

Abstract
Ribonuclease (RNase) P and RNase MRP are closely related catalytic ribonucleoproteins involved in the metabolism of a wide range of RNA molecules, including tRNA, rRNA, and some mRNAs. The catalytic RNA component of eukaryotic RNase P retains the core elements of the bacterial RNase P ribozyme; however, the peripheral RNA elements responsible for the stabilization of the global architecture are largely absent in the eukaryotic enzyme. At the same time, the protein makeup of eukaryotic RNase P is considerably more complex than that of the bacterial RNase P. RNase MRP, an essential and ubiquitous eukaryotic enzyme, has a structural organization resembling that of eukaryotic RNase P, and the two enzymes share most of their protein components. Here, we present the results of the analysis of interactions between the largest protein component of yeast RNases P/MRP, Pop1, and the RNA moieties of the enzymes, discuss structural implications of the results, and suggest that Pop1 plays the role of a scaffold for the stabilization of the global architecture of eukaryotic RNase P RNA, substituting for the network of RNA–RNA tertiary interactions that maintain the global RNA structure in bacterial RNase P.
Keywords: RNase P, RNase MRP, Pop1, ribonucleoprotein, RNA folding, Saccharomyces cerevisiae
INTRODUCTION
RNase P is a ribonucleoprotein complex that is almost universally found in all three domains of life. RNase P is primarily responsible for the maturation of 5′-ends of tRNA (for review, see Evans et al. 2006; Marvin and Engelke 2009; Altman 2010; Esakova and Krasilnikov 2010; Jarrous and Gopalan 2010). The phylogenetically conserved RNA component of RNase P is the catalytic moiety of the enzyme (Guerrier-Takada et al. 1983; Chen and Pace 1997; Pannucci et al. 1999; Thomas et al. 2000; Kikovska et al. 2007; Li et al. 2009). In a number of cases, the ribonucleoprotein RNase P is supplemented or altogether replaced by unrelated protein-only enzymes termed PRORPs (Pinker et al. 2013).
RNase MRP is a ubiquitous eukaryotic ribonucleoprotein closely related to RNase P (Chang and Clayton 1987a,b; Karwan et al. 1991). RNase MRP is mainly localized to the nucleolus (Chang and Clayton 1987a; Reimer et al. 1988; Kiss and Filipowicz 1992) and the cytosol (Gill et al. 2006). While small amounts of RNase MRP can also be found in the mitochondria, the mitochondrial enzyme has a distinct composition and specificity (Lu et al. 2010) and will not be further discussed in this work. RNase MRP is an essential enzyme involved in the maturation of rRNA (Schmitt and Clayton 1993; Chu et al. 1994; Lygerou et al. 1996; Lindahl et al. 2009), regulation of the cell cycle through cleavage of specific mRNAs (Cai et al. 2002; Gill et al. 2004; Aulds et al. 2012), as well as in the metabolism of a variety of other RNA molecules (Mattijssen et al. 2010; Jaag et al. 2011; Aulds et al. 2012; Saito et al. 2014). While the specificity of RNase MRP differs from that of eukaryotic RNase P, the two enzymes share most of their protein components (Chamberlain et al. 1998; Walker et al. 2010), as well as many RNA features (Fig. 1A,B, 2), and demonstrate similarities in their structural organizations (Esakova et al. 2008; Khanova et al. 2012; Hipp et al. 2012).
FIGURE 1.

(A) Secondary structure of Saccharomyces cerevisiae RNase P RNA and results of footprinting analysis. Highlighted nucleotides: Fe(II)-EDTA assays of the quaternary RNP (RNA + Pop1 + Pop6/Pop7). (Green) No protection; (orange) partial protection; (red) complete protection of RNA. Solid lines indicate RNA regions protected from bulky probes (RNase A and RNase V1). (Brown) Protection by the Pop6/Pop7 heterodimer; (magenta) protection by Pop1 alone; (blue) the additional regions protected when Pop1 is present in combination with Pop6/Pop7. Gray arrows indicate positions of previously identified UV-induced protein–RNA crosslinks in the context of the holoenzymes (Khanova et al. 2012). (B) Secondary structure of S. cerevisiae RNase MRP RNA and results of footprinting analysis. The results of footprinting analysis are marked as in panel A. Diagrams A and B are based on Esakova and Krasilnikov (2010). (C) Regions of RNase P RNA that become hypersensitive to the cleavage by RNase A (arrows) and RNase V1 (solid lines), color-coded as indicated on the right. (D) Regions of RNase MRP RNA that become hypersensitive to the cleavage by RNase A (arrows) and RNase V1 (solid lines), color-coded as indicated on the left.
FIGURE 2.

List of protein components of yeast RNase P and RNase MRP.
Bacterial RNase P consists of a large RNA and a small protein (Stark et al. 1978; Kole and Altman 1979). Judging by the available structural information (Kazantsev et al. 2005; Torres-Larios et al. 2005; Reiter et al. 2010), the protein appears to play little, if any, role in the stabilization of the global tertiary structure of bacterial RNase P RNA. Instead, the structure is stabilized by a network of tertiary interactions between variable auxiliary RNA elements (Brown et al. 1996; Chen and Pace 1997; Brown 1999; Krasilnikov et al. 2004; Kazantsev et al. 2005; Torres-Larios et al. 2005; Reiter et al. 2010; Mondragón 2013).
Archaeal RNase P consists of an RNA component and typically four or five proteins (Lai et al. 2010 and references therein). The RNA component closely resembles bacterial RNase P RNA and usually possesses auxiliary elements that are likely to be involved in the stabilization of its tertiary structure (Esakova and Krasilnikov 2010). Archaeal RNase P proteins are not related to the bacterial RNase P protein (for review, see Evans et al. 2006; Marvin and Engelke 2009; Esakova and Krasilnikov 2010; Jarrous and Gopalan 2010); their role, if any, in the stabilization of the RNA structure is not clear.
The closely related eukaryotic RNase P and RNase MRP are more complex than bacterial and archaeal RNases P. In Saccharomyces cerevisiae, the enzymes are composed of one large RNA (Fig. 1A,B) and nine (in RNase P) or 10 (in RNase MRP) essential proteins (Fig. 2). The proteins range in size from 15 to 100 kDa and constitute the bulk of the enzymes (Evans et al. 2006; Marvin and Engelke 2009; Esakova and Krasilnikov 2010; Jarrous and Gopalan 2010). The eukaryotic RNase P RNA component retains several core regions (CR-I to CR-V, Fig. 1A) that are implicated in pre-tRNA recognition and catalysis (Chen and Pace 1997; Brown 1999; Marquez et al. 2006); however, the auxiliary elements involved in the stabilization of the tertiary RNA structure in bacteria and archaea are missing and it can be expected that their role has been delegated to proteins (Evans et al. 2006; Marvin and Engelke 2009; Esakova and Krasilnikov 2010; Jarrous and Gopalan 2010 and references therein).
Several eukaryotic RNase P/MRP proteins are related to the proteins found in archaeal RNase P (Hall and Brown 2002; Evans et al. 2006; Marvin and Engelke 2009; Esakova and Krasilnikov 2010; Jarrous and Gopalan 2010). In yeast, six of the nine RNase P proteins (Pop3 [Dichtl and Tollervey 1997], Pop4 [Chu et al. 1997], Pop5, Pop8 [Chamberlain et al. 1998], Rpp1 [Stolc and Altman 1997], and Rpr2 [Chamberlain et al. 1998]) are homologs of archaeal RNase P proteins, whereas three proteins (Pop1 [Lygerou et al. 1994], Pop6, and Pop7 [Chamberlain et al. 1998]) are strictly eukaryotic (Evans et al. 2006; Marvin and Engelke 2009; Esakova and Krasilnikov 2010; Jarrous and Gopalan 2010). Considering that the addition of Pop1, Pop6, and Pop7 in eukaryotes was accompanied by the loss of auxiliary RNA elements that stabilize RNase P RNA structure in bacteria and archaea, these three exclusively eukaryotic protein components can potentially be seen as primary candidates for the role of the global structural scaffold in eukaryotic RNase P (and, by inference, RNase MRP), replacing the missing auxiliary elements found in bacterial and archaeal enzymes.
In yeast, proteins Pop6 and Pop7 were shown to form a heterodimer that binds to the P3 RNA domain (Fig. 1A,B; Perederina et al. 2007, 2010a,b); human homologs of Pop6 and Pop7 were shown to behave similarly to their yeast counterparts (Welting et al. 2007; Hands-Taylor et al. 2010). The helix–loop–helix P3 RNA domain is a conserved feature that is specific to the eukaryotic enzymes of the RNase P/MRP family (Lindahl et al. 2000; Ziehler et al. 2001; Piccinelli et al. 2005). It was suggested that this RNA domain serves as a protein-binding hub in eukaryotic RNase P and RNase MRP (Ziehler et al. 2001; Perederina and Krasilnikov 2010; Perederina et al. 2010b) and thus may be indirectly involved in the stabilization of the tertiary structure of RNA (Perederina and Krasilnikov 2010; Perederina et al. 2010b). In RNase P, the Pop6/Pop7 heterodimer was shown to mediate a tertiary interaction between the P3 RNA domain and P15 RNA stem (Esakova et al. 2008; Khanova et al. 2012). However, this interaction appears to be too localized to fulfill the role of the structural scaffold that is played by tertiary RNA–RNA interactions in bacteria.
Pop1 (Lygerou et al. 1994), the third of the exclusively eukaryotic protein components, is the largest (100.5 kDa in yeast, Fig. 2) RNase P/MRP protein (Marvin and Engelke 2009; Esakova and Krasilnikov 2010; Jarrous and Gopalan 2010). Like other RNase P/MRP proteins, Pop1 is essential for the viability of the cell (Lygerou et al. 1994; Chamberlain et al. 1998). Pop1 mutations affect the holoenzyme assembly and result in destabilization of the RNA component in the cell as well as in substrate processing defects in both RNase P and RNase MRP (Lygerou et al. 1994; Chamberlain et al. 1998; Xiao et al. 2006). In humans, certain Pop1 mutations result in a severe skeletal dysplasia (Glazov et al. 2011), similar to the dysplasias caused by mutations in the RNase MRP RNA or by defects in RNase MRP RNA promoter region (Ridanpaa et al. 2001; Mattijssen et al. 2011).
Yeast three-hybrid studies (Ziehler et al. 2001; Houser-Scott et al. 2002) indicated interactions between Pop1 and the P3 RNA domain. Mutations of certain conserved residues of the loop region of the P3 RNA domain disrupted observed Pop1–RNA interactions (Ziehler et al. 2001); however, the mutated residues were later shown to be directly involved in interactions with the Pop6/Pop7 heterodimer, and it was suggested that Pop1 binding to the P3 RNA domain might be mediated or facilitated by Pop6/Pop7 (Pluk et al. 1999; Perederina et al. 2007, 2010b). The results of the RNase P/MRP holoenzyme footprinting studies (Tranguch et al. 1994; Esakova et al. 2008) indicate that most of the internal loop region of the P3 RNA domain and its proximal helical stem are protected in the presence of the proteins in the holoenzyme. This degree of protection cannot be explained solely by Pop6/Pop7 binding (Perederina and Krasilnikov 2010; Perederina et al. 2007, 2010b), suggesting that another protein(s) may be binding to the P3 domain in addition to Pop6/Pop7.
UV-crosslinking analysis of RNase P/MRP holoenzymes (Khanova et al. 2012) shows that interactions of Pop1 with RNA components involve direct interactions with the CR-IV/mCR-IV regions (Fig. 1A,B). This result positions a part of Pop1 in the immediate vicinity of the RNA catalytic core. At the same time, cryo-EM studies (Hipp et al. 2012) indicate that the N terminus of Pop1 is positioned in a distal part of RNase P, in the vicinity of the P12 stem.
The size of Pop1, its presence exclusively in the eukaryotic RNases P/MRP (Evans et al. 2006; Marvin and Engelke 2009; Esakova and Krasilnikov 2010; Jarrous and Gopalan 2010), the effects of Pop1 mutations on the holoenzymes’ assembly (Lygerou et al. 1994; Chamberlain et al. 1998; Xiao et al. 2006), and the apparent spread of the RNase P/MRP RNA regions involved (directly or not) in interactions with Pop1 (Ziehler et al. 2001; Houser-Scott et al. 2002; Hipp et al. 2012; Khanova et al. 2012), are all consistent with a potential central role played by Pop1 in the stabilization of the global structure of the RNA components of eukaryotic RNases P/MRP. However, insolubility of individually expressed Pop1 (Ziehler et al. 2001; Xiao et al. 2006) has previously hindered direct studies of this protein and its interactions with other RNase P/MRP components.
RESULTS AND DISCUSSION
Coexpression of Pop1 with Pop4 results in soluble Pop1
Yeast Pop1 (Lygerou et al. 1994), like many other RNase P/MRP proteins, is not soluble when expressed individually (Ziehler et al. 2001; Xiao et al. 2006). However, we previously showed that coexpression of several insoluble RNase P/MRP proteins resulted in the formation of soluble and structurally homogeneous complexes (Perederina et al. 2007, 2010b, 2011). Yeast two-hybrid studies (Houser-Scott et al. 2002), pull-down experiments (Welting et al. 2004; Aspinall et al. 2007), as well as our data (Khanova et al. 2012; E Khanova, AS Krasilnikov, unpubl.) indicated interactions between Pop1 and another RNase P/MRP protein component, Pop4, prompting us to consider coexpression of Pop1 and Pop4 as a possible solution to the insolubility of Pop1.
Coexpression of Pop1 and Pop4 in Escherichia coli resulted in soluble Pop1 that was purified (Fig. 3A) using standard biochemical approaches (Materials and Methods). Unexpectedly, while the initial presence of Pop4 was evidently essential for the solubility of Pop1, the two proteins did not copurify under the used conditions and, once soluble, isolated Pop1 remained soluble and essentially aggregate-free (as judged by dynamic light scattering) even when Pop4 was removed early in the Pop1 purification. Subsequent attempts to reconstitute a stable complex between Pop1 and isolated Pop4 did not yield positive results (data not shown). Further experimentation involving Pop4 will require production of structurally homogeneous Pop4.
FIGURE 3.
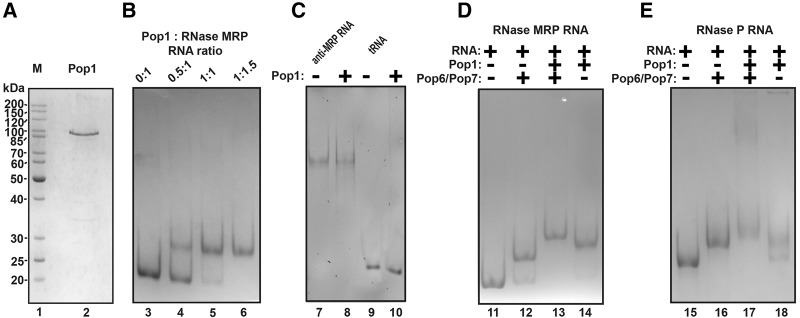
(A) Purified recombinant Pop1. (Lane 1) Protein size marker (the sizes are shown on the left); (lane 2) Pop1 preparation. Twelve percent SDS-polyacrylamide gel stained with Coomassie Blue. (B) Gel mobility shift analysis of the stoichiometry of the Pop1–RNase MRP RNA complex. The protein and RNA were taken at the molar ratios indicated above the gel. The concentrations of the components were considerably higher than the dissociation constant. (Lane 3) RNase MRP RNA only; (lane 4) 0.5:1 Pop1: RNA molar ratio; (lane 5) 1:1 Pop1: RNA molar ratio; (lane 6) 1.5:1 Pop1: RNA molar ratio. (C) Gel mobility shift assay of the interactions between Pop1 and control “antisense” RNase MRP RNA (lane 7: RNA only; lane 8: RNA and Pop1, 1:1 molar ratio) and between Pop1 and control tRNA (lane 9: RNA only; lane 10: RNA and Pop1, 1:1 molar ratio). (D) Gel mobility shift assay of interactions between Pop1, the Pop6/Pop7 heterodimer and the RNA component of RNase MRP. (Lane 11) RNase MRP RNA only; (lane 12) a ternary complex between the Pop6/Pop7 heterodimer and RNase MRP RNA (1:1 molar ratio); (lane 13) a quaternary complex between the Pop6/Pop7 heterodimer, Pop1, and RNase MRP RNA (1:1:1 molar ratio); (lane 14) a complex between Pop1 and RNase MRP RNA (1:1 molar ratio). (E) Gel mobility shift assay of the interactions between Pop1, the Pop6/Pop7 heterodimer and the RNA component of RNase P. (Lane 15) RNase P RNA only; (lane 16) a ternary complex between the Pop6/Pop7 heterodimer and RNase P RNA (1:1 molar ratio); (lane 17) a quaternary complex between the Pop6/Pop7 heterodimer, Pop1, and RNase P RNA (1:1:1 molar ratio); lane 18: a complex between the Pop1 and RNase P RNA (1:1 molar ratio). RNA was resolved on 6% native polyacrylamide gel and stained with toluidine blue.
The Pop1 purification procedure (Materials and Methods) relied on the binding of the protein to a metal affinity resin in the presence of Zn2+ ions. Given that Pop1 had no purification tags fused to it, Pop1 binding to the metal affinity resin suggests that Pop1 might be a metal-binding protein.
Pop1 binds the RNA components of RNase P and RNase MRP
Incubation of Pop1 with the RNA components of RNase P and RNase MRP resulted in the formation of RNA–protein complexes, where RNA and Pop1 bound at a 1:1 stoichiometric ratio (Fig. 3B,D,E). (The complexes were formed at the concentrations of RNA and proteins that were considerably higher than the dissociation constants [below], thus allowing the estimation of the stoichiometry of RNA–protein complexes.)
Incubation of Pop1 with control RNAs, including an RNA that was complementary to the RNase MRP RNA (“antisense” RNase MRP RNA [Perederina et al. 2011]) and yeast tRNA, did not result in the formation of RNA–protein complexes as judged by gel mobility shift assays (Fig. 3C), indicating that interactions of Pop1 with RNA require specific structural and/or sequence determinants.
The apparent dissociation constants (Kd) for the Pop1 complexes with RNase P and RNase MRP RNAs estimated using filter-binding assays in the presence of a 100-fold excess of competitor yeast tRNA (Materials and Methods) were found to be 145 ± 40 nM and 150 ± 40 nM, respectively. These numbers are comparable to the dissociation constants observed for complexes of RNase P/MRP RNAs with Pop5/Rpp1 (60 ± 20 nM for RNase MRP [Perederina et al. 2011]) and Pop6/Pop7 (120 ± 40 nM and 150 ± 40 nM for RNase P and RNase MRP, respectively [Perederina et al. 2007]).
Incubation of Pop1 with the preformed complexes of the RNA components of RNase P/MRP and Pop6/Pop7 heterodimers (Perederina et al. 2007, 2010a,b) resulted in the formation of quaternary ribonucleoprotein complexes containing Pop1, Pop6, Pop7, and the corresponding RNA component (Fig. 3D,E). The presence of Pop6/Pop7 appeared to have stabilized the Pop1–RNase P/MRP RNA complexes: The dissociation constants were estimated to be 25 ± 8 nM and 20 ± 5 nM for RNase P and RNase MRP.
Complexes of Pop1 and Pop1/Pop6/Pop7 with RNase P and RNase MRP RNAs as well as the RNA components alone did not demonstrate reliable (above the background) catalytic activity under various conditions of moderate ionic strengths (0.2–0.6 M of NH4Cl in the presence of 3–100 mM of MgCl2 and 0–1 mM of spermidine, using yeast pre-tRNATyr and the A3 site-containing fragment of the pre-rRNA ITS1 as substrates [Esakova et al. 2011; data not shown]). It should be noted that while a weak catalytic activity of eukaryotic RNase P RNA has been reported (Kikovska et al. 2007), this activity was observed for different RNAs at a high ionic strength that precludes the formation of complexes of RNase P/MRP RNAs with Pop1 and Pop6/Pop7.
Footprinting analysis of interactions involving Pop1 and the RNA components of RNase P and RNase MRP
To examine interactions between Pop1 and the RNA components of RNases P/MRP, we performed footprinting analysis using hydroxyl ions produced in a Fenton-type reaction [Fe(II)-EDTA, cleaving accessible RNA backbone], as well as RNase A (cleaving RNA at accessible pyrimidines preferentially located in single-stranded regions) and RNase V1 (cleaving accessible double-stranded or stacked RNA) as probes. In addition, the effects of the presence of the Pop6/Pop7 heterodimer (Perederina et al. 2007, 2010a,b) on interactions of Pop1 with RNA were examined.
The analysis was performed for both RNase P and RNase MRP, and included the refolded RNA components alone, binary complexes between Pop1 and RNA, quaternary complexes between Pop1, Pop6, Pop7 and RNA, as well as ternary Pop6/Pop7–RNA complexes. In all assays, equimolar amounts of the protein(s) and RNA were used; protein and RNA concentrations were considerably higher than corresponding dissociation constants. To cover the entire length of the RNA components, both 5′-end- and 3′-end-labeled RNA were used. Typical results of footprinting experiments are shown in Figures 4–6. To facilitate a reliable identification of distant RNA regions, all standard gel runs were supplemented by longer runs; typical results are shown in Figure 7. The results of our footprinting analysis are summarized in Figure 1A,B.
FIGURE 4.

Footprinting assays for RNase MRP RNA, typical gels. 5′-end 32P-labeled RNase MRP RNA alone or in complexes with equimolar amounts of Pop1 and the Pop6/Pop7 heterodimer (as indicated above the gels) was partially digested with RNase V1 (A), RNase A (B), and hydroxyl radicals (C). (Lanes 1,20,28,47,52) RNase MRP RNA digested with RNase T1 (sequence ladder, identifies positions of guanines, shown on the left); (lanes 2,19,29,46) alkali hydrolysis of RNase MRP RNA (ladder); (lanes 3–6,30–33,53) digestion of RNase MRP RNA; (lanes 7–10,34–37,54) digestion of the complex of RNase MRP RNA with Pop1; (lanes 11–14,38–41,55) digestion of the quaternary complex of RNase MRP RNA with proteins Pop1, Pop6, and Pop7; (lanes 15–18,42–45,56) digestion of the complex of RNase MRP RNA with the Pop6/Pop7 heterodimer; (lanes 21–23,57–59) controls (RNA incubated with proteins as indicated above the gels). Secondary structure elements are marked on the left of the gels: helical regions are shown by thick solid lines, terminal loops are shown by thin solid lines, and the internal loop in the P3 RNA domain is shown by dotted lines. Gel traces are shown on the right of the gels (lanes 24–27,47–51,60–63) as marked on top.
FIGURE 6.

Footprinting assays for RNase P RNA, typical gels. 5′-end 32P-labeled RNase P RNA alone or in complexes with equimolar amounts of Pop1 and the Pop6/Pop7 heterodimer (as indicated above the gels) was partially digested with RNase V1 (A) and hydroxyl radicals (B). (Lanes 1,20,25) RNase P RNA digested with RNase T1 (sequence ladder, identifies positions of guanines, shown on the left); (lanes 2,19) alkali hydrolysis of RNase P RNA (ladder); (lanes 3–6,26) digestion of RNase P RNA; (lanes 7–10,27) digestion of the complex of RNase P RNA with Pop1; (lanes 11–14,28) digestion of the quaternary complex of RNase P RNA with proteins Pop1, Pop6, and Pop7; (lanes 15–18,29) digestion of the complex of RNase P RNA with the Pop6/Pop7 heterodimer; (lanes 30–32) controls (RNA incubated with proteins as indicated above the gels). Secondary structure elements are marked on the left of the gels: Helical regions are shown by thick solid lines, terminal loops by thin solid lines, and the internal loop in the P3 RNA domain by dotted line. Gel traces are shown on the right of the gels (lanes 21–24,33–36) as marked on top.
FIGURE 7.

Footprinting assays for RNase P RNA, typical gels. 3′-end 32P-labeled RNase P RNA alone or in complexes with equimolar amounts of Pop1 and the Pop6/Pop7 heterodimer (as indicated above the gels) was partially digested with RNase A. (A) Standard gel run. (B) Long gel run. (Lanes 2,3,20,28,47) RNase P RNA digested with RNase T1 (sequence ladder, identifies positions of guanines, shown on the left); (lanes 1,29,46) alkali hydrolysis of RNase P RNA (ladder); (lanes 4–7,30–33) digestion of RNase P RNA; (lanes 8–11,34–37) digestion of the complex of RNase P RNA with Pop1; (lanes 12–15,38–41) digestion of the quaternary complex of RNase P RNA with proteins Pop1, Pop6, and Pop7; (lanes 16–19,42–45) digestion of the complex of RNase P RNA with the Pop6/Pop7 heterodimer; (lanes 21–23,48–50) controls (RNA incubated with proteins as indicated above the gels). Secondary structure elements are marked on the left of the gels: Helical regions are shown by thick solid lines, terminal loops by thin solid lines. Gel traces are shown on the right of the gels (lanes 24–27,51–54) as marked on top.
The simultaneous binding of Pop1 and Pop6/Pop7 extends the protected areas of RNAs beyond the combined areas protected by these proteins individually in both RNase P and RNase MRP (Fig. 1A,B). This result points to a synergy in interactions of Pop1 and Pop6/Pop7 with RNase P/MRP RNA components, consistent with the increased stability of the quaternary Pop1–Pop6/Pop7–RNA complexes (above).
Protein binding results in protection of multiple RNA regions. Pop1 protection of the P10/11-CR-II-P12 region of RNase P is consistent with the results of cryo-EM studies that placed the N terminus of Pop1 in that region of the holoenzyme (Hipp et al. 2012); the protection of the CR-IV/mCR-IV regions is consistent with the results of UV crosslinking studies of RNase P/MRP holoenzymes (Khanova et al. 2012). There are clear parallels between the patterns of the protection observed for the “catalytic” (C) domain of RNase P and the corresponding part of RNase MRP, Domain 1 (Esakova and Krasilnikov 2010), in agreement with their previously observed similarities (Esakova et al. 2008; Hipp et al. 2012; Khanova et al. 2012).
Besides protection, the binding of proteins results in an increased sensitivity of several RNA regions to RNase A and RNase V1 cleavage (Fig. 1C,D). The hypersensitivity becomes more pronounced upon the addition of extra protein components. Similar hypersensitivity effects were previously seen upon the binding of the Pop6/Pop7 and Pop5/Rpp1 protein complexes to refolded RNase P/MRP RNAs, even though the hypersensitive RNA regions were not involved in interactions with the proteins and had no peculiarities (Perederina et al. 2007, 2011). We suggested that this hypersensitivity was a result of a more complete secondary structure folding of the in vitro refolded RNA in the presence of protein components (Perederina et al. 2011): Protein binding may force misfolded RNA molecules into the proper secondary structure, thus providing additional molecules suitable for cleavage. The locations of the observed hypersensitive RNase A and RNase V1 cleavage sites are consistent with the previously established secondary structure models of RNase P and RNase MRP RNAs (Esakova and Krasilnikov 2010 and references therein).
In the presence of divalent ions, bacterial RNase P RNA forms a compact three-dimensional structure that is stabilized by a network of tertiary interactions between variable auxiliary RNA elements; addition of Mg2+ results in a compaction of bacterial RNase P RNA (Fang et al. 2000; Krasilnikov et al. 2003, 2004; Kazantsev et al. 2005; Torres-Larios et al. 2005; Baird et al. 2010; Reiter et al. 2010). The results of Fe(II)-EDTA footprinting analysis of bacterial RNase P RNA reveal regions that are completely protected from hydroxyl radicals in the presence of magnesium due to a tight packing of RNA (Pan 1995; Westhof et al. 1996). Importantly, upon folding of the bacterial RNase P RNA, key phylogenetically conserved regions involved in the formation of the catalytic core of the ribozyme (CR-I, CR-IV, CR-V) as well as the phylogenetically conserved part of the CR-II/CR-III region (that is directly involved in pre-tRNA substrate recognition [Krasilnikov et al. 2003, 2004; Kazantsev et al. 2005; Torres-Larios et al. 2005; Reiter et al. 2010]) become protected from the cleavage in Fe(II)-EDTA assays (Pan 1995); the locations of the protected areas are consistent with crystallographic data (Krasilnikov et al. 2003, 2004; Kazantsev et al. 2005; Torres-Larios et al. 2005; Reiter et al. 2010). Given their phylogenetic and, by inference, functional conservation, one can expect similar structural organizations of these regions from bacteria to eukaryotes. For example, in bacteria, the conserved part of the CR-II/CR-III region adopts a complex and dense fold that consists of two interleaving T-loop motifs that recognizes T- and D-loops in the substrate pre-tRNA (Krasilnikov and Mondragon 2003; Krasilnikov et al. 2003, 2004; Torres-Larios et al. 2005; Reiter et al. 2010). In the footprinting assays, this region should be entirely protected due to RNA packing alone, the only exception being a phylogenetically conserved purine corresponding to G245 in S. cerevisiae that has its base exposed to the solvent. Indeed, footprinting analysis of the yeast RNase P holoenzyme revealed exactly this pattern of protection (Esakova et al. 2008).
However, in striking contrast with the bacterial RNase P results obtained using the same methodology (Pan 1995; Westhof et al. 1996), hydroxyl radical probing of refolded RNase P and RNase MRP RNAs revealed little if any evidence of robust tertiary structures in the absence of proteins (Figs. 4–7), and the phylogenetically conserved regions that (in their folded form) are expected to be protected due to RNA packing alone (above) are exposed. The addition of Pop1 and, especially, Pop1 in combination with Pop6/Pop7, results in protection of large portions of RNase P/RNase MRP RNAs in Fe(II)-EDTA footprinting assays. The protection observed upon binding of Pop1–Pop6/Pop7 to refolded RNA components is comparable to that observed in the context of the holoenzymes with all protein components present (Esakova et al. 2008).
The binding of Pop1–Pop6/Pop7 results in the protection of the catalytic core in the reconstituted complexes; this protection most likely results from a combination of direct Pop1 binding (as Pop1 crosslinks to the CR-IV/mCR-IV region [Khanova et al. 2012]) and structural rearrangements in the RNA. Interestingly, while in most regions the inclusion of Pop6/Pop7 results in expansion of the protected area compared with Pop1 alone, in the catalytic domain of RNase MRP the addition of Pop6/Pop7 results in exposure of localized RNA regions located far away from the Pop6/Pop7 binding site, consistent with structural rearrangements in RNA (272–274 and 293–296 regions in lane 30 versus lane 29, Fig. 5B). At the same time, weak protection of the conserved part of the CR-III region of RNase P (which is fully protected in both the bacterial RNase P RNA [Pan 1995] and the yeast RNase P holoenzyme [Esakova et al. 2008]) indicates that folding of this key substrate-binding region requires additional factors (Fig. 8).
FIGURE 5.

Footprinting assays for RNase MRP RNA, typical gels. 3′-end 32P-labeled RNase MRP RNA alone or in complexes with equimolar amounts of Pop1 and the Pop6/Pop7 heterodimer (as indicated above the gels) was partially digested with RNase V1 (A) and hydroxyl radicals (B). (Lanes 1,19,27) RNase MRP RNA digested with RNase T1 (sequence ladder, identifies positions of guanines, shown on the left); (lanes 2,18) alkali hydrolysis of RNase MRP RNA (ladder); (lanes 3–6,28) digestion of RNase MRP RNA; (lanes 7–9,29) digestion of the complex of RNase MRP RNA with Pop1; (lanes 10–13,30) digestion of the quaternary complex of RNase MRP RNA with proteins Pop1, Pop6, and Pop7; (lanes 14–17,31) digestion of the complex of RNase MRP RNA with the Pop6/Pop7 heterodimer; (lanes 20–22,32–34) controls (RNA incubated with proteins as indicated above the gels). Secondary structure elements are marked on the left of the gels: Helical regions are shown by thick solid lines, terminal loops by thin solid lines. Gel traces are shown on the right of the gels (lanes 23–26,35–38) as marked on top.
FIGURE 8.
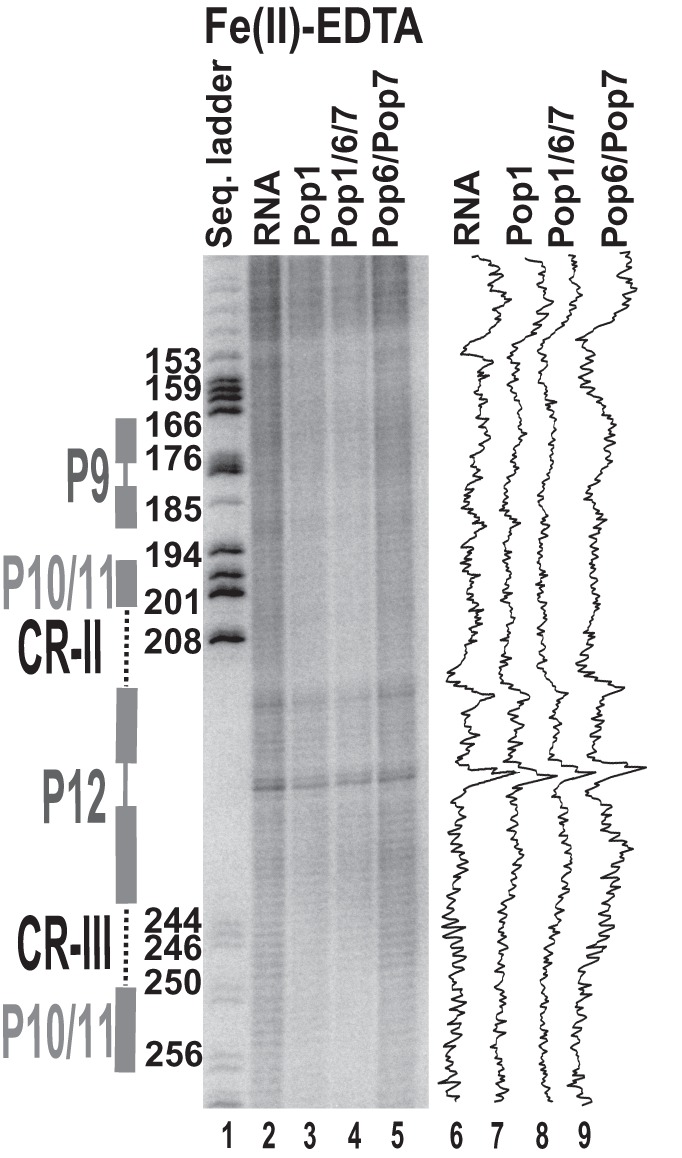
Fe(II)-EDTA footprinting assay for the CR-II/CR-III region of RNase P RNA. 3′-end 32P-labeled RNase P RNA alone or in complexes with equimolar amounts of Pop1 and the Pop6/Pop7 heterodimer (as indicated above the gel). (Lane 1) RNase P RNA digested with RNase T1 (sequence ladder, identifies positions of guanines, shown on the left); (lane 2) digestion of RNase P RNA alone; (lane 3) digestion of the complex of RNase P RNA with Pop1; (lane 4) digestion of the quaternary complex of RNase P RNA with proteins Pop1, Pop6, and Pop7; (lane 5) digestion of the complex of RNase P RNA with the Pop6/Pop7 heterodimer. Secondary structure elements are marked on the left of the gel: Helical regions are shown by thick solid lines, terminal loops by thin solid lines, and internal loops by dotted lines. Gel traces are shown on the right of the gel (lanes 6–9) as marked on top.
The binding of the Pop6/Pop7 heterodimer alone results in the protection of parts of the P3 RNA domain (Figs. 1A,B, 4, 6). The protection of the RNase MRP P3 RNA domain matches the results that were obtained previously (Perederina et al. 2007) and is consistent with the crystal structure of the Pop6/Pop7–P3 RNA domain complex (Perederina et al. 2010b), as well as with the results of the holoenzyme crosslinking (Khanova et al. 2012). The protection of the P3 RNA domain of RNase P in the presence of Pop6/Pop7 is similar to that observed for RNase MRP (Perederina et al. 2007), consistent with the similarities between the P3 domains of RNases P and MRP (Lindahl et al. 2000; Perederina et al. 2010b; Hipp et al. 2012). It should be noted that the efficiency of the protection of the P3 domain RNA by Pop6/Pop7 varies considerably depending on the probe used in the footprinting assays: The affected regions are nearly completely protected from the bulky RNase A and RNase V1 (Figs. 4, 6; Perederina et al. 2007), whereas the protection from hydroxyl radicals, while observable, is rather weak (Figs. 4, 6). This difference likely reflects a local susceptibility of Pop6/Pop7–RNA complexes to thermal fluctuations.
The simultaneous binding of Pop1 and the Pop6/Pop7 heterodimer (Perederina and Krasilnikov 2010; Perederina et al. 2007, 2010b) results in a considerable expansion of the area protected from all probes, including most of the P3 RNA domain and surrounding regions, well beyond the combined areas protected by Pop1 and Pop6/Pop7 bound separately (Figs. 1A,B, 4–6). In the Fe(II)-EDTA assays, the protection of RNA regions known to be involved in direct interactions with Pop6/Pop7 (Perederina et al. 2010b) considerably improves in the presence of Pop1 (Figs. 4, 6); this likely reflects the stabilizing effect of the Pop1 binding on the Pop6/Pop7 interactions with RNA.
The area protected in the presence of the Pop6/Pop7 heterodimer alone matches the available crystallographic data, and, given the structure of the Pop6/Pop7–RNA complex (Perederina et al. 2010b), it is highly unlikely that a simple strengthening of the Pop6/Pop7–RNA interactions can account for the additional protected area that is observed when the Pop6/Pop7 heterodimer is present together with Pop1. Thus, the extension of the protection of the P3 RNA domain observed upon the addition of Pop1 must be either due to direct interactions between the P3 domain RNA and Pop1, or due to the formation of a compact RNA fold protecting most of the P3 RNA domain from both bulky probes and hydroxyl radicals upon Pop1 binding, or due to a combinations of both factors.
The crystal structure of the Pop6/Pop7–P3 RNA domain complex shows that the Pop6/Pop7 heterodimer mostly interacts with the bottom strand of the P3 RNA loop region, whereas a large portion of the top strand remains exposed to the solvent (Perederina et al. 2007, 2010b). The subsequent binding of Pop1 protects the remainder of the top strand as well as the proximal helical stem of the P3 domain (Fig. 1A,B) in a manner similar to the protection of this region observed in the context of the holoenzymes (Esakova et al. 2008). Given the extent of the protection of the P3 RNA domain region, the apparent synergy of the Pop6/Pop7 and Pop1 binding to RNA, the sizes of the involved proteins, and the available structural information (Perederina et al. 2010b), it seems most likely that Pop1 directly interacts with both the Pop6/Pop7 heterodimer and the P3 domain RNA. It should be noted that we did not detect the formation of Pop1–Pop6/Pop7 complexes in the absence of RNA, and that our attempts to form ternary complexes of Pop1/Pop6/Pop7 with separated P3 RNA domains (as opposed to the whole-length RNase P/MRP RNAs) resulted in protein precipitation (data not shown).
As a result of the Pop6/Pop7 binding, the exposed part of the top strand of the P3 RNA loop adopts a highly specific and unusual conformation that requires the presence of the Pop6/Pop7 heterodimer (Perederina et al. 2010b). Thus, the involvement of Pop6/Pop7 in the binding of Pop1 can be twofold. First, the Pop6/Pop7 binding changes the conformation of the top P3 RNA loop strand. This change can make the RNA conformation suitable for the RNA structure-specific interactions with Pop1 and thus facilitate Pop1 binding to RNA. Second, the bound Pop6/Pop7 heterodimer may provide a specific interface for protein–protein interactions that stabilize Pop1 binding. Interestingly, Pop7, a basic protein (pI 9.3), has a highly acidic distal loop region that is not involved in the stabilization of the protein structure or interactions with RNA, but is still phylogenetically conserved (Perederina and Krasilnikov 2010). This acidic loop may in principle serve as an anchoring point for the basic Pop1. While the elucidation of the exact nature of interactions of Pop1 with the P3 RNA domain and Pop6/Pop7 will require results from ongoing crystallographic studies, it seems likely that the Pop6/Pop7 heterodimer facilitates Pop1 recruitment by creating a specific binding interface. The binding of the Pop6/Pop7 involves roughly the bottom part of the P3 RNA domain (Perederina and Krasilnikov 2010; Perederina et al. 2010b); Pop1 is likely to cover the top part, sandwiching the P3 RNA between the three proteins. A less likely alternative involves interactions between Pop1 and the Pop6/Pop7 heterodimer, but not the P3 domain RNA; in this scenario, the observed extension of the protected region in the P3 domain RNA is solely due to compactization of the RNA upon Pop1 binding. In any case, our results support the hypothesis that the P3 RNA domain serves as a protein-binding hub in the eukaryotic RNase P/MRP (Ziehler et al. 2001; Perederina and Krasilnikov 2010; Perederina et al. 2010b), providing an anchoring point for both the Pop6/Pop7 heterodimer and Pop1.
The effects of Pop1 binding on RNA sensitivity in footprinting assays are profound. The binding of Pop1 protects multiple regions that are dispersed over most of the major structural elements of RNase P and MRP RNAs. This is in sharp contrast to the more localized protection that was previously observed upon the binding of smaller RNase P/MRP components, the complexes of proteins Pop6 and Pop7 (Perederina et al. 2007) and Pop5/Rpp1 (Perederina et al. 2011). Pop1 protection of a particular RNA region does not necessarily indicate a direct protein binding to the protected part of RNA as protein-induced changes of RNA conformation may result in RNA protection even if the protein is bound elsewhere; however, generally, the protein-binding site is located in the geometric vicinity of the affected RNA region. The observed protection of multiple scattered regions of RNase P/MRP RNAs implies interactions of Pop1 with anchor points in the RNA that are spread over multiple RNA regions, enabling Pop1 to affect the global RNA fold, consistent with the potential role of Pop1 in the stabilization of the global RNA architecture in eukaryotic RNases P/MRP.
Our results provide support to the hypothesis that Pop1, one of the three eukaryotic RNase P proteins that have no homologs in bacteria and archaea, serves as a scaffold for the stabilization of the global architecture of eukaryotic RNase P RNA, replacing the network of tertiary RNA–RNA interactions that stabilize the structures of bacterial and most of the archaeal RNase P RNAs, while the role of the remaining two uniquely eukaryotic protein components, Pop6 and Pop7, is to bind a specialized eukaryotic RNA element (the helix–loop–helix P3 domain) and provide an additional specific anchoring point for Pop1.
MATERIALS AND METHODS
Expression and purification of proteins
The S. cerevisiae Pop6/Pop7 heterodimer was expressed in E. coli and purified as previously described (Perederina et al. 2007). The purified complex was stored either at −20°C in a buffer containing 5 mM Tris–HCl pH 7.8, 50 mM NaCl, 25 mM KCl, 2.5 mM DTT, 0.05 mM EDTA, and 50% (v/v) glycerol for gel mobility shift and RNase A/RNase V1 footprinting assays, or at 4°C in a buffer containing 10 mM Tris–HCl pH 7.8, 100 mM NaCl, 50 mM KCl, 5 mM DTT, 0.1 mM EDTA for Fe(II)-EDTA footprinting assays.
Protein component Pop1 was produced by a coexpression with another RNase P/MRP protein component, Pop4, in E. coli. The coexpression of the two proteins was essential to obtain soluble Pop1; the putative Pop1/Pop4 complex was separated during Pop1 purification.
POP1 sequence was PCR-amplified from S. cerevisiae strain W303 and inserted into the pET-21R plasmid (see Supplemental Information). pET-21R was produced by the removal of the β-lactamase gene-containing DraI/DraIII fragment of pET21b plasmid (Novagen), followed by the reinsertion of the β-lactamase gene in the inverted orientation. The open reading frame of the cloned POP1 gene contained no purification tags (see Supplemental Information).
An artificial POP4 gene was initially inserted into the NdeI site of the pET21R plasmid (above). The open reading frame of the cloned POP4 was codon-optimized for expression in E. coli with no amino acid changes compared with the wild-type S. cerevisiae POP4 gene and did not include any tags (see Supplemental Information). The POP4-containing MluI–XhoI fragment of the original pET-21R-based POP4 plasmid was subsequently subcloned into the vector that was produced by the digestion of the pCOLADuet-1 plasmid (Novagen) with MluI–XhoI. All protein sequences were verified by sequencing.
For the coexpression, E. coli strain BL21(DE3) was cotransformed with the pET-21R-based POP1 plasmid (above), pCOLADuet-1-based POP4 plasmid (above), and pRARE (a plasmid encoding a number of rare E. coli tRNA codons, Novagen). The cells were grown at 32°C in LB media supplemented with 50 μg/mL ampicillin, 25 μg/mL kanamycin, 17 μg/mL chloramphenicol, and 0.2% (w/v) glucose. The expression was induced by the addition of IPTG to 1 mM at OD600 = 0.6, and continued for 4 h.
All purification procedures were performed at 4°C unless stated otherwise; all buffers and reagents were RNase-free.
Cells from 12 L of culture were pelleted by centrifugation at 4000g, resuspended in 120 mL of lysis buffer (50 mM Tris–HCl pH 7.5, 1 M NaCl, 50 mM KCl, 2 µM ZnCl2, 2 mM MgCl2, 100 mM DTT, 10 mM EDTA, and 1 mM PMSF), and incubated on ice for 15 min. The cells were then disrupted by sonication, and the lysate was cleared by centrifugation for 30 min at 16,000g. Tween-20 (0.002% v/v) and polyethylenimine (0.5% v/v) were added to the clarified extract, the mix was incubated on ice for 10 min, and then centrifuged for 10 min at 16,000g. Supernatant was mixed with 50% (w/v) ammonium sulfate and incubated overnight. The protein precipitate was collected by centrifugation for 30 min at 16,000g. The pellet was dissolved in 40 mL of buffer A (50 mM MES-NaOH pH 6.5, 600 mM NaCl, 50 mM KCl, 2 µM ZnCl2, 2 mM MgCl2, 10 mM β-mercaptoethanol, 0.002% (v/v) Tween 20, 10 mM Na-imidazole pH 8.0). The resulting solution was incubated with 10 mL of preequilibrated Ni-NTA affinity resin for 1 h with gentle agitation; Pop4 did not bind the affinity resin. (It should be noted that neither Pop1 nor Pop4 had any affinity purification tags attached, thus the binding of Pop1 to Ni-NTA resin resulted from intrinsic properties of the protein.) The slurry was packed into a column, washed with 100 mL of buffer A, followed by a wash with 40 mL of buffer A supplemented with 30 mM Na-imidazole pH 8.0. Pop1 was eluted with 30 mL of buffer A supplemented with 400 mM Na-imidazole pH 8.0. The eluted fraction was diluted 1.5-fold with buffer B (50 mM MES-NaOH pH 6.5, 50 mM KCl, 2 µM ZnCl2, 2 mM MgCl2, 10 mM DTT, 0.002% (v/v) Tween-20), and the sample was loaded onto a 1 mL SP-Sepharose column (Amersham) preequilibrated in buffer B that was supplemented with 400 mM NaCl. Protein was eluted from the column with a 60 mL gradient of 0.4–1 M NaCl in the starting buffer. Fractions containing Pop1 were combined, concentrated using an Amicon Ultra-50 concentrator (50,000 MWCO), and then fractionated using a Superdex 200 gel-filtration column (Amersham) that was preequilibrated with gel-filtration buffer (50 mM MES-NaOH pH 6.5, 600 mM NaCl, 50 mM KCl, 2 µM ZnCl2, 2 mM MgCl2, 10 mM DTT, 0.002% (v/v) Tween-20). The fractions containing Pop1 were pooled and concentrated using an Amicon Ultra-50 concentrator (50,000 MWCO) to ∼1.0 mg/mL. Protein concentration was determined using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific) based on absorbance at 280 nm. The extinction coefficient of Pop1 (1.11 × 105 M−1cm−1) was calculated using ExPASy ProtParam tool (http://www.expasy.org); measurement errors were estimated to be <5%. UV spectra did not reveal any presence of nucleic acids in the protein preparations. The purification procedure yielded ∼350 µg of RNase-free Pop1. The protein was aliquoted, frozen in liquid nitrogen, and stored at −76°C.
The identity of Pop1 was confirmed by mass spectrometry analysis. The protein band at ∼100 kDa was excised from an SDS-polyacrylamide gel and treated with trypsin. Tandem mass spectrometry was performed using a LTQ Orbitrap Velos ETD mass spectrometer with a Dionex Ultimate 3000 nano-LC system (Thermo Scientific). The data were analyzed using Scaffold application (Proteome Software Inc.). Unambiguous identification of Pop1 was achieved with 67% sequence coverage.
Production of RNA
The RNA component of RNase MRP was produced by run-off in vitro transcription with T7 RNA polymerase using a traditional approach with linearized plasmids as templates (Milligan and Uhlenbeck 1989). To produce RNase MRP RNA with homogeneous 3′-ends that was used in the footprinting assays involving 3′-end radiolabeled RNase MRP RNA, linearized plasmid pHST7*NME1* (a generous gift from Johanna Avis) that contained the RNase MRP RNA sequence flanked by hammerhead and hepatitis delta virus ribozymes (Walker and Avis 2004) was used as the template; in all other cases, linearized p31/51 plasmid (Perederina et al. 2007) was used.
In vitro transcribed S. cerevisiae RNase P RNA misfolds (Perederina et al. 2007, 2011). Misfolding of in vitro transcribed RNA has been traced to the elongation of the P2 helical stem beyond the 7-bp length found in yeast RNase P/MRP holoenzymes (Esakova et al. 2008). Accordingly, in this work, to facilitate RNase P RNA folding, nucleotides A24 and U25 located in the nonconserved region between P2 and P3 stems of the RNase P RNA (Tranguch and Engelke 1993) were deleted.
The RNA component of RNase P was produced by T7 RNA polymerase-driven in vitro transcription (Milligan and Uhlenbeck 1989) using a linearized plasmid pYRP2 as the template. Plasmid pYRP2 was produced from RNase P RNA-encoding plasmid pYRP (Perederina et al. 2007) by the removal of nucleotides A24 and U25 (above).
Following transcription, RNase P and RNase MRP RNAs were purified using 6% denaturing (8 M urea) polyacrylamide gels as previously described (Perederina et al. 2007). RNA concentration was determined with a NanoDrop ND-1000 spectrophotometer (Thermo Scientific) based on absorbance at 260 nm and using the absorbance coefficient 0.025 (μg/mL)−1/cm−1; measurement errors were estimated to be under 15%.
Immediately before use, RNase P and RNase MRP RNAs were refolded as follows. RNase P RNA was incubated at 70°C for 10 min in 25 mM Tris–HCl pH 7.5, cooled to 25°C in a styrofoam rack, then MgCl2 (to 2.2 mM) and, lastly, KCl (to 200 mM) were added. The RNA component of RNase MRP was folded by incubation at 70°C for 10 min in 25 mM Tris–HCl pH 7.5, cooling to 25°C in a styrofoam rack, incubation at 50°C for 10 min in the presence of 2.2 mM MgCl2, cooling to 25°C in a styrofoam rack, followed by the addition of 200 mM KCl. The final buffer concentrations were 20 mM Tris–HCl pH 7.5, 2 mM MgCl2, and 200 mM KCl.
Control “antisense” RNase MRP RNA was produced using T7 RNA polymerase-driven in vitro transcription (Milligan and Uhlenbeck 1989) with a linearized plasmid pINV (Perederina et al. 2011) as the template, and subsequently purified using denaturing (8 M urea) polyacrylamide gels as previously described (Perederina et al. 2011). Control yeast tRNA was prepared by five rounds of phenol extraction followed by ethanol precipitation from a commercially available (Ambion, AM7119) yeast tRNA preparation.
Gel mobility shift assays
The RNA components of RNases P and MRP were refolded as described above. For the assays involving only Pop1, 9 pmol of RNA was mixed with 4.5–13.5 pmol of Pop1 (depending on the experiment; the amounts and volumes are given per gel lane) and incubated 15 min at 28°C, followed by 15 min at 25°C. The binding was carried out in 11.5 μL of a buffer containing 30 mM MES-NaOH pH 6.5, 8 mM Tris–HCl pH 7.5, 100 mM NaCl, 50 mM KCl, 1 µM ZnCl2, 2 mM MgCl2, 0.001% (v/v) Tween-20, 10% (v/v) glycerol, and 6 mM DTT. The samples were loaded on 6% native polyacrylamide 1× TBE gels and fractionated at 4°C; RNA in the gels was visualized with toluidine blue stain. Assays involving Pop1 and control “antisense” RNase MRP RNA and control tRNA were performed as described above, except for the omission of the RNA refolding step; Pop1 and RNA were taken at a 1:1 molar ratio.
The assays involving both Pop1 and the Pop6/Pop7 heterodimer were performed as described above with the following exceptions. The amounts of the RNA components were increased to 1.5 μg (12.5 pmol of RNase P RNA or 13.5 pmol of RNase MRP RNA). The complex formation was performed in two steps: first, an equimolar amount of the Pop6/Pop7 heterodimer was added to RNA and the mix was incubated at 28°C and 25°C as described above, then an equimolar amount of Pop1 was added and the two incubation steps were repeated. The final volumes of the binding reactions (per gel lane) were 10 μL; the final buffer comprised 30 mM MES-NaOH pH 6.5, 8 mM Tris–HCl pH 7.5, 120 mM NaCl, 50 mM KCl, 1 µM ZnCl2, 2 mM MgCl2, 0.001% (v/v) Tween-20, 10% (v/v) glycerol, 6 mM DTT, and 0.01 mM EDTA.
Dissociation constant estimation
Dissociation constants were roughly estimated using filter-binding assays as previously described (Perederina et al. 2007, 2011) in the presence of a 100-fold molar excess of unlabeled competitor yeast tRNA. RNase P/MRP RNA was 5′-end labeled with 32P, gel purified, and refolded as described above. The RNA–protein complexes were formed following the same protocol that was used in the gel mobility shift assays (above) except for the addition of acetylated bovine serum albumin (BSA) to the final buffer (to 0.1 mg/mL, to reduce protein sorption on plastic ware), final concentration of the radiolabeled RNA (1 nM), and concentrations of the proteins of interest (which ranged from 4 to 800 nM). The solution containing RNA–protein complexes was loaded on and filtered through a stack of BA83 nitrocellulose membrane (protein-binding; Whatman), followed by Hybond-N+ membrane (RNA-binding; GE Healthcare), then the membranes were washed and dried. The radioactivity of the resulting spots on the membranes was quantified using a PhosphorImager (Molecular Dynamics). The intensity of the spots on the nitrocellulose membrane reflected the amount of loaded labeled RNA that formed complexes with protein, while the Hybond-N+ membrane captured the remaining RNA that did not form complexes with protein; the sum of the intensities of spots on the two membranes corresponded to the total amount of labeled RNA loaded. All assays were repeated three times with two parallel runs for each assay to the total of six runs. To estimate dissociation constants, the fraction of RNA that formed complexes with protein was plotted as a function of the protein concentration (the protein was always present in high access), and the graphs (see Supplemental Fig. S1) were interpolated to 50%, which corresponded to the estimated Kd.
Footprinting analysis
For the experiments involving 5′-end-labeled RNase P or RNase MRP RNAs, in vitro transcribed RNA was dephosphorylated using calf intestinal alkaline phosphatase (CIAP), 5′-end labeled with 32P using T4 polynucleotide kinase (PNK), and subjected to an additional round of purification on a 6% denaturing (8 M urea) polyacrylamide gel.
For the footprinting assays involving 3′-end-labeled RNase MRP RNA, in vitro transcribed RNA was treated with PNK (to remove the 2′,3′-cyclophosphate left by the ribozyme, above), 3′-end-labeled using 32P pCp and T4 RNA ligase 1, and subjected to an additional round of purification on a 6% denaturing (8 M urea) polyacrylamide gel.
For the footprinting assays involving 3′-end-labeled RNase P RNA, in vitro transcribed RNA was 3′-end-labeled using 32P pCp and T4 RNA ligase 1. The ligation time was limited so that the incorporation of the radiolabel did not exceed 20% of the saturation level. This limited the appearance of additional bands caused by nonhomogeneous 3′-ends of the in vitro transcribed RNase P RNA to an acceptable level.
For RNase A and RNase V1 footprinting assays, radiolabeled RNA (1 pmol per reaction) was mixed with 8 pmol of “cold” RNA, and RNA–protein complexes of interest were formed using equimolar amounts of proteins in 10 μL of the buffer containing 30 mM MES-NaOH pH 6.5, 8 mM Tris–HCl pH 7.5, 90 mM NaCl, 40 mM KCl, 1 µM ZnCl2, 2 mM MgCl2, 0.001% Tween 20, 10% (v/v) glycerol, 6 mM DTT, and 0.01 mM EDTA as described above. Partial digestions with RNase A and RNase V1 were performed for 10 min on ice and for 15 min at 25°C, respectively, using varying concentrations of the enzymes. The reactions were stopped by phenol/chloroform extraction, which was followed by ethanol precipitation.
For Fe(II)-EDTA footprinting assays, the use of DEPC-treated water and glycerol in samples were avoided. One picomole (per reaction) of radiolabeled RNA was mixed with 8 pmol of “cold” RNA of interest; RNA was refolded, and RNA–protein complexes were formed following the general procedure used for gel mobility shift assays (above). Proteins and RNA were taken at a 1:1 molar ratio, and the final reaction conditions were adjusted to 30 mM MES-NaOH pH 6.5, 4 mM Tris–HCl pH 7.5, 90 mM NaCl, 42 mM KCl, 1.0 µM ZnCl2, 2.0 mM MgCl2, 0.0002% v/v Tween-20, 6 mM DTT, 12 µM EDTA, final volume 10 μL per reaction. The Fe(II)-EDTA footprinting reactions were performed for 15 min on ice and were stopped by the addition of thiourea, followed by phenol/chloroform extraction and ethanol precipitation as previously described (Pan 1995; Esakova et al. 2008).
The reference ladders were produced by partial alkaline hydrolysis and by partial digestion with RNase T1 as previously described (Perederina et al. 2011).
Samples were analyzed on denaturing (8 M urea) polyacrylamide gels; the radioactive bands were visualized using a PhosphorImager (Molecular Dynamics).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank the staff of the Proteomics Core Facility at the Huck Institutes of Life Sciences (PSU) for their help with mass spectrometry analysis. This work was supported by National Institutes of Health grant GM085149 and American Heart Association grant 12GRNT10590001 to A.S.K.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.049007.114.
REFERENCES
- Altman S. 2010. History of RNase P and overview of its catalytic activity. In Ribonuclease P protein reviews (ed. Liu F, Altman S), Vol. 10, pp. 1–15. Springer, New York. [Google Scholar]
- Aspinall TV, Gordon JMB, Bennett HJ, Karahalios P, Bukowski J-P, Walker SC, Engelke DR, Avis JM. 2007. Interactions between subunits of Saccharomyces cerevisiae RNase MRP support a conserved eukaryotic RNase P/MRP architecture. Nucleic Acids Res 35: 6439–6450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulds J, Wierzbicki S, McNairn A, Schmitt ME. 2012. Global identification of new substrates for the yeast endoribonuclease, RNase mitochondrial RNA processing (MRP). J Biol Chem 287: 37089–37097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird NJ, Gong H, Zaheer SS, Freed KF, Pan T, Sosnick TR. 2010. Extended structures in RNA folding intermediates are due to nonnative interactions rather than electrostatic repulsion. J Mol Biol 397: 1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JW. 1999. The Ribonuclease P database. Nucleic Acids Res 27: 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JW, Nolan JM, Haas ES, Rubio MAT, Major F, Pace NR. 1996. Comparative analysis of ribonuclease P RNA using gene sequences from natural microbial populations reveals tertiary structural elements. Proc Natl Acad Sci 93: 3001–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai T, Aulds J, Gill T, Cerio M, Schmitt ME. 2002. The Saccharomyces cerevisiae RNase Mitochondrial RNA Processing is critical for cell cycle progression at the end of mitosis. Genetics 161: 1029–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain JR, Lee Y, Lane WS, Engelke DR. 1998. Purification and characterization of the nuclear RNase P holoenzyme complex reveals extensive subunit overlap with RNase MRP. Genes Dev 12: 1678–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DD, Clayton DA. 1987a. A novel endoribonuclease cleaves at a priming site of mouse mitochondrial DNA replication. EMBO J 6: 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DD, Clayton DA. 1987b. A mammalian mitochondrial RNA processing activity contains nucleus-encoded RNA. Science 235: 1178–1184. [DOI] [PubMed] [Google Scholar]
- Chen J-L, Pace NR. 1997. Identification of the universally conserved core of ribonuclease P RNA. RNA 3: 557–560. [PMC free article] [PubMed] [Google Scholar]
- Chu S, Archer RH, Zengel JM, Lindahl L. 1994. The RNA of RNase MRP is required for normal processing of ribosomal RNA. Proc Natl Acad Sci 91: 659–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S, Zengel JM, Lindahl L. 1997. A novel protein shared by RNase MRP and RNase P. RNA 3: 382–391. [PMC free article] [PubMed] [Google Scholar]
- Dichtl B, Tollervey D. 1997. Pop3p is essential for the activity of the RNase MRP and RNase P ribonucleoproteins in vivo. EMBO J 16: 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esakova O, Krasilnikov AS. 2010. Of proteins and RNA: the RNase P/MRP family. RNA 16: 1725–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esakova O, Perederina A, Quan C, Schmitt ME, Krasilnikov AS. 2008. Footprinting analysis demonstrates extensive similarity between eukaryotic RNase P and RNase MRP holoenzymes. RNA 14: 1558–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esakova O, Perederina A, Quan C, Berezin I, Krasilnikov AS. 2011. Substrate recognition by ribonucleoprotein ribonuclease MRP. RNA 17: 356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans D, Marquez SM, Pace NR. 2006. RNase P: interface of the RNA and protein worlds. Trends Biochem Sci 31: 333–341. [DOI] [PubMed] [Google Scholar]
- Fang X, Littrell K, Yang XJ, Henderson SJ, Siefert S, Thiyagarajan P, Pan T, Sosnick TR. 2000. Mg2+-dependent compaction and folding of yeast tRNAPhe and the catalytic domain of the B. subtilis RNase P RNA determined by small-angle X-ray scattering. Biochemistry 39: 11107–11113. [DOI] [PubMed] [Google Scholar]
- Gill T, Cai T, Aulds J, Wierzbicki S, Schmitt ME. 2004. RNase MRP cleaves the CLB2 mRNA to promote cell cycle progression: novel method of mRNA degradation. Mol Cell Biol 24: 945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill T, Aulds J, Schmitt ME. 2006. A specialized processing body that is temporally and asymmetrically regulated during the cell cycle in Saccharomyces cerevisiae. J Cell Biol 173: 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazov EA, Zankl A, Donskoi M, Kenna TJ, Thomas GP, Clark GR, Duncan EL, Brown MA. 2011. Whole-exome re-sequencing in a family quartet identifies POP1 mutations as the cause of a novel skeletal dysplasia. PLoS Genet 7: e1002027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. 1983. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35: 849–857. [DOI] [PubMed] [Google Scholar]
- Hall TA, Brown JW. 2002. Archaeal RNase P has multiple protein subunits homologous to eukaryotic nuclear RNase P proteins. RNA 8: 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hands-Taylor KL, Martino L, Tata R, Babon JJ, Bui TT, Drake AF, Beavil RL, Pruijn GJ, Brown PR, Conte MR. 2010. Heterodimerization of the human RNase P/MRP subunits Rpp20 and Rpp25 is a prerequisite for interaction with the P3 arm of RNase MRP RNA. Nucleic Acids Res 38: 4052–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp K, Galani K, Batisse C, Prinz S, Böttcher B. 2012. Modular architecture of eukaryotic RNase P and RNase MRP revealed by electron microscopy. Nucleic Acids Res 40: 3275–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser-Scott F, Xiao S, Millikin CE, Zengel JM, Lindahl L, Engelke DR. 2002. Interactions among the protein and RNA subunits of Saccharomyces cerevisiae nuclear RNase P. Proc Natl Acad Sci 99: 2684–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaag HM, Lu Q, Schmitt ME, Nagy PD. 2011. Role of RNase MRP in viral RNA degradation and RNA recombination. J Virol 85: 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrous N, Gopalan V. 2010. Archaeal/eukaryal RNase P: subunits, functions and RNA diversification. Nucleic Acids Res 38: 7885–7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwan R, Bennett JL, Clayton DA. 1991. Nuclear RNase MRP processes RNA at multiple discrete sites: interaction with an upstream G box is required for subsequent downstream cleavages. Genes Dev 5: 1264–1276. [DOI] [PubMed] [Google Scholar]
- Kazantsev AV, Krivenko AA, Harrington DJ, Holbrook SR, Adams PD, Pace NR. 2005. Crystal structure of a bacterial ribonuclease P RNA. Proc Natl Acad Sci 102: 13392–13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanova E, Esakova O, Perederina A, Berezin I, Krasilnikov AS. 2012. Structural organizations of yeast RNase P and RNase MRP holoenzymes as revealed by UV-crosslinking studies of RNA–protein interactions. RNA 18: 720–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikovska E, Svard SG, Kirsebom LA. 2007. Eukaryotic RNase P RNA mediates cleavage in the absence of protein. Proc Natl Acad Sci 104: 2062–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss T, Filipowicz W. 1992. Evidence against a mitochondrial location of the 7-2/MRP RNA in mammalian cells. Cell 70: 11–16. [DOI] [PubMed] [Google Scholar]
- Kole R, Altman S. 1979. Reconstitution of RNase P activity from inactive RNA and protein. Proc Natl Acad Sci 76: 3795–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasilnikov AS, Mondragón A. 2003. On the occurrence of the T-loop RNA folding motif in large RNA molecules. RNA 9: 640–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasilnikov AS, Yang X, Pan T, Mondragón A. 2003. Crystal structure of the specificity domain of ribonuclease P. Nature 421: 760–764. [DOI] [PubMed] [Google Scholar]
- Krasilnikov AS, Xiao Y, Pan T, Mondragón A. 2004. Basis for structural diversity in homologous RNAs. Science 306: 104–107. [DOI] [PubMed] [Google Scholar]
- Lai LB, Vioque A, Kirsebom LA, Gopalan V. 2010. Unexpected diversity of RNase P, an ancient tRNA processing enzyme: challenges and prospects. FEBS Lett 584: 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Willkomm DK, Hartmann RK. 2009. Minor changes largely restore catalytic activity of archaeal RNase P RNA from Methanothermobacter thermoautotrophicus. Nucleic Acids Res 37: 231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl L, Fretz S, Epps N, Zengel JM. 2000. Functional equivalence of hairpins in the RNA subunits of RNase MRP and RNase P in Saccharomyces cerevisiae. RNA 6: 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl L, Bommankanti A, Li X, Hayden L, Jones A, Khan M, Oni T, Zengel JM. 2009. RNase MRP is required for entry of 35S precursor rRNA into the canonical processing pathway. RNA 15: 1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Wierzbicki S, Krasilnikov AS, Schmitt ME. 2010. Comparison of mitochondrial and nucleolar RNase MRP reveals identical RNA components with distinct enzymatic activities and protein components. RNA 16: 529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lygerou Z, Mitchell P, Petfalski E, Séraphin B, Tollervey D. 1994. The POP1 gene encodes a protein component common to the RNase MRP and RNase P ribonucleoproteins. Genes Dev 8: 1423–1433. [DOI] [PubMed] [Google Scholar]
- Lygerou Z, Allmang C, Tollervey D, Séraphin B. 1996. Accurate processing of a eukaryotic precursor ribosomal RNA by Ribonuclease MRP in vitro. Science 272: 268–270. [DOI] [PubMed] [Google Scholar]
- Marquez SM, Chen JL, Evans D, Pace NR. 2006. Structure and function of eukaryotic Ribonuclease P RNA. Mol Cell 24: 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin MC, Engelke DR. 2009. Broadening the mission of an RNA enzyme. J Cell Biochem 108: 1244–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattijssen S, Welting TJ, Prijin GJ. 2010. RNase MRP and disease. Wiley Interdiscip Rev RNA 1: 102–116. [DOI] [PubMed] [Google Scholar]
- Mattijssen S, Hinson ER, Onnekink C, Hermanns P, Zabel B, Cresswell P, Prijin GJ. 2011. Viperin mRNA is a novel target for the human RNase MRP/RNase P endoribonuclease. Cell Mol Life Sci 68: 2469–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan JF, Uhlenbeck OC. 1989. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol 180: 51–62. [DOI] [PubMed] [Google Scholar]
- Mondragón A. 2013. Structural studies of RNase P. Annu Rev Biophys 42: 537–557. [DOI] [PubMed] [Google Scholar]
- Pan T. 1995. Higher order folding and domain analysis of the ribozyme from Bacillus subtilis ribonuclease P. Biochemistry 34: 902–909. [DOI] [PubMed] [Google Scholar]
- Pannucci JA, Haas ES, Hall TA, Harris JK, Brown JW. 1999. RNase P RNAs from some Archaea are catalytically active. Proc Natl Acad Sci 96: 7803–7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perederina A, Krasilnikov AS. 2010. The P3 domain of eukaryotic RNases P/MRP: making a protein-rich RNA-based enzyme. RNA Biol 7: 534–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perederina A, Esakova O, Koc H, Schmitt ME, Krasilnikov AS. 2007. Specific binding of a Pop6/Pop7 heterodimer to the P3 stem of the yeast RNase MRP and RNase P RNAs. RNA 13: 1648–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perederina A, Esakova O, Quan C, Khanova E, Krasilnikov AS. 2010a. Crystallization and preliminary X-ray diffraction analysis of the P3 RNA domain of yeast ribonuclease MRP in a complex with RNase P/MRP protein components Pop6 and Pop7. Acta Crystallogr Sect F Struct Biol Cryst Commun 66: 76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perederina A, Esakova O, Quan C, Khanova E, Krasilnikov AS. 2010b. Eukaryotic ribonucleases P/MRP: the crystal structure of the P3 domain. EMBO J 29: 761–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perederina A, Khanova E, Quan C, Berezin I, Esakova O, Krasilnikov AS. 2011. Interactions of a Pop5/Rpp1 heterodimer with the catalytic domain of RNase MRP. RNA 17: 1922–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccinelli P, Rosenblad MA, Samuelsson T. 2005. Identification and analysis of Ribonuclease P and MRP RNA in a broad range of eukaryotes. Nucleic Acids Res 33: 4485–4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinker F, Bonnard G, Gobert A, Gutmann B, Hammani K, Sauter C, Gegenheimer PA, Giegé P. 2013. PPR proteins shed a new light on RNase P biology. RNA Biol 10: 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluk H, van Eenennaam H, Rutjes SA, Pruijn GJ, van Venrooij WJ. 1999. RNA-protein interactions in the human RNase MRP ribonucleoprotein complex. RNA 5: 512–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer G, Raska I, Scheer U, Tan EM. 1988. Immunolocalization of 7-2-ribonucleoprotein in the granular component of the nucleolus. Exp Cell Res 176: 117–128. [DOI] [PubMed] [Google Scholar]
- Reiter NJ, Osterman A, Torres-Larios A, Swinger KK, Pan T, Mondragón A. 2010. Structure of a bacterial ribonuclease P holoenzyme in complex with tRNA. Nature 468: 784–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridanpaa M, Eenennaam H, Pelin K, Chadwick R, Johnson C, Yuan B, van Venrooij W, Pruijn G, Salmela R, Rockas S, et al. 2001. Mutations in the RNA component of RNase MRP cause a pleiotropic human disease, cartilage-hair hypoplasia. Cell 104: 195–203. [DOI] [PubMed] [Google Scholar]
- Saito Y, Takeda J, Adachi K, Nobe Y, Kobayashi J, Hirota K, Oliveira DV, Taoka M, Isobe T. 2014. RNase MRP cleaves pre-tRNASer-Met in the tRNA maturation pathway. PLoS One 9: e112488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt ME, Clayton DA. 1993. Nuclear RNase MRP is required for correct processing of pre-5.8S rRNA in Saccharomyces cerevisiae. Mol Cell Biol 13: 7935–7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark BC, Kole R, Bowman EJ, Altman S. 1978. Ribonuclease P: an enzyme with an essential RNA component. Proc Natl Acad Sci 75: 3717–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolc V, Altman S. 1997. Rpp1, an essential protein subunit of nuclear RNase P required for processing of precursor tRNA and 35S precursor rRNA in Saccharomyces cerevisiae. Genes Dev 11: 2926–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas BC, Chamberlain J, Engelke DR, Gegenheimer P. 2000. Evidence for an RNA-based catalytic mechanism in eukaryotic nuclear ribonuclease P. RNA 6: 554–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Larios A, Swinger KK, Krasilnikov AS, Pan T, Mondragón A. 2005. Crystal structure of the RNA component of bacterial ribonuclease P. Nature 437: 584–587. [DOI] [PubMed] [Google Scholar]
- Tranguch AJ, Engelke DR. 1993. Comparative structural analysis of nuclear RNase P RNAs from yeast. J Biol Chem 268: 14045–14055. [PubMed] [Google Scholar]
- Tranguch AJ, Kindelberger DW, Rohlman CE, Lee JY, Engelke DR. 1994. Structure-sensitive RNA footprinting of yeast nuclear ribonuclease P. Biochemistry 33: 1778–1787. [DOI] [PubMed] [Google Scholar]
- Walker SC, Avis JM. 2004. A conserved element in the yeast RNase MRP RNA subunit can participate in a long-range base-pairing interaction. J Mol Biol 341: 375–388. [DOI] [PubMed] [Google Scholar]
- Walker SC, Marvin MC, Engelke DR. 2010. Eukaryote RNase P and RNase MRP. In Ribonuclease P, protein reviews (ed. Liu F, Altman S), Vol. 10, pp. 173–202. Springer, New York. [Google Scholar]
- Welting TJM, van Venrooij WJ, Pruijn GJ. 2004. Mutual interactions between subunits of the human RNase MRP ribonucleoprotein complex. Nucleic Acids Res 32: 2138–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welting TJ, Peters FM, Hensen SM, van Doorn NL, Kikkert BJ, Raats JM, van Venrooij WJ, Pruijn GJ. 2007. Heterodimerization regulates RNase MRP/RNase P association, localization, and expression of Rpp20 and Rpp25. RNA 13: 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhof E, Wesolowski D, Altman S. 1996. Mapping in three dimensions of regions in a catalytic RNA protected from attack by an Fe(II)-EDTA reagent. J Mol Biol 258: 600–613. [DOI] [PubMed] [Google Scholar]
- Xiao S, Hsien J, Nugent RL, Coughlin DJ, Fierke CA, Engelke DR. 2006. Functional characterization of the conserved amino acids in Pop1p, the largest common protein subunit of yeast RNases P and MRP. RNA 12: 1023–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziehler WA, Morris J, Scott FH, Millikin C, Engelke DR. 2001. An essential protein-binding domain of nuclear RNase P RNA. RNA 7: 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.