

Background: Foxp3 activity is regulated by various posttranslational modifications.
Results: Pim-2 kinase phosphorylates the Foxp3 N-terminal domain and influences the Foxp3 level in vivo.
Conclusion: Pim-2 is a negative regulator of Foxp3 activity.
Significance: Phosphorylation of Foxp3 by Pim-2 kinase negatively regulates Treg cell suppressive function and stability.
Keywords: autoimmunity, colitis, immunosuppression, phosphorylation, posttranslational modification (PTM), Foxp3, Pim2 kinase, regulatory T cell
Abstract
Regulation of the extent of immune responses is a requirement to maintain self-tolerance and limit inflammatory processes. CD4+CD25+Foxp3+ regulatory T (Treg) cells play a role in regulation. The Foxp3 transcription factor is considered a dominant regulator for Treg cell development and function. Foxp3 function itself is directly regulated by multiple posttranslational modifications that occur in response to various external stimuli. The Foxp3 protein is a component of several dynamic macromolecular regulatory complexes. The complexes change constituents over time and through different signals to regulate the development and function of regulatory T cells. Here we identified a mechanism regulating Foxp3 level and activity that operates through discrete phosphorylation. The Pim-2 kinase can phosphorylate Foxp3, leading to decreased suppressive functions of Treg cells. The amino-terminal domain of Foxp3 is modified at several sites by Pim-2 kinase. This modification leads to altered expression of proteins related to Treg cell functions and increased Treg cell lineage stability. Treg cell suppressive function can be up-regulated by either pharmacologically inhibiting Pim-2 kinase activity or by genetically knocking out Pim-2 in rodent Treg cells. Deficiency of Pim-2 activity increases murine host resistance to dextran sodium sulfate-induced colitis in vivo, and a Pim-2 small molecule kinase inhibitor also modified Treg cell functions. Our studies define a pathway for limiting the regulation of Foxp3 function because the Pim-2 kinase represents a potential therapeutic target for modulating the Treg cell suppressive activities in controlling immune responses.
Introduction
CD4+CD25+FOXP3+ regulatory T (Treg) cells2 represent a subset of T cells and mediate suppressive function and play a role in the regulation of self-tolerance and the maintenance of normal immune homeostasis (1–6). Treg cells can be categorized into two major types: natural Treg cells, which are generated in the thymus, and induced Treg cells, which appear to be converted from conventional T cells by the action of cytokines such as TGF-β in the periphery. The development and function of both types of Treg cells can be regulated by FOXP3, a member of the forkhead family of transcriptional regulators. Dysfunction or mutations of FOXP3 may cause fatal autoimmune diseases, such as the scurfy phenotype in mice and immune dysregulation polyendocrinopathy enteropathy X-linked syndrome in human (7, 8).
The function of FOXP3 is regulated at various levels. FOXP3 has a unique proline rich amino-terminal domain that is required for its repressive transcriptional activity. Structural alterations of this domain are important. GFP insertion at the Foxp3 N-terminal domain alters Foxp3 functions and renders rodents more susceptible to autoimmune diabetes but, inexplicably, more resistant to antibody-mediated arthritis (9, 10). The Foxp3 N-terminal domain also contributes to Foxp3 nuclear transport (11). FOXP3 interacts with other partners, including Tip60, HDAC1, HDAC7, and HDAC9, enzymes involved in acetylation. Many of these interactions occur at the N-terminal domain (12–14). FOXP3 transcriptional activity is also regulated by posttranslational modifications (15) such as ubiquitination. Foxp3 phosphorylation modification has been noted previously in our laboratory (16). It has been suggested that phosphatase PP1 activated by TNFα can dephosphorylate FOXP3 at Ser-418, which may then influence Treg cell suppressive function (17), although the kinase itself responsible for FOXP3 phosphorylation was not characterized. Recently, cyclin-dependent kinase 2 (CDK2) has been reported to phosphorylate Ser-19 and Thr-175 of Foxp3 and regulate Foxp3 stability (18), although the consistency of phosphorylation is unclear.
Pim kinases are serine/threonine kinases that play an essential role in T cell development and differentiation. Three members have been identified so far: Pim-1, Pim-2, and Pim-3. Among them, Pim-1 and Pim-2 have been identified as oncogenes in murine T cell lymphomas induced by leukemia proviral integration (19, 20).
Pim kinases function in several aspects of lymphocyte function, such as survival, cell cycle progression, and transcription gene expression (21). Cytokines such as IL-4 and IL-7 can induce Pim-1 and Pim-2 expression in T cells and promote T cell growth and survival in a rapamycin-insensitive manner (22). In addition, Pim-2 induced by FOXP3 in Treg cells is permissive for Treg cell expansion in the presence of rapamycin (23). Animals deficient in all Pim kinases (pim1−/−pim2−/−pim3−/−) display no significant differences in comparison with wild-type mice except for reduced body size (24). In a previous study, PIM1 has been found able to phosphorylate Ser-422 in the carboxyl terminus of human FOXP3 (25). However, Ser-422 is not evolutionally conserved and is not present in the mouse Foxp3 protein sequence, precluding preclinical studies in rodents.
In this study, we found that Foxp3 serves as a substrate of Pim-2. Mass spectrometry data indicated that Pim-2 can phosphorylate multiple sites of the Foxp3 N-terminal domain and that it negatively regulates Treg cell suppressive function. Inhibition of Pim-2 activity increases the suppressive function of Treg cells in vitro. Deficiency of Pim-2 in vivo increased Treg lineage stability. Pim-2 knockout mice have increased resistance to DSS-induced colitis. These observations may contribute to new strategies to modulate Treg functions for human autoimmune diseases.
Experimental Procedures
Mice
Wild-type FVB mice were purchased from The Jackson Laboratory. pim2−/− FVB mice were provided by Dr. Anton Berns (Netherland Cancer Institute) and Dr. Paul Rothman (Johns Hopkins Medicine). Mice, including wild-type and pim2−/− knockout mice, were bred and maintained at the University of Pennsylvania Research Animal Facility. The animal protocol was approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
DNA Constructs, Antibodies, and Reagents
The HA-Foxp3-expressing plasmid was constructed as described previously (14). Human and mouse Pim-2 cDNAs were purchased from Open Biosystems and subcloned into pCDNA3.1, which already contained a FLAG tag, resulting in the FLAG-Pim-2 expressing plasmid and pGEX-5X-3 vector, resulting in the GST-Pim-2 expressing plasmid. FLAG-Pim-2 K61M (kinase-dead (KD) mutant) and various site mutants of HA-Foxp3 were constructed using site-directed mutagenesis (Stratagene) and verified by sequencing.
The following antibodies were used: anti-FLAG (HRP) M2 and anti-β-actin (HRP) from Sigma-Aldrich; anti-HA (HRP), anti-Pim-2 (1D12), and HRP-conjugated secondary antibody from Santa Cruz Biotechnology; anti-phosphoserine (HRP) from Novus; and anti-CD4 phycoerythrin-Cy7, anti-Foxp3 Alexa Fluor 488, anti-CD25 PE, anti-GITR PE, and anti-CD45RB allophycocyanin from BD Biosciences.
λ-Phosphatase was ordered from New England Biolabs. The Pim-2 inhibitor (Z)-5-(4-propoxyben-zylidene) thiazolidine-2,4-dione (526524) was ordered from Calbiochem. Protein inhibitor mixture tablets and phosphatase inhibitor mixture tablets were purchased from Roche.
Cell Culture and Western Blot Analysis
Jurkat cells and HEK 293T cells were maintained in RPMI 1640 medium (Invitrogen) supplemented with 10% heat-inactivated FBS, 1% penicillin/streptomycin (Invitrogen) at 37 °C in a humidified incubator with 5% CO2 (v/v). FOXP3-expressing Jurkat cells were generated as described previously (26). HEK 293T cells were transfected with plasmid DNA and FuGENE6 reagent (Roche). After 24-h transfection, cells were washed twice with cold PBS buffer and lysed for protein sample preparation.
For Western blot analysis, protein was separated by SDS-PAGE and transferred to nitrocellulose membrane. Membranes were subsequently blocked with 5% nonfat dry milk in PBS buffer for 1 h at room temperature or otherwise blocked with 5% BSA in TBST buffer (Tris-buffered saline, 0.1% Tween-20) for phosphorylation detection and incubated with antibody at optimized dilution overnight in a cold room. Membranes were then washed and treated with chemiluminescent HRP substrate (Millipore) and exposed to Hyblot CL autoradiography film (Denville Scientific Inc.).
Quantitative Real-time PCR
RNA isolation and cDNA preparation were performed according to the protocols of the manufacturer (Qiagen and Invitrogen, respectively). Primer sets used for PCR detection were as follows: β-actin, 5′-GGACTTCGAGCAAGAGATGG-3′ and 5′-AGCACTGTGTTGGCGTACAG-3′; FOXP3, 5′-TCCCAGAGTTCCTCCACAAC-3′ and 5′-ATTGAGTGTCCGCTGCTTCT-3′; Pim-1, 5′-GCTTCGGCTCGGTCTACTC-3′ and 5′-GCCTCTCGAACCAGTCCAG-3′; Pim-2, 5′-CGCACTGCTATGGAAAGTGG-3′ and 5′-GGAATGGCAGTGCTGGATGG-3′; and Pim-3, 5′-AAGCTCATCGACTTCGGTTC-3′ and 5′-AGGATCTCCTCGTCCTGC TC-3′.
In Vitro Kinase Assay
The GST-Pim-2 kinase WT and GST-Pim-2 KD fusion proteins were expressed in Escherichia coli and affinity-purified with glutathione-Sepharose chromatography resins (GE Healthcare) according to the instructions of the manufacturer. The kinase proteins were dialyzed against Tris buffer prior to use. Mouse HA-tagged Foxp3 was transfected into the 293T cell line and purified through immunoprecipitation with anti-HA-agarose beads.
For the kinase assay analyzed by 32P incorporation, each bead-bound HA-Foxp3 substrate protein (1 μg) was incubated with 0.2 μg of GST-Pim-2 (WT or KD) in 50 μl of kinase buffer (25 mm Tris (pH 7.4), 150 mm NaCl, 10 mm MgCl2, 10 mm MnCl2, 0.2 mm NaF, 0.1 mm Na3VO4, 1 mm DTT, and 20 μm ATP) containing 10 μCi of [γ-32P]ATP. The reactions were carried out at 30 °C for 30 min. Equal volumes of 2× Laemmli buffer were added and boiled for 5 min to terminate the reactions. Samples were loaded onto SDS-PAGE gels that were dried prior to exposure to Hyblot CL autoradiography film.
Mass Spectrometry
HEK 293T cells were cotransfected with Foxp3 and Pim-2-expressing plasmids. Foxp3 protein was immunoprecipitated from cell lysates and separated on 8% SDS-PAGE. For phosphorylation site mapping, the Foxp3 band was excised from the gel, digested with Trypsin enzyme, and then submitted to mass spectrum analysis. Identification of phosphopeptides was performed by nano liquid chromatography (nanoLC)/nanospray/linear ion trap mass spectrometry. Sites of phosphorylation within the peptides were determined by a combination of mass spectrometry and solid-phase Edman sequencing.
In Vitro Suppression Assay of Treg Cells
Spleens were removed from healthy wild-type and pim2−/− knockout mice after euthanasia with CO2. CD4+ T cells were enriched from splenocytes using MACS separation (Miltenyi), and CD4+CD25−CD45RBhigh Teff cells and CD4+CD25+ CD45RBlow Treg cells were separated from CD4+ cells by FACS Aria II, yielding a purity of ∼97% for both cell types.
To determine suppression of Teff cell proliferation by measuring carboxyfluorescein succinimidyl ester (CFSE, Invitrogen) dilution, freshly isolated Teff cells were labeled with 10 μm CFSE for 10 min at 37 °C. The labeled Teff cells were cocultured with different ratios of Treg cells in a 96-well plate under stimulation of anti-CD3 and anti-CD28 beads at a 1:0.5 Teff:bead ratio in RPMI medium supplemented with 10% FBS, 1× non-essential amino acids (Invitrogen), 2 mm sodium pyruvate (Invitrogen), and 50 μm β-mercaptoethanol (Sigma). For the analysis of the effect of the Pim-2 inhibitor, splenocytes from wild-type female C57BL/6 mice were subjected to a suppression assay as described above, and Pim-2 inhibitor was added as indicated. After 3 days of coculture, cells were harvested, and their proliferations were analyzed by FACS Canto flow cytometry (BD Biosciences).
Induction of Colitis
Acute colitis was induced in FVB wild-type mice and pim2−/− mice by administration of 5% DSS (molecular weight 5000 Da; Affimetrix Inc., Cleveland, OH) in the drinking water ad libitum for 6 days. The development of colitis was assessed daily by measurement of body weight until day 12.
Statistical Analysis
The means of each data set were analyzed using Student's t test, with a one-tailed distribution assuming equal sample variance for the colitis models and a two-tailed distribution for the others.
Results
Pim-2 Is Highly Expressed and Interacts with Foxp3 in Human Treg Cells
The Pim kinases are highly expressed and active in certain tumor cells (21, 27, 28). We analyzed Pim kinase expression patterns in human Treg cells by real-time quantitative RT-PCR and noted that Pim-2 was the most highly expressed form within the Pim kinase family in Treg cells (Fig. 1A). Negligible expression of Pim-1 and of Pim-3 was noted. This result extends a previous study suggesting that Pim-2 is expressed in Treg cells (23).
FIGURE 1.

Pim-2 is highly expressed in Treg cells and physically associates with Foxp3. A, human CD4+CD25+ Treg cells were sorted from human peripheral blood mononuclear cells. The expression profile of PIM kinases in Treg cells was assessed by real-time qRT-PCR analysis. B, representative immunoblotting data showing the endogenous association between Pim-2 and Foxp3 in mouse splenocytes. Cell lysates of splenocytes were subjected to immunoprecipitation (IP) with antibody to Pim-2 or control IgG and then subjected to Western blot (WB) analysis with antibodies to Foxp3 and Pim-2. Bottom panel, Foxp3 protein in whole lysates. C, the in vitro association between Pim-2 and Foxp3 was analyzed in HEK293T cells by coimmunoprecipitation assay. The lysates from 293T cells, cotransfected with HA-Foxp3 and FLAG-Pim-2 expression plasmids, were immunoprecipitated with HA-agarose (top panel) and FLAG-agarose (bottom panel) and then analyzed by Western blotting with anti-FLAG-HRP and anti-HA-HRP as indicated. D, Foxp3 could be pulled down by the GST-Pim-2 WT/KD fusion protein in vitro. E and F, both the N-terminal domain and the zinc finger and leucine zipper domain of Foxp3 were required for the association between Foxp3 and Pim-2. Top panel, schematic of the Foxp3 fragments. N, N-terminal domain; ZL, zinc finger and leucine zipper domain; FK, forkhead domain; ΔC, forkhead truncated fragment; ΔN, N-terminal domain truncated fragment; FL, full-length Foxp3.
Because both Pim-2 and Foxp3 are highly expressed in mammalian Treg cells, we investigated whether Pim-2 interacts with Foxp3. To test this hypothesis, we first employed coimmunoprecipitations between Pim-2 and Foxp3 present in murine splenocytes. Endogenous Foxp3 was immunoprecipitated by antibody against Pim-2 but not control IgG (Fig. 1B). The interaction of Pim-2 and Foxp3 was then studied using tagged human proteins in transfected human cells. 293T cells were cotransfected with plasmids encoding FLAG-tagged Pim-2 and HA-tagged Foxp3. 24 h after transfection, cell lysates were subjected to coimmunoprecipitation analysis. Immunoprecipitation with HA-conjugated agarose led to the coprecipitation of FLAG-Pim-2, as revealed by Western blot analysis (Fig. 1C, top panel). Reciprocally, HA-Foxp3 was coimmunoprecipitated by FLAG-agarose that targeted FLAG-Pim-2 (Fig. 1C, bottom panel). In addition, Foxp3 was also pulled down by WT and KD Pim-2 kinases in GST pulldown assays (Fig. 1D). These studies revealed that Pim-2 kinase interacts with Foxp3 in a manner independent of its intrinsic kinase domain activity.
Foxp3 contains three major domains: the N-terminal domain, the zinc finger and leucine zipper domain, and the highly conserved forkhead domain located at the C terminus. To map the specific domain in Foxp3 that regulates its association with Pim-2, 293T cells were cotransfected with plasmids expressing various fragments of Foxp3 and Pim-2 and subjected to coimmunoprecipitation analysis. As seen in Fig. 1E, Foxp3 lacking the C-terminal forkhead domain (ΔC) as well as full-length of Foxp3 interacted with Pim-2. However, truncation of the N terminus of Foxp3 (ΔN) abolished its interaction with Pim-2. Both the N-terminal domain and the zinc finger and leucine zipper domain were required for Foxp3 to associate with Pim-2 (Fig. 1F). Taken together, these results indicate that Foxp3 interacts with Pim-2 in vitro and in vivo and that the N-terminal domain of Foxp3 is required for its interaction with Pim-2 in human cells.
Pim-2 Phosphorylates Foxp3 in Vitro and in Vivo
On the basis of the interaction that occurs between Pim-2 kinase and Foxp3, we next determined whether Foxp3 acts as a substrate of Pim-2. To address this question, we performed in vitro kinase assays using 32P incorporation. First, the recombinant GST-Pim-2 WT and GST-Pim-2 KD mutant species were expressed in E. coli and then purified with glutathione-Sepharose beads (Fig. 2A). HA-Foxp3 was also purified from transfected 293T cells and used as the substrate. As shown in Fig. 2B, the purified recombinant GST-Pim-2 WT kinase could be autophosphorylated and readily phosphorylated Foxp3. These studies support a direct physical interaction of the kinase and FOXP3.
FIGURE 2.

Foxp3 was phosphorylated by Pim-2 in vitro and in vivo. A, recombinant GST, GST-Pim-2 WT, and GST-Pim-2 KD were expressed in E. coli cells and purified with glutathione-Sepharose beads. B, the kinase assay was analyzed with 32P incorporation. The agarose-bound HA-Foxp3 was immunoprecipitated from the cell lysates of Foxp3-transfected HEK293T cells and used as substrate for the kinase assay in vitro. Experiment details are described under “Experimental Procedures.” C, phosphorylation of Foxp3 by Pim-2 in transfected HEK293T cells was detected by Western blot (WB) analysis. D, phosphorylation of Foxp3 by Pim-2 can be reversed by λ-phosphatase in vitro. The agarose-bound HA-Foxp3 was immunoprecipitated from Foxp3- and Pim-2-cotransfected 293T cell lysates and then incubated with different concentrations of λ-phosphatase as indicated. The reactions were carried out at 30 °C for 30 min, terminated by adding 2× loading buffer, and then subjected to 8% SDS-PAGE and Western blot analysis.
To investigate the phosphorylation of Foxp3 by Pim-2 in vivo, 293T cells were cotransfected with HA-Foxp3- and FLAG-Pim-2-expressing plasmids. Foxp3 was immunoprecipitated from the cell lysates and subjected to Western blot analysis. As revealed in Fig. 2C, wild-type Pim-2 but not the Pim-2 KD mutant could phosphorylate Foxp3. When we treated the HA-agarose bound HA-Foxp3 with λ-phosphatase, phosphorylation of Foxp3 by Pim-2 decreased in a dose-dependent manner (Fig. 2D). Therefore, Pim-2 can phosphorylate Foxp3 both in vitro and in vivo, and phosphorylation can be reversed by λ-phosphatase treatment.
NanoLC/nanospray/mass spectrometry permits the rapid identification of phosphopeptides and subsequent localization of the phosphorylation sites. We sought to determine the phosphorylation sites of Foxp3 by Pim-2 with MS/MS analysis. After cotransfection of 293T cells with Pim-2 and Foxp3, Foxp3 was isolated by immunoprecipitation and subjected to MS/MS analysis. Three phosphorylated peptides were identified. Ser-33 and Ser-41 were identified to be phosphorylation sites by Pim-2 in the first two phosphorylated peptides (Fig. 3, A and B). The actual phosphorylation site identified in the third phosphorylated peptide was not identified precisely because MS/MS analysis suggested that this represented a mixture of phosphorylation on either serine or threonine residues (Fig. 3C). These results indicate that Foxp3 acts as a substrate of the Pim-2 kinase and is phosphorylated at multiple sites in the Foxp3 N-terminal domain.
FIGURE 3.

MS/MS analysis identified three phosphorylated peptides of mouse Foxp3. Foxp3 protein was purified from Pim-2- and Foxp3-cotransfected 293T cells. A, MS/MS spectrum of the phosphorylated peptide, 28TAPKGS*ELLGTRG40. S* indicates that serine residue 33 is phosphorylated by Pim-2. B, MS/MS spectrum of the phosphorylated peptide, 32GSELLGTRGS*GGPFQGRD49. S* indicates that serine residue 41 is phosphorylated by Pim-2. C, comparison of the MS/MS spectra of m/z 1103.9255 (unphosphorylated) and m/z 1130.5815 (phosphorylated) peptide, 52S*GAHT*S*S*S*LNPLPPSQLQLPTVPLVMVAPSGAR84. S* and T* suggest the phosphorylation site to be either serine or threonine among 52, 56, 57, 58, or 59 residues. In the phosphorylation assignment (panel A spectrum), the phosphorylation site could be at either Thr-28, Ser-33, or Thr-38 in this peptide sequence. Nevertheless, the observation of the y11/y12 ions eliminated the Thr-28 possibility (could only at either Ser-33 or Thr-38 sites). Furthermore, the observation of the y4 ion eliminated the Thr-38 possibility, and the observation of y9 ion further confirmed the phosphorylation at the Ser-33 site. The same approach of using key MS2 spectrum ions was conducted for the phosphorylation site assignments for panels B and C.
Pim-2 Inhibitor Increases Treg Cell Suppressive Function
We identified a role of Pim-2 kinase in the regulation of Treg cell function. We used the Pim-2 inhibitor (Z)-5-(4-propoxybenzylidene) thiazolidine-2,4-dione, which dominantly (but not exclusively) suppresses Pim-2 activity (Calbiochem, IC50 = 150 nm and 20 nm against Pim-1 and Pim-2, respectively).
Primary CD4+CD25−CD45RBhigh Teff cells and CD4+CD25+CD45RBlow Treg cells were sorted from wild-type FVB mouse spleens. Teff cells were stained with CFSE as described under “Experimental Procedures” and then cocultured with different ratios of Treg cells in the presence of different concentrations of Pim-2 inhibitor for 3 days. Subsequently, the proliferation of Teff cells was analyzed in a CFSE-based suppression assay. As shown (Fig. 4A), the proliferation of Teff was decreased with the increasing ratio of Treg/Teff cells.
FIGURE 4.

Deficiency of Pim-2 activity increased Treg suppressive function. A, the Pim-2 inhibitor enhanced Treg cell suppressive function. Teff and Treg cells were cocultured in different ratios and treated with or without different concentrations of the Pim-2 inhibitor as indicated. Teff cell proliferation was analyzed by measuring the CFSE profile. B and C, Treg cell suppressive function was enhanced in Pim-2 knockout mice. Data are mean ± S.D. of three independent experiments. *, p < 0.05; **, p < 0.01 as determined by two-tailed Student's t test.
It should be noted that the Pim-2 inhibitor did not influence the proliferation of Teff cells. However, when Teff cells were cocultured with Treg cells, the suppressive function of Treg cells was found to have increased significantly with the treatment with Pim-2 inhibitor compared with dimethyl sulfoxide, indicating that the Pim-2 inhibitor influenced only Treg cell rather than Teff cell function in vitro. The effects are significant because when the Teff:Treg cell ratio was 1:0.5, Treg cell suppressive function was increased ∼5-fold in the presence of 12.5 μm inhibitor.
Elimination of Endogenous Pim-2 Enhances the Suppressive Function of Treg Cells
Because the small molecule Pim-2 inhibitor promotes the suppressive function of Treg cells but has no discernable effect on T effector cells, we speculated that the suppressive function of Treg might be up-regulated by the elimination of endogenous Pim-2 kinase. To verify this hypothesis, we analyzed suppressive functions using Treg cells from Pim-2 knockout mice.
Primary CD4+CD25−CD45RBhigh Teff cells and CD4+CD25+CD45RBlow Treg cells were sorted from wild-type and Pim-2 knockout mouse spleens and used in the same CFSE assay. Wild-type Teff cells were cocultured with wild-type or Pim-2 knockout Treg cells at ratios of 1:0.5 or 1:1, and the proliferation of Teff cells was measured after 3 days of culture. As expected, compared with wild-type Treg cells, Treg cells from Pim-2 knockout mice were able to suppress the proliferation of Teff cells more efficiently (Fig. 4, B and C). Taken together, these studies indicate that phosphorylation of the Foxp3 N-terminal domain by Pim-2 kinase negatively regulates Treg function.
Effect of Pim-2 Deficiency on Molecules Relevant to Treg Cell Lineage Stability
The transcriptional factor Foxp3 is the master regulator for the Treg cell lineage. Dysfunction of Foxp3 could cause fatal autoimmune diseases in humans and mice. Foxp3 level and activity contribute to Treg cell suppressive function. CD25 is the α chain of the IL-2 receptor. Treg cells highly express CD25, which competes with Teff cells for IL-2 consumption and contributes to Teff cell proliferation suppression (29). However, signaling from glucocorticoid-induced tumor necrosis factor receptor-related protein (GITR) may reverse the immunosuppressive effects of Treg cells (30). We next determined the expression of Foxp3, CD25, and GITR in Pim-2-deficient Treg cells.
Fresh splenocytes were isolated from age-matched WT and Pim-2 KO mice. Expression of Foxp3, CD25, and GITR of CD4+Foxp3+ Treg cells was analyzed by flow cytometry (Fig. 5A). Consistent with the result of a previous suppressive assay of Treg cells, the absence of Pim-2 resulted in increased CD25 expression. The expression of GITR, which negatively regulates Treg function, was slightly down-regulated (Fig. 5B). Most importantly, Foxp3 expression was increased significantly (p = 0.0399) in Pim-2 KO Treg cells compared with the WT counterparts. Taken together, altered expression of Foxp3 and other molecules that are critical for Treg cell suppressive function in Pim-2 KO mice could contribute to the enhanced suppressive function and lineage stability of Treg cells.
FIGURE 5.

Deficiency of Pim-2 promotes Foxp3 and CD25 expression in Treg cells in vivo. A, splenocytes were isolated from 13-week-old age-matched WT (n = 3) and Pim-2 KO (n = 4) mice. Cells were Fc-blocked prior to extracellular (CD4, CD25, and GITR) and intracellular (Foxp3) staining. Treg cells were gated on live CD4+Foxp3+ singlets. B, representative histogram (left panels) and combined data (right panels) of expressions of Foxp3, CD25, and GITR. Solid gray areas, WT; black lines, Pim-2 KO. C, frequency and numbers of Treg cells increased in Pim-2 KO mice. Statistical analysis was calculated on unpaired two-tailed Student's t test. *, p < 0.05.
Pim-2 KO Mice Increase Resistance in DSS-induced Colitis
Reduced Treg cell activity is closely linked to exacerbated intestinal inflammation. Previous studies have shown that exogenous Treg cell injection decreased DSS-induced colitis (46). We therefore investigated whether the deficiency of Pim-2 kinase influenced Treg cell function in DSS-induced colitis. Colitis was induced in both wild-type and Pim-2 knockout mice by giving 5% DSS ad libitum for 6 days, and colitis was monitored indirectly by measuring body weight through day 12 (Fig. 6A). As shown in Fig. 6B, wild-type mice were more susceptible to weight loss from DSS exposure than Pim-2 KO mice. It is noteworthy that half of the Pim-2 KO mice showed only around 5% weight loss and that the other half of the group developed the same severity of colitis as wild-type mice (around 20% weight loss). The absence of Pim-2 kinase activity increases rodent resistance to DSS-induced colitis in vivo but does not entirely correct the lesion in some animals.
FIGURE 6.
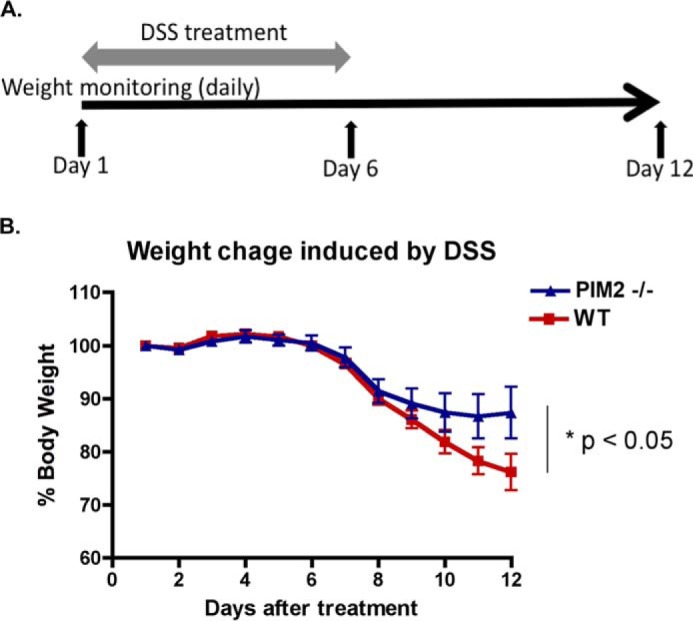
Increased resistance to DSS-induced colitis in Pim-2 knockout mice. A, schematic protocol for the DSS-induced colitis experiment. B, weight loss was measured every day and expressed as the percentage change from day 1. Numbers of animals in each group: WT, n = 8; Pim-2−/−, n = 7. *, p < 0.05.
Discussion
FOXP3 function is influenced by various posttranslational modifications. We have reported previously that TIP60 can acetylate FOXP3 and regulate FOXP3-mediated transcriptional repression by forming a complex containing histone acetyltransferase/histone deacetylase molecules (14, 31). In addition, we have observed that Tip60 and p300 modify distinct residues of the Foxp3 protein (32, 33). Dephosphorylation of Ser-418 by PP1 at the C-terminal domain of FOXP3 may negatively regulate Treg cell function (17). Protein phosphorylation is a common posttranslational modification to regulate signal transduction pathways and cellular processes. It has been reported that Foxp3 could be phosphorylated by cyclin-dependent kinase 2 (CDK2) and lymphocyte-specific protein tyrosine kinase (Lck) (18, 34). Additionally, PIM1 kinase negatively regulates human FOXP3 activity through phosphorylating its C-terminal serine 422 residue (25). In this study, we demonstrated that phosphorylation of the Foxp3 N-terminal domain by Pim-2 kinase negatively regulates Treg cell suppressive function by influencing the Foxp3 level and expression of Treg cell-associated surface markers, including CD25 and GITR.
Pim kinases are constitutively active serine/threonine protein kinases (35). Although Pim kinases appear to be relevant for certain transformation events, the roles of Pim kinases in T cell development and function remain largely undefined. Mice deficient for all Pim kinases (pim1−/−pim2−/−pim3−/−) displayed subtle changes in hematopoietic differentiation but impaired T-cell proliferation in response to TCR and IL-2 because of a reduced capacity to undergo cell division (24). Pim-2 can complement altered AKT/the mammalian target of rapamycin activity and confer T cell survival abilities in the presence of rapamycin (22, 36).
In this study, we analyzed the expression profile of Pim kinases in human Treg cells and found that Pim-2 is a highly expressed member in Treg cells. Indeed, we found that the suppressive function of Treg cells was increased significantly in pim-2−/− mice and, consequently, in the presence of a specific Pim-2 inhibitor, pim-2−/− mice increased resistance to DSS-induced colitis in vivo (Fig. 6), demonstrating that Pim-2 regulates Treg cell function in a negative manner.
Crellin et al. (37, 38) have reported that Akt, a kinase that is involved in the survival signaling pathways in parallel to Pim-2, negatively affects the suppressive function of Treg cells. Although there is no clear evidence that AKT phosphorylates Foxp3, activation of Akt in CD4+CD25+ cells enhanced the expression of some cytokines (e.g. IFN-γ, TNFα, IL-10, and IL-4) while having no effect on FOXP3, CD25, and IL-2 levels (37, 38). Our studies define an active kinase and demonstrate a direct interaction between Pim-2 and Foxp3.
Treg cells are thought to suppress immune responses through direct cell contact-dependent mechanisms as well as by the production of anti-inflammatory cytokines (39). Persistent elevated expression of CD25 on Treg cells may exhaust local levels of IL-2, which are necessary for Teff cell proliferation (40). Foxp3, together with other transcriptional factors, regulates the expression of CD25 and GITR by binding to the promoter regions (26). In this study, we demonstrated that knockout of Pim-2 in vivo enhanced Treg cell suppressive function and stability through altered expression of CD25 and Foxp3. On the other hand, the expression of GITR, which limits the function of Treg cells, was down-regulated slightly. These results indicated that deficiency of Pim-2 kinase increased some of the functional properties of Foxp3.
Structurally, FOXP3 has a unique proline-rich N-terminal domain compared with the glutamine-rich N-terminal domain in FOXP1, FOXP2, and FOXP4 (41, 42). The N-terminal domain of FOXP3 can be considered a repressive element (43–45). Disrupting the Foxp3 N-terminal domain affects the association of Foxp3 with other cofactors like Eos, Tip60, and HDAC7. The N-terminal structural modification also influences certain Treg functions, including the regulation of susceptibility to diabetes (9, 10).
This set of observations indicates that Foxp3 level and activity can be regulated through phosphorylation of its N-terminal domain and helps to define Foxp3 regulation mechanisms in Treg cell development and function. Our studies also indicate that Pim-2 represents a therapeutic target to modulate Treg cell suppressive activities and to control pathological immune responses.
Acknowledgments
We thank Anton Berns and Paul Rothman for Pim-2 deficient mice, James L. Riley for human Treg cells; and Taku Kambayashi and Amanda Schmid for the suppression assay protocol.
This study was supported, in whole or in part, by National Institutes of Health Grant PO1 AI073489-03 (to M. I. G.).
- Treg cell
- regulatory T cell
- KD
- kinase-dead
- GITR
- glucocorticoid-induced tumor necrosis factor receptor-related protein
- DSS
- dextran sodium sulfate
- CFSE
- carboxyfluorescein succinimidyl ester
- Teff cell
- T effector cell.
References
- 1. Wing K., Sakaguchi S. (2010) Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat. Immunol. 11, 7–13 [DOI] [PubMed] [Google Scholar]
- 2. Josefowicz S. Z., Rudensky A. (2009) Control of regulatory T cell lineage commitment and maintenance. Immunity 30, 616–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barnes M. J., Powrie F. (2009) Regulatory T cells reinforce intestinal homeostasis. Immunity 31, 401–411 [DOI] [PubMed] [Google Scholar]
- 4. Taylor P. A., Noelle R. J., Blazar B. R. (2001) CD4(+)CD25(+) immune regulatory cells are required for induction of tolerance to alloantigen via costimulatory blockade. J. Exp. Med. 193, 1311–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sakaguchi S., Fukuma K., Kuribayashi K., Masuda T. (1985) Organ-specific autoimmune diseases induced in mice by elimination of T cell subset: I: evidence for the active participation of T cells in natural self-tolerance: deficit of a T cell subset as a possible cause of autoimmune disease. J. Exp. Med. 161, 72–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Qin S., Cobbold S. P., Pope H., Elliott J., Kioussis D., Davies J., Waldmann H. (1993) “Infectious” transplantation tolerance. Science 259, 974–977 [DOI] [PubMed] [Google Scholar]
- 7. Brunkow M. E., Jeffery E. W., Hjerrild K. A., Paeper B., Clark L. B., Yasayko S. A., Wilkinson J. E., Galas D., Ziegler S. F., Ramsdell F. (2001) Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 27, 68–73 [DOI] [PubMed] [Google Scholar]
- 8. Chatila T. A., Blaeser F., Ho N., Lederman H. M., Voulgaropoulos C., Helms C., Bowcock A. M. (2000) JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J. Clin. Invest. 106, R75–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bettini M. L., Pan F., Bettini M., Finkelstein D., Rehg J. E., Floess S., Bell B. D., Ziegler S. F., Huehn J., Pardoll D. M., Vignali D. A. (2012) Loss of epigenetic modification driven by the Foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity 36, 717–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Darce J., Rudra D., Li L., Nishio J., Cipolletta D., Rudensky A. Y., Mathis D., Benoist C. (2012) An N-terminal mutation of the Foxp3 transcription factor alleviates arthritis but exacerbates diabetes. Immunity 36, 731–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hancock W. W., Ozkaynak E. (2009) Three distinct domains contribute to nuclear transport of murine Foxp3. PLoS ONE 4, e7890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang H., Xiao Y., Zhu Z., Li B., Greene M. I. (2012) Immune regulation by histone deacetylases: a focus on the alteration of FOXP3 activity. Immunol. Cell Biol. 90, 95–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xiao Y., Li B., Zhou Z., Hancock W. W., Zhang H., Greene M. I. (2010) Histone acetyltransferase mediated regulation of FOXP3 acetylation and Treg function. Curr. Opin. Immunol. 22, 583–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li B., Samanta A., Song X., Iacono K. T., Bembas K., Tao R., Basu S., Riley J. L., Hancock W. W., Shen Y., Saouaf S. J., Greene M. I. (2007) FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc. Natl. Acad. Sci. U.S.A. 104, 4571–4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van Loosdregt J., Coffer P. J. (2014) Post-translational modification networks regulating FOXP3 function. Trends Immunol. 35, 368–378 [DOI] [PubMed] [Google Scholar]
- 16. Samanta A., Li B., Song X., Bembas K., Zhang G., Katsumata M., Saouaf S. J., Wang Q., Hancock W. W., Shen Y., Greene M. I. (2008) TGF-beta and IL-6 signals modulate chromatin binding and promoter occupancy by acetylated FOXP3. Proc. Natl. Acad. Sci. U.S.A. 105, 14023–14027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nie H., Zheng Y., Li R., Guo T. B., He D., Fang L., Liu X., Xiao L., Chen X., Wan B., Chin Y. E., Zhang J. Z. (2013) Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-α in rheumatoid arthritis. Nat. Med. 19, 322–328 [DOI] [PubMed] [Google Scholar]
- 18. Morawski P. A., Mehra P., Chen C., Bhatti T., Wells A. D. (2013) Foxp3 protein stability is regulated by cyclin-dependent kinase 2. J. Biol. Chem. 288, 24494–24502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Breuer M. L., Cuypers H. T., Berns A. (1989) Evidence for the involvement of pim-2, a new common proviral insertion site, in progression of lymphomas. EMBO J. 8, 743–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Selten G., Cuypers H. T., Berns A. (1985) Proviral activation of the putative oncogene Pim-1 in MuLV induced T-cell lymphomas. EMBO J. 4, 1793–1798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Isaac M., Siu A., Jongstra J. (2011) The oncogenic PIM kinase family regulates drug resistance through multiple mechanisms. Drug Resist. Updates 14, 203–211 [DOI] [PubMed] [Google Scholar]
- 22. Fox C. J., Hammerman P. S., Thompson C. B. (2005) The Pim kinases control rapamycin-resistant T cell survival and activation. J. Exp. Med. 201, 259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Basu S., Golovina T., Mikheeva T., June C. H., Riley J. L. (2008) Cutting edge: Foxp3-mediated induction of pim 2 allows human T regulatory cells to preferentially expand in rapamycin. J. Immunol. 180, 5794–5798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mikkers H., Nawijn M., Allen J., Brouwers C., Verhoeven E., Jonkers J., Berns A. (2004) Mice deficient for all PIM kinases display reduced body size and impaired responses to hematopoietic growth factors. Mol. Cell. Biol. 24, 6104–6115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li Z., Lin F., Zhuo C., Deng G., Chen Z., Yin S., Gao Z., Piccioni M., Tsun A., Cai S., Zheng S. G., Zhang Y., Li B. (2014) PIM1 kinase phosphorylates the human transcription factor FOXP3 at serine 422 to negatively regulate its activity under inflammation. J. Biol. Chem. 289, 26872–26881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen C., Rowell E. A., Thomas R. M., Hancock W. W., Wells A. D. (2006) Transcriptional regulation by Foxp3 is associated with direct promoter occupancy and modulation of histone acetylation. J. Biol. Chem. 281, 36828–36834 [DOI] [PubMed] [Google Scholar]
- 27. Hogan C., Hutchison C., Marcar L., Milne D., Saville M., Goodlad J., Kernohan N., Meek D. (2008) Elevated levels of oncogenic protein kinase Pim-1 induce the p53 pathway in cultured cells and correlate with increased Mdm2 in mantle cell lymphoma. J. Biol. Chem. 283, 18012–18023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen X. P., Losman J. A., Cowan S., Donahue E., Fay S., Vuong B. Q., Nawijn M. C., Capece D., Cohan V. L., Rothman P. (2002) Pim serine/threonine kinases regulate the stability of Socs-1 protein. Proc. Natl. Acad. Sci. U.S.A. 99, 2175–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Suzuki H., Kündig T. M., Furlonger C., Wakeham A., Timms E., Matsuyama T., Schmits R., Simard J. J., Ohashi P. S., Griesser H. (1995) Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor β. Science 268, 1472–1476 [DOI] [PubMed] [Google Scholar]
- 30. Shevach E. M., Stephens G. L. (2006) The GITR-GITRL interaction: co-stimulation or contrasuppression of regulatory activity? Nat. Rev. Immunol. 6, 613–618 [DOI] [PubMed] [Google Scholar]
- 31. Song X., Li B., Xiao Y., Chen C., Wang Q., Liu Y., Berezov A., Xu C., Gao Y., Li Z., Wu S. L., Cai Z., Zhang H., Karger B. L., Hancock W. W., Wells A. D., Zhou Z., Greene M. I. (2012) Structural and biological features of FOXP3 dimerization relevant to regulatory T cell function. Cell Rep. 1, 665–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Du T., Nagai Y., Xiao Y., Greene M. I., Zhang H. (2013) Lysosome-dependent p300/FOXP3 degradation and limits Treg cell functions and enhances targeted therapy against cancers. Exp. Mol. Pathol. 95, 38–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xiao Y., Nagai Y., Deng G., Ohtani T., Zhu Z., Zhou Z., Zhang H., Ji M. Q., Lough J. W., Samanta A., Hancock W. W., Greene M. I. (2014) Dynamic interactions between TIP60 and p300 regulate FOXP3 function through a structural switch defined by a single lysine on TIP60. Cell Rep. 7, 1471–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nakahira K., Morita A., Kim N. S., Yanagihara I. (2013) Phosphorylation of FOXP3 by LCK downregulates MMP9 expression and represses cell invasion. PLoS ONE 8, e77099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alvarado Y., Giles F. J., Swords R. T. (2012) The PIM kinases in hematological cancers. Expert Rev. Hematol. 5, 81–96 [DOI] [PubMed] [Google Scholar]
- 36. Hammerman P. S., Fox C. J., Birnbaum M. J., Thompson C. B. (2005) Pim and Akt oncogenes are independent regulators of hematopoietic cell growth and survival. Blood 105, 4477–4483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. White E. (2003) The pims and outs of survival signaling: role for the Pim-2 protein kinase in the suppression of apoptosis by cytokines. Genes Dev. 17, 1813–1816 [DOI] [PubMed] [Google Scholar]
- 38. Crellin N. K., Garcia R. V., Levings M. K. (2007) Altered activation of AKT is required for the suppressive function of human CD4+CD25+ T regulatory cells. Blood 109, 2014–2022 [DOI] [PubMed] [Google Scholar]
- 39. Shevach E. M. (2009) Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 30, 636–645 [DOI] [PubMed] [Google Scholar]
- 40. Pandiyan P., Zheng L., Ishihara S., Reed J., Lenardo M. J. (2007) CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat. Immunol. 8, 1353–1362 [DOI] [PubMed] [Google Scholar]
- 41. Deng G., Xiao Y., Zhou Z., Nagai Y., Zhang H., Li B., Greene M. I. (2012) Molecular and biological role of the FOXP3 N-terminal domain in immune regulation by T regulatory/suppressor cells. Exp. Mol. Pathol. 93, 334–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li B., Greene M. I. (2007) FOXP3 actively represses transcription by recruiting the HAT/HDAC complex. Cell Cycle 6, 1432–1436 [PubMed] [Google Scholar]
- 43. Lopes J. E., Torgerson T. R., Schubert L. A., Anover S. D., Ocheltree E. L., Ochs H. D., Ziegler S. F. (2006) Analysis of FOXP3 reveals multiple domains required for its function as a transcriptional repressor. J. Immunol. 177, 3133–3142 [DOI] [PubMed] [Google Scholar]
- 44. Holmes D., Knudsen G., Mackey-Cushman S., Su L. (2007) FoxP3 enhances HIV-1 gene expression by modulating NFκB occupancy at the long terminal repeat in human T cells. J. Biol. Chem. 282, 15973–15980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bettelli E., Dastrange M., Oukka M. (2005) Foxp3 interacts with nuclear factor of activated T cells and NF-κ B to repress cytokine gene expression and effector functions of T helper cells. Proc. Natl. Acad. Sci. U.S.A. 102, 5138–5143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huber S., Schramm C., Lehr H. A., Mann A., Schmitt S., Becker C., Protschka M., Galle P. R., Neurath M. F., Blessing M. (2004) Cutting edge: TGF-β signaling is required for the in vivo expansion and immuno-suppressive capacity of regulatory CD4+CD25+ T cells. J. Immunol. 173, 6526–6531 [DOI] [PubMed] [Google Scholar]