

Background: Plasmodium tryptophan-rich antigens are involved in host-parasite interaction.
Results: Plasmodium vivax tryptophan-rich antigen PvTRAg38 interacts with three exofacial loops of Band 3 through its peptide domain KWVQWKNDKIRSWLSSEW to facilitate parasite growth.
Conclusion: A novel receptor-ligand interaction between host and parasite has been defined.
Significance:The study will help in understanding the host-parasite interaction and development of therapeutics for vivax malaria.
Keywords: erythrocyte, malaria, membrane protein, parasite, peptides, protein-protein interaction, receptor
Abstract
Plasmodium tryptophan-rich proteins are involved in host-parasite interaction and thus potential drug/vaccine targets. Recently, we have described several P. vivax tryptophan-rich antigens (PvTRAgs), including merozoite expressed PvTRAg38, from this noncultivable human malaria parasite. PvTRAg38 is highly immunogenic in humans and binds to host erythrocytes, and this binding is inhibited by the patient sera. This binding is also affected if host erythrocytes were pretreated with chymotrypsin. Here, Band 3 has been identified as the chymotrypsin-sensitive erythrocyte receptor for this parasite protein. Interaction of PvTRAg38 with Band 3 has been mapped to its three different ectodomains (loops 1, 3, and 6) exposed at the surface of the erythrocyte. The binding region of PvTRAg38 to Band3 has been mapped to its sequence, KWVQWKNDKIRSWLSSEW, present at amino acid positions 197–214. The recombinant PvTRAg38 was able to inhibit the parasite growth in in vitro Plasmodium falciparum culture probably by competing with the ligand(s) of this heterologous parasite for the erythrocyte Band 3 receptor. In conclusion, the host-parasite interaction at the molecular level is much more complicated than known so far and should be considered during the development of anti-malarial therapeutics.
Introduction
Malaria caused by Plasmodium vivax is very common in Southeast Asian countries and South America, where it affects millions of people each year, leading to a huge economic loss. Unlike Plasmodium falciparum, this parasite is noncultivable in the laboratory. Earlier, it used to cause benign malaria, but recent reports indicate that this parasite is also causing severe malaria in humans leading to deaths (1). Potential drug and vaccine targets need to be identified considering the fact that there is no effective malaria vaccine available, and this parasite has also started showing resistance toward anti-malarial drugs. Parasite ligands interacting with host receptor molecules are considered good targets to develop anti-malarial therapeutics.
The malaria parasite invades the host erythrocytes through a very complex and multistep process where merozoites come in contact with uninfected erythrocytes. This red cell invasion by the merozoites requires interaction between host and parasite molecules. In case of P. falciparum, several parasite ligands and some of their corresponding host receptors, involved in red cell invasion, have been identified, whereas others are yet to be discovered (2). For P. vivax, a single pathway to invade the host erythrocytes through the Duffy antigen had been suggested in the literature (3). However, the P. vivax infections among the Duffy-negative population indicate that there could be yet another pathway or some other erythrocyte receptors involved in assisting host cell invasion by this parasite (4). The scanty information on host receptors and parasite ligands involved in erythrocyte invasion by the P. vivax warrants further investigation.
The tryptophan-rich antigens expressed by the merozoites of different Plasmodium species have been implicated in erythrocyte invasion. For example, the P. falciparum tryptophan-threonine-rich antigen (PfTryThrA) has a role in invasion of the host erythrocyte by the parasite (5). Similarly, tryptophan-rich proteins of murine malaria have shown partial protection against Plasmodium yoelli infection in mice (6, 7). The orthologous proteins of this family are also reported from P. vivax (8), where several of them bind to the uninfected human erythrocytes (9, 10). Some of these binder proteins are being expressed at schizont/merozoite stages (11). However, the erythrocyte receptors for tryptophan-rich proteins of any of the Plasmodium species, including P. vivax, are yet to be identified. Our earlier findings have revealed that one of the erythrocyte binding P. vivax tryptophan-rich antigens, called PvTRAg382 (accession no. PVX_088820) expressed during merozoites (11), binds to the chymotrypsin-sensitive erythrocyte receptor (9). Here, we have identified Band 3 as the chymotrypsin-sensitive erythrocyte receptor for this parasite protein, defined the receptor and ligand binding domains, and show parasite growth inhibition in in vitro P. falciparum culture by PvTRAg38.
Experimental Procedures
Ethics Statement
Heparinized blood was collected from healthy individuals. Written consent was obtained from the individuals prior to their blood collection, following the institutional ethical guidelines. Ethics committee of the All India Institute of Medical Sciences (New Delhi, India) had approved the study via Approval Numbers IEC/NP-342/2012 and RP-11/2012.
Materials
The purified histidine-tagged PvTRAg38, bacterial thioredoxin, and rabbit antibodies against PvTRAg38 were available in the lab (9, 12, 13). Octaethylene glycol mono dodecyl ether (C12E8), a nonionic detergent; o-phenyldiamine; RPMI 1640; hypoxanthine; penicillin-streptomycin; and monoclonalantibodies against Band 3, His6, and GST (Sigma-Aldrich) pGEX4T-1, glutathione-SepharoseTM 4B resin, CM5 chips, and other coupling reagents (GE Healthcare), pPROEX HT, Pfx polymerase, fetal calf serum, Lipofectamine® 2000, AcTEV protease, and Alexa Flour 488-conjugated goat anti-mouse or anti-rabbit IgG (Invitrogen Life Technologies) were obtained commercially.
Methods
PCR Amplification, Cloning, Expression, and Purification of Recombinant Proteins
Exon 2 of PvTRAg38, encoding majority of the protein, was divided into three parts: the N-terminal region (amino acids 58–142), the middle (M) part (amino acids 143–231), and the C-terminal region (amino acids 231–316). The DNA fragments covering these regions were amplified from the original pvtrag38 clone (9) using specific primers: 5′-ACATAGGATCCAGCTATAGACCA-3′ and 5′-TTTAAAGCTTTTTTCACTATAGTG-3′ for the N-terminal coding region, 5′-AAAAGGATCCAAGAACACAAAAGT-3′ and 5′-TTTTGAAGCTTTAGCGCGCTCTAC-3′ for middle coding region, and 5′-GGATCCAAATGGTTGCAAGAAGCA-3′ and 5′-CTAAACCTCGAGTGCAGTATTTGC-3′ for the C-terminal coding region. The DNA was denatured at 94 °C for 10 min followed by 35 cycles as follows: denaturation at 94 °C, annealing at 55 °C, and extension at 68 °C for 1 min each. The final extension at 68 °C was carried out for 15 min. The PCR products were cloned into the pGEM-T Easy vector (Promega, Madison, WI) and then subcloned into pPROEX HT expression vector. The expression and purification of histidine-tagged recombinant proteins was carried out as described by Bora et al. (14).
To perform GST pulldown assay, exon 2 coding region of pvtrag38 was amplified from the original clone (9) using primers 5′-ACATAGGGATCCGCTAATAGACCA-3′ and 5′-CTAAACCTCGAGTGCAGTATTTGC-3′ under the same PCR conditions as described above except the annealing temperature used was 52 °C. The PCR product was cloned in pGEMT-T Easy and then subcloned in pGEX4T-1 vector to express PvTRAg38 with N-terminal GST tag. The recombinant GST-PvTRAg38 was purified on glutathione-SepharoseTM 4B resin following the manufacturer's instructions.
The DNA encoding each of the six extracellular loop regions of Band 3 (15) was amplified from the cDNA (prepared from the human blood RNA) using following primers: 5′-GGATCCGGCGGCCTCCTGGGAGAA-3′ and 5′-CTCGAGCGACCACAAGCAGGGGCT-3′ for loop1 (B3F1), 5′-GGATCCCCTGCTTGTGGTCGGCTT-3′ and 5′-CTCGAGAAGCCGATCCACACGCGGC-3′ for loop2 (B3F2), 5′-GGATCCTCCAAGCTGATCAAGATCTT-3′ and 5′-CTCGAGTGTGTTGGGCAGGGGGC-3′ for loop3 (B3F3), 5′-GGATCCTTCATTCAGGATACCTAC-3′ and 5′-CTCGAGCATCCAGATGGGAAACT-3′ for loop4 (B3F4), 5′-GGATCCGGGATGCCCTGGCTCAG-3′ and 5′-CTCGAGGATCCGCTGCTCTTTGAC-3′ for loop5 (B3F5), and 5′-GGATCCGACCGCATCTTGCTTCTG-3′ and 5′-CTCGAGGGGCAGGGCCAGGGAG-3′ for loop6 (B3F6). The PCR conditions were the same as above except the annealing temperature was 59 °C for B3F1 and B3F2; 57 °C for B3F3, B3F5, and B3F6; and 50 °C for B3F4. The PCR products were cloned in to pET32a to express histidine-tagged recombinant proteins, which were purified on Ni2+-nitrilotriacetic acid column as described in Ref. 14. The proofreading Pfx polymerase enzyme was used to amplify the DNA. The restriction sites introduced in the primers are underlined.
The 1851-bp coding region of exon 2 of pftrythrA (PlamoDB accession no. PF3D7_0830500) was amplified from the P. falciparum parasite DNA using primers 5′-TAT TTA TAA AAT CGT CTC ATC CAG-3′ and 5′-CCA TAC ATT CAT ACA ATT TAA ATG-3′ and nested primers 5′-GGA TCC GCT CTC AAA GAG AAG AAG-3′ and 5′-CTC GAG TTA AAC ATT AAT TTC ATT TCC-3′. The DNA was denatured at 94 °C for 10 min followed by 35 cycles as follows; denaturation at 94 °C, annealing at 41 °C for primary PCR and 46 °C for nested PCR, and extension at 60 °C for 3 min each. The final extension at 60 °C was carried out for 15 min. The PCR products were cloned into the pGEM-T Easy and then subcloned into pGEX4T-1 vector to express PfTryThrA with N-terminal GST tag. The recombinant GST-PfTryThrA was purified on glutathione-SepharoseTM 4B resin as above.
Cloning and Expression of PvTRAg38 and Its Fragments in CHO-K1 Cells
For surface expression, exon 2 of PvTRAg38 and its fragments were PCR-amplified and cloned, in frame, to PvuII and ApaI sites of pRE4 vector (kindly gifted by Gary Cohn and Roselyn Eisenberg) so as to produce a fusion protein containing the signal sequence and transmembrane region of the Herpes simplex glycoprotein D (HSVgD) at the N-terminal and C-terminal regions, respectively, of the recombinant protein (16). This recombinant DNA was then used to transfect the CHO-K1 cells (American Type Culture Collection), which were cultured in RPMI 1640 medium with 10% fetal calf serum, 4 mm glutamine, 1× penicillin-streptomycin at pH 7.4 in a humidified 5% CO2 incubator at 37 °C. The transfection was done with cationic lipid Lipofectamine® 2000, following the manufacturer's protocol. Transfected CHO-K1 cells were incubated for 1 h with mouse monoclonal antibodies DL6 (Santa Cruz, Dallas, TX) directed against the C terminus of the H. simplex glycoprotein D sequences or rabbit polyclonal anti-sera directed against PvTRAg38. Alexa Flour 488-conjugated goat anti-mouse or anti-rabbit IgG were used as secondary antibodies. DAPI was used for nuclei staining. Images were captured under UV light at 400× magnification using Nikon eclipse 80i microscope (Nikon Corporation, Tokyo, Japan).
Synthetic Peptides
The 18-amino acid-long nonoverlapping peptides, derived from M-PvTRAg38 sequences, were commercially synthesized (Thermo Fisher Scientific) with or without six histidine residues attached to their C terminus.
Erythrocyte Binding Assay by Cell-ELISA
ELISA-based erythrocyte binding assay was performed as described earlier (9, 17). Briefly, ∼1 million human erythrocytes were added to each well of a 96-well microtiter plate. After overnight incubation at 4 °C, the erythrocytes were fixed with 0.3% glutaraldehyde at 25 °C for 30 min. Plates were blocked with 5% BSA and then incubated with different concentrations (0–2 μm) of histidine-tagged PvTRAg38, recombinant fragments of PvTRAg38, or synthetic peptides for 4 h at room temperature. Recombinant thioredoxin from Desulfovibrio desulfuricans was used as negative control (12). Plates were washed with PBST (PBS containing 0.05% Tween 20) and incubated with 1:2000 dilution of the primary mouse monoclonal anti-His6 antibody, followed by HRP-conjugated anti-mouse IgG secondary antibody (Pierce). Finally, plates were developed with o-phenyldiamine substrate, and OD was measured at 490 nm.
For competition assay, a mixture of histidine-tagged recombinant M-PvTRAg38 (200 nm) and variable amounts (0–4 μm) of untagged PvTRAg38 or histidine-tagged (200 nm) and untagged (0–4 μm) peptides were added to each well of the ELISA plate containing ∼1 million RBCs. The plates were developed with the anti-His6 monoclonal antibodies as above.
Erythrocyte Binding Assay by Rosetting
This assay was done according to the method described by Chitnis and Miller (18) and adopted by Zeeshan et al. (10) using CHO-K1 cell line instead of COS-7 cells. Briefly, erythrocytes were collected in 10% citrate phosphate dextrose and washed three times in RPMI 1640 medium (pH 7.4) containing 0.36 mm hypoxanthine. Hematocrit at 1% was added to CHO-K1 cells expressing PvTRAg38 or its fragments and incubated for 1 h in a humidified 5% CO2 incubator at 37 °C. After 1 h of incubation, the cells were washed four times with incomplete RPMI 1640 medium, pH 7.4, and the numbers of rosettes (more than five erythrocytes bound to each transfected CHO-K1 cell) were scored in 20 fields at 200× magnification using Nikon Eclipse TS 100 inverted microscope (Nikon Corporation, Tokyo, Japan) and were normalized to transfection efficiency of 15%. CHO-K1 cells transfected with plasmid pHVDR22 (containing PvRII region of P. vivax Duffy binding protein) (18) and nonbinder PvTRAg53.7 (10) were used as positive and negative controls, respectively.
For competition assays, erythrocytes at 1% hematocrit were preincubated with different concentrations of PvTRAg38, M-PvTRAg38, or peptide P-1, P-2, P-3, P-4, or P-5 (0–20 μm, in incomplete RPMI 1640, pH 7.4) for 1 h at 37 °C. Erythrocytes, preincubated with these recombinant proteins or peptides, were added to transfected CHO-K1 cells, and an erythrocyte binding assay was performed. In another set, RE4-PvTRAg38-transfected CHO-K1 cells were preincubated with different concentrations (0–10 μm) of Band 3 fragments (B3F1, B3F3, or B3F6) for 1 h at 37 °C in a humidified 5% CO2 incubator and then allowed to interact with human erythrocytes as above.
Erythrocyte Binding Assay by Flow Cytometry
The flow cytometry-based erythrocyte binding assay was performed according to Tran et al. (19) as described earlier (9). Briefly, ∼1 million human erythrocytes were incubated with 1 μm of histidine-tagged recombinant PvTRAg38, its fragments, or synthetic peptides for 4 h at room temperature. Cells were pelleted by centrifugation at 2000 × g for 5 min. Pellets were washed two times with 1% BSA in PBS and incubated with mouse anti-pentahistidine Alexa Fluor 647-conjugated monoclonal antibody (Qiagen) for 1 h at 4 °C in dark. Samples were again pelleted and washed three times with 1% BSA in PBS. After washing, the pellet was suspended in 1% BSA (in PBS) for acquiring erythrocytes. Two hundred thousand total events were acquired per sample using a BDLSRII flow cytometer (Becton-Dickinson) and Facs Diva software.
Pulldown Assay
Erythrocyte membrane was prepared by hypotonic lysis of the erythrocytes using the method of Dodge et al. (20). To identify the host erythrocyte membrane proteins interacting with PvTRAg38, the pulldown assay was performed as described by Yajima et al. (21). Briefly, the above mentioned erythrocyte membrane preparation in 5 mm phosphate buffer (pH 7.5) containing protease inhibitor mixture was dissolved in 1% C12E8 by incubating it on ice for 20 min. After centrifugation at 20,000 × g for 30 min at 4 °C, the solubilized erythrocyte membrane preparation was incubated with 1:1 glutathione-Sepharose 4B resin for 1 h. 10 μg of GST-PvTRAg38 or GST alone were incubated with 100 μl of 50% glutathione-Sepharose 4B resin for 1 h at 4 °C, followed by washing with Tris-buffered saline (25 mm Tris, 150 mm NaCl, 2 mm KCl, pH 7.5) containing 0.1% C12E8. The GST-PvTRAg38 (or GST alone) bound to glutathione resin was then incubated overnight at 4 °C with 500 μg of solubilized erythrocyte membrane proteins. To remove unbound material, resin was washed five times with Tris-buffered saline containing 0.1% C12E8, and finally with 10 mm Tris-HCl, pH 7.5. The GST-glutathione complexes of the pulldown were dissociated with 2× loading dye and then separated on SDS-PAGE. After electrophoresis, the gel was stained with Coomassie Blue, and protein bands were identified by mass spectroscopy.
To free the erythrocyte's membrane from the cytoskeleton peripheral membrane proteins, the membrane was treated with 0.1 m NaOH as described by Casey et al. (22). The stripped erythrocyte membrane was solubilized in 1% C12E8 and used in pulldown assay as above.
Purification of Human Erythrocyte Membrane Protein Band 3
The Band 3 was isolated from RBC according to the method described by Casey et al. (22). First, the erythrocyte membrane was prepared by hypotonic lysis of erythrocytes, and then peripheral proteins were stripped from membrane by resuspending it in 10 volumes of ice-cold 2 mm EDTA, pH 12. The stripped membrane was solubilized in 1% (v/v) C12E8 in 5 mm phosphate buffer (pH 8), and Band 3 protein was purified by affinity chromatography on aminoethyl-agarose resin (ABT Beads, Madrid, Spain). The purity and identity of the purified protein was confirmed by SDS-PAGE, Western blot analysis using monoclonal anti-Band 3 antibody, and mass spectrometry.
Solid Phase Binding Assay
This assay was performed by coating each well of 96-well ELISA plate with 50 nm of Band 3 protein in carbonate buffer, pH 9.6. Before coating the protein, each well of the plate was pretreated with gluteraldehyde and poly-l-lysine (Sigma-Aldrich) as described by Sorette et al. (23). The coated ELISA plates were blocked with 5% BSA in PBS for 2 h at 37 °C and then washed with 0.02% PBST followed by incubation with different concentration (0–2 μm) of histidine-tagged PvTRAg38, its fragments, synthetic peptides, or bacterial thioredoxin and developed with monoclonal anti-His6 antibody as above.
For binding of Band 3 fragments to PvTRAg38 or synthetic peptides, each well of a 96-well ELISA plate was coated with 0.5 μm of untagged PvTRAg38 or peptides. After blocking with 5% BSA, the plates were incubated with different concentrations (0–2 μm) of histidine-tagged Band 3 fragments and developed with monoclonal anti-His6 antibody as above.
For competition assay, the histidine tag of the recombinant PvTRAg38 was removed by the treatment of AcTEV protease according to the manufacturer's protocol. Increasing concentration (0–4 μm) of untagged PvTRAg38 was mixed with 0.5 μm of histidine-tagged PvTRAg38, M-PvTRAg38, or peptide P-4. This mixture was added to Band3 (50 nm) coated ELISA plate and incubated for 4 h at 37 °C. Plates were developed with monoclonal anti-His6 antibody as above.
For competitive binding of Band 3 fragments with PvTRAg38 or its peptides, the Band 3 fragments (0.5 μm) were mixed with increasing concentrations (0–4 μm) of untagged PvTRAg38 or peptide P-2 or P-4. Then this mixture was added to GST-tagged PvTRAg38-coated ELISA plate and incubated for 4 h at 37 °C. Plates were developed with monoclonal anti-His6 antibody as above.
Surface Plasmon Resonance Analysis
Real time interactions of PvTRAg38, its fragment M-PvTRAg38, or peptide P-4 with Band 3 or with Band 3 fragments (B3F1, B3F3, and B3F6) were analyzed by surface plasmon resonance studies under flow condition using a Biacore 2000 instrument (Biacore, Uppsala, Sweden) as described by Bhalla et al. (24). The carboxymethylated-dextran surfaces of Biacore CM5 sensor chips were activated by injection of 1:1 mixture of N-hydroxysuccinimide and 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride for 7 min at a flow rate of 10 μl/min. Purified Band 3 protein (0.06 μg/μl) and recombinant Band 3 fragments (0.01 μg/μl) in 10 mm sodium acetate buffer, pH 4.5, were injected over the activated surface of different sensor chip's flow cell at a flow rate of 10 μl/min. A nonspecific recombinant protein (bacterial recombinant thioredoxin from D. desulfuricans) at a concentration of 0.03 μg/μl was immobilized on surface of another flow cell in a similar way and used as a negative control (12). A third sensor chip flow cell was blocked by injection of ethanolamine at a flow rate of 10 μl/min for 5 min and was used as blank. Kinetic binding analysis for interaction of PvTRAg38, M-PvTRAg38, and P-4 was carried out by injecting the different analytes (0.1–1.5 μm) over the immobilized Band 3 and its fragments, separately, at a flow rate of 30 μl/min in independent experiments. The peptide or recombinant proteins were injected for 240 s at a flow rate of 30 μl/min with the running buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 3 mm EDTA, and 0.005% P20 surfactant). The dissociation was monitored for 240 s with the running buffer at the same flow rate. To correct for refractive index changes caused by running buffer and instrument noise, running buffer was injected at a flow rate of 30 μl/min in the blank flow cells, and the observed response difference was subtracted from the experimental data. The flow rate of the peptide and the recombinant proteins was varied from 10 to 75 μl/min to assess whether the binding interactions were limited by mass transfer. The experimental surface was regenerated by injecting regeneration solution (10 mm NaOH) at a flow rate of 30 μl/min for 1 min. The binding constant, Kd, was calculated as kd1/ka1 using the data analysis program BIAevaluation 3.2RC1 (Biacore). Kinetic rate constants were determined by fitting the corrected response data to a simple 1:1 Hill-Langmuir binding isotherm model using BIAevaluation 3.2RC1 software.
P. falciparum Culture and Growth Inhibition Assay
P. falciparum 3D7 was cultured in complete RPMI 1640 medium containing 27.2 mg/liter hypoxanthine, 0.5 g/liter AlbuMAX I (Gibco), and 2 g/liter sodium bicarbonate, using O+ human erythrocytes (4% hematocrit) under mixed gas (5% O2, 5% CO2, and 90% N2). Growth inhibition assay was performed as described by Persson et al. (25). To assess the effect of PvTRAg38 on P. falciparum growth, parasite culture was synchronized by sorbitol treatment and synchronous culture at late schizont stage with parasitemia of 1% were treated with the PvTRAg38, PfTryThrA, and PvTRAg53.7 at different concentrations (1–25 μm) in a 96-well culture plate in triplicate. Uninfected erythrocytes, infected erythrocytes alone and infected erythrocytes with PBS were taken as controls. Parasites were maintained for 40 h and stained with ethidium bromide. One hundred thousand total events were acquired per sample, using FACSDiva software on a BDLSRII flow cytometer.
Results
Defining the Erythrocyte-binding Domains of PvTRAg38
Earlier, we described erythrocyte binding activity of PvTRAg38 by Cell-ELISA and flow cytometry assay (9). Here, we have further confirmed it by rosetting assay where exon 2 encoded PvTRAg38 was expressed on the surface of the transfected CHO-K1 cells and allowed to bind to human erythrocytes. The surface expression of the proteins was detected by the DL6 antibody against vector-derived protein (Fig. 1, upper panels). The surface expression of PvTRAg38 was also confirmed by the anti-PvTRAg38 antibody raised in rabbits. Several human erythrocyte cells (more than five) were found attached with the PvTRAg38 expressing CHO-K1 cell (Fig. 1, lower panels). This rosette formation was similar to that of the positive control PVRII of Duffy binding protein of P. vivax (18). The CHO-K1 cells expressing non-erythrocyte-binding protein PvTRAg53.7 (10) did not show rosette formation with erythrocytes. Numbers of CHO-K1 cells with rosettes, counted in 20 fields at 200× magnification, were 19.6 ± 2.6 and 19.95 ± 4 for PvTRAg38 and PvRII, respectively. Only 1.1 ± 1.17 CHO-K1 cells transfected with PvTRA53.7 showed rosetting. The transfection efficiency for each experiment was normalized to 15%. The mean ± S.D. value is derived from three different experiments.
FIGURE 1.
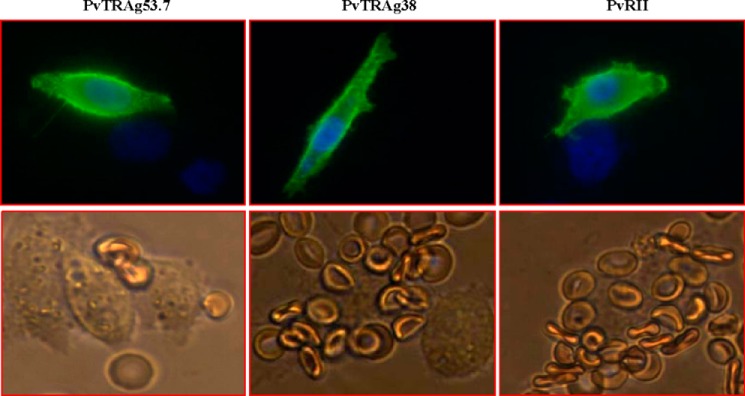
PvTRAg38 expressed on the CHO-K1 cell surface binds to human erythrocytes. The transfected CHO-K1 cells were incubated with mouse monoclonal antibody DL6 directed against the C terminus of the H. simplex glycoprotein D sequences and then stained with Alexa Fluor 488-conjugated secondary antibodies (green fluorescence). The nuclei were stained with DAPI (blue color). Merged images of blue and green florescence are shown in the upper panels. The lower panels show the rosette formation where transfected CHO-K1 cell is binding to more than five erythrocytes. Positive and negative controls were pHVDR22 clone containing region II of P. vivax Duffy binding protein (PvRII) in RE4 vector (18) and nonbinder PvTRAg53.7 (10), respectively.
Exon 2 encoded PvTRAg38 was divided in to three parts (Fig. 2A). Each fragment was expressed either as histidine-tagged fusion protein in Escherichia coli for Cell-ELISA or HSVgD fusion protein on the surface of CHO-K1 cells for rosetting assay. Only one of these three fragments, i.e. M-PvTRAg38, showed erythrocyte binding activity by both assays (Fig. 2, B–E). The specificity of binding was confirmed by competition data in both the assays (Fig. 2, C and E). These results indicate that the erythrocyte binding activity of PvTRAg38 resides in the region of 143–231 amino acids.
FIGURE 2.

Binding of PvTRAg38 fragments to human erythrocytes. A, PvTRAg38 encoded by exon 2 was divided in to three (N-terminal, middle, and C-terminal) parts. These DNA fragments were PCR-amplified, cloned, and expressed in E. coli for Cell-ELISA or in mammalian cell line CHO-K1 for rosetting assay. B, erythrocyte binding activity of PvTRAg38 fragments by Cell-ELISA. Each well of the ELISA plate was coated with ∼1 million erythrocytes and reacted with different concentrations (0–2 μm) of histidine-tagged recombinant PvTRAg38 and its fragments. The plate was developed with mouse anti-His6 monoclonal antibody as described in the text. C, specificity of erythrocyte binding to M-PvTRAg38 fragment by competition assay. For this, a mixture of histidine-tagged recombinant M-PvTRAg38 (200 nm) and variable amounts (0–4 μm) of untagged PvTRAg38 were added to ∼1 million RBCs coated in each well of the ELISA plate. The plate was developed with mouse anti-His6 monoclonal antibody as described in the text. Binding in the absence of untagged PvTRAg38 was taken as percentage control for rest of the concentrations. D, erythrocyte binding activity of PvTRAg38 fragments by rosetting assay. PvTRAg38 fragments were expressed on the surface of the transfected CHO-K1 cells, detected by the mouse monoclonal antibody DL6 as in Fig. 1 (green fluorescence). The nuclei were stained with DAPI (blue color). Merged images of blue and green fluorescence are shown in the upper panels. The lower panels show the rosette formation where transfected CHO-K1 cell is binding to more than five erythrocytes. E, specificity of binding of PvTRAg38 and its fragment M-PvTRAg38 expressed on CHO-K1 cells to erythrocytes by competition assay. Erythrocytes (1% hematocrit) were preincubated with different concentrations of histidine-tagged PvTRAg38 or M-PvTRAg38 (0–20 μm) for 1 h at 37 °C. They were then allowed to bind to RE4-PvTRAg38 transfected CHO-K1 cells at 37 °C for 1 h. The results were expressed as relative binding to positive control (binding of PvTRAg38 transfected cells with untreated RBCs). The mean and ± standard deviation of three experiments is reported.
To further define the erythrocyte-binding domains within the M-PvTRAg38 (amino acid positions 143–231), the nonoverlapping peptides of 18 amino acids, each having six histidine residues at its C-terminal end, were synthesized (Table 1) and tested for their erythrocyte binding activity. Peptides P-2 (amino acid positions 161–178) and P-4 (amino acid positions 197–214) were found to contain this activity (Fig. 3A). This binding was specific as evident by the competition assay in Cell-ELISA (Fig. 3B). Similarly, only these two peptides were able to compete for erythrocyte binding in rosetting assay (Fig. 3C).
TABLE 1.
Amino acid sequence of synthetic peptides derived from M-PvTRAg38, used in erythrocyte binding assay
Peptides were synthesized with and without six histidine residues attached at their C-terminal end.
| Name of peptide | Amino acids position in PvTRAg38 | Amino acid sequence of peptides |
|---|---|---|
| P-1 | 143–160 | MLKEHKSNVMEKSANWND |
| P-2 | 161–178 | TQWGNWIKTEGRKILEAQ |
| P-3 | 179–196 | WEKWIKKGDDQLQKLILD |
| P-4 | 197–214 | KWVQWKNDKIRSWLSSEW |
| P-5 | 215–231 | KTEEDYYWANVERATTA |
FIGURE 3.
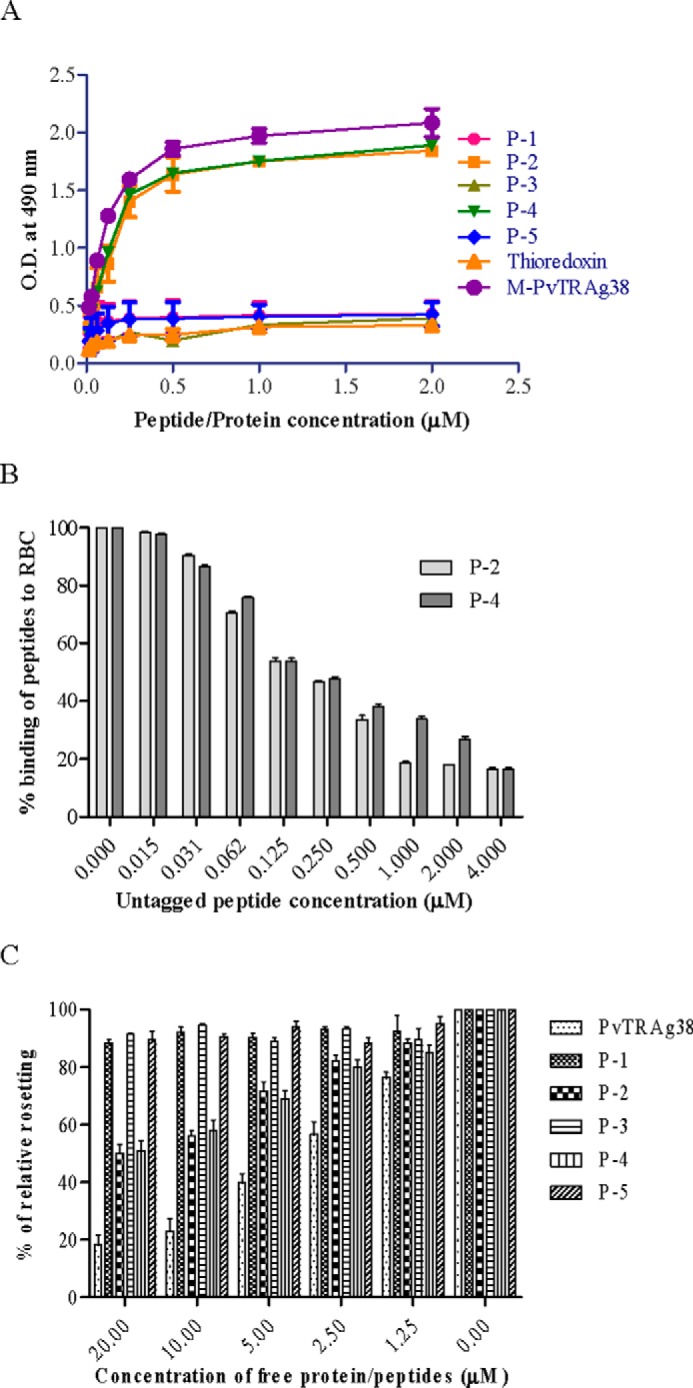
Erythrocyte binding activity of the synthetic peptides derived from middle fragment of PvTRAg38. A, the peptides were tested for their erythrocyte binding activity by Cell-ELISA where the ELISA plate was coated with ∼1 million erythrocytes and reacted with different concentrations (0–2 μm) of histidine-tagged peptides. The plate was developed with mouse anti-His6 monoclonal antibody as described in the text. B, specificity of binding of peptides to erythrocytes by competition assay in Cell-ELISA. For this, a mixture of histidine-tagged (200 nm) and untagged (0–4 μm) peptide was added to ∼1 million RBCs. The plate was developed with mouse anti-His6 monoclonal antibody as described in the text. Binding in the absence of respective untagged peptides was taken as percentage control for rest of the concentrations. C, specificity of binding of PvTRAg38 and its peptides to erythrocytes by competition in rosetting assay. Erythrocytes (1% hematocrit) were preincubated with different concentrations of recombinant PvTRAg38 or peptide P-1, P-2, P-3, P-4, or P-5 (0–20 μm) for 1 h at 37 °C. They were then allowed to bind to RE4-PvTRAg38 transfected CHO-K1 cells at 37 °C for 1 h. The results are expressed as relative binding to positive control (binding of PvTRAg38 transfected cells with untreated RBCs). The mean and ± standard deviation of three experiments is reported.
The erythrocyte binding activity of these two peptides along with middle fragment of PvTRAg38 was also confirmed by flow cytometry data shown in Fig. 4. Thus there were two erythrocyte-binding domains in PvTRAg38 located at amino acid positions 161–178 and 197–214.
FIGURE 4.

Erythrocyte binding activity of PvTRAg38 and its peptides by flow cytometry. A, representative dot plots for binding of PvTRAg38 and its fragments/peptides to human erythrocytes. Normal human RBCs (∼1 million) were incubated with 1 μm of recombinant proteins or synthetic peptides and then labeled with an anti-pentahistidine mAb Alexa Fluor 647 conjugate. PvTRAg38 and its fragments/peptides bound to erythrocytes are shown as red dots. The number in the red box refers to percentage of erythrocyte bound by proteins or peptides respective to total erythrocyte population. Thioredoxin and PvTRAg53.7 were taken as negative control. B, bar diagram shows the percent binding of PvTRAg38 or its fragments/peptides with the erythrocytes. The data shown are the means ± S.D. of at least two independent experiments.
Band 3 of Human Erythrocyte Binds to PvTRAg38
The pulldown assay with PvTRAg38 captured four erythrocyte membrane proteins (Fig. 5A). The mass spectroscopy analysis of these captured protein bands identified them as Spectrin, Ankyrin, Band 3, and Band 4.2. Among these four proteins, only Band 3 is exposed at the surface of RBC. Association of Ankyrin and Band 4.2 with Band 3 in addition to interaction of beta chain of Spectrin with Ankyrin (26–28) in erythrocyte cytoskeleton meshwork could have captured them in pulldown assay. Indeed, these associated proteins were removed by treating the erythrocyte membrane by NaOH and pulldown assay captured only Band 3 protein (Fig. 5B). For further analysis, we purified Band 3 protein from the human erythrocyte membrane. Purity and identity of Band 3-purified protein was confirmed by SDS-PAGE followed by Western blot using anti-Band 3 monoclonal antibody (Fig. 6A). The identity of the purified Band 3 protein was also confirmed by mass spectrometry analysis. This purified Band 3 protein showed dose-dependent binding to PvTRAg38 in a solid phase ELISA in comparison with the recombinant bacterial thioredoxin (Fig. 6B). The specificity of binding of PvTRAg38 with Band 3 was further confirmed by the competition data in solid phase ELISA (Fig. 6C). The SPR analysis also confirmed this interaction between PvTRAg38 and Band 3 (Fig. 6D) with a dissociation constant value of 5.29 ± 0.43 × 10−8 m.
FIGURE 5.
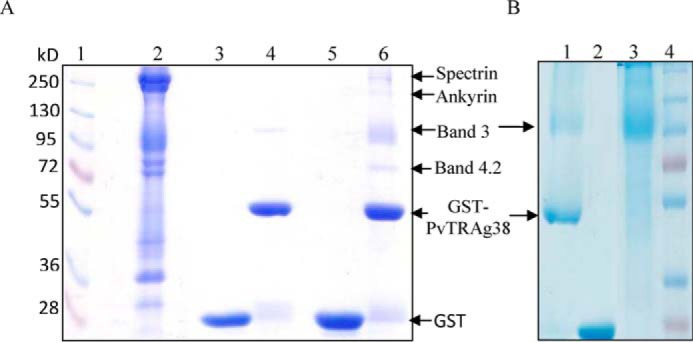
Identification of erythrocyte membrane proteins interacting with recombinant PvTRAg38 by pulldown assay. A, erythrocyte membrane proteins extracted with 1% C12E8 were incubated with recombinant GST-tagged PvTRAg38 bound to glutathione-Sepharose 4B column, washed, and resolved on 10% SDS-PAGE. Lane 1, molecular weight marker; lane 2, erythrocyte membrane proteins; lane 3, GST; lane 4, GST-PvTRAg38; lane 5, erythrocyte membrane extract incubated with GST; lane 6, erythrocyte membrane extract incubated with GST-PvTRAg38. Arrows indicate the position of proteins in lane 6. B, pulldown assay with NaOH-treated erythrocyte membrane. Lane 1, NaOH-treated erythrocyte membrane extract incubated with GST-PvTRAg38; lane 2, NaOH-treated erythrocyte membrane extract incubated with GST; lane 3, erythrocyte membrane treated with 0.1 m NaOH; lane 4, molecular weight markers.
FIGURE 6.

Binding of PvTRAg38 with purified Band 3 protein. A, Band 3 was purified from human erythrocytes using the protocol of Casey et al. (22) by affinity chromatography on aminoethyl-agarose resin. RBC membrane preparations and purified Band 3 were analyzed by SDS-PAGE (panel a) followed by Western blot analysis using anti-Band 3 monoclonal antibodies (panel b). Lane 1, molecular weight marker; lane 2, RBC membrane; lane 3, purified Band 3. The monoclonal anti-Band 3 antibody reacted with full-length Band 3, as well as with truncated Band 3. Arrows indicate the position of Band 3. B, binding of recombinant PvTRAg38 to the purified Band 3 by solid phase ELISA. Increasing concentrations (0–2 μm) of histidine-tagged PvTRAg38 or bacterial thioredoxin from D. desulfuricans (negative control) were added to the wells of an ELISA plate already coated with 50 nm of Band 3. The plate was developed with mouse anti-His6 monoclonal antibody as described in the text. The mean ± S.D. value of absorbance from three experiments is plotted. C, specificity of binding of PvTRAg38 to Band 3 by competition assay. Increasing concentrations of untagged PvTRAg38 was mixed with fixed concentration (0. 5 μm) of histidine-tagged PvTRAg38 and allowed to interact with 50 nm of immobilized Band 3 for 4 h at 37 °C. Bound recombinant histidine-tagged PvTRAg 38 was detected with mouse anti-His6 monoclonal antibody as described in the text. Binding in the absence of untagged PvTRAg38 was taken as percentage control for the rest of the concentrations. The mean value of three independent experiments is plotted with S.D. D, SPR analysis of Band 3 interaction with PvTRAg38. Band 3 was immobilized on the cell of CM5 chip. Three different concentrations of PvTRAg38 (0.2, 0.8, and 1.4 μm) were injected at flow rate of 30 μl/min over the surface of immobilized Band 3. Sensogram curves show dose-dependent response of PvTRAg38 binding with Band 3.
Band 3 Binds to Peptide P-4 of PvTRAg38
The solid phase binding assay results showed that only middle fragment M-PvTRAg38 and not the other two fragments of this parasite protein bind to Band 3 (Fig. 7A). The specificity of this binding of M-PvTRAg38 to Band 3 was confirmed by competition assay results of solid phase ELISA (Fig. 7B). The SPR analysis further confirmed this interaction between Band 3 and the M-PvTRAg38 fragment with a KD value of 7.39 ± 1.24 × 10−8 m (Fig. 7C). This interaction is similar to that of the Cell-ELISA results where the same fragment showed binding to erythrocytes (Fig. 2B). Because synthetic peptides P-2 and P-4 showed binding to erythrocytes in Cell-ELISA (Fig. 3A), we investigated their binding capacity to Band 3. Only peptide P-4 showed interaction with Band 3 protein in solid phase ELISA (Fig. 7D). The specificity of binding was confirmed by competition assay results in solid phase ELISA (Fig. 7E) and by SPR with KD value of 1.8 ± 0.65 × 10−9 m (Fig. 7F).
FIGURE 7.

Binding of PvTRAg38 fragments and synthetic peptides to Band 3. A, binding of PvTRAg38 fragments with purified Band 3 by solid phase ELISA. Each well of the plate was coated with 50 nm of Band 3 and then allowed to react with increasing concentrations (0–2 μm) of histidine-tagged PvTRAg38 or its fragments. The plates were developed with mouse anti-His6 monoclonal antibody as described in the text. Recombinant bacterial thioredoxin was taken as negative control. B, specificity of M-PvTRAg38 binding to Band 3 was determined by competition assay. Increasing concentrations of untagged PvTRAg38 was mixed with fixed concentration (0. 5 μm) of histidine-tagged M-PvTRAg38 and allowed to react with 50 nm immobilized Band 3 for 4 h at 37 °C. Bound recombinant histidine-tagged M-PvTRAg38 was detected with mouse anti-His6 monoclonal antibody as described in the text. Binding in the absence of untagged PvTRAg38 was taken as percentage control for rest of the concentrations. The data shown are the means ± S.D. of three independent experiments. C, SPR analysis of Band 3 interaction with M-PvTRAg38. Three different concentrations of M-PvTRAg38 (0.2, 0.8, and 1.4 μm) were injected at flow rate of 30 μl/min over the immobilized Band 3 on the cell of CM5 chip. Sensogram curves show dose-dependent response of M-PvTRAg38 binding with Band 3. D, binding of PvTRAg38 peptides to Band 3 by solid phase ELISA. The plate was coated with purified Band 3 (50 nm) protein and reacted with different concentrations (0–2 μm) of histidine-tagged PvTRAg38, P-2, or P-4. Plate was developed with mouse anti-His6 monoclonal antibody as described in the text. Histidine-tagged bacterial thioredoxin from D. desulfuricans was used as negative control. The data shown are the means ± S.D. of three independent experiments. E, specificity of binding of Band 3 to peptide P-4 by competition assay. Recombinant GST-PvTRAg38 (0.5 μm) was mixed with increasing concentrations (0–4 μm) of peptide P-4. Then this mixture was added to Band 3 (50 nm) coated ELISA plate and incubated for 4 h at 37 °C. The plate was then incubated with mouse anti-GST monoclonal antibody and HRP-conjugated secondary antibody as described in the text. Binding in the absence of respective untagged peptides was taken as percentage control for rest of the concentrations. F, SPR analysis of Band 3 interaction with P-4. Three different concentrations of P-4 (0.2, 0.6, and 1.2 μm) were injected at flow rate of 30 μl/min over the immobilized Band 3 on the cell of CM5 chip. Sensogram curves show dose-dependent response of P-4 binding with Band 3.
Three Ectodomains of Band 3 Bind to PvTRAg38 and Its Peptide P-4
We have cloned and expressed all the six extracellular loop regions of Band 3 as histidine-tagged fusion proteins in E. coli and evaluated them for their binding ability to PvTRAg38 and its peptide P-4. Band 3 fragment 1 (B3F1) covered 424–462 amino acid residues, fragment 2 (B3F2) covered 458–494 amino acid residues, fragment 3 (B3F3) covered 538–570 amino acid residues, fragment 4 (B3F4) covered 623–663 amino acid residues, fragment 5 (B3F5) covered 720–761 amino acid residues, and fragment 6 (B3F6) covered 807–860 amino acid residues of this protein. The results of solid phase ELISA showed that PvTRAg38 binds to B3F1, B3F3, and B3F6 of Band 3 in a dose-dependent manner (Fig. 8A). Other fragments of Band 3 and bacterial thioredoxin showed insignificant binding to PvTRAg38. The competition assay results of solid phase ELISA showed that the binding of Band 3 fragments with the PvTRAg38 was specific (Fig. 8B). Interaction of these three Band 3 fragments (B3F1, B3F3, and B3F) with PvTRAg38 was also evident from the rosetting experiment where these fragments competed with erythrocytes to bind to CHO-K1 cells expressing PvTRAg38 protein (Fig. 8C). The SPR analysis further confirmed that PvTRAg38 interacts with these three fragments of Band 3 (Fig. 8, D--F). The dissociation constants for PvTRAg38 interaction with B3F1, B3F3, and B3F6 were 2.11 ± 0.55 × 10−9 m, 9.2 ± 0.88 × 10−9 m, and 1.8 ± 0.74 × 10−9 m, respectively.
FIGURE 8.

Binding of Band 3 fragments with PvTRAg38. A, solid phase ELISA. The plate was coated with fixed concentration (0.5 μm) of untagged PvTRAg38 and reacted with increasing concentrations (0–2 μm) of histidine-tagged recombinant Band 3 fragments. The plate was developed with mouse anti-His6 monoclonal antibody as described in the text. Histidine-tagged bacterial thioredoxin from D. desulfuricans was used as negative control. B, specificity of binding of Band 3 fragments to PvTRAg38 by competition in solid phase ELISA. Increasing concentrations of untagged PvTRAg38 were mixed with a fixed concentration (0.5 μm) of histidine-tagged Band 3 fragments and allowed to react with immobilized GST-PvTRAg38 (50 nm) for 4 h at 37 °C. Bound recombinant histidine-tagged Band 3 fragments were detected with mouse anti-His6 monoclonal antibody as described in the text. Binding in the absence of untagged PvTRAg38 was taken as percentage control for rest of the concentrations. The mean ± S.D. value of absorbance from three experiments is plotted. C, specificity of binding of Band 3 fragments to PvTRAg38 by competition in rosetting assay. The RE4-PvTRAg38 transfected CHO-K1 cells were preincubated with different concentrations (0–10 μm) of Band 3 fragments and then allowed to interact with human erythrocytes. The results are expressed as relative binding to positive control (binding of untreated PvTRAg38 transfected cells with RBCs). The mean and ± standard deviation of three experiments are reported. D–F, SPR analysis of PvTRAg38 interaction with Band 3 fragments B3F1 (D), B3F3 (E), and B3F6 (F). These Band 3 fragments (B3F1, B3F3, and B3F6) were immobilized on the different cells of CM5 chip surface. Three different concentrations of PvTRAg38 (0.4, 0.8, and 1.3 μm) were injected at flow rate of 30 μl/min over the surface of immobilized B3F1, B3F3, and B3F6 in independent experiments. Sensogram curves show dose-dependent responses of PvTRAg38 binding with these Band 3 fragments.
Because we have shown above that peptide P-4 of PvTRAg38 binds to Band 3, we studied here the interaction of this peptide with Band 3 fragments by solid phase ELISA and SPR. The results of solid phase ELISA showed that P-4 was able to bind to all three extracellular loops (B3F1, B3F3, and B3F6) of Band 3 (Fig. 9A). This binding was specific as evident from the competition assay data (Fig. 9B). Interaction between P-4 and Band 3 fragments was further confirmed by the SPR results (Fig. 9, C–E). The dissociation constants for interaction of P-4 with B3F1, B3F3, and B3F6 were 9.8 ± 0.64 × 10−9 m, 8.36 ± 0.59 × 10−9 m, and 1.33 ± 0.66 × 10−8 m, respectively.
FIGURE 9.

Binding of PvTRAg38 peptide P-4 to Band 3 fragments. A, in solid phase ELISA, the plate was coated with (0.5 μm) untagged peptide P-4 and reacted with increasing concentrations (0–2 μm) of histidine-tagged Band 3 fragments (B3F1, B3F3, and B3F6). Plates were developed with mouse anti-His6 monoclonal antibody as described in the text. Histidine-tagged bacterial thioredoxin from D. desulfuricans was used as negative control. The mean ± S.D. value of absorbance from three experiments is plotted. B, specificity of binding of Band 3 fragments to peptide P-4 by competition assay. Increasing concentrations of untagged P-4 was mixed with a fixed concentration (0. 5 μm) of histidine-tagged Band 3 fragments and allowed to react with immobilized GST-PvTRAg38 (50 nm) for 4 h at 37 °C. Bound recombinant histidine-tagged Band 3 fragments were detected with mouse anti-His6 monoclonal antibody as described in the text. Binding in the absence of untagged P-4 was taken as percentage control for rest of the concentrations. The mean ± S.D. value of absorbance from three experiments is plotted. C–E, SPR analysis of peptide P-4 interaction with Band 3 fragments B3F1 (C), B3F3 (D), and B3F6 (E). These Band 3 fragments (B3F1, B3F3, and B3F6) were immobilized on different cells of CM5 chip. Peptide P-4 at three different concentrations (0.2, 0.4, and 0.6 μm) was injected at flow rate of 30 μl/min over the surface of immobilized B3F1, B3F3, and B3F6 in independent experiments. Sensogram curves show dose-dependent response of P-4 binding with Band 3 fragments.
PvTRAg38 Inhibits Parasite Growth in in Vitro P. falciparum Culture
To evaluate the role of PvTRAg38 and Band 3 interaction on parasite growth, we used the heterologous parasite P. falciparum culture system because P. vivax is difficult to culture. We used the P. falciparum model based on the fact that Band 3 acts as receptor for P. falciparum MSP 1 protein on merozoite surface and blockade of Band 3 on human erythrocytes by monoclonal antibodies had been shown to affect the parasite invasion (29, 30). Our results on P. falciparum (3D7) culture at 1% parasitemia treated with different concentrations of PvTRAg38 showed a parasite growth inhibition in a dose-dependent manner in comparison to culture treated with nonbinder PvTRAg53.7 (Fig. 10). The maximum growth inhibition was ∼35% at 20 μm of PvTRAg38. Similarly, the positive control PfTryThrA also showed the parasite growth inhibition. This inhibition suggests the sharing of common receptor Band 3 by P. vivax and P. falciparum merozoite molecules involved in invasion process.
FIGURE 10.

Inhibition of P. falciparum growth by PvTRAg38. Parasite culture at late schizont stage was incubated with PvTRAg38, PvTRAg53.7, PfTryThrA, and other controls. After 40 h, parasitemia was determined by ethidium bromide staining measured by flow cytometry for treated cultures as compared with the control. A, representative dot plots showing percent infection. Panel a, unstained erythrocytes; panel b, uninfected erythrocytes; panel c, infected erythrocytes; panel d, infected erythrocytes treated with PBS; panel e, infected erythrocytes treated with PvTRAg38; panel f, infected erythrocytes treated with PfTryThrA; panel g, infected erythrocytes treated with PvTRAg53.7. B, bar graph shows the percentage of parasite growth inhibition in a dose-dependent manner. The data shown are the means ± S.D. for two triplicate experiments.
Discussion
Earlier, we have described that 10 PvTRAg proteins, including PvTRAg38, bind to human erythrocytes, which were affected by natural antibodies produced during the P. vivax infection (9, 10). Binding of the PvTRAg38 to erythrocytes was also affected by chymotrypsin treatment, indicating that the receptor molecule must be sensitive to this enzyme (9). Here, we have identified this chymotrypsin-sensitive erythrocyte membrane protein, which binds to PvTRAg38. To identify this host protein, the pulldown assay was performed with PvTRAg38, where it captured four erythrocyte proteins: Band 3, Band 4.2, Spectrin, and Ankyrin (Fig. 5A). Because Band 3 is exposed at the erythrocyte surface and also can be cleaved by chymotrypsin, this host protein could act as receptor for parasite ligand PvTRAg38. The other three erythrocyte proteins could have been captured in pulldown because they show association with one another in cytoskeleton meshwork (26–28). This was confirmed by the results of pulldown assay performed on the NaOH-treated erythrocyte membrane, where only Band 3 was captured, whereas other proteins were removed by this treatment (Fig. 5B). Because PvTRAg38 interacts with two erythrocyte receptors (9), we were able to capture here only Band 3, which binds to PvTRAg38 through peptide P-4 domain and were unable to capture the other erythrocyte receptor, which is resistant to chymotrypsin treatment and binds to this parasite ligand through its P-2 domain. This could be because Band 3 is present in larger quantity in erythrocytes, whereas the other receptor may be present in a very low quantity and thusis not captured here in a pulldown assay and remains unidentified.
Band 3, which is defined here as potential erythrocyte receptor for PvTRAg38, is also known to interact with a number of known and unknown P. falciparum proteins and plays a crucial role in host cell invasion (29, 31–36). This is based on the fact that the monoclonal antibodies against Band 3 blocked the invasion of rhesus monkey erythrocytes by the Plasmodium knowlesi merozoites in a primate model (37). Similarly, the in vitro invasion of human erythrocytes by P. falciparum merozoites was also blocked by the monoclonal antibodies against extracellular epitopes of Band 3 (30). Furthermore, the erythrocytes from the ovalocytosis patients having genetically defective Band 3 protein were poor host cell for P. falciparum invasion and growth (38). Thus it is possible that similar host receptor and parasite ligand interaction between Band 3 and PvTRAg38 is taking place during the host cell invasion by the P. vivax merozoites. This is supported by the fact that its orthologous tryptophan-rich proteins of human malaria parasite P. falciparum are involved in host cell invasion (5). This interaction between PvTRAg38 and Band 3 may be involved in the initial reversible attachments of merozoites to the erythrocytes as in case of P. falciparum (MSP1-Band 3), or it may act in association with other receptors/proteins as an alternate pathway of red cell invasion by P. vivax. Binding of erythrocytes to the CHO-K1 cells expressing PvTRAg38 at its surface (Fig. 1) could be nearly mimicking the natural phenomenon where merozoite expressing PvTRAg38 interacts with erythrocytes. However, a question can arise that this erythrocyte binding activity of PvTRAg38 could be due to its glycosylated form because there are two potential N-linked glycosylation sites (at amino acid positions 109–111 and 159–161) in this exon 2 encoded sequence, which can be glycosylated in mammalian cells (CHO-K1) but not in parasite. We can rule out this possibility because the first potential glycosylation site falls in the N-PvTRAg38 fragment, which does not bind to erythrocytes, and the erythrocyte binding activity of M-PvTRAg38, which contains the other glycosylation site, is competed out by the E. coli expressed nonglycosylated M-PvTRAg38 form in rosetting assay (Fig. 2). Furthermore, the nonglycosylated M-PvTRAg38 and its synthetic peptides P-2 and P-4 were binding to erythrocytes in Cell-ELISA (Figs. 2B and 3B).
The molecular interaction between PvTRAg38 and Band 3 seems to have an important biological role for the parasite survival and growth. This is evident from the experimental results where PvTRAg38 interferes with the parasite growth in P. falciparum culture (Fig. 10). This may be because PvTRAg38 could be competing with the P. falciparum merozoites for Band 3 receptor on erythrocytes, and it is known that Band 3 blockade inhibits invasion of red cells by P. falciparum merozoites (30, 37). The question arises regarding which of the P. falciparum merozoite molecules are competing with PvTRAg38 in this assay. Because it is known that MSP 1 of P. falciparum merozoites recognize the same Band 3 on erythrocyte surface as PvTRAg38, it is possible that these two cross-species parasite proteins are competing with each other for the same host receptor molecule. However, competition of PvTRAg38with its homologues on P. falciparum merozoites such as PfTryThrA and PfMaTrA (merozoite-associated tryptophan-rich antigen) cannot be ruled out at this stage. Although Band 3 and PfMSP 1 interaction is well known, the same need to be investigated for these two P. falciparum tryptophan-rich homologues because their erythrocyte receptors are not yet known. PvTRAg38 shares 31.96% (58% homology) and 27.44% (52% homology) amino acid sequence identity with PfTryThrA and PfMaTrA of P. falciparum, respectively. PfTryThrA contains erythrocyte binding activity, which resides in the nonhomologous flanking regions, and peptides from this region were able to inhibit merozoite invasion in P. falciparum cultures (5). Role of PfMaTrA in merozoite invasion is not known (39). Further work is required to find the molecular interaction that takes place during merozoite invasion in P. vivax involving PvTRAg38 and its other host receptor, as well as its interaction with other parasite proteins in the process.
It will be interesting to find out whether P. vivax MSP 1 also binds to Band 3 via same extracellular region as PfMSP 1. It is known that PfMSP1 forms complex with RhopH3, as well as with PfMSP9, to interact with 5ABC (defined here as B3F5 fragment) and 6A of Band 3 (29, 31, 34). In that case, the cooperative binding of several parasite proteins (the PvTRAg38, MSP 1, MSP9, and maybe other additional proteins) to the same host erythrocyte receptor through its different exofacial regions may be providing the parasite an opportunity to tighten its interaction with the host cell. This phenomenon may indicate the complexity of molecular interactions needed for the successful invasion process (40). Thus multiple parasite ligands recognizing the same erythrocyte receptor, as well as the same parasite ligand recognizing more than one erythrocyte receptor, are an indication that the parasite must be using multiple pathways to invade the host cell (9, 10, 34). This could be a backup strategy for red cell invasion or the strategy to invade different erythrocyte types to widen its invasion capacity. In either case, the parasite has developed a well organized strategy to invade and establish its growth and development inside the host cell.
Molecular interactions between host and parasite are important events for the survival and multiplication strategy of the parasite within its host. The disruption of these molecular interactions might prove lethal to the parasite. This has generated a great deal of interest among workers so as to develop drug molecules or immunotherapeutics. Here, we have identified an erythrocyte receptor, which is utilized by P. vivax tryptophan-rich antigen expressed by the merozoites (11). This is indeed for the first time when a receptor for any of the tryptophan-rich protein of any Plasmodium species is being identified, although such parasite ligands on merozoites of human malaria parasite P. falciparum have been identified that are involved in red cell invasion (5). Further, we have defined here the binding domains of both the receptor (Band 3) and ligand (PvTRAg38) molecules to unravel the molecular mechanisms involved in receptor-ligand binding during host-parasite interaction. Nevertheless, further work is required so as to localize these PvTRAgs, using monoclonal antibodies because polyclonal antibodies cross-react, and to investigate whether these antibodies are able to affect the parasite growth and invasion phenomenon. These well defined sequences of host receptor and parasite ligand molecules and further work on the other erythrocyte receptors and parasite ligands might lead to the development of therapeutics against P. vivax malaria.
Author Contributions
Y. D. S. and M. S. A. designed the study and wrote the paper. M. S. A. performed cloning and expression of PvTRAg38 and PfTryThrA, erythrocyte binding assay by rosetting and flow cytometry, pulldown assay, purified Band 3, SPR, and solid phase ELISA. V. C. expressed and purified Band 3 fragments and performed solid phase ELISA and SPR. M. Z. performed erythrocyte binding by flow cytometry and parasite growth inhibition assay. R. K. T. performed cloning of PvTRAg38 fragments, erythrocyte binding assay by ELISA, and flow cytometry. S. R. performed SPR. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
Plasmid pRE4 was from Gary Cohn and Roselyn Eisenberg, and pHVDR22 was provided by Chetan Chitnis. We thank S. S. Chauhan, Pawan Malhotra, and Sharmishtha Day for help and discussion and Shalini Narang for preparing the manuscript. We acknowledge the Protein Purification Facility of the Biophysics Department and the Biotechnology Information System of the Biotechnology Department.
This work was supported by Department of Biotechnology Grant BT/PR9800/MED/29/44/2007, by Indian Council for Medical Research Grant 61/4/2012-BMS; by a DST-INSA INSPIRE faculty fellowship (to S. R.) from the Department of Science and Technology; by senior research fellowships from the Council of Scientific and Industrial Research (to M. S. A. and V. C.), and by funds from the Department of Biotechnology (to M. Z. and R. K. T.). The authors declare that they have no conflicts of interest with the contents of this article.
- PvTRAg
- P. vivax tryptophan-rich antigen.
References
- 1. Naing C., Whittaker M. A., Nyunt Wai V., Mak J. W. (2014) Is Plasmodium vivax malaria a severe malaria?: a systematic review and meta-analysis. PLoS Negl. Trop. Dis. 8, e3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cowman A. F., Berry D., Baum J. (2012) The cellular and molecular basis for malaria parasite invasion of the human red blood cell. J. Cell Biol. 198, 961–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miller L. H., Mason S. J., Clyde D. F., McGinniss M. H. (1976) The resistance factor to Plasmodium vivax in blacks: the Duffy-blood-group genotype, FyFy. N. Engl. J. Med. 295, 302–304 [DOI] [PubMed] [Google Scholar]
- 4. Ménard D., Barnadas C., Bouchier C., Henry-Halldin C., Gray L. R., Ratsimbasoa A., Thonier V., Carod J. F., Domarle O., Colin Y., Bertrand O., Picot J., King C. L., Grimberg B. T., Mercereau-Puijalon O., Zimmerman P. A. (2010) Plasmodium vivax clinical malaria is commonly observed in Duffy-negative Malagasy people. Proc. Natl. Acad. Sci. U.S.A. 107, 5967–5971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Curtidor H., Ocampo M., Rodríguez L. E., López R., García J. E., Valbuena J., Vera R., Puentes A., Leiton J., Cortes L. J., López Y., Patarroyo M. A., Patarroyo M. E. (2006) Plasmodium falciparum TryThrA antigen synthetic peptides block in vitro merozoite invasion to erythrocytes. Biochem. Biophys. Res. Commun. 339, 888–896 [DOI] [PubMed] [Google Scholar]
- 6. Burns J. M., Jr., Adeeku E. K., Dunn P. D. (1999) Protective immunization with a novel membrane protein of Plasmodium yoelii-infected erythrocytes. Infect. Immun. 67, 675–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burns J. M., Jr., Dunn P. D., Russo D. M. (1997) Protective immunity against Plasmodium yoelii malaria induced by immunization with particulate blood-stage antigens. Infect. Immun. 65, 3138–3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carlton J. M., Adams J. H., Silva J. C., Bidwell S. L., Lorenzi H., Caler E., Crabtree J., Angiuoli S. V., Merino E. F., Amedeo P., Cheng Q., Coulson R. M., Crabb B. S., Del Portillo H. A., Essien K., Feldblyum T. V., Fernandez-Becerra C., Gilson P. R., Gueye A. H., Guo X., Kang'a S., Kooij T. W., Korsinczky M., Meyer E. V., Nene V., Paulsen I., White O., Ralph S. A., Ren Q., Sargeant T. J., Salzberg S. L., Stoeckert C. J., Sullivan S. A., Yamamoto M. M., Hoffman S. L., Wortman J. R., Gardner M. J., Galinski M. R., Barnwell J. W., Fraser-Liggett C. M. (2008) Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature 455, 757–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tyagi R. K., Sharma Y. D. (2012) Erythrocyte binding activity displayed by a selective group of Plasmodium vivax tryptophan rich antigens is inhibited by patients' antibodies. PLoS One 7, e50754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zeeshan M., Tyagi R. K., Tyagi K., Alam M. S., Sharma Y. D. (2015) Host-parasite interaction: selective numbers of ‘Pv-fam-a’ family proteins of Plasmodium vivax bind to a restricted number of human erythrocyte receptors. J. Infect. Dis. 211, 1111–1120 [DOI] [PubMed] [Google Scholar]
- 11. Bozdech Z., Mok S., Hu G., Imwong M., Jaidee A., Russell B., Ginsburg H., Nosten F., Day N. P., White N. J., Carlton J. M., Preiser P. R. (2008) The transcriptome of Plasmodium vivax reveals divergence and diversity of transcriptional regulation in malaria parasites. Proc. Natl. Acad. Sci. U.S.A. 105, 16290–16295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sarin R., Sharma Y. D. (2006) Thioredoxin system in obligate anaerobe Desulfovibrio desulfuricans: identification and characterization of a novel thioredoxin 2. Gene 376, 107–115 [DOI] [PubMed] [Google Scholar]
- 13. Zeeshan M., Bora H., Sharma Y. D. (2013) Presence of memory T cells and naturally acquired antibodies in Plasmodium vivax malaria-exposed individuals against a group of tryptophan-rich antigens with conserved sequences. J. Infect. Dis. 207, 175–185 [DOI] [PubMed] [Google Scholar]
- 14. Bora H., Garg S., Sen P., Kumar D., Kaur P., Khan R. H., Sharma Y. D. (2011) Plasmodium vivax tryptophan-rich antigen PvTRAg33.5 contains alpha helical structure and multidomain architecture. PLoS One 6, e16294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fujinaga J., Tang X. B., Casey J. R. (1999) Topology of the membrane domain of human erythrocyte anion exchange protein, AE1. J. Biol. Chem. 274, 6626–6633 [DOI] [PubMed] [Google Scholar]
- 16. Cohen G. H., Wilcox W. C., Sodora D. L., Long D., Levin J. Z., Eisenberg R. J. (1988) Expression of Herpes simplex virus type 1 glycoprotein D deletion mutants in mammalian cells. J. Virol. 62, 1932–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koganei A., Tsuchiya T., Samura K., Nishikibe M. (2007) Use of whole sheep red blood cells in ELISA to assess immunosuppression in vivo. J. Immunotoxicol. 4, 77–82 [DOI] [PubMed] [Google Scholar]
- 18. Chitnis C. E., Miller L. H. (1994) Identification of the erythrocyte binding domains of Plasmodium vivax and Plasmodium knowlesi proteins involved in erythrocyte invasion. J. Exp. Med. 180, 497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tran T. M., Moreno A., Yazdani S. S., Chitnis C. E., Barnwell J. W., Galinski M. R. (2005) Detection of a Plasmodium vivax erythrocyte binding protein by flow cytometry. Cytometry A 63, 59–66 [DOI] [PubMed] [Google Scholar]
- 20. Dodge J. T., Mitchell C., Hanahan D. J. (1963) The preparation and chemical characteristics of hemoglobin-free ghosts of human erythrocytes. Arch. Biochem. Biophys. 100, 119–130 [DOI] [PubMed] [Google Scholar]
- 21. Yajima A., Urano-Tashiro Y., Shimazu K., Takashima E., Takahashi Y., Konishi K. (2008) Hsa, an adhesin of Streptococcus gordonii DL1, binds to alpha2–3-linked sialic acid on glycophorin A of the erythrocyte membrane. Microbiol. Immunol. 52, 69–77 [DOI] [PubMed] [Google Scholar]
- 22. Casey J. R., Lieberman D. M., Reithmeier R. A. (1989) Purification and characterization of Band 3 protein. Methods Enzymol. 173, 494–512 [DOI] [PubMed] [Google Scholar]
- 23. Sorette M. P., Galili U., Clark M. R. (1991) Comparison of serum anti-Band 3 and anti-Gal antibody binding to density-separated human red blood cells. Blood 77, 628–636 [PubMed] [Google Scholar]
- 24. Bhalla K., Chugh M., Mehrotra S., Rathore S., Tousif S., Prakash Dwivedi V., Prakash P., Kumar Samuchiwal S., Kumar S., Kumar Singh D., Ghanwat S., Kumar D., Das G., Mohmmed A., Malhotra P., Ranganathan A. (2015) Host ICAMs play a role in cell invasion by Mycobacterium tuberculosis and Plasmodium falciparum. Nat. Commun. 6, 6049. [DOI] [PubMed] [Google Scholar]
- 25. Persson K. E., Lee C. T., Marsh K., Beeson J. G. (2006) Development and optimization of high-throughput methods to measure Plasmodium falciparum-specific growth inhibitory antibodies. J. Clin. Microbiol. 44, 1665–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bennett V., Stenbuck P. J. (1980) Human erythrocyte ankyrin. Purification and properties. J. Biol. Chem. 255, 2540–2548 [PubMed] [Google Scholar]
- 27. Ipsaro J. J., Huang L., Gutierrez L., MacDonald R. I. (2008) Molecular epitopes of the ankyrin-spectrin interaction. Biochemistry 47, 7452–7464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Korsgren C., Cohen C. M. (1986) Purification and properties of human erythrocyte band 4.2. Association with the cytoplasmic domain of Band 3. J. Biol. Chem. 261, 5536–5543 [PubMed] [Google Scholar]
- 29. Goel V. K., Li X., Chen H., Liu S. C., Chishti A. H., Oh S. S. (2003) Band 3 is a host receptor binding merozoite surface protein 1 during the Plasmodium falciparum invasion of erythrocytes. Proc. Natl. Acad. Sci. U.S.A. 100, 5164–5169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Clough B., Paulitschke M., Nash G. B., Bayley P. M., Anstee D. J., Wilson R. J., Pasvol G., Gratzer W. B. (1995) Mechanism of regulation of malarial invasion by extraerythrocytic ligands. Mol. Biochem. Parasitol. 69, 19–27 [DOI] [PubMed] [Google Scholar]
- 31. Baldwin M., Yamodo I., Ranjan R., Li X., Mines G., Marinkovic M., Hanada T., Oh S. S., Chishti A. H. (2014) Human erythrocyte Band 3 functions as a receptor for the sialic acid-independent invasion of Plasmodium falciparum: role of the RhopH3-MSP1 complex. Biochim. Biophys. Acta 1843, 2855–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kushwaha A., Perween A., Mukund S., Majumdar S., Bhardwaj D., Chowdhury N. R., Chauhan V. S. (2002) Amino terminus of Plasmodium falciparum acidic basic repeat antigen interacts with the erythrocyte membrane through Band 3 protein. Mol. Biochem. Parasitol. 122, 45–54 [DOI] [PubMed] [Google Scholar]
- 33. Li X., Chen H., Oh S. S., Chishti A. H. (2008) A Presenilin-like protease associated with Plasmodium falciparum micronemes is involved in erythrocyte invasion. Mol. Biochem. Parasitol. 158, 22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li X., Chen H., Oo T. H., Daly T. M., Bergman L. W., Liu S. C., Chishti A. H., Oh S. S. (2004) A co-ligand complex anchors Plasmodium falciparum merozoites to the erythrocyte invasion receptor Band 3. J. Biol. Chem. 279, 5765–5771 [DOI] [PubMed] [Google Scholar]
- 35. Okoye V. C., Bennett V. (1985) Plasmodium falciparum malaria: Band 3 as a possible receptor during invasion of human erythrocytes. Science 227, 169–171 [DOI] [PubMed] [Google Scholar]
- 36. Roggwiller E., Bétoulle M. E., Blisnick T., Braun Breton C. (1996) A role for erythrocyte Band 3 degradation by the parasite gp76 serine protease in the formation of the parasitophorous vacuole during invasion of erythrocytes by Plasmodium falciparum. Mol. Biochem. Parasitol. 82, 13–24 [DOI] [PubMed] [Google Scholar]
- 37. Miller L. H., Hudson D., Rener J., Taylor D., Hadley T. J., Zilberstein D. (1983) A monoclonal antibody to rhesus erythrocyte Band 3 inhibits invasion by malaria (Plasmodium knowlesi) merozoites. J. Clin. Invest. 72, 1357–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jarolim P., Palek J., Amato D., Hassan K., Sapak P., Nurse G. T., Rubin H. L., Zhai S., Sahr K. E., Liu S. C. (1991) Deletion in erythrocyte Band 3 gene in malaria-resistant Southeast Asian ovalocytosis. Proc. Natl. Acad. Sci. U.S.A. 88, 11022–11026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ntumngia F. B., Bouyou-Akotet M. K., Uhlemann A. C., Mordmüller B., Kremsner P. G., Kun J. F. (2004) Characterisation of a tryptophan-rich Plasmodium falciparum antigen associated with merozoites. Mol. Biochem. Parasitol. 137, 349–353 [DOI] [PubMed] [Google Scholar]
- 40. Sahar T., Reddy K. S., Bharadwaj M., Pandey A. K., Singh S., Chitnis C. E., Gaur D. (2011) Plasmodium falciparum reticulocyte binding-like homologue protein 2 (PfRH2) is a key adhesive molecule involved in erythrocyte invasion. PLoS One 6, e17102. [DOI] [PMC free article] [PubMed] [Google Scholar]