

Background: Threonylcarbamoyl-AMP synthase catalyzes formation of the biosynthetic intermediate of a critical tRNA modification, t6A37.
Results: Structural analyses provide insight into the interaction between the substrates ATP and l-threonine and the E. coli enzyme.
Conclusion: The threonylcarbamoyl-AMP synthase binds l-threonine and ATP cooperatively; l-threonine is required for positioning of ATP.
Significance: Mechanistic insights into t6A37 biosynthesis provide an understanding of this complex enzymatic pathway.
Keywords: Escherichia coli (E. coli), ligand-binding protein, nuclear magnetic resonance (NMR), protein structure, RNA modification, transfer RNA (tRNA)
Abstract
The hypermodified nucleoside N6-threonylcarbamoyladenosine (t6A37) is present in many distinct tRNA species and has been found in organisms in all domains of life. This post-transcriptional modification enhances translation fidelity by stabilizing the anticodon/codon interaction in the ribosomal decoding site. The biosynthetic pathway of t6A37 is complex and not well understood. In bacteria, the following four proteins have been discovered to be both required and sufficient for t6A37 modification: TsaC, TsaD, TsaB, and TsaE. Of these, TsaC and TsaD are members of universally conserved protein families. Although TsaC has been shown to catalyze the formation of l-threonylcarbamoyl-AMP, a key intermediate in the biosynthesis of t6A37, the details of the enzymatic mechanism remain unsolved. Therefore, the solution structure of Escherichia coli TsaC was characterized by NMR to further study the interactions with ATP and l-threonine, both substrates of TsaC in the biosynthesis of l-threonylcarbamoyl-AMP. Several conserved amino acids were identified that create a hydrophobic binding pocket for the adenine of ATP. Additionally, two residues were found to interact with l-threonine. Both binding sites are located in a deep cavity at the center of the protein. Models derived from the NMR data and molecular modeling reveal several sites with considerable conformational flexibility in TsaC that may be important for l-threonine recognition, ATP activation, and/or protein/protein interactions. These observations further the understanding of the enzymatic reaction catalyzed by TsaC, a threonylcarbamoyl-AMP synthase, and provide structure-based insight into the mechanism of t6A37 biosynthesis.
Introduction
An essential aspect of RNA maturation is the post-transcriptional modification of nucleosides, which adds chemical complexity to the four major nucleosides and permits a greater range of functionality. Modified nucleosides are particularly abundant in transfer RNA (tRNA), where over 90 distinct modifications are found across all phylogenetic domains (1). One of the sites with the highest frequency of modification in tRNA and some of the most chemically complex modifications found in any RNA is that of the conserved purine 3′-adjacent to the anticodon at position 37 (2). More than 70% of tRNA species are modified at this site (3). Of these, the universally conserved N6-threonylcarbamoyladenosine (t6A37)3 modification and its derivatives, 2-methylthio-N6-threonylcarbamoyladenosine (ms2t6A37) and N6-methyl-N6-threonylcarbamoyladenosine, are found at the 37-position in nearly all tRNAs that read ANN codons (Fig. 1, A and B) (3, 4). Each of these tRNAs contains a strictly conserved sequence of 36UAA38 in the anticodon stem-loop (3). Both U36 and A37 are essential for modification and A38 enhances the rate of the modification reaction (5). The t6A37 modification has been shown to have an important role in ribosome-mediated codon binding for several tRNA species (6–8), mainly due to its ability to enhance the stability of the anticodon-codon base pairing by creating cross-strand base-stacking interactions with the first position of the codon, as depicted in structural analyses of tRNALysUUU decoding at the ribosome A-site (9, 10). This stabilization is necessary to overcome the low enthalpy of binding for U-A base pairs and tRNALysUUU in particular with three U-A base pairs (11). The size and location of t6A37 on the Watson-Crick face of the nucleoside negate intra-loop base pairing with the invariant U33, which is critical for the U-turn backbone structure of tRNA. This creates an open structured loop that is more favorable for entry into the ribosome and the binding of the mRNA codon (9, 12). By strengthening the codon/anticodon interaction and ribosome entry, t6A37 is believed to participate in the maintenance of the translational reading frame. This is supported by the increase in translational frameshifts in cells lacking the ability to form t6A37 (13–15).
FIGURE 1.
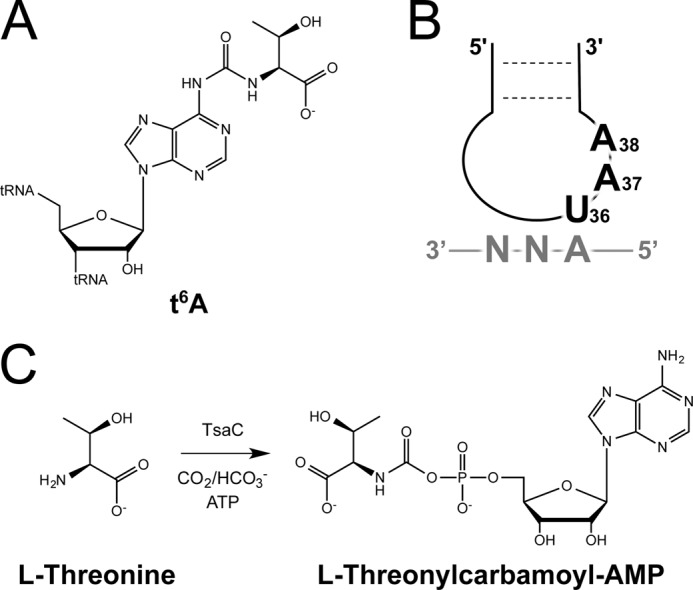
Requirements for threonylcarbamoylation of tRNA. A, structure of t6A. B, consensus sequence for t6A-modifed tRNA species, which all recognize ANN codons in mRNA. C, TC-AMP formation requires l-threonine, CO2/HCO3−, ATP, and TsaC.
Recent studies of the t6A37 biosynthesis pathway have identified the following four proteins in Escherichia coli that are necessary and sufficient for in vitro formation of the modification: TsaC, TsaD, TsaE, and TsaB, also known as YrdC, YgjD, YjeE, and YeaZ, respectively (16). Two of the four proteins essential to the enzyme complex are unique to bacteria, TsaB and TsaE. The other two, TsaC and TsaD, are members of universal families and are associated with the t6A pathway in several organisms. The biosynthesis of t6A37 has been reconstituted in vitro in the bacterial species E. coli and Bacillus subtilis (17) and the eukaryotic and archaeal species Saccharomyces cerevisiae and Pyrococcus abyssi, respectively (18, 19). The biosynthesis in S. cerevisiae and P. abyssi requires l-threonine, ATP, and CO2/HCO3−, analogous to bacteria, but five proteins are necessary as follows: Sua5 (the TsaC homolog), Kae1 (the TsaD homolog), Bud32, Pcc1, and Cgi121 (18). The latter four proteins compose the KEOPS complex, a protein complex associated with a variety of physiological phenomena (15, 20–23). While both divalent and monovalent cations are suspected to be essential for this reaction, no systematic analysis has determined their requirement.
Although the proteins involved in t6A37 biosynthesis have been identified, and experimental evidence suggests that they function together in a heteromultimeric complex, little is known about the structure of the complexes or the mechanistic contributions of each protein. TsaC, which is central to the biosynthesis of t6A37, is a member of the TsaC/Sua5 protein family. This family has been found in all sequenced genomes to date. However, the essentiality varies between organisms. TsaC is essential in E. coli, but Sua5 is not essential in S. cerevisiae (24), even though both appear to have similar roles in t6A37 biosynthesis. E. coli TsaC selectively binds both ATP and l-threonine (24, 25) and is the sole protein of the t6A-synthase complex capable of l-threonine-dependent conversion of ATP to AMP (16). More recently, both TsaC and its B. subtilis homolog, YwlC, have been shown to catalyze the formation of the activated precursor l-threonylcarbamoyl-AMP (TC-AMP) from l-threonine, ATP, and CO2/HCO3− (Fig. 1C) (17). This strongly suggests that TsaC is the enzyme responsible for the first step in the mechanism. Subsequently, TC-AMP is transferred to the A37 of the tRNA substrate by TsaB, TsaD, and TsaE (YeaZ, YgjD, and YjeE). Consistent with this model is the observation that the B. subtilis homologs of TsaBDE are capable of t6A37 biosynthesis in the presence of TC-AMP and tRNA and the absence of YwlC, the TsaC homolog (17). The argument is strengthened by the ability of P. abyssi Kae1, the TsaD homolog, to bind TC-AMP and catalyze the transfer to tRNA (26). However, several studies have shown that E. coli TsaC selectively binds hypomodified tRNA (24, 25, 27), indicating TsaC may have a more substantial, diverse role in bacteria than in eukaryotes because S. cerevisiae Sua5 does not exhibit RNA binding activity (18).
Within the TsaC/Sua5 family, four structures have been solved by x-ray crystallography, including E. coli YciO (28), Sulfolobus tokodaii Sua5 (29), E. coli HypF (30), and E. coli TsaC (27). Each of these structures contains a TsaC domain with a unique folding pattern and the ability to catalyze similar chemistries. However, neither HypF nor YciO are associated with the t6A37 pathway. Unfortunately, the TsaC crystal structure lacks several of the C-terminal residues (presumably due to low electron density). In addition, the crystal packing-induced dimerization appears to alter the conformation of functionally relevant regions in the binding of l-threonine, ATP, and protein/protein interactions (27). Here, we sought to characterize the dynamics and structural interactions of TsaC with its substrates to provide insight into the mechanism of action of TsaC. Therefore, structural characterization through solution nuclear magnetic resonance (NMR) was employed to observe full-length TsaC under native reaction conditions. As such, we report the high-resolution structure of the monomer and interaction studies with l-threonine and ATP in solution using NMR. We were able to identify several key residues in TsaC that interact with l-threonine and ATP using chemical shift perturbation analyses. These analyses were used to guide molecular docking to provide the first models of interactions between E. coli TsaC and ATP and l-threonine. These studies provide insight into how TsaC catalyzes the formation of TC-AMP.
Experimental Procedures
Protein Expression and Purification
Protein samples were expressed and purified as described previously (16). For isotope-labeled TsaC, M9 minimal medium was supplemented with 15NH4Cl and [13C]glucose (ISOTEC) (31). The TsaC construct lacks an N-terminal Met-1 due to a Factor Xa cleavage site used to remove the His6 tag. All TsaC samples were prepared in a buffer of 90:10% H2O/2H2O, 20 mm potassium phosphate, pH 7.0, 100 mm NaCl unless otherwise noted.
Nuclear Magnetic Resonance
All NMR experiments were performed at 298 K on either a Varian Inova 600 MHz or a Bruker Avance II 700 MHz spectrometer, both equipped with cryoprobes. Backbone chemical shifts were assigned in a sequential manner from the following experiments: two-dimensional 1H-15N-HSQC, HNCA, HN(CO)CA, HN(CO)CACB, HNCACB, HNCO, and HN(CA)CO. TALOS was used to determine coupling constants for assigning backbone ψ and ϕ angles (32). Side chain proton and carbon chemical shifts were assigned using the following experiments: CC(CO)NH (nCyc2 and -3), H(CCO)NH (nCyc3 and -4), and a 15N-total correlation spectroscopy-HSQC with a 50-ms mixing time. NOEs were obtained from three-dimensional 15N-NOESY-HSQC experiments with 120- and 150-ms mixing times and three-dimensional 13C-NOESY-HSQC experiments with 100- and 120-ms mixing times. For residual dipolar couplings, 15N-labeled TsaC was aligned in bacteriophage Pf1 (ASLA Biotech) to a final concentration of 12 mg/ml. 1DHN couplings were obtained from a two-dimensional 1H-15N HSQC-IPAP (in-phase/antiphase) experiment (33). Data were processed using NMRPipe (34) and analyzed using SPARKY (35) and NMRViewJ (36).
Protein backbone dynamics were determined using steady-state heteronuclear 1H-15N NOE experiments as described previously (37) at 298 K. Values were measured from the peak heights in two-dimensional 1H-15N-HSQC spectra, normalized for background noise, and graphed by Isat/Iunsat.
Structure Calculation
CYANA 2.1 was first used for automatic NOE assignment, and 100 structures were calculated with the standard simulated annealing protocol, using NOE and hydrogen bonding distance restraints and ψ and ϕ dihedral angle restraints from TALOS+ predictions (38). The final round of 100 structural calculations was performed in XPLOR-NIH using the same NOE, hydrogen bonding, and dihedral restraints together with residual dipolar coupling restraints (39, 40). The ensemble of the 20 lowest energy structures was assessed using the Protein Structure Validation Software (PSVS) Suite (41). Molecules were visualized and aligned with PyMOL (42).
Substrate Titrations Observed by NMR
The binding of l-threonine and ATP by TsaC were observed at 298 K on a Bruker Avance III 500 MHz spectrometer equipped with an ultrasensitive triple resonance cryoprobe capable of applying pulsed field gradients along the z axis. The TsaC protein sample was concentrated to 100 μm in a buffer of 90:10% H2O/2H2O, 50 mm potassium phosphate, pH 7.5, 150 mm KCl. ATP (Sigma) was titrated into samples that provided the following molar ratios of ligand to protein: 0:1, 0.5:1, 1:1, 2:1, 4:1, and 8:1. l-Threonine (Sigma) was added to the protein in ligand to protein ratios of 0:1, 1:1, 2:1, and 4:1. A two-dimensional 1H-15N-HSQC experiment was collected at each titration point. Data were processed using NMRPipe (34) and analyzed using SPARKY (35). The total shift change, ΔδN-H, for each peak was calculated by , where ΔδHN and ΔδN are the chemical shift differences for 1H and 15N, respectively. NMR spectra of the protein in the absence of any ligands were the same although collected under buffer conditions for structural determinations (90:10% H2O/2H2O, 20 mm potassium phosphate, pH 7.0, 100 mm NaCl) that were slightly different in pH and ionic strength from conditions for the protein's titration with ATP and l-threonine followed by NMR and ITC (90:10% H2O/2H2O, 50 mm potassium phosphate, pH 7.5, 150 mm KCl).
Isothermal Titration Calorimetry
The interaction of TsaC with ATP was characterized by ITC experiments conducted at 4 °C (MicroCal VP-ITC). TsaC was prepared at 100 μm in 50 mm potassium phosphate, pH 7.5, 150 mm KCl. ATP at a concentration of 1 mm in the same buffer was titrated in 10 μl injection volumes into the experimental cell containing TsaC. Titration curves were baseline-subtracted and analyzed by nonlinear least-squares fitting using the MicroCal Origin 5.0 software.
HADDOCK Docking Procedure
Default HADDOCK (high ambiguity driven docking) (43) parameters were used throughout all docking procedures. Active and passive residues (Table 3) with solvent accessibility >50% calculated by NACCESS (44) were assigned from TsaC NMR titration studies. Passive residues were defined as all other residues with 50% solvent accessibility. The adenosine moiety of the ATP molecule was defined as the active portion, and the phosphate closest to the adenosine moiety was defined as passive. The remaining phosphate moieties were not actively involved in the interaction between ATP and TsaC. One thousand structures were generated per iteration, and the 200 lowest energy structures were water refined. Each docking attempt was performed 10 times, and the solution with the lowest HADDOCK score was retained. The root-mean-square deviation (r.m.s.d.) values of the complexes were calculated using the McLachlan algorithm (45) as implemented in ProFit (46). A cluster analysis was performed on the final docking solutions using a minimum cluster size of four. The cutoff for clustering was manually determined for each docking run. The r.m.s.d. matrix was calculated over the backbone atoms of the interface residues.
TABLE 3.
Active and passive residues for all docking
| Liganda |
TsaC |
|||
|---|---|---|---|---|
| Active | Passive | Active | Passive | |
| TsaC/l-threonine | 1 | NA | 27, 176 | 25, 28–31, 59, 91–93, 121–123, 167–170, 174, 175, 177, 187–190 |
| TsaC/ATP | Adenosine moiety | Phosphate group closest to the adenosine moiety | 59, 109, 114, 115, 188 | 25, 28–31, 59, 91–93, 121–123, 167–170, 174, 175, 177, 187, 189, 190 |
| TsaC/l-threonine/ATP | Adenosine moiety | Phosphate group closest to the adenosine moiety | 59, 109, 114, 115, 188 | 38, 10, 14, 17, 20, 28, 30, 38, 40, 41, 44, 48, 51, 52, 64, 66, 69–70, 73, 75, 77–80, 83, 86–87, 91, 100–103, 110–111, 120–121, 125, 129, 131–132, 139, 141, 143–146, 149–150, 152, 155–158, 161–162, 167–168, 170–173, 175, 180, 182–183, 185–187, 189, 190 |
a The molecule defined as the ligand is shown in boldface.
Results
TsaC has been shown to interact with three small substrates, l-threonine (25), ATP (24), and CO2/HCO3− (17), as well as hypomodified tRNA (24, 25, 27), TsaD and TsaB (16). The synthesis of TC-AMP, the central role of TsaC in t6A37 biosynthesis, requires a number of intermolecular interactions and enzymatic activity. These interactions have yet to be fully characterized structurally and mechanistically, and consequently, the precise details of this enzymatic biosynthesis are unknown. To provide some understanding of the mechanism of TC-AMP synthase, we characterized the structure and dynamics of the functional TsaC monomer and its substrate interactions. Based on this structural information, molecular docking was performed between TsaC and ATP and l-threonine.
Structure of E. coli TsaC
The monomeric structure of E. coli TsaC in solution was verified by NMR because there was the possibility of dimerization as had been observed in the crystal structure but contrary to monomers observed in solution by dynamic light scattering (27). The application of a pulsed field gradient method for analysis of diffusion (47) confirmed that the protein was a monomer under NMR conditions (data not shown). The 15N-13C-TsaC displayed excellent spectral dispersion of resonances for a 20.6-kDa protein in a two-dimensional 1H-15N-transverse relaxation optimized spectroscopy spectrum (Fig. 2). Therefore, conventional heteronuclear multidimensional NMR methods were utilized to assign 1H, 15N, and 13C backbone and side chain resonances of isotope-labeled E. coli TsaC. Most amide peaks were present in the 1H-15N-HSQC and were assigned with the exception of Asn-2, Asn-3, Val-118, and Ser-139. In total, the molecule was 87% assigned with 98% assignment of the backbone, including HN, N, Cα, and C′, and 79% assignment of the side chains. Using these assignments, sequential connectivities and distances between intramolecular protons were determined for construction of a high-resolution solution structure of TsaC.
FIGURE 2.

E. coli TsaC 1H-15N-transverse relaxation optimized spectroscopy spectrum with amino acid amide assignments. All expected amides were present and assigned with the exception of Asn-2, Asn-3, Val-118, and Ser-139. The two unlabeled downfield peaks are the Trp-89 and Trp-106 NHϵ resonances.
The 20 lowest energy, water-refined structures were determined using a total of 1869 distance restraints with 656 sequential, 444 medium range, and 379 long range restraints. The structures contained zero NOE and hydrogen bond violations (Table 1; Fig. 3). The TsaC structures have an average backbone r.m.s.d. of 0.79 Å and an average heavy atom r.m.s.d. of 1.35 Å for residues comprising regions of secondary structure. The Ramachandran analysis indicates the reported structure of TsaC is of good quality; of all the residues in the ensemble of 20 structures, 95.3% are in the allowed Ramachandran space or better. The residues that are in the generously allowed (4.1%) or the disallowed (0.6%) regions reside either in loop areas of the protein or regions of no RDC restraints due to spectral overlap (Table 1).
TABLE 1.
TsaC NMR and refinement statistics
| NMR experimental restraints | |
| Distance restraints | |
| Total NOE | 1869 |
| Intra-residue | 390 |
| Inter-residue | 1479 |
| Sequential (|i − j| = 1) | 656 |
| Medium range (|i − j| <4) | 444 |
| Long range (|i − j| >5) | 379 |
| Hydrogen bonds | 99 |
| Total dihedral angle restraints | |
| ϕ | 98 |
| ψ | 89 |
| 15N-1H residual dipolar couplings | 66 |
| Structural statistics | |
| r.m.s.d. from solution NMR restraints | |
| Distance restraints (Å) | 0.061 ± 0.001 |
| Dihedral angle restraints (°) | 0.599 ± 0.066 |
| RDC restraints (Hz) | 0.111 ± 0.020 |
| r.m.s.d. from idealized covalent geometry | |
| Bonds (Å) | 0.006 ± 0.0001 |
| Angles (°) | 0.746 ± 0.007 |
| Impropers (°) | 0.504 ± 0.010 |
| RDC quality factora | 0.182 ± 0.006 |
| Precision of atomic coordinates over secondary structureb | |
| Backbone (N, Cα, C′, O) (Å) | 0.792 ± 0.148 |
| Heavy atoms (Å) | 1.355 ± 0.132 |
| Ramachandran analysisc | |
| Most favored | 77.4% |
| Additionally allowed | 17.9% |
| Generously allowed | 4.1% |
| Disallowed | 0.6% |
a RDC quality factor was calculated according to the method of Clore and Garrett (48).
b The secondary elements used were as follows: residues 4–19, 40–48, 65–71, 79–86, 105–109, 123–131, 151–158, 23–25, 32–35, 59–62, 94–97, 114–117, 135–138, and 177–180.
c Ramachandran statistics were calculated for the ensemble using PROCHECK-NMR (49).
FIGURE 3.
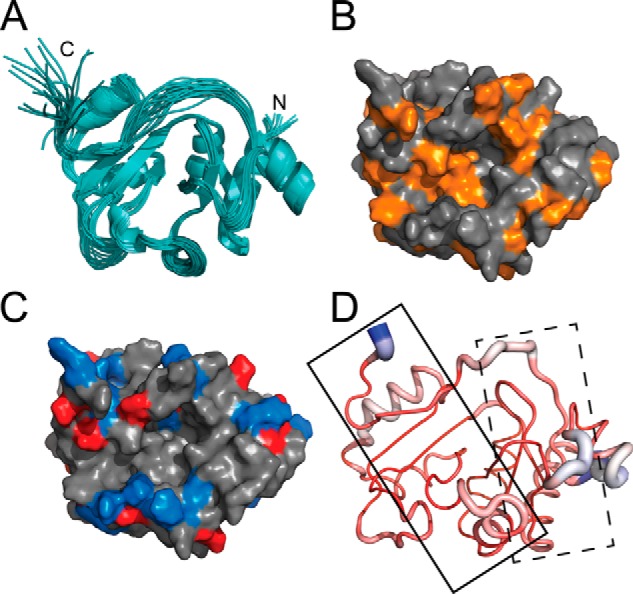
TsaC NMR solution structure. A, 20 lowest energy structures of E. coli TsaC characterized by NMR. B, hydrophobic residues (orange) of the TsaC NMR solution structure. C, electrostatic potential of the TsaC NMR solution structure, where blue and red denote positive and negative charges, respectively. D, comparison of the TsaC crystal (Protein Data Bank code 1HRU) and NMR-solved TsaC structures. The loop width is directly proportional to the differences in Cα r.m.s.d. between the two structures. Small to large differences in Cα r.m.s.d. are colored red to white to blue. Spheres denote ATP- (solid box) and l-threonine (dotted box)- binding sites. The solid box also represents the crystal packing dimerization interface of the crystal structure. All structures are in the same orientation.
The E. coli TsaC structure has an α/β twisted open-sheet structure with parallel and antiparallel adjacent β-strands, consistent with other members of the family (Fig. 3A). The structure includes seven α-helices and seven β-strands connected by 13 loop regions. The β-strands are aligned in the center of the protein and helically twist 180° from β1 to β7. The C terminus exhibits considerable conformational flexibility, suggesting an enzymatically relevant role. At the center of the protein there is a deep cavity of hydrophobic character lined with positive surface potential, providing potential binding surfaces (Fig. 3, B and C) (27). Comparison of the crystal structure (27) and the NMR solution structure reveals the areas of greatest difference based on Cα r.m.s.d. (Fig. 3D). There is considerable divergence between the two structures in the malformed dimerization region from the crystal structure (Fig. 3D, dashed box), suggesting that crystal packing greatly affected the structure in this region, which is important for substrate/protein and protein/protein interactions.
l-Threonine-binding Site
E. coli TsaC and S. tokodaii Sua5 have been shown to exhibit specificity for l-threonine over other amino acids (25, 50), and E. coli TsaC has been shown to catalyze TC-AMP formation (17). Therefore, we sought to characterize the TsaC/l-threonine binding interface to provide insight into structural aspects of the enzymatic mechanism. Using the fully assigned 1H-15N-HSQC spectrum (Fig. 2), amide chemical shift changes within the protein were monitored by 1H-15N-HSQC experiments with protein/ligand ratios of 1:0, 1:1, 1:2, and 1:4. We observed two distinct and significant changes in the spectra for Thr-27 and Ser-176. There were no pronounced up- or downfield movements in any resonance. However, the amide peaks for Thr-27 and Ser-176 broaden to the point of disappearance (Fig. 4, A and B). This is likely due to the line broadening caused by intermediate exchange between the free and bound forms and is indicative of l-threonine interaction at these sites. Thr-27 is located in the loop between β1 and β2 strands, and Ser-176 is within the loop connecting α7 and β7. Both are situated in close proximity to each other within the putative ligand-binding active site and are likely to coordinate l-threonine during TC-AMP formation.
FIGURE 4.
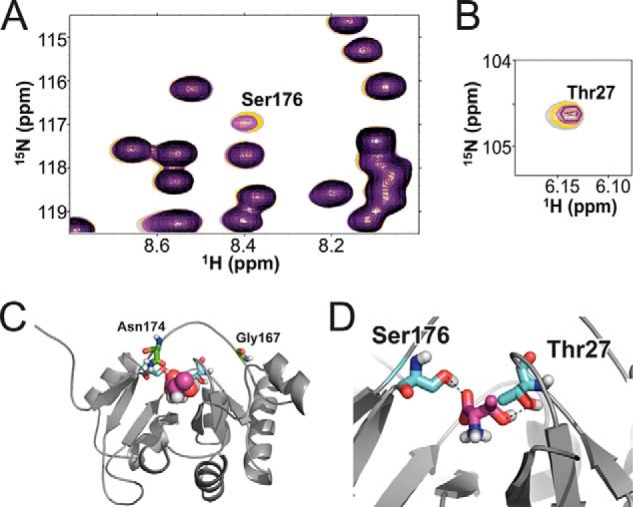
Binding of l-threonine and TsaC in 1H-15N-HSQC spectra. Spectra of the various protein/ligand ratios are overlaid as follows: 1:0 (gray), 1:1 (yellow), 1:2 (purple), and 1:4 (black). A, 1H-15N-HSQC region containing Ser-176. At a 1:4 ratio, the resonance has completely disappeared. B, 1H-15N-HSQC displaying the signal for Thr-27 weakens due to exchange with each addition of l-threonine. C, lowest energy modeled structure of l-threonine (magenta) with the E. coli TsaC structure from cluster 1. Residues Thr-27 and Ser-176 (cyan) and Asn-174 and Gly-167 (green) are highlighted. D, local structure of the l-threonine binding site with Thr-27 and Ser-176 (cyan).
Data from titration of the protein with l-threonine were incorporated into molecular modeling studies for the interaction between l-threonine and TsaC. HADDOCK (43) was used to develop a structural model of the binding of l-threonine from the NMR structure of TsaC, and active and passive residues with solvent accessibility >50% were assigned from TsaC NMR titration studies (Fig. 4, C and D; Table 3). A plot of the Einter (interaction energy) and the sum of restraint, van der Waals, and electrostatic energy terms, as a function of backbone r.m.s.d. from the lowest energy model, reveal that the models converge to a Cα (protein) and Cα (l-threonine) r.m.s.d. of 0.5 ± 0.7 Å at the defined protein/l-threonine interface with an average buried surface area of 215 ± 79 Å2. Three clusters of structures with low r.m.s.d. and energy were obtained for all calculated models based on a minimum cluster size of four models and a Cα r.m.s.d. of 7.5 Å. The lowest energy structure displays direct interactions between TsaC Ser-176 and Thr-27 and l-threonine (Fig. 4D). The distances between the heavy atoms of the hydroxyls of Ser-176 and Thr-27 and the substrate are suggestive of hydrogen bonding. The hydroxyl oxygen of Ser-176 is 2.7 Å from the carboxyl oxygen of l-threonine. The distance between the hydroxyl oxygen of Thr-27 and the hydroxyl oxygen of the substrate threonine is 3.3 Å.
TsaC Interaction with ATP
E. coli TsaC has been shown to bind ATP and l-threonine to produce TC-AMP (16, 24, 25). The interaction of TsaC with ATP was characterized by isothermal titration calorimetry (ITC; Fig. 5A). The binding was shown to have a 1:1 stoichiometry and considerable affinity with a dissociation constant (Kd) of 14.8 ± 2.0 μm (Table 2). The Gibbs free energy (ΔG) of the binding event was −6.12 ± 0.01 kcal/mol, indicating a favorable interaction. These ITC results could represent a nonproductive mode of binding for ATP in the absence of the other substrates.
FIGURE 5.

Interaction of TsaC with ATP. A, ITC of TsaC with ATP. The baseline-corrected data of the exothermic reaction (top) and the integrated heat changes (bottom) were obtained upon the titration of ATP into TsaC. The solid line represents the data fit to a nonlinear least squared isotherm fitting using a 1:1 binding model. B, overlaid 1H-15N-HSQC spectra of TsaC titrated with ATP. The chemical shifts with the greatest changes are labeled and marked with colored arrows corresponding to the chemical shift perturbation profile. C, chemical shift perturbation profile of the combined amide chemical shift changes for each TsaC residue at the final titration point (1:8 molar ratio of TsaC/ATP). D, local structure of E. coli TsaC with residues Ile-59, Gly-109, Leu-114, Ala-115, Arg-188, and Gln-189 highlighted in colors corresponding to the graph.
TABLE 2.
Thermodynamic parameters of TsaC binding
| ATP | |
|---|---|
| Stoichiometry (n) | 1.07 ± 0.022 |
| Ka (m−1) | (6.83 ± 0.92) × 104 |
| Kd (μm) | 14.8 ± 2.0 |
| ΔH (cal/mol) | −440.6 ± 13.6 |
| ΔS (cal/(K·mol)) | 20.5 |
| TΔS (kcal/mol) | 5.678 |
| ΔG (kcal/mol) | −6.12 ± 0.01 |
To identify the amino acids involved in the binding of ATP, amide chemical shift changes of 15N-labeled TsaC were monitored by observing 1H-15N-HSQC spectra. Spectra were collected at six different concentrations of ATP resulting in protein/ligand ratios of 1:0 to 1:8 (Fig. 5B). The amino acid resonances found to be the most strongly affected by the addition of ATP were those of Arg-188, Gly-109, Ile-59, Leu-114, and Ala-115. They exhibited maximum total chemical shift changes of ΔδN,H of 1.03, 0.96, 0.59, and 0.58 ppm, respectively, at a protein/ATP ratio of 1:8 (Fig. 5C). These resonances among others of the most affected residues are located in three distinct regions of the TsaC structure. Arg-188 is at the C terminus; Gly-109 is found in the loop between β4 and β5; Ile-59 is located in β3, and Leu-114 and Ala-115 are within β5, but they are all situated on one side of the central protein cavity (Fig. 5D). The perturbations of these chemical shifts with the addition of ATP suggest that these amino acids are in direct contact with ATP, or are indirectly affected by the environmental changes produced by ATP binding or through conformational rearrangement of the protein.
As with the TsaC·l-threonine modeled complex, the ATP titration data were used to direct molecular modeling of the NMR-derived structure of E. coli TsaC with ATP using HADDOCK (Table 3). The Einter was plotted as a function of backbone r.m.s.d. from the lowest energy model. The models converged to a Cα (protein) and C + P (ATP) r.m.s.d. of 0.6 ± 1.3 Å at the defined protein/ATP interface with an average buried surface area of 481 ± 168 Å2. Nine clusters of structures with low r.m.s.d. and energy were obtained for all calculated models based on a minimum cluster size of four models and a Cα (TsaC) and C + P (ATP) r.m.s.d. of 7.5 Å. Of the resulting models in each cluster, the first cluster contained 62% of the total structures calculated indicating a high degree of convergence. The lowest energy structure in this cluster depicts the ATP-bound TsaC in direct contact only with the adenosine, with the phosphates outside of the binding pocket. As seen in the 1H-15N-HSQC experiments, the docking results depict aliphatic residues in the region of β3 and β5 strands creating a hydrophobic pocket for adenosine.
Finally, the TsaC·l-threonine·ATP complex was modeled using the lowest energy TsaC·l-threonine structure and docking ATP (Table 3). In plotting the Einter as a function of backbone r.m.s.d. (Fig. 6) from the lowest energy model, the models converged to a Cα (protein) and C + P (ATP) r.m.s.d. of 0.3 ± 0.5 Å at the defined protein/ATP interface with an average buried surface area of 644 ± 74 Å2. Three clusters of structures (Fig. 6) with low r.m.s.d. and energy were obtained for all calculated models based on a minimum cluster size of four models and a Cα (TsaC) and C + P (ATP) r.m.s.d. of 7.5 Å. Of the resulting models in each cluster, the lowest energy structure in the third cluster best characterizes the interaction (Fig. 7A). However, this model differs noticeably from the TsaC/ATP structure in the positioning of the ligand. The adenosine of ATP is in a similar conformation, but the phosphates are now flipped into the binding pocket. So, it appears that the presence of l-threonine provides a more favorable environment for the phosphates to enter the binding pocket, indicating that l-threonine may be required for ATP binding and AMP formation. In the comparison of the TsaC·l-threonine·ATP-modeled complex to the co-crystal structure of S. tokodaii Sua5 with l-threonine and AMP-PNP (Fig. 7B), several similarities and differences can be observed. For instance, the location of ATP in the binding pocket is similar and agrees quite well, but the orientation is slightly different. The difference in orientation could be the result of a comparison between AMP-PNP in the crystal and ATP of the NMR study. In both structures, the functionally important amino group of the substrate l-threonine is positioned near ATP in the same orientation.
FIGURE 6.
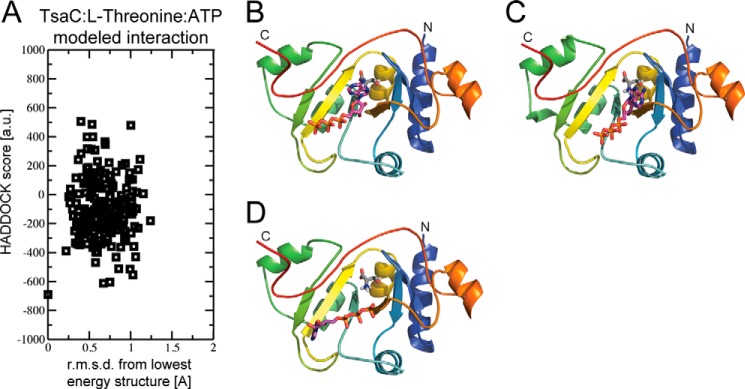
HADDOCK was used to develop a structural model of l-threonine/ATP and TsaC using the NMR structure of TsaC and the NMR titration data. A, plot of the Einter (sum of restraint, van der Waals, and electrostatic energy terms) as a function of backbone r.m.s.d from the lowest energy model. B–D, lowest energy structure from each cluster, cluster 1–3, respectively.
FIGURE 7.
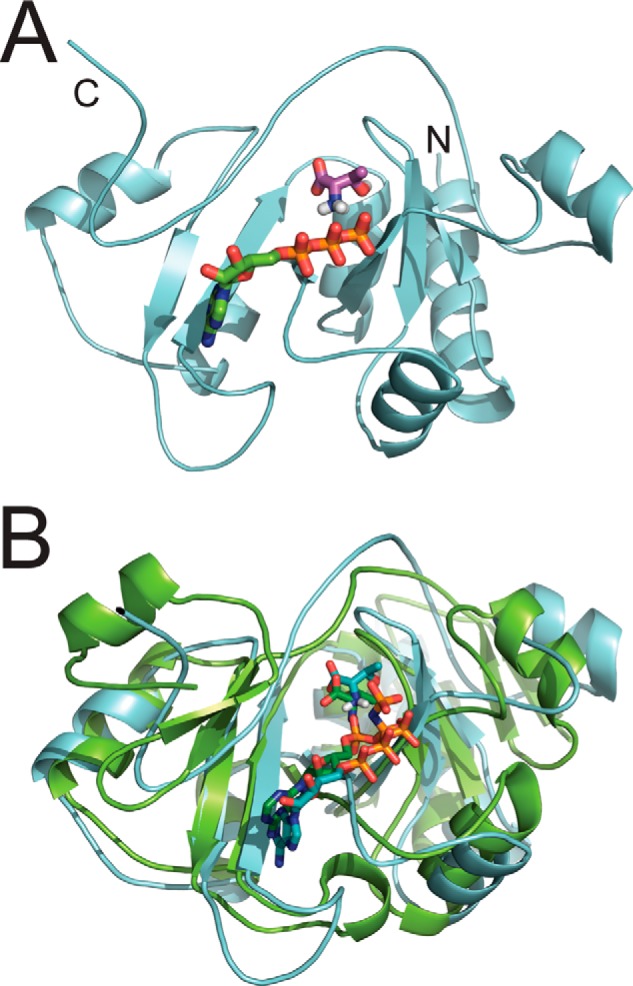
Data-driven molecular docking of E. coli TsaC with l-threonine and ATP. A, lowest energy modeled structure of the most populated cluster for the modeled interaction of TsaC with l-threonine (magenta) and ATP (green). B, for comparison, the co-crystal structure of S. tokodaii Sua5 (green, Protein Data Bank code 3AJE) with l-threonine and AMP-PNP is aligned with the E. coli TsaC structure shown in A (cyan).
Backbone Dynamics of TsaC
The potential for the protein's binding of l-threonine and ATP to include conformational changes prompted us to investigate the structural dynamics of TsaC. Backbone r.m.s.d. values were calculated for individual residues among the 10 lowest energy structures using MOLMOL (51) to identify regions of localized high and low r.m.s.d. values (Fig. 8A). Several regions of high local r.m.s.d. were observed. Loop regions and both termini exhibited high r.m.s.d. values compared with the average, which is common for all protein structures due to the range of motion at the termini. However, residues 186–190, at the C terminus, had considerably higher values than typically observed. This is reflected in the ensemble of lowest energy structures and the sparse data for the C terminus in the NMR spectra (Fig. 3A). Pro-165–Glu-177, located in the long loop that connects the α7 helix to β7 strand, had the highest individual r.m.s.d. values of the internal residues. This region contains the putative binding site for l-threonine and is one of the regions that displays the largest difference between the crystal and NMR structures (Fig. 3D). This difference is suggestive of a dynamic role in binding that is observable by NMR.
FIGURE 8.
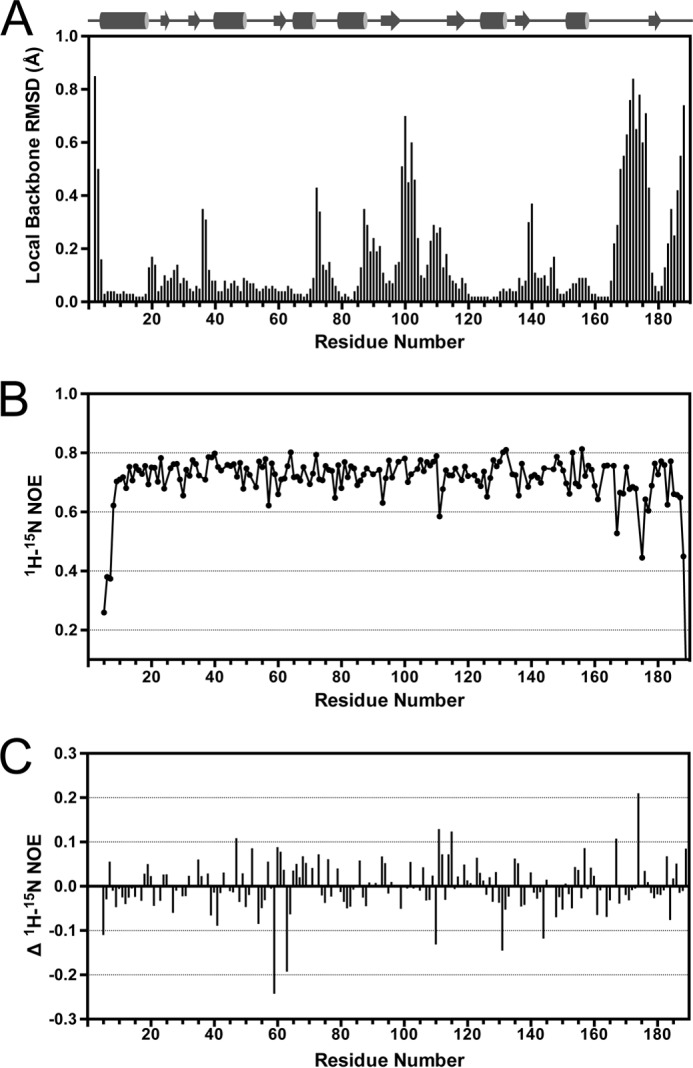
Backbone dynamics of TsaC. A, backbone r.m.s.d. values per residue calculated using the 10 lowest energy structures. B, 1H-15N heteronuclear NOE relaxation data. C, difference in 1H-15N heteronuclear NOE relaxation values with and without ATP present. Negative and positive values represent more and less dynamic residues, respectively.
To verify that the high local r.m.s.d. values result from inherent dynamics and are not due to sparse restraints, TsaC backbone dynamics were determined using steady-state heteronuclear 1H-15N-NOE experiments (35). On the nanosecond time scale, the heteronuclear NOE values were indicative of a lack of backbone motion for most regions (Fig. 8B). The C terminus exhibited the greatest range of motion with residues Arg-188 and Gln-189 having two of the lowest NOE values. The Gly-190 signal completely disappeared resulting in a negative NOE. The N-terminal residues Leu-5–Arg-7 also had a lower average NOE, indicative of backbone dynamics. Additionally, residues Gly-167 and Asn-174 exhibited increased conformational flexibility. Both of these are located in the long loop of TsaC that connects α7 to β7, flanking the location of the l-threonine-binding site (Fig. 4C).
The backbone dynamics correlate with the r.m.s.d. values that were calculated for the NMR-derived structures. This indicates that the most dynamic regions, specifically the C-terminal residues and the α7-β7 loop, may be important for protein function such as the coordination of l-threonine binding with ATP binding. To test this experimentally, a heteronuclear NOE experiment was conducted in which TsaC was titrated with ATP (Fig. 8C). The addition of ATP induced noteworthy changes to a few amino acids. Overall, the C terminus remained dynamic upon the addition of ATP, but several other resonances were affected. Ile-59, Ala-63, Tyr-131, Arg-110, and Gly-144 exhibited greater conformational dynamics, whereas Asn-174, Phe-111, Ala-115, Glu-47, and Gly-167 became more structured in the presence of ATP. Of these, Ile-59 and Ala-115 were the two most affected by the titration with ATP in this experiment. This is consistent with the amide chemical shift changes observed in the 1H-15N-HSQC spectra. Combining the results of these datasets provides evidence that Ile-59 and Ala-115 could be essential for the binding of ATP by TsaC.
Discussion
The t6A nucleoside modification, found 3′-adjacent to the anticodon in ANN-decoding tRNAs across all domains of life, is essential to translational fidelity. Of the four proteins in E. coli that have been found to be required for the biosynthesis of t6A, TsaC has been the most extensively studied in the context of this modification pathway, and it is essential to t6A37 formation and cell viability (24). More recently, TsaC has been shown to function as a threonylcarbamoyl-AMP synthase by catalyzing TC-AMP formation from l-threonine, CO2/HCO3−, and ATP (17). Here, insights into the mechanism of TsaC function have been achieved by utilizing an NMR-derived structure, biochemical data, and molecular modeling.
The high-resolution solution structure of the full-length protein is present in solution as a monomer with a large hydrophobic binding pocket (Fig. 3). The structure is consistent with all protein structures solved to date in the TsaC/Sua5 family, such as YciO (28), Sua5 (29), and HypF (30). They all have an α/β twisted open-sheet topology with parallel and antiparallel adjacent β-strands and a central concave cavity lined with positive electrostatic potential. In relation to the TsaC crystal structure, the NMR-derived structure of E. coli TsaC is generally homologous to one subunit of the homodimer (27). Some differences, likely caused by the crystal packing, are observed and obscure structural information in the substrate-binding regions in the crystal structure (Fig. 3D). In particular, the dimerization interface affects the C terminus of TsaC, which appears to be important for substrate/protein and protein/protein interactions.
The mechanism by which TsaC catalyzes the formation of TC-AMP appears to include conformational changes in the protein with the binding of the substrates. Using NMR titration analyses, we were able to observe and track these changes upon binding of l-threonine and ATP. The titration data with TsaC and l-threonine suggest that Thr-27 and Ser-176 in E. coli TsaC are involved in substrate binding. Both of these residues are conserved in the TsaC/Sua5 protein family (Fig. 9), suggesting that they may be important for function. In the co-crystal structure of S. tokodaii Sua5 with l-threonine, the corresponding residues, Thr-34 and Ser-182, both interact with l-threonine (50). S. tokodaii Sua5 Thr-34 forms a hydrophobic environment for the γ-carbon of l-threonine, and Ser-182 forms two hydrogen bonds with the carboxyl oxygen (50). From our molecular docking studies, we observe Ser-176 coordinating l-threonine in a similar fashion. However, TsaC Thr-27 appears to be making a hydrogen bond with the hydroxyl of the substrate (Fig. 4). This could be significant to differences in coordination of l-threonine or possibly an artifact of the molecular docking process.
FIGURE 9.

TsaC/Sua5 conserved amino acids. Alignment of E. coli TsaC with E. coli HypF, S. tokodaii Sua5, B. subtilis YwlC, and E. coli YciO was completed using the ClustalW2 sequence alignment server. l-Threonine-binding residues, Thr-27 and Ser-175 (orange), are moderately conserved. Adenosine-binding residues (blue), Ile-59, Leu-114, Ala-115,and Leu-178, are conserved, with Arg-188 highly conserved; however, Gly-109 is not conserved. The KXR/SXN ATP-binding motif is highlighted in purple. Gray and black indicate moderately and highly conserved residues, respectively.
The binding of ATP by TsaC observed in the 1H-15N-HSQC spectra indicates a binding site within the putative catalytic pocket. The residues with the greatest changes in amide chemical shifts were the hydrophobic amino acids Gly-109, Leu-114, Ala-115, and Ile-59 (Fig. 5). The E. coli TsaC residues Lys-56, Leu-58–Ile-59–Leu-60 and Ser-113–Leu-114–Ala-115–Val-116–Arg-117 are conserved across this family (Fig. 9). These residues have been shown to surround the adenine-binding site in co-crystal structures of both E. coli HypF (middle domain, residues 188–378) and S. tokodaii Sua5 with the nonhydrolysable ATP analog AMP-PNP (30, 50). For HypF, the adenine is buried in a hydrophobic environment between Leu-277 and Pro-249, and it forms weak hydrogen bonds with Glu-296 and Arg-372 (30). Pro-249 near the ATP-binding site in HypF is not conserved in this family of proteins and is not present in TsaC. Sua5 coordinates the adenosine with hydrophobic residues Ile-66, Val-101, Ala-120, and Ile-184 (50). When TsaC is aligned with the HypF and Sua5 structures, Ile-59, Ala-115, and Arg-188 (resonances that were all greatly affected during the titration with ATP) correspond to the residues in the other two proteins that are important for the binding of the adenosine. To examine this further, HADDOCK was used to dock ATP to the E. coli TsaC structure to model the binding mode. The lowest energy TsaC:ATP structure resulting from the docking simulation placed the adenosine in the hydrophobic region of Leu-114, Ala-115, and Ile-59. Thus, the observation that these residues in TsaC are affected by titration with ATP appears to be caused by the binding of adenosine. Gly-109 in TsaC was also affected by ATP binding, yet it is located in the loop between β4 and β5, a region of the protein not in direct contact with ATP. Therefore, we postulate that ATP binding may cause a conformational change in this loop. However, no significant conformational change is observed in the lowest energy docking model.
It is possible that the TsaC forms contacts with ligands in addition to those observable by NMR using the 1H-15N-HSQC analysis. The KXR/SXN ATP-binding motif present in TsaC is conserved throughout the TsaC/Sua5 family (24). This motif is important for the coordination of the phosphates of ATP in S. tokodaii Sua5 (50), but no significant changes in chemical shifts were detected for Lys-50, Arg-52, or Asn-141 (Ser-139 was unobservable) in our experiments with E. coli TsaC. ATP hydrolysis, dynamics, or H2O exchange are reasonable explanations for not observing the binding of the phosphates. Therefore, we repeated the titration using the nonhydrolysable ATP analog AMP-PNP used in the crystallography of S. tokodaii Sua5 and E. coli HypF. However, TsaC residues Lys-50, Arg-52, and Asn-141 remained unaffected in our NMR studies (data not shown). This indicates that the inability to observe interactions of TsaC with the phosphates of ATP was not due to hydrolysis of the phosphates. To further investigate the interaction of TsaC with the ATP phosphates, the TsaC/ATP interaction was modeled, and it was revealed that the phosphate atoms were not in contact with any part of TsaC. The lack of contact between the two entities explains the unaffected KXR/SXN residues in the 1H-15N-HSQC experiments.
Perhaps TsaC requires the l-threonine to properly coordinate ATP in the active site. To test this idea and to further understand the mechanism employed by TsaC for the synthesis of TC-AMP, we added l-threonine to the TsaC/ATP NMR sample. No additional 1H-15N-HSQC chemical shift changes were observed (data not shown). Therefore, the ability of TsaC to bind both substrates simultaneously, and possibly cooperatively, was unclear. The binding of ATP could require the binding of l-threonine to occur first, providing the contact surface area for ATP to bind in the correct conformation. As seen in the Sua5 structure (50), contacts are observed between ATP and the l-threonine with the l-threonine buried further into the Sua5 binding cavity (Fig. 7B). To investigate this possibility, the TsaC/l-threonine modeled structure was docked to ATP (Fig. 7A). The lowest energy structure of this modeled interaction is remarkably similar to that seen in the Sua5 crystal structure (Fig. 7) (45). In fact, the phosphates of ATP are positioned in close enough proximity to Lys-50 and Ser-139 of the KXR/SXN ATP-binding motif to suggest coordination. This compellingly suggests that the recognition of ATP is dependent on the presence of l-threonine. Indeed, it has been shown that TsaC forms AMP only in the presence of l-threonine and bicarbonate (16). However, the binding order and kinetics of this reaction are unknown, and more investigation into this is necessary.
E. coli TsaC contains sites that are highly dynamic, particularly the C terminus. Both the heteronuclear NOEs and the r.m.s.d. values from the 10 lowest energy structures depict considerable flexibility for Gly-167–Gly-190 (Fig. 8). The Gly-167–Gly-190 region may function in protein/protein interactions with the other t6A-synthase subunits, TsaD, TsaE, and TsaB, or as an arm capable of folding into the concave binding pocket. The presence of two consecutive glycines at 170–171 indicates that the Gly-167–Gly-190 region may function as a hinge enabling the C terminus to act as a gate during substrate binding. Both Arg-188 and Gln-189 display a high degree of conformational motion relative to the other residues and also are affected by ATP binding. The conformational dynamics of Arg-188 and Gln-189 are consistent with the hypothesis that the C terminus acts a flexible linker or arm that may participate in the binding of ATP or interact with other proteins. HypF is composed of domains homologous to TsaC and TsaD/Kae1. In fact, the TsaC domain of HypF (residues 188–378) is linked directly to the TsaD domain (residues 379–746) (30) suggesting a direct interaction of the C terminus of TsaC with TsaD in t6A37 biosynthesis. There are clear structural differences between the TsaC crystal and NMR structures with the greatest difference at this exact location (Fig. 3). Because we wished to visualize the C terminus of TsaC in our studies, it was important to use the NMR-derived structure of TsaC and to perform a blind docking of the TsaC/l-threonine/ATP modeled interaction.
In conclusion, the study presented here provides valuable information about the mechanism of TsaC in t6A37 biosynthesis. The identification of TsaC residues that are affected by the binding of ATP and l-threonine provides sites at which a future in-depth mutational analysis coupled to ITC and NMR experiments could confirm the importance of individual amino acids, and/or their properties, for binding. E. coli TsaC binds both ATP and l-threonine adjacent to each other with conserved residues located within the large concave cavity at the center of the structure. The adenosine of ATP is inserted into a hydrophobic environment created by residues within β3 and β5. The proper coordination of ATP in the binding pocket seems to require the presence of l-threonine, suggesting a cooperative binding event between these substrates, in which the binding of l-threonine occurs first. The C-terminal amino acids appear to be important for ATP binding and possibly catalytic activity. A central and compelling issue in the mechanism of TC-AMP formation, not addressed in this work, is whether TsaC utilizes CO2 or HCO3−, and how it reacts with l-threonine to form intermediate l-threonine carbamate. Further analyses of TsaC with CO2/HCO3−, and the structural interactions of the binding of TsaC with tRNA, TsaD, TsaB, and TsaE, especially in revealing the protein/RNA and protein/protein interfaces, will provide additional and significant insight into this complex enzymatic process.
Author Contributions
P. F. A. and D. I. R. designed the study. K. A. H. and K. L. S. performed NMR experiments, analyzed data, and solved the structure. K. A. H. and A. F. S. conducted binding experiments and analyzed data. B. G. B. performed in silico docking experiments. B. G. B., Y. B., and C. D. provided technical assistance in NMR spectroscopy and protein expression. K. A. H., K. L. S., and P. F. A. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Drs. Manal Swairjo and Kyla Frohlich for insightful discussions, Dr. Alex Shekhtman for NMR assistance, and the Research IT team at the University at Albany.
This work was supported, in whole or in part, by National Institutes of Health Grant 5R01-GM23037-25 (to P. F. A.) and NIH Grant R01 GM110588 (to P. F. A.; Manal Swairjo, P.I.). This work was also supported by National Science Foundation Grants MCB-0548602 and MCB-1101859 (to P. F. A.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 2MX1) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The chemical shift assignments are available at the Biological Magnetic Resonance Data Bank under accession number 25381.
- t6A37
- N6-threonylcarbamoyladenosine
- HSQC
- heteronuclear single quantum coherence
- r.m.s.d.
- root-mean-square deviation
- ITC
- isothermal titration calorimetry
- AMP-PNP
- adenosine-5′-(β,γ-imido)triphosphate
- TC-AMP
- l-threonylcarbamoyl-AMP
- RDC
- residual dipolar coupling.
References
- 1. Cantara W. A., Crain P. F., Rozenski J., McCloskey J. A., Harris K. A., Zhang X., Vendeix F. A., Fabris D., Agris P. F. (2011) The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res. 39, D195–D201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Auffinger P., Westhof P. (1998) in Modification and Editing of RNA (Grosjean H., Benne R., eds) pp. 569–576, American Society for Microbiology, Washington, D. C. [Google Scholar]
- 3. Jühling F., Mörl M., Hartmann R. K., Sprinzl M., Stadler P. F., Pütz J. (2009) tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 37, D159–D162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grosjean H., Sprinzl M., Steinberg S. (1995) Posttranscriptionally modified nucleosides in transfer RNA: their locations and frequencies. Biochimie 77, 139–141 [DOI] [PubMed] [Google Scholar]
- 5. Morin A., Auxilien S., Senger B., Tewari R., Grosjean H. (1998) Structural requirements for enzymatic formation of threonylcarbamoyladenosine (t6A) in tRNA: an in vivo study with Xenopus laevis oocytes. RNA 4, 24–37 [PMC free article] [PubMed] [Google Scholar]
- 6. Miller J. P., Hussain Z., Schweizer M. P. (1976) The involvement of the anticodon adjacent modified nucleoside N-(9-(β-d-ribofuranosyl)purine-6-ylcarbamoyl)-threonine in the biological function of E. coli tRNAIle. Nucleic Acids Res. 3, 1185–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weissenbach J., Grosjean H. (1981) Effect of threonylcarbamoyl modification (t6A) in yeast tRNA Arg III on codon-anticodon and anticodon-anticodon interactions. A thermodynamic and kinetic evaluation. Eur. J. Biochem. 116, 207–213 [DOI] [PubMed] [Google Scholar]
- 8. Yarian C., Townsend H., Czestkowski W., Sochacka E., Malkiewicz A. J., Guenther R., Miskiewicz A., Agris P. F. (2002) Accurate translation of the genetic code depends on tRNA modified nucleosides. J. Biol. Chem. 277, 16391–16395 [DOI] [PubMed] [Google Scholar]
- 9. Murphy F. V., 4th, Ramakrishnan V., Malkiewicz A., Agris P. F. (2004) The role of modifications in codon discrimination by tRNA(Lys)UUU. Nat. Struct. Mol. Biol. 11, 1186–1191 [DOI] [PubMed] [Google Scholar]
- 10. Vendeix F. A., Murphy F. V., 4th, Cantara W. A., Leszczyńska G., Gustilo E. M., Sproat B., Malkiewicz A., Agris P. F. (2012) Human tRNA(Lys3)UUU is pre-structured by natural modifications for cognate and wobble codon binding through Keto–Enol tautomerism. J. Mol. Biol. 416, 467–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Agris P. F. (1991) Wobble position modified nucleosides evolved to select transfer RNA codon recognition: a modified-wobble hypothesis. Biochimie 73, 1345–1349 [DOI] [PubMed] [Google Scholar]
- 12. Nishimura S. (1972) Minor components in transfer RNA: their characterization, location, and function. Prog. Nucleic Acids Res. Mol. Biol. 12, 49–85 [PubMed] [Google Scholar]
- 13. Lin C. A., Ellis S. R., True H. L. (2010) The Sua5 protein is essential for normal translational regulation in yeast. Mol. Cell. Biol. 30, 354–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. El Yacoubi B., Hatin I., Deutsch C., Kahveci T., Rousset J.-P., Iwata-Reuyl D., Murzin A. G., de Crécy-Lagard V. (2011) A role for the universal Kae1/Qri7/YgjD (COG0533) family in tRNA modification. EMBO J. 30, 882–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Daugeron M. C., Lenstra T. L., Frizzarin M., El Yacoubi B., Liu X., Baudin-Baillieu A., Lijnzaad P., Decourty L., Saveanu C., Jacquier A., Holstege F. C., de Crécy-Lagard V., van Tilbeurgh H., Libri D. (2011) Gcn4 misregulation reveals a direct role for the evolutionary conserved EKC/KEOPS in the t6A modification of tRNAs. Nucleic Acids Res. 39, 6148–6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Deutsch C., El Yacoubi B., de Crécy-Lagard V., Iwata-Reuyl D. (2012) Biosynthesis of threonylcarbamoyl adenosine (t6A), a universal tRNA nucleoside. J. Biol. Chem. 287, 13666–13673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lauhon C. T. (2012) Mechanism of N6-threonylcarbamoyladenonsine (t6A) biosynthesis: isolation and characterization of the intermediate threonylcarbamoyl-AMP. Biochemistry 51, 8950–8963 [DOI] [PubMed] [Google Scholar]
- 18. Perrochia L., Crozat E., Hecker A., Zhang W., Bareille J., Collinet B., van Tilbeurgh H., Forterre P., Basta T. (2013) In vitro biosynthesis of a universal t6A tRNA modification in Archaea and Eukarya. Nucleic Acids Res. 41, 1953–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wan L. C., Mao D. Y., Neculai D., Strecker J., Chiovitti D., Kurinov I., Poda G., Thevakumaran N., Yuan F., Szilard R. K., Lissina E., Nislow C., Caudy A. A., Durocher D., Sicheri F. (2013) Reconstitution and characterization of eukaryotic N6-threonylcarbamoylation of tRNA using a minimal enzyme system. Nucleic Acids Res. 41, 6332–6346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Downey M., Houlsworth R., Maringele L., Rollie A., Brehme M., Galicia S., Guillard S., Partington M., Zubko M. K., Krogan N. J., Emili A., Greenblatt J. F., Harrington L., Lydall D., Durocher D. (2006) A genome-wide screen identifies the evolutionarily conserved KEOPS complex as a telomere regulator. Cell 124, 1155–1168 [DOI] [PubMed] [Google Scholar]
- 21. Kisseleva-Romanova E., Lopreiato R., Baudin-Baillieu A., Rousselle J. C., Ilan L., Hofmann K., Namane A., Mann C., Libri D. (2006) Yeast homolog of cancer-testis antigen defines a new transcription complex. EMBO J. 25, 3576–3585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mao D. Y., Neculai D., Downey M., Orlicky S., Haffani Y. Z., Ceccarelli D. F., Ho J. S., Szilard R. K., Zhang W., Ho C. S., Wan L., Fares C., Rumpel S., Kurinov I., Arrowsmith C. H., Durocher D., Sicheri F. (2008) Atomic structure of the KEOPS complex: an ancient protein kinase-containing molecular machine. Mol. Cell 32, 259–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Srinivasan M., Mehta P., Yu Y., Prugar E., Koonin E. V., Karzai A. W., Sternglanz R. (2011) The highly conserved KEOPS/EKC complex is essential for a universal tRNA modification, t6A. EMBO J. 30, 873–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. El Yacoubi B., Lyons B., Cruz Y., Reddy R., Nordin B., Agnelli F., Williamson J. R., Schimmel P., Swairjo M. A., de Crécy-Lagard V. (2009) The universal YrdC/Sua5 family is required for the formation of threonylcarbamoyladenosine in tRNA. Nucleic Acids Res. 37, 2894–2909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harris K. A., Jones V., Bilbille Y., Swairjo M. A., Agris P. F. (2011) YrdC exhibits properties expected of a subunit for a tRNA threonylcarbamoyl transferase. RNA 17, 1678–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Perrochia L., Guetta D., Hecker A., Forterre P., Basta T. (2013) Functional assignment of KEOPS/EKC complex subunits in the biosynthesis of the universal t6A tRNA modification. Nucleic Acids Res. 41, 9484–9499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Teplova M., Tereshko V., Sanishvili R., Joachimiak A., Bushueva T., Anderson W. F., Egli M. (2000) The structure of the yrdC gene product from Escherichia coli reveals a new fold and suggests a role in RNA binding. Protein Sci. 9, 2557–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jia J., Lunin V. V., Sauvé V., Huang L. W., Matte A., Cygler M. (2002) Crystal structure of the YciO protein from Escherichia coli. Proteins 49, 139–141 [DOI] [PubMed] [Google Scholar]
- 29. Agari Y., Sato S., Wakamatsu T., Bessho Y., Ebihara A., Yokoyama S., Kuramitsu S., Shinkai A. (2008) X-ray crystal structure of a hypothetical Sua5 protein from Sulfolobus tokodaii strain 7. Proteins 70, 1108–1111 [DOI] [PubMed] [Google Scholar]
- 30. Petkun S., Shi R., Li Y., Asinas A., Munger C., Zhang L., Waclawek M., Soboh B., Sawers R. G., Cygler M. (2011) Structure of hydrogenase maturation protein HypF with reaction intermediates shows two active sites. Structure 19, 1773–1783 [DOI] [PubMed] [Google Scholar]
- 31. Sivashanmugam A., Murray V., Cui C., Zhang Y., Wang J., Li Q. (2009) Practical protocols for production of very high yields of recombinant proteins using Escherichia coli. Protein Sci. 18, 936–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ottiger M., Delaglio F., Bax A. (1998) Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reson. 131, 373–378 [DOI] [PubMed] [Google Scholar]
- 34. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 35. Goddard T. D., Kneller D. G. (2008) SPARKY 3. University of California, San Francisco, CA [Google Scholar]
- 36. Johnson B. A. (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 278, 313–352 [DOI] [PubMed] [Google Scholar]
- 37. Farrow N. A., Muhandiram R., Singer A. U., Pascal S. M., Kay C. M., Gish G., Shoelson S. E., Pawson T., Forman-Kay J. D., Kay L. E. (1994) Backbone dynamics of a free and phosphopeptide-complexed Src Homology 2 domain studied by 15N NMR relaxation. Biochemistry 33, 5984–6003 [DOI] [PubMed] [Google Scholar]
- 38. Güntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol 278, 353–378 [DOI] [PubMed] [Google Scholar]
- 39. Schwieters C. D., Kuszewski J. J., Tjandra N., Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn Reson. 160, 66–74 [DOI] [PubMed] [Google Scholar]
- 40. Schwieters C. D., Kuszewski J. J., Clore G. M. (2006) Using Xplor-NIH for NMR molecular structure determination. Progr. NMR Spectroscopy 48, 47–62 [Google Scholar]
- 41. Bhattacharya A., Tejero R., Montelione G. T. (2007) Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795 [DOI] [PubMed] [Google Scholar]
- 42. DeLano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.3, Schrodinger, LLC, New York [Google Scholar]
- 43. Dominguez C., Boelens R., Bonvin A. M. (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 44. Hubbard S. J., Thornton J. M. (1993) NACCESS, Version 2.1.1, University College; London [Google Scholar]
- 45. McLachlan A. D. (1982) Rapid Comparison of Protein Structures. Acta Crystallogr. Sect. A 38, 871–873 [Google Scholar]
- 46. Martin A. C., Porter C. T. (1996) ProFit, Version 3.1, University College; London [Google Scholar]
- 47. Ferrage F., Zoonens M., Warschawski D. E., Popot J.-L., Bodenhausen G. (2003) Slow diffusion of macromolecular assemblies by a new pulsed field gradient NMR method. J. Am. Chem. Soc. 125, 2541–2545 [DOI] [PubMed] [Google Scholar]
- 48. Clore G. M., Garrett D. S. (1999) R-factor, free R, and complete cross-validation for dipolar coupling refinement of NMR structures. J. Am. Chem. Soc. 121, 9008–9012 [Google Scholar]
- 49. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 50. Kuratani M., Kasai T., Akasaka R., Higashijima K., Terada T., Kigawa T., Shinkai A., Bessho Y., Yokoyama S. (2011) Crystal structure of Sulfolobus tokodaii Sua5 complexed with l-threonine and AMPPNP. Proteins 79, 2065–2075 [DOI] [PubMed] [Google Scholar]
- 51. Koradi R., Billeter M., Wüthrich K. (1996) MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55 [DOI] [PubMed] [Google Scholar]