscles of 3-mo-old and 27-mo-old mice to induce muscle regeneration following a previously described method (Conboy and Rando 2012). Briefly, TA muscles were injected with 50 µL of 20 µM CTX. To deliver miR-431 and siSmad4 into TA muscles, we made mixtures of miRNA mimics or siRNAs in RNAiMAX reagent (Invitrogen) as described earlier (Dey et al. 2014). Three microliters of 1 mM miRNA mimic or siRNA and 37 μL of nuclease-free water were mixed with 40 μL of RNAiMAX reagents (Invitrogen) and incubated for 30 min. A 50-μL aliquot of the mixture was injected into TA muscle at 2 and 5 d after CTX injection.
Immunohistochemistry and immunofluorescence
Skeletal muscle tissues were fixed in 4% paraformaldehyde, and frozen section samples or paraffin-embedded sections were prepared. The frozen sections (10 µm thick) were stained with DAPI and antibody according to standard protocols. Antibodies for immunohistochemical analysis recognized laminin (Sigma-Aldrich), PAX7 (Developmental Studies Hybridoma Bank), or SMAD4 (Abcam). The paraffin sections were stained with H&E according to standard protocols. The measurement of cross-section areas (CSAs) was performed using NIH ImageJ software (http://rsb.info.nih.gov/ij). For immunofluorescence, primary myoblasts were grown on laminin-coated (Invitrogen) coverslips in either growth medium or differentiation medium. Cells were fixed in 4% paraformaldehyde, washed with PBS buffer, and permeabilized with 0.3% Triton X-100 in PBS buffer. Cells were blocked with 3% bovine serum albumin in PBS buffer and then incubated with primary antibody diluted in blocking solution. Cells were then washed in PBS buffer and incubated with secondary antibody. After further washes with PBS, the coverslips were mounted on glass slides with VectoLab mounting medium (with DAPI). Antibodies for immunofluorescence were directed to MYHC (Santa Cruz Biotechnology), MYOD (Santa Cruz Biotechnology), or PAX7 (Developmental Studies Hybridoma Bank). The fusion index was calculated as the ratio of the number of multinucleated MYHC-positive cells to the number of total MYHC-positive cells.
Statistical analysis
Quantitative data are presented as the means ± SD unless indicated otherwise. Differences between means were evaluated using Student's unpaired t-test. A P-value of <0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank B. Vogelstein for providing the 4xSBE-luc plasmid. This study was supported by grants from the Bio and Medical Technology Development Program (20110030133, 2013M3A9B6076413, and 20120009022 to K.-S.K.) of the National Research Foundation, which is funded by the Korean government (Ministry of Science, ICT, and Future Planning), and the Korea Research Institute of Bioscience and Biotechnology Research Initiative Program. A.C.P., K.A., and M.G. were supported by the National Institute on Aging Intramural Research Program of the National Institutes of Health.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.263574.115.
References
- Ambros V. 2004. The functions of animal microRNAs. Nature 431: 350–355. [DOI] [PubMed] [Google Scholar]
- Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297. [DOI] [PubMed] [Google Scholar]
- Bernet JD, Doles JD, Hall JK, Kelly Tanaka K, Carter TA, Olwin BB. 2014. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat Med 20: 265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack AS, Bildsoe H, Hughes SM. 2005. Evidence that satellite cell decrement contributes to preferential decline in nuclear number from large fibres during murine age-related muscle atrophy. J Cell Sci 118: 4813–4821. [DOI] [PubMed] [Google Scholar]
- Callis TE, Chen JF, Wang DZ. 2007. MicroRNAs in skeletal and cardiac muscle development. DNA Cell Biol 26: 219–225. [DOI] [PubMed] [Google Scholar]
- Carlson ME, Hsu M, Conboy IM. 2008. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature 454: 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson ME, Conboy MJ, Hsu M, Barchas L, Jeong J, Agrawal A, Mikels AJ, Agrawal S, Schaffer DV, Conboy IM. 2009. Relative roles of TGF-β1 and Wnt in the systemic regulation and aging of satellite cell responses. Aging Cell 8: 676–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charge SB, Rudnicki MA. 2004. Cellular and molecular regulation of muscle regeneration. Physiol Rev 84: 209–238. [DOI] [PubMed] [Google Scholar]
- Cheung TH, Quach NL, Charville GW, Liu L, Park L, Edalati A, Yoo B, Hoang P, Rando TA. 2012. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature 482: 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CA, Zammit PS, Ruiz AP, Morgan JE, Partridge TA. 2007. A population of myogenic stem cells that survives skeletal muscle aging. Stem Cells 25: 885–894. [DOI] [PubMed] [Google Scholar]
- Conboy IM, Rando TA. 2012. Heterochronic parabiosis for the study of the effects of aging on stem cells and their niches. Cell Cycle 11: 2260–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, Llewellyn ME, Delp SL, Blau HM. 2014. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med 20: 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey BK, Gagan J, Yan Z, Dutta A. 2012. miR-26a is required for skeletal muscle differentiation and regeneration in mice. Genes Dev 26: 2180–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey BK, Pfeifer K, Dutta A. 2014. The H19 long noncoding RNA gives rise to microRNAs miR-675-3p and miR-675-5p to promote skeletal muscle differentiation and regeneration. Genes Dev 28: 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg I, Eran A, Nishino I, Moggio M, Lamperti C, Amato AA, Lidov HG, Kang PB, North KN, Mitrani-Rosenbaum S, et al. 2007. Distinctive patterns of microRNA expression in primary muscular disorders. Proc Natl Acad Sci 104: 17016–17021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frontera WR, Hughes VA, Lutz KJ, Evans WJ. 1991. A cross-sectional study of muscle strength and mass in 45- to 78-yr-old men and women. J Appl Physiol 71: 644–650. [DOI] [PubMed] [Google Scholar]
- Fulle S, Di Donna S, Puglielli C, Pietrangelo T, Beccafico S, Bellomo R, Protasi F, Fano G. 2005. Age-dependent imbalance of the antioxidative system in human satellite cells. Exp Gerontol 40: 189–197. [DOI] [PubMed] [Google Scholar]
- Goodpaster BH, Park SW, Harris TB, Kritchevsky SB, Nevitt M, Schwartz AV, Simonsick EM, Tylavsky FA, Visser M, Newman AB. 2006. The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. J Gerontol A Biol Sci Med Sci 61: 1059–1064. [DOI] [PubMed] [Google Scholar]
- Gopinath SD, Rando TA. 2008. Stem cell review series: aging of the skeletal muscle stem cell niche. Aging Cell 7: 590–598. [DOI] [PubMed] [Google Scholar]
- Hughes VA, Frontera WR, Wood M, Evans WJ, Dallal GE, Roubenoff R, Fiatarone Singh MA. 2001. Longitudinal muscle strength changes in older adults: influence of muscle mass, physical activity, and health. J Gerontol A Biol Sci Med Sci 56: B209–B217. [DOI] [PubMed] [Google Scholar]
- Jang YC, Sinha M, Cerletti M, Dall'Osso C, Wagers AJ. 2011. Skeletal muscle stem cells: effects of aging and metabolism on muscle regenerative function. Cold Spring Harb Symp Quant Biol 76: 101–111. [DOI] [PubMed] [Google Scholar]
- Khanna N, Ge Y, Chen J. 2014. MicroRNA-146b promotes myogenic differentiation and modulates multiple gene targets in muscle cells. PLoS One 9: e100657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Lee YC, Kim C. 2010. Direct inhibition of Pumilo activity by Bam and Bgcn in Drosophila germ line stem cell differentiation. J Biol Chem 285: 4741–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Park YK, Lee KP, Lee SM, Kang TW, Kim HJ, Dho SH, Kim SY, Kwon KS. 2014. Genome-wide profiling of the microRNA-mRNA regulatory network in skeletal muscle with aging. Aging 6: 524–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal A, Thomas MP, Altschuler G, Navarro F, O'Day E, Li XL, Concepcion C, Han YC, Thiery J, Rajani DK, et al. 2011. Capture of microRNA-bound mRNAs identifies the tumor suppressor miR-34a as a regulator of growth factor signaling. PLoS Genet 7: e1002363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Lee YS, Zimmers TA, Soleimani A, Matzuk MM, Tsuchida K, Cohn RD, Barton ER. 2010. Regulation of muscle mass by follistatin and activins. Mol Endocrinol 24: 1998–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeper NJ, Raiesdana A, Kojima Y, Chun HJ, Azuma J, Maegdefessel L, Kundu RK, Quertermous T, Tsao PS, Spin JM. 2011. MicroRNA-26a is a novel regulator of vascular smooth muscle cell function. J Cell Physiol 226: 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Black BL, Derynck R. 2001. TGF-β inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev 15: 2950–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Kang JS, Derynck R. 2004. TGF-β-activated Smad3 represses MEF2-dependent transcription in myogenic differentiation. EMBO J 23: 1557–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Williams AH, Maxeiner JM, Bezprozvannaya S, Shelton JM, Richardson JA, Bassel-Duby R, Olson EN. 2012. microRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. J Clin Invest 122: 2054–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JF, Li L, Olson EN. 1992. Repression of myogenin function by TGF-β1 is targeted at the basic helix-loop-helix motif and is independent of E2A products. J Biol Chem 267: 10956–10960. [PubMed] [Google Scholar]
- Massague J. 2012. TGFβ signalling in context. Nat Rev Mol Cell Biol 13: 616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J, Cheifetz S, Endo T, Nadal-Ginard B. 1986. Type β transforming growth factor is an inhibitor of myogenic differentiation. Proc Natl Acad Sci 83: 8206–8210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy JJ. 2011. The myomiR network in skeletal muscle plasticity. Exerc Sport Sci Rev 39: 150–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy JJ, Esser KA. 2007. MicroRNA-1 and microRNA-133a expression are decreased during skeletal muscle hypertrophy. J Appl Physiol 102: 306–313. [DOI] [PubMed] [Google Scholar]
- McCarthy JJ, Esser KA, Peterson CA, Dupont-Versteegden EE. 2009. Evidence of MyomiR network regulation of β-myosin heavy chain gene expression during skeletal muscle atrophy. Physiol Genomics 39: 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherron AC, Lawler AM, Lee SJ. 1997. Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature 387: 83–90. [DOI] [PubMed] [Google Scholar]
- Moren A, Imamura T, Miyazono K, Heldin CH, Moustakas A. 2005. Degradation of the tumor suppressor Smad4 by WW and HECT domain ubiquitin ligases. J Biol Chem 280: 22115–22123. [DOI] [PubMed] [Google Scholar]
- Narici MV, Maffulli N. 2010. Sarcopenia: characteristics, mechanisms and functional significance. Br Med Bull 95: 139–159. [DOI] [PubMed] [Google Scholar]
- O'Rourke JR, Georges SA, Seay HR, Tapscott SJ, McManus MT, Goldhamer DJ, Swanson MS, Harfe BD. 2007. Essential role for Dicer during skeletal muscle development. Dev Biol 311: 359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando TA, Blau HM. 1994. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J Cell Biol 125: 1275–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabourin LA, Rudnicki MA. 2000. The molecular regulation of myogenesis. Clin Genet 57: 16–25. [DOI] [PubMed] [Google Scholar]
- Sartorelli V, Caretti G. 2005. Mechanisms underlying the transcriptional regulation of skeletal myogenesis. Curr Opin Genet Dev 15: 528–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz E, Lipton BH. 1982. Skeletal muscle satellite cells: changes in proliferation potential as a function of age. Mech Ageing Dev 20: 377–383. [DOI] [PubMed] [Google Scholar]
- Shefer G, Van de Mark DP, Richardson JB, Yablonka-Reuveni Z. 2006. Satellite-cell pool size does matter: defining the myogenic potency of aging skeletal muscle. Dev Biol 294: 50–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardi M, Ballestar E, Gonzalez S, Serrano AL, et al. 2014. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506: 316–321. [DOI] [PubMed] [Google Scholar]
- Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, et al. 2007. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation 116: 258–267. [DOI] [PubMed] [Google Scholar]
- Townley-Tilson WH, Callis TE, Wang D. 2010. MicroRNAs 1, 133, and 206: critical factors of skeletal and cardiac muscle development, function, and disease. Int J Biochem Cell Biol 42: 1252–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagers AJ, Conboy IM. 2005. Cellular and molecular signatures of muscle regeneration: current concepts and controversies in adult myogenesis. Cell 122: 659–667. [DOI] [PubMed] [Google Scholar]
- Wang YX, Rudnicki MA. 2012. Satellite cells, the engines of muscle repair. Nat Rev Mol Cell Biol 13: 127–133. [DOI] [PubMed] [Google Scholar]
- Yoon JH, Abdelmohsen K, Srikantan S, Yang X, Martindale JL, De S, Huarte M, Zhan M, Becker KG, Gorospe M. 2012. LincRNA-p21 suppresses target mRNA translation. Mol Cell 47: 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, Kern SE. 1998. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell 1: 611–617. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.