

Background: Topoisomerase II-disabling antibacterials and anticancer drugs exist but not antimalarials.
Results: Engineered Plasmodium falciparum topoisomerase II (PfTopoII) led to a functional enzyme and to early inhibitors with antimalarial properties in cell-based assays.
Conclusion: Pure, stable PfTopoII is now ready for advanced HTS and lead optimization studies.
Significance: This biochemical platform should help accelerate antimalarial drug discovery.
Keywords: DNA enzyme, drug discovery, fluorescence, plasmodium, translation, cell-free, wheat germ
Abstract
Historically, type II topoisomerases have yielded clinically useful drugs for the treatment of bacterial infections and cancer, but the corresponding enzymes from malaria parasites remain understudied. This is due to the general challenges of producing malaria proteins in functional forms in heterologous expression systems. Here, we express full-length Plasmodium falciparum topoisomerase II (PfTopoII) in a wheat germ cell-free transcription-translation system. Functional activity of soluble PfTopoII from the translation lysates was confirmed through both a plasmid relaxation and a DNA decatenation activity that was dependent on magnesium and ATP. To facilitate future drug discovery, a convenient and sensitive fluorescence assay was established to follow DNA decatenation, and a stable, truncated PfTopoII was engineered for high level enzyme production. PfTopoII was purified using a DNA affinity column. Existing TopoII inhibitors previously developed for other non-malaria indications inhibited PfTopoII, as well as malaria parasites in culture at submicromolar concentrations. Even before optimization, inhibitors of bacterial gyrase, GSK299423, ciprofloxacin, and etoposide exhibited 15-, 57-, and 3-fold selectivity for the malarial enzyme over human TopoII. Finally, it was possible to use the purified PfTopoII to dissect the different modes by which these varying classes of TopoII inhibitors could trap partially processed DNA. The present biochemical advancements will allow high throughput chemical screening of compound libraries and lead optimization to develop new lines of antimalarials.
Introduction
With 300–500 million clinical cases, each year, malaria causes more than 0.5 million deaths worldwide (1, 2). Among the five known human malaria parasites, Plasmodium falciparum and Plasmodium vivax are the major agents of pathology. Repeated emergence of drug-resistant parasites demands a continual search for new targets and new antimalarials (3–7). Malaria parasites proliferate through rapid asexual cell division in humans. Starting with an estimated 10–100 sporozoites, each incoming parasite expands exponentially to about 105 cells in the liver and 1013 in red blood cells (8). All such parasite transformations and growth involve continual but varying transcription and require smooth replication of the three parasite genomes (nuclear, mitochondrial, and apicoplast).
In all living cells, DNA topoisomerases play a key role in proper DNA transcription, DNA replication, DNA repair, and overall cell division (9–15). These enzymes are responsible for relieving DNA topological constraints that are generated during cellular activity on genomes. The obligatory requirement of topoisomerases for cell function has been exploited to treat bacterial infections and cancer (10–14). The fluoroquinolone class of inhibitors, like ciprofloxacin, target bacterial DNA gyrase and topoisomerase IV (10, 11). Newer piperidinylalkylquinolines, like GSK299423, target bacterial gyrases (11, 14). Human topoisomerase IIs are targeted by etoposide and teniposide (derivatives of podophyllotoxins) and doxorubicin and daunorubicin (derivatives of anthracyclines) (12, 13).
Topoisomerases are broadly classified as type I or as type II enzymes based on their mode of action on DNA (9, 15). Type I topoisomerases are monomeric, metal ion-dependent, and ATP-independent. They cleave a single-strand of DNA and, by swivel mechanisms, help to relax both negative and positive DNA supercoils. Type II DNA topoisomerases are dimeric or heterotetrameric, ATP and metal ion-dependent. They bind DNA and cleave both strands to create a gate through which a neighboring duplex DNA can pass. Thus, type II enzymes can help to relax both negative and positive supercoils and can also decatenate and unknot tangled DNA (9, 15).
The malaria parasite nuclear genome codes for both type I and type II topoisomerases. P. falciparum has two putative type I enzymes (topoisomerase I and III) and three putative type II enzymes (topoisomerases II and VI and DNA gyrase) (from the PlasmoDB: Plasmodium Genomics Resource and Ref. 17). The functional unit of each topoisomerase I, II, and III is encoded by single gene, and each of topoisomerase VI and DNA gyrase is encoded by two gene products (from the PlasmoDB: Plasmodium Genomics Resource and Ref. 17). Even a decade after sequencing of the malarial genome (17), biochemical functions for the majority of malarial topoisomerase are unrealized. This is directly related to the previous unavailability of dependable heterologous systems to express functional malarial enzymes (18, 19). Except for the malarial DNA gyrase B (20), a subunit of gyrase, and topoisomerase I (21), the remaining five malarial topoisomerase genes have not been expressed successfully in heterologous expression systems. Further, gel-based assays that are commonly used to study topoisomerase inhibitors are tedious and not quantitative. Thus, in addition to obtaining pure, active enzyme, development of high throughput fluorescent assays for topoisomerases would greatly facilitate target-based drug development against malaria parasites (22, 23).
Recently, we demonstrated the broad utility of a wheat cell-free protein expression system to successfully express several malarial enzymes in functional form (24, 25). When malaria gene codons are altered for expression in the wheat system, one sees improvement in the quality of functional protein and also increased expression. In this report, we express milligram quantities of functional P. falciparum topoisomerase II, using a wheat cell-free system and subsequently purifying the stable active protein. A simple, sensitive fluorescent assay for topoisomerase II was also established and, based on early work with existing topoisomerase II inhibitors against other pathogens, we demonstrate that malarial topoisomerase II offers an opportunity to identify and ultimately optimize species-specific malarial topoisomerase inhibitors.
Experimental Procedures
Bioinformatics
Topoisomerase II proteins from P. falciparum (PlasmoDB ID PF14_0316; new PlasmoDB ID PF3d7_1433500), P. vivax (PlasmoDB ID PVX_084855), Homo sapiens (TopoIIA: GenBankTM accession no. AAC77388.1 and TopoIIB: GenBankTM accession no. NM_001068.3), and Saccharomyces cerevisiae (GenBankTM accession no. AAB36610.1) were aligned by ClustalW method on Genius software (Biomatters, Ltd., Auckland, New Zealand). A model structure of P. falciparum topoisomerase II (PfTopoII)2 was built based on the available crystal structure of human and yeast topoisomerase IIs using Modeler and Rosetta tools (26, 27).
Subcloning of PfTopoII Gene into Cell-free Expression Vector
Codons of the PfTopoII gene (PlasmoDB ID PF3d7_1433500) were optimized for wheat expression system and chemically synthesized with desired restriction sites on each end of the gene (Life Technologies, Inc.). The PfTopoII synthetic gene and wheat cell-free plasmids were digested with restriction enzymes and purified through gel extraction (Qiagen). Ligated DNA samples were transformed into E. coli DH5α chemically competent cells (Life Technologies) and grown overnight on ampicillin-containing agar plates at 37 °C. Full-length gene insertion into cell-free expression plasmid was verified by colony PCR (25). The active site mutant (Y829A) of PfTopoII (PfTopoII-M) was constructed using the following primers: P1, 5′-AAGGACGCCTCCGCTGCGCGGGCTATCTTTACCAAGCTCGCCTCCAGC-3′ and P2, 5′-GCTGGAGGCGAGCTTGGTAAAGATAGCCCGCGCAGCGGAGGCGTCCTT-3′ and a QuikChange mutagenesis kit (Agilent Technologies, Santa Clara, CA). The underlined bases in the primer sequence represent the mutagenic site. Various truncated forms of PfTopoII gene were also constructed based on domain assignments (see Fig. 4A) using primers listed in Table 1. Plasmids were isolated using plasmid isolation kits (Qiagen), and their quality was checked and confirmed by DNA sequencing. These isolated plasmids were used for cell-free transcription and translation. A GFP-expressing plasmid (24, 25) was used as a positive control in wheat germ expression studies.
FIGURE 4.

Molecular dissection of full-length PfTopoII to isolate a pure, stable functional PfTopoII construct with full ATP and Mg2+-dependent DNA decatenation activity. A, five new combinations of the three predicted PfTopoII domains were constructed (individually and in pairs) and tested for full topoisomerase functions. B, an autoradiogram demonstrating soluble expression of single-sized, radiolabeled truncated derivatives of PfTopoII. Solubility of the expressed proteins was confirmed by showing good representation in the soluble (S) fractions compared with the total (T) fraction in the wheat germ expression system. C, demonstration of decatenation activity in the PfTopoII-ΔCTD construct carrying NAD-CRD but lacking CTD. The decatenation function of PfTopoII-ΔCTD domain was stimulated by 50–100 mm NaCl, slightly less than that required for full-length PfTopoII (100–150 mm). D, demonstration of purity of PfTopoII-ΔCTD construct by SDS-PAGE, dependent DNA affinity chromatography (see “Experimental Procedures”).
TABLE 1.
Primers used to construct various truncated PfTopoIIs
| Constructs (amino acid positions) | Nucleotide positions | Sequence (5′ → 3′)a |
|---|---|---|
| NAD (1–474) | 1–1422 | Forward: ATACCTCGAGATGGCCAAGAACAAGACCATCGAG |
| Reverse: GATACGGCCGTCATCAGATGATGCGCTCCCGGGC | ||
| CRD (469–1212) | 1405–3636 | Forward: ATACCTCGAGATGGCCCGGGAGCGCATCATC |
| Reverse: GTATCGGCCGTCATCAGTTGGACTCCTCGCGGTTGGA | ||
| CTD (1206–1472) | 3616–4416 | Forward: ATACCTCGAGATGTCCAACCGCGAGGAGTCCAAC |
| Reverse: ATAGCGGCCGTCATCAGATGTTGTAGCT | ||
| NAD-CRD (1–1212) | 1–3636 | Forward: ATACCTCGAGATGGCCAAGAACAAGACCATCGAG |
| Reverse: GTATCGGCCGTCATCAGTTGGACTCCTCGCGGTTGGA | ||
| CRD-CTD (469–1472) | 1405–4416 | Forward: ATACCTCGAGATGGCCAAGAACAAGACCATCGAG |
| Reverse: ATAGCGGCCGTCATCAGATGTTGTAGCT |
a Restriction sites are shown in italics, and start and stop sites are underlined.
Wheat Germ Cell-free Transcription and Translation
Wheat germ lysate, an essential component of the wheat cell-free protein expression system, was prepared in-house (25). Cell-free transcription, translation, and radiolabeling of proteins by a batch method and translation by a dialysis method were described earlier (25). Before purifying PfTopoII-ΔCTD (construct IV as shown in Fig. 4) on a double-stranded DNA-cellulose column, fresh protocols for scaled up cell-free transcription and translations were established (see below).
A typical scaled up transcription reaction was carried out in 40 ml with 3 mg of PfTopoII-ΔCTD gene-carrying plasmid, 80 mm HEPES-KOH, pH 7.8, 16 mm magnesium acetate, 2 mm spermidine, 50 mm β-mercaptoethanol, 1 kilounits of ribonuclease inhibitor (New England Biolabs), 30 kilounits of SP6 RNA polymerase (New England Biolabs, Ipswich, MA), and 3 mm each of GTP, ATP, CTP, and UTP. The transcription mixture was incubated at 37 °C for 4 h. Turbidity appears during the transcription process because of pyrophosphate released from nucleotide triphosphates and β-mercaptoethanol from the transcription mixture. At the end of transcription, such a precipitate was removed by centrifuging at 8,000 × g for 2 min, and the supernatant was collected. To eliminate the PfTopoII-ΔCTD coding plasmid from mRNA, the supernatant was treated with 1000 units of TURBO DNase and 2 ml of 10× reaction buffer (Life Technologies, Inc.) at 37 °C for 1 h. This DNase treatment was to digest the internal DNA transcription templates that could compete with DNA-cellulose for binding to PfTopoII-ΔCTD. After the treatment, DNase from the PfTopoII-ΔCTD mRNA was denatured by phenol chloroform extraction, and the mRNA was recovered in the aqueous phase on Phase Lock Gel (5 Prime, Inc., Gaithersburg, MD). The aqueous phase contained PfTopoII-ΔCTD DNA-free mRNA, and this was ethanol-precipitated, washed with 70% ethanol, and solubilized in 15 ml of autoclaved distilled water.
Cell-free translation of purified PfTopoII-ΔCTD mRNA was carried out using a dialysis reaction method (25). Starting with 15 ml of PfTopoII-ΔCTD, 40 mg of mRNA, 1600 units of wheat germ lysate (280 mg of protein), 8 mg of creatine kinase, 1000 units of ribonuclease inhibitor, and 8 ml of 5× protein expression buffer (1- protein expression buffer: 30 mm HEPES-KOH, pH 7.8, 100 mm potassium acetate, 2.7 mm magnesium acetate, 5 mm DTT, 0.4 mm spermidine, 0.3 mm each of 20 amino acids, 1.2 mm ATP, 0.25 mm GTP, 16 mm creatine phosphate), the volume was adjusted to 40 ml with autoclaved distilled water. Each 20-ml portion of translation reaction mixture was placed in a 2.5 × 75-cm cellulose membrane tube (12,000–14,000 molecular weight cutoff; Spectrum Laboratories, Inc., Rancho Dominguez, CA). The dialysis tubes were sealed, and the reaction mixture was distributed evenly along the length of the dialysis tube. It was then immersed in 200 ml of 1× protein expression buffer and incubated at 26 °C for 36 h. At the end, translated samples were spun at 20,000 × g for 15 min at 4 °C, and the soluble fraction was used for enzyme purification.
Gel-based Topoisomerase II Assay
The catalytic function of cell-free translated full-length PfTopoII and its truncated variations were tested based on eukaryotic type II topoisomerase protocols (28). A typical 40-μl reaction contained topoisomerase II assay buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 10 mm MgCl2, 5 mm DTT, and 2 mm ATP), 180 ng of kinetoplast catenated DNA (kDNA) from the insect parasite Crithidia fasciculate (Topogen, Inc., Port Orange, FL), or 180 ng of supercoiled pUC18 plasmid and appropriate quantities of PfTopoII, PfTopoII-M, or GFP translated lysates. At the end of 30 min of incubation at 26 °C, the reactions were stopped by adding 5 μl of 0.25 m EDTA. Topoisomerase II-catalyzed reaction products were resolved from their reaction substrates on 1% agarose gel in TAE buffer (40 mm Tris acetate with 1 mm EDTA at pH 8.3) and visualized by ethidium bromide. The supercoiled pUC18 plasmid for relaxation assay and as a marker was prepared using Qiagen plasmid isolation kits followed by dissolving plasmid DNA in Tris-EDTA buffer (10 mm Tris-HCl, pH 8.0, and 1 mm EDTA) and stored at 4 °C. The relaxed plasmid marker was prepared in 0.5 ml of relaxation buffer (50 mm Tris-HCl, pH 7.5, 100 mm NaCl, and 1 mm DTT) containing 100 μg of supercoiled pUC18 plasmid and 200 units of vaccinia virus topoisomerase I (Epicenter, Madison, WI) followed by incubation at 37 °C for 1 h. Topoisomerase I from the reaction mixture was denatured by phenol chloroform extraction, and the relaxed plasmid was recovered in aqueous phase on the phase lock gel. The relaxed plasmid in the aqueous phase was ethanol-precipitated, dissolved in Tris-EDTA buffer, and stored at 4 °C.
Fluorescence Assay for Topoisomerase II
The quick and convenient fluorescence decatenation assay is based on selective precipitation of large starting kDNA networks by centrifugation (34), followed by fluorescent quantification of released decatenated DNA products in supernatant solutions using SYBR Green I (Life Technologies, Inc.). The topoisomerase II assays were set up as described for the gel-based decatenation assay (see above). After stopping the reactions with EDTA, samples were spun at 13,000 × g for 10 min to precipitate unreacted kDNA. Supernatants of 30 μl were mixed with 60 μl of 13,000-fold, TAE-diluted SYBR Green I and fluorescence was measured on a multi-mode microplate reader, Synarge 4 (BioTek Instruments, Inc., Winooski, VT). Decatenated DNA products from the supernatants were quantified using a DNA calibration curve. In anticipation of using DMSO with inhibitors, various concentrations (v/v) of DMSO were added at the start of decatenation assay to follow any changes in activity. Direct effects of changes in DNA fluorescence caused by solvent changes were assessed separately.
Purification of Cell-free Expressed PfTopoII-ΔCTD
Wheat cell-free translated untagged PfTopoII-ΔCTD was purified on dsDNA-cellulose column. To a 40-ml supernatant of PfTopoII-ΔCTD translated lysate, 200 ml of binding buffer (50 mm Tris-HCl pH 8.0, 100 mm NaCl, 1 mm EDTA and 5 mm DTT) and 300 mg of dsDNA cellulose (Sigma-Aldrich) were added and magnetically stirred for 3 h. Sample was then loaded onto a column and washed with 50 ml of wash buffer (50 mm Tris-HCl, pH 8.0, 125 mm NaCl, 1 mm EDTA, and 5 mm DTT). DNA-bound PfTopoII-ΔCTD was eluted in 15 ml of high salt buffer (50 mm Tris-HCl, pH 8.0, 200 mm NaCl, 1 mm EDTA, and 5 mm DTT). Eluted samples were concentrated and further purified on an AKTA system connected to a Sephadex G-200 column (GE Healthcare) equilibrated with high salt buffer. Column-eluted PfTopoII-CTD-containing samples were pooled, concentrated, and equilibrated with binding buffer containing 10 mm MgCl2 and 2 mm ATP. Concentrated PfTopoII-ΔCTD protein was quantified using the Bradford method (29) (Bio-Rad). From a 40-ml translation, 0.3 mg of pure PfTopoII-ΔCTD protein was collected with a specific activity of 100 mg of decatenated DNA h−1 mg−1.
Primary Structure of the Purified PfTopoII-ΔCTD
Enzyme eluted from the Sephadex G-200 column above was analyzedby mass spectrophotometry to validate its makeup. Purified PfTopoII-ΔCTD (100 μg) was dissolved in a denaturation buffer (8 m urea, 0.1 m ammonium bicarbonate, pH 8.0, and 12 mm DTT). After incubation at 37 °C for 30 min, iodoacetamide was added to 40 mm and incubated at room temperature for 1 h. The denatured protein was then diluted 10 times with 0.1 m ammonium bicarbonate, followed by overnight digestion at 37 °C with 1 μg of trypsin (Promega, Madison, WI). Proteolysis was stopped by adding formic acid to 0.1%. The peptides were then analyzed with Finnigan LTQ ion trap mass spectrometer ((Thermo Fisher Scientific) with Agilent 1100 capillary liquid chromatography inlet (Santa Clara, CA). Data were analyzed with Proteome Discoverer 1.4 from Thermo Fisher Scientific.
Synthesis of GSK299423
GSK299423 synthesis was carried out using three key intermediates (14). Oxathiolo-pyridine-carbaldehyde was derived from commercially available kojic acid (Sigma-Aldrich), and cyano-quinoline-piperidine amine was synthesized from 4-ethenyl-6-(methyloxy)-3-quinolinecarbonitrile (Sigma-Aldrich). Finally, the target GSK299423 was derived from reductive amination of the above aldehyde and the amine provided, and its structure was confirmed by 1H NMR and electrospray ionization-MS.
Inhibitor Binding Studies
Select type II topoisomerase inhibitors were tested on PfTopoII, PfTopoII-ΔCTD, and human TopoIIα using the newly established fluorescence assay (see above). Inhibitory activities of test compounds on PfTopoII-ΔCTD were tested in the presence of 100 mm NaCl in the assay buffer. For full-length PfTopoII and human TopoIIα assays, NaCl concentration was set at 150 mm. Various concentrations of etoposide (Sigma-Aldrich), ciprofloxacin (Enzo Life Sciences, Inc., Farmingdale, NY), and GSK299423 were prepared in 60% DMSO. Aliquots (2 μl) of concentrated inhibitors in 60% DMSO were added to the decatenation reaction mixture, and the assays were initiated by adding 0.5 μg of PfTopoII, or 1 unit of human TopoIIα (Topogen, Inc., Port Orange, FL). At the end of 30 min of incubation at 26 °C, reactions were stopped by adding EDTA, and fluorescence of decatenated DNA was measured as described (see above). Half-maximal inhibitory concentrations (IC50) for all inhibitors were calculated on GraphPad Prism (GraphPad Software, Inc., La Jolla, CA).
DNA Cleavage Assay
Plasmid DNA cleavage assays were carried out as reported (14) with a few modifications. Topoisomerase II-catalyzed plasmid relaxation assays used solvents and buffers described above for the gel-based topoisomerase II assays. A total volume of 20 μl, with various quantities of inhibitors in DMSO (or plain 3% DMSO), was added to 30 ng of purified PfTopoII-ΔCTD. After 30 min at 26 °C, reactions were stopped by adding SDS to 0.2% followed by proteinase K (Life Technologies, Inc.) treatment for 30 min at 37 °C. Samples were resolved overnight by 1% agarose gel electrophoresis in TAE buffer, at 1 V/cm and visualized using ethidium bromide at 0.1%.
Testing of Inhibitors on Malarial Parasites
Inhibition of proliferation of P. falciparum clones, 3D7, Dd2, and HB3 (3, 4) or mouse L1210 cells (22) by select type II topoisomerase inhibitors was measured by following the uptake of radioactive hypoxanthine (30).
Results
Bioinformatics Analysis of PF3d7_1433500
A putative 4419-base pair DNA sequence of P. falciparum DNA topoisomerase II (PfTopoII) was identified on PlasmoDB (PF3d7_1433500; old ID PF14_0316). The predicted 1472-amino acid protein sequence of PfTopoII was aligned to topoisomerase IIs of S. cerevisiae (ScTopoII) and H. sapiens (HsTopoIIα and HsTopoIIβ) (Fig. 1A). Including an insertion of a 67-amino acid asparagine-rich sequence (between amino acids 306 and 375), PfTopoII had 40 and 36% identity to human and yeast topoisomerase IIs, respectively. Three distinct functional domains within PfTopoII were identified based on available biochemical information from yeast (28, 31) and human topoisomerase II (32) (Fig. 1B): a N-terminal ATPase domain (NAD), a central DNA cleavage reunion domain (CRD), and a C-terminal domain (CTD) with uncertain function. An essential catalytic tyrosine that is directly involved in cleavage and reunion of DNA, Tyr-805 of HsTopoIIα, Tyr-821 of HsTopoIIβ, and Tyr-783 of ScTopoII, aligned to Tyr-829 of PfTopoII.
FIGURE 1.

Protein sequence of P. falciparum topoisomerase II (PF3d7_1433500). A, confidence in the annotated DNA and predicted protein function of PF3d7_1433500 comes in part from the highly conserved nature of this gene compared with PvTopoII, HsTopoIIα, HsTopoIIβ, and ScTopoII. Dark boxes represent identical amino acids, and gaps represent insertions in some but not all species. B, overall organization of three typical functional organization of eukaryotic topoisomerase IIs. See Fig. 4 for more details.
PF3d7_1433500 Gene Product Is a Type II Topoisomerase
A systematic effort was devoted to conclusively demonstrate topoisomerase function from PF3d7_1433500. Just as expression of functional malarial proteins in heterologous cell-based systems has been difficult for the malaria community (18, 19), attempts to express PfTopoII have also been difficult (33). Our recent success in expression of a large set of active malaria enzymes in a wheat cell-free expression system (24, 25) encouraged us to subclone a wheat-optimized, chemically synthesized PfTopoII gene into a cell-free expression vector for wheat-based translation. In parallel, a vector was prepared to express an active-site mutant (Y829A) form of PfTopoII. This served as a control for potential background topoisomerase catalytic activity from wheat extract. Finally, a GFP-expressing vector served as a control to monitor the quality of the translation preparations.
First, expression of full-length PfTopoII and PfTopoII-M (the active-site mutant) was confirmed by autoradiography upon protein synthesis in the presence of radioactive amino acids (Fig. 2A). The protein products were seen as single entities with the expected mass of 169 kDa (Fig. 2A). Next, catalytic function of PfTopoII was tested using scaled up, cell-free translation lysates and nonradioactive amino acids. In a simple direct assay, relaxed plasmid product was produced from supercoiled DNA in the presence of PfTopoII translated lysate, and only when both magnesium and ATP were present (Fig. 2B). The PfTopoII function was further confirmed by a more demanding decatenation assay (Fig. 2C). Time-dependent release of decatenated products from kDNA was seen from PfTopoII translated lysates, but not from the active site mutant PfTopoII-M nor from GFP translated lysates (Fig. 2D). This decatenation of kDNA by PfTopoII was dependent on both magnesium and ATP. Based on these data, we conclude that the 169-kDa full-length protein product of PF3d7_1433500 is a functionally active type II topoisomerase.
FIGURE 2.
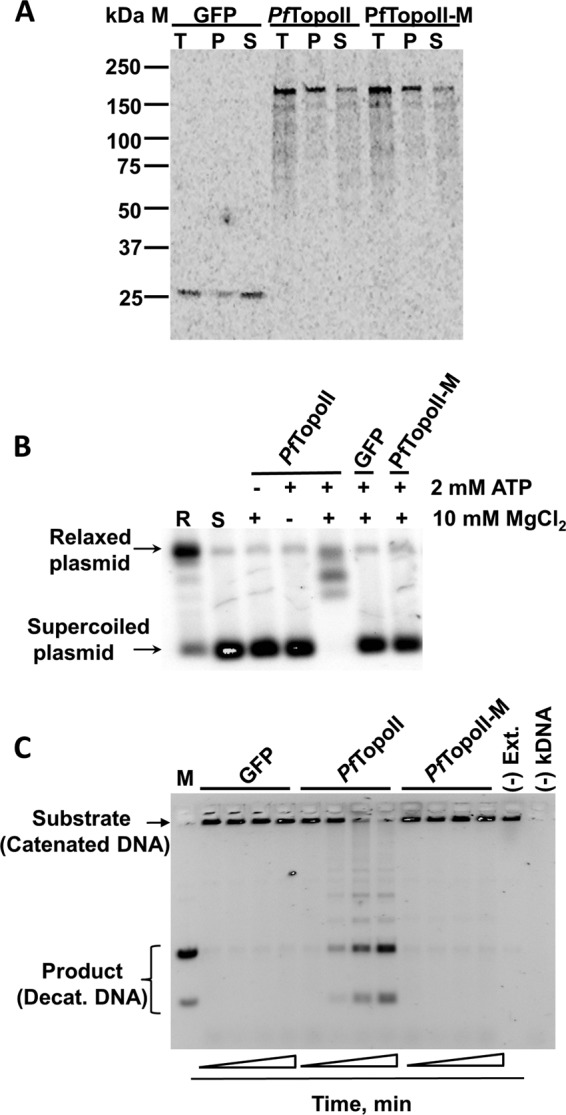
Wheat cell-free expression and functional characterization of PfTopoII. A, demonstration of full-length PfTopoII protein production using an autoradiogram of freshly translated radiolabeled GFP (translation system control), PfTopoII, and PfTopoII-M (an active site mutant) using a wheat germ cell-free expression system. Total (T), pellet (P), and soluble (S) fractions were prepared as detailed under “Experimental Procedures.” B, demonstration of ATP-dependent and Mg2+-dependent topoisomerase activity from PfTopoII using a gel-based plasmid relaxation assay. Electrophoretic mobility of relaxed plasmid product is shown in first lane marked R and that of the supercoiled substrate is shown in the second lane marked S. Successful relaxation of supercoiled plasmid DNA occurred when both ATP and Mg2+ were present with PfTopoII (lane 5) but neither with GFP translated lysate (lane 6) nor with in vitro translated PfTopoII with an active site mutation (lane 7). C, demonstration of full decatanation (Decat.) of kinetoplast DNA by PfTopoII. Release of low molecular weight, high mobility decatenated DNA was followed by electrophoresis. Time-dependent decatenation was observed when the assay included aliquots of freshly translated PfTopoII, but not with proportional amounts of freshly translated GFP or PfTopoII-M or if translation extract was left out ((−) Ext.). M, decatenated DNA marker mobility positions.
Fluorescence-based Decatenation Assay for Topoisomerase II
Type II topoisomerase gel-based assays, although informative about the state of the DNA substrates and products, are not well suited for screening of many chemical inhibitors. Others have tried alternatives such as tracking the ATPase function of type II topoisomerase using coupled enzymes (34), supercoil-specific triplex DNA formation (35), flow injection-based decatenation (36), and dual color fluorescence cross-correlation spectroscopy (37). We sought a simple, direct, selective, and sensitive fluorescence assay that tracked the decatenation activity of TopoII. Ideally, such an assay would be sensitive to the full natural catalytic cycle of type II topoisomerase enzymes and not give false signals with enzymes that nick single strands of DNA. Below, we describe a fluorescence-based decatenation assay that relies on selective precipitation of kDNA substrate networks, by centrifugation, away from TopoII-catalyzed decatenated products (38).
During method validation, decatenation reactions were initiated with PfTopoII, PfTopoII-M, and GFP translated lysates and then stopped at various time intervals using EDTA (see “Experimental Procedures”). The aliquots were first analyzed on agarose gel before and after centrifugation to remove catenated kDNA substrate. Before centrifugation, both starting kDNA and decatenated DNA product were seen in reactions run with PfTopoII, but only the starting kDNA substrate was seen in control reactions with PfTopoII-M or GFP (Fig. 3A). After centrifugation, kDNA substrate was absent in all samples (Fig. 3A). Because only the PfTopoII catalyzed reactions showed decatenated DNA products, the stage was set for a rapid method to selectively detect decatenated DNA products. To visualize products as a fluorescent signal, the supernatant samples of decatenation reactions were mixed with the DNA binding dye, SYBR Green I. Initially, at time 0, fluorescence in samples containing kDNA, SYBR Green I, and translated lysates of PfTopoII, PfTopoII-M, or GFP was nearly identical. After centrifugation, as the kDNA pelleted out, more than 95% of the fluorescence decreased (Fig. 3B), which is consistent with the data from the agarose gel (Fig. 3A). In a time-dependent analysis of potential decatenation involving PfTopoII translated lysates, there was a linear increase in fluorescence in the supernatant after centrifugation in the presence of PfTopoII for up to 30 min. Such a response was absent in PfTopoII-M and GFP initiated reactions. The large increase in fluorescent signal in PfTopoII catalyzed reactions, but not in PfTopoII-M or GFP, validates the sensitivity and selectivity of this fluorescence decatenation assay.
FIGURE 3.
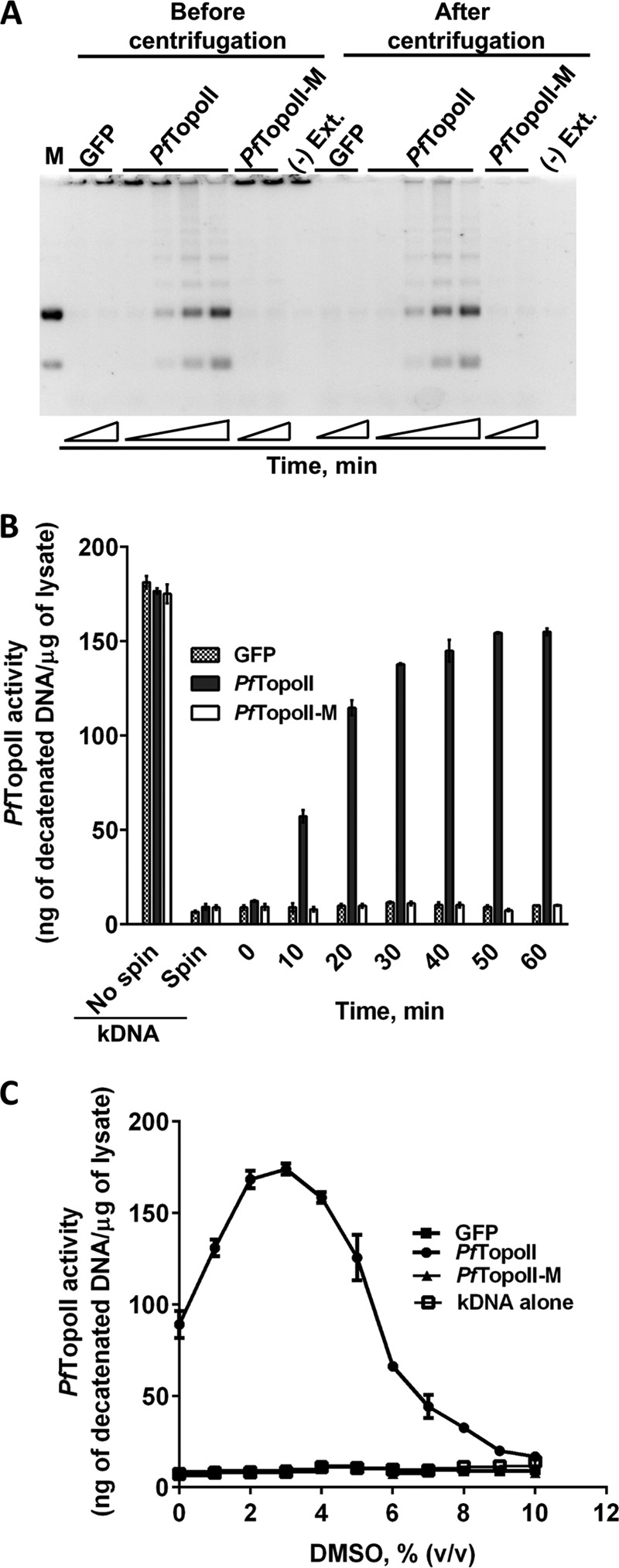
A simple, sensitive, and selective fluorescence-based decatenation assay for topoisomerase II. A, gel-based separation of catenated DNA substrates and decatenated DNA before and after centrifugation. DNA decatenated products formed by incubation with PfTopoII (lanes 5–7 and 13–15) but not control lysates (GFP, mutant PfTopoII; lanes 2–3, 8–10, 16, and 17). The decatenated product remained in solution after centrifugation (lanes 13–15). Catenated starting DNA was visible before centrifugation but not after centrifugation (lanes 11–17). B, time-dependent, PfTopoII-generated decatenated DNA products was measured by fluorescence in the supernatant using a DNA binding dye. Control lysates (GFP or mutant PfTopoII-m) failed to generate fluorescent decatenated DNA in the supernatant. C, stimulation of malarial PfTopoII activity by DMSO.
Next, in anticipation of using this convenient assay with nonpolar potential inhibitors of PfTopoII, the effect of DMSO on PfTopoII activity was tested. As the DMSO concentration increased from 0 to 2%, PfTopoII decatenation activity increased by 2-fold compared with reactions without DMSO (Fig. 3C). The stimulation peaked at 3% DMSO, after which there was a steep decrease. Complete inhibition of PfTopoII was seen when DMSO exceeded 9% (v/v). Controls showed no change in fluorescent DNA quantities with kDNA alone, GFP, or PfTopoII-M at all tested DMSO concentrations (Fig. 3C).
Stabilization and Purification of Functional PfTopoII, Lacking CTD
In preliminary studies, the full PfTopoII enzyme became inactive within a few hours after early purification steps. The native enzyme is expected to be a dimer of a single polypeptide, each carrying a NAD, a CRD in the middle, and a CTD of unknown function (Fig. 4A). Gene fragments, corresponding to NAD, CRD, and CTD, and a combined NAD-CRD or CRD-CTD were cloned separately into a cell-free expression vector (Table 1 and Fig. 4A). Cell-free expression from these truncated gene sequences yielded soluble protein (Fig. 4B). In the presence of 0 and 50 mm NaCl, none of constructs, including full-length PfTopoII, displayed detectable decatenation activity (Fig. 4C). At 150 mm NaCl, maximum decatenation activity was seen in full-length PfTopoII, and at 100 mm NaCl, the ATPase-cleavage reunion domain (NAD-CRD) showed maximum decatenation activity.
Decatenation activity in NAD-CRD translated lysate indicated that the CTD of PfTopoII was dispensable for the in vitro catalytic function of PfTopoII. The ATPase and the cleavage reunion domain were necessary and sufficient for the catalytic function of topoisomerase II. Hereafter, the ATPase-cleavage reunion domain (NAD-CRD) is named PfTopoII-ΔCTD. Incidentally, the NAD domain could not complement TopoII function in trans to the CRD-CTD unit: when mRNAs derived from plasmids harboring NAD and CRD-CTD gene fragments were co-expressed in a cell-free system (sample NAD + CRD-CTD in Fig. 4B), there was no decatenation activity using 0–200 mm NaCl (NAD + CRD-CTD in Fig. 4C).
Deletion of CTD from PfTopoII improved functional stability and permitted purification of PfTopoII-ΔCTD with full decatenation activity. We exploited the enzymatic properties of PfTopoII to facilitate purification. As described above, optimal salt concentrations were required for turnover of DNA by PfTopoII. It is possible that, during the catalytic cycle, at high salt concentrations, PfTopoII is unable to bind DNA to initiate the cleavage reaction, and at low salt concentrations, PfTopoII is not released from religated DNA. We further hypothesized that PfTopoII-ΔCTD would bind to DNA in a magnesium-free environment but not cleave DNA and that such binding could be reversed by higher salt concentrations. To test this “bind and release” strategy for enzyme purification, it was necessary to modify the cell-free transcription protocol to minimize the amount of residual DNA plasmid/template to help promote binding of PfTopoII-ΔCTD protein on DNA-cellulose. Thus, after transcription but before initiation of cell-free PfTopoII-ΔCTD synthesis, DNA template was digested with Turbo DNase. The DNase was subsequently denatured and removed from mRNA transcripts by phenol-chloroform extraction. After translation, upon binding of PfTopoII-ΔCTD to DNA-cellulose in a magnesium-free environment, decatenation activity in total and flow through samples was quantified. The flow through fractions contained only 20% of the initial activity, indicating efficient binding of PfTopoII-ΔCTD to the DNA-cellulose column. Washing with up to 125 mm salt concentration did not release PfTopoII-ΔCTD from the DNA column. Further increase in salt concentration to 150–200 mm resulted in complete release of PfTopoII-ΔCTD from the DNA column. Final purification on a gel filtration column yielded more than 90% pure dimeric PfTopoII-ΔCTD (Fig. 4D). The identity and amino acid sequence of purified PfTopoII-ΔCTD was confirmed by mass spectroscopy (see “Experimental Procedures”). Storage of purified PfTopoII-ΔCTD for more than 3 weeks at 4 °C in the presence of magnesium and ATP resulted in no loss in decatenation activity.
Inhibition of PfTopoII by Type II Topoisomerase Inhibitors
Type II topoisomerases are targeted by many antibacterial (10, 11, 14) and anticancer agents (12, 13). Different classes of inhibitors bind differently to type II topoisomerases with different mode of inhibition (see Fig. 6B). After purifying functional PfTopoII-ΔCTD and establishing a fluorescent assay, the potency and selectivity of known type II topoisomerase inhibitors against the malaria enzyme were tested. Representative inhibitors from each of three classes of inhibitors were chosen: GSK299423 (a piperidinylalkylquinoline), ciprofloxacin (a fluoroquinolone), and etoposide (a epipodophyllotoxin). GSK299423 was synthesized (Fig. 5A) as reported (14), and the other two inhibitors were from a commercial source.
FIGURE 6.
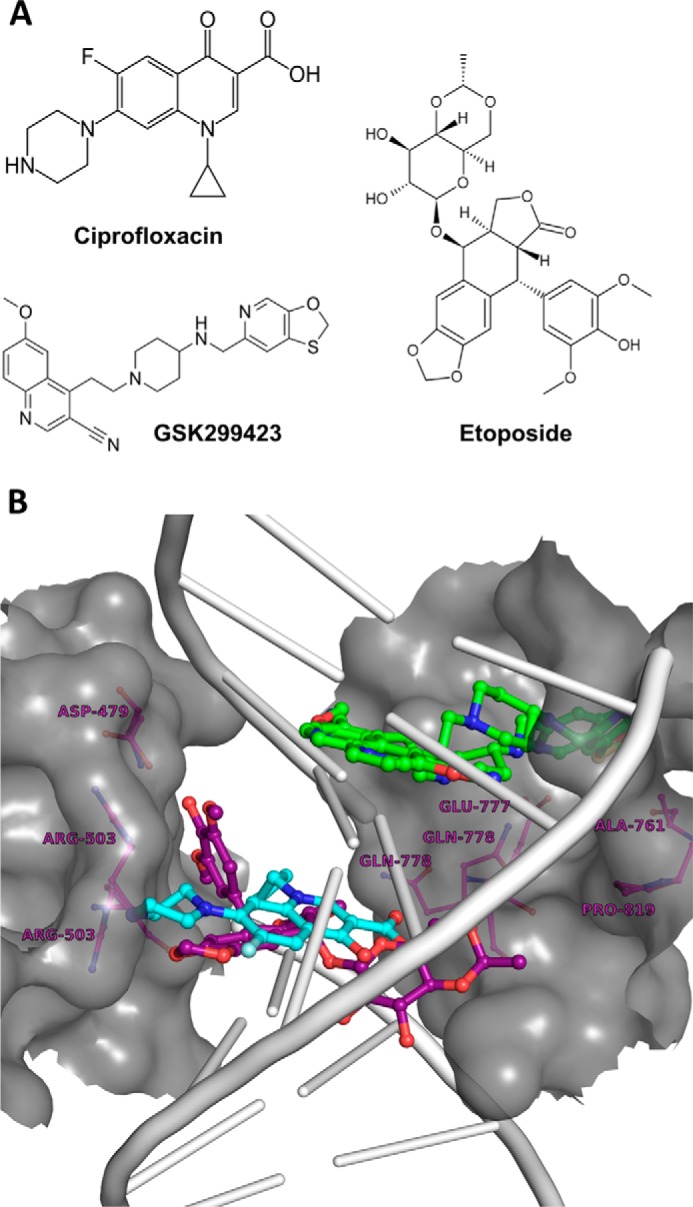
A molecular model highlights the different opportunities for inhibitors to bind PfTopoII. A, varied structures of well known potent inhibitors that target type II topoisomerases. B, illustration of the interplay between TopoII protein, its affinity for DNA, and different niches preferred by different TopoII inhibitors, as illustrated by a molecular model depicting where DNA (gray), ciprofloxacin (cyan), etoposide (purple), and GSK299423 (green) would choose to bind hsTopoIIβ (dark gray). Conservation and potential variations in amino acids in these inhibitor binding sites in PfTopoII are discussed in the text.
FIGURE 5.

Inhibition of PfTopoII by chemical scaffolds known to inhibit TopoII from other cell types. A, chemical scheme for the synthesis of bacterial gyrase inhibitor, GSK299423. B, selective inhibition of PfTopoII and PfTopoII-ΔCTD over HsTopoIIα by GSK299423. C, modes of action of different topoisomerase functions by examining the fates of supercoiled DNA in contact with inhibited PfTopoII. Etoposide and GSK299423 captured single- as well as double-stranded DNA breaks, as represented by appearance of nicked and linearized plasmid in PfTopoII catalyzed reactions.
Initially, we confirmed that full-length PfTopoII from translated lysate and the truncated, purified PfTopoII-ΔCTD had comparable inhibitor binding properties (Table 2). To test potential selectivity of GSK299423, ciprofloxacin, and etoposide, parallel inhibition studies were carried out on HsTopoIIα. GSK29942, a bacterial gyrase inhibitor, inhibited decatenation function of PfTopoII and PfTopoII-ΔCTD with a half-maximal inhibitory concentrations (IC50) of 0.6 ± 0.2 and 2.5 ± 0.3 μm, respectively. This compound displayed less potency toward HsTopoIIα with an IC50 of 10 ± 2 μm (Fig. 5B). GSK299423 inhibited growth of 3D7, Dd2, and HB3 clones of P. falciparum with IC50 of 2.0 ± 0.1, 6.4 ± 1.7, and 2.1 ± 0.7 μm respectively, while displaying at least 50-fold less potency (EC50 = >100) against mouse lymphocytic leukemia cells (L1210) (Table 2). Ciprofloxacin, another bacterial gyrase inhibitor, was 50-fold selective toward PfTopoII over HsTopoIIα. It displayed 41-fold selectivity toward proliferating parasite cells over mouse cell lines (Table 2). Etoposide, a HsTopoIIα inhibitor, displayed similar inhibition activity on both PfTopoII and hTopoIIα (Table 2). PfTopoII inhibitor binding pockets appear to be similar to bacterial Type II topoisomerases because of similar amino acids at inhibitor binding pockets and possibly because of similar effects from distant sites in the large, complex enzyme system.
TABLE 2.
Type II topo inhibitors selectively inhibit malarial enzyme
| GSK 299423 | Ciprofloxacin | Etoposide | |
|---|---|---|---|
| μm | μm | μm | |
| IC50 of target enzymes | |||
| PfTopoII | 0.6 ± 0.2 | 35 ± 9 | 20 ± 4 |
| PfTopoII-ΔCTD | 2.5 ± 0.3 | 63 ± 13 | 17 ± 1 |
| hTopoII | 10 ± 2 | 199957 | 68 |
| Selectivity | >15 | >50 | >3 |
| EC50 of cell lines | |||
| 3D7 | 2.0 ± 0.1 | 2454 | 40 |
| Dd2 | 6.4 ± 1.7 | ||
| HB3 | 2.1 ± 0.7 | ||
| L1210 | >100 | 174056a | 0.016 |
| Selectivity | >50 | >70 | - |
a EC50 values are for human macrophages.
Next, we explored the mechanisms by which each of these compounds blocked the complex catalytic cycle of PfTopoII. Etoposide is known to make double-stranded DNA breaks in HsTopoII-catalyzed reactions (12). On the other hand, GSK299423 creates single-stranded DNA breaks in gyrase-catalyzed reactions (14). To determine whether and which types of DNA breaks are generated in malarial topoisomerase II-catalyzed reactions, we tested these two compounds in a plasmid relaxation assay with purified PfTopoII-ΔCTD. Relaxation reactions were stopped by SDS and treated with proteinase K to digest DNA bound PfTopoII-ΔCTD. DNA was resolved on 1% agarose gel and visualized by ethidium bromide. In a control reaction with PfTopoII-ΔCTD alone, all supercoiled plasmid was converted into relaxed plasmid (Fig. 5C). Upon addition of the highest concentration (50 μm) etoposide to the relaxation reaction, plasmid not only relaxed, but also created an additional DNA band (arrow), which was of similar size to a linearized plasmid. This suggests that etoposide-treated PfTopoII freezes the enzyme after making double-stranded DNA breaks, just as seen with the mammalian enzymes. As the concentration of etoposide decreased, the corresponding linear plasmid band intensity decreased. In contrast, such double-strand DNA breaks were not generated in the presence of GSK299423 or danofloxacin (a control). Just as seen with the purified enzyme, etoposide-mediated TopoII-cleaved nuclear genomic DNA was previously reported in P. falciparum cells (39). Together, these results confirm that etoposide can target nuclear localized PfTopoII and cause cell death through double-stranded DNA breaks.
Inhibition by the highest concentration (25 μm) of GSK299423, as viewed by plasmid relaxation assay, showed complete inhibition of PfTopoII-ΔCTD but arrested PfTopoII function differently compared with etoposide (Fig. 5C). Reducing GSK299423 concentration down to 5 μm allowed the supercoiled DNA plasmid substrate to relax, but this created a DNA product with a size characteristic of a plasmid with a single-stranded DNA break. The absence of similar breaks in the presence of danofloxacin or etoposide (Fig. 5C) indicates that GSK299423 allows PfTopoII to generate asymmetric single-stranded DNA breaks at the DNA binding site. Overall, the availability of pure fully functional PfTopoII will not only allow discovery of new antimalarial leads and their optimization but also inform on their potentially varying modes of action.
Discussion
With potentially decreasing efficacy of artemisinin-based antimalarials (5, 7), there is an urgent need to identify safe, effective drugs against malaria that work through novel mechanisms for this disease. The current global antimalarial drug portfolio is rich in artemisinin derivatives and related synthetic endoperoxides, but with few new chemical warheads with new modes of action (Medicines for Malaria Venture). The current portfolio of new, upstream targets is still very limited, there is a high attrition rate of candidate inhibitors, and many of these compounds will have to be deployed jointly as part of novel combinations.
One important way to develop new antimalarials is to identify new targets based on successes with other infectious pathogens and based on a priori knowledge of biology. Topoisomerase II, the target against many bacterial diseases, plays an obligatory role in cell growth. During all stages of the malaria life cycle, the processes of cell maintenance, cell division, cell repair, and cellular mating are expected to generate entangled chromatids that must be separated for normal function and growth. Transcription and replication around antiparallel double-stranded DNA creates topological constraints and supercoiling tension both in front of and behind DNA sequences engaged in replication and transcription (9–13, 15). Without fully functional topoisomerase II, malaria parasites would suffer from stalled transcription and replication and would experience rapid cell death. In recent years, large scale genomic studies have further affirmed the value of topoisomerase II as a potential drug target.
PfTopoII is a single copy gene on chromosome 14, and its expression is seen across all stages of the parasite life cycle (from the PlasmoDB: Plasmodium Genomics Resource and Refs. 41 and 42). Within malarial parasite cells, PfTopoII protein is localized to the nucleus (41–44). Extensive proteomic studies also identified PfTopoII expression across all asexual and sexual stages of parasite (44–47). Pioneering studies with partially purified parasite lysate have suggested that ATP and magnesium-dependent decatenation activities in parasite lysates are sensitive to both prokaryotic and eukaryotic type II topoisomerase inhibitors (48, 49). Further progress in understanding PfTopoII function and inhibitor binding properties and in exploiting newer tools in drug discovery such as high throughput screens, structure-based drug discovery, and modern iterative lead optimization campaigns are hampered by lack of an active malaria target protein in large quantities.
As an extension of our long time interest in exploiting host-parasite differences in nucleotide and DNA metabolism for antimalarial discovery (24, 25, 30, 50–52), we sought to develop a robust platform for developing species-specific malarial topoisomerase II inhibitors. By utilizing a wheat cell-free system to produce functional malaria enzymes (24, 25), it was possible to express PfTopoII in functionally active form. A topoisomerase II fluorescent assay was developed that is simple, sensitive, and selective, and we identified optimum assay solvents to get maximum activity from the recombinant protein. Through construction of various truncated forms of PfTopoII gene followed by expressing in cell-free system, we learned that double-strand DNA cleavage, ATP-dependent strand passage of a different DNA through the break and re-ligation of cleaved DNA by PfTopoII require a covalently connected NAD domain and CRD domain, but that the C terminus domain is expendable. Finally, a salt-dependent DNA affinity chromatography step helped purify stable malarial PfTopoII-ΔCTD that had full decatenation activity.
Existing topoisomerase inhibitors developed for other indications can both serve as an inspiration for developing inhibitors tailored for treating malaria and provide a starting framework for developing Plasmodium TopoII inhibitors. Representatives of the three prototype bacterial and human type II topoisomerase inhibitor classes discussed here are ciprofloxacin, GSK299423, and etoposide (Fig. 6A). All three type II DNA topoisomerases were active against purified PfTopoII (Table 2 and Fig. 5B). Etoposide, often used for treating human cancers, displayed no significant selective inhibition of PfTopoII over the human enzyme. However, importantly, mechanistic studies showed that etoposide-inhibited PfTopoII formed double-stranded DNA breaks as seen in the plasmid relaxation assay (Fig. 5C). This in vitro result with purified enzyme is consistent with an earlier demonstration that etoposide treatment generates genome-wide DNA breaks in P. falciparum (39). These results are consistent with the idea that the etoposide target is the nuclear-localized PfTopoII, although the challenge is to understand how this particular class of TopoII inhibitors may be optimized and deployed with selectivity. Ciprofloxacin, heavily used as an antibacterial, has previously been tested as an experimental inhibitor of P. falciparum (53–56), Trypanosoma brucei (57), and Leishmania panamensis (16, 58). In those studies, without structural optimization, the compound showed more than 70-fold selective inhibition of parasite cells over human macrophage cells (Table 2). In our current work, ciprofloxacin inhibited purified PfTopoII with 50-fold selectively over the human enzyme, raising the possibility that the cellular selectivity arises from an intrinsic difference in species-specific target binding that can be improved through lead optimization. Even more encouraging, the bacterial gyrase inhibitor GSK299423 showed 50-fold selective growth inhibition of parasite cells over mammalian cells, and a significant component seems to arise from the 15-fold intrinsic selectivity for the malarial PfTopoII over the human enzyme. In our mode of action studies, unlike etoposide, GSK299423-inhibited PfTopoII generated single-stranded DNA breaks (Fig. 5C), as seen with treatment of bacterial gyrase with GSK299423 (14). Although the inhibitory activity of ciprofloxacin and GSK299423 correlates well between purified PfTopoII and malaria parasite, additional studies are needed to solve crystal structures of inhibitor-bound PfTopoII and to biochemical characterize the remaining topoisomerases and genetically manipulated parasites to rule out alternate possibility.
To help visualize the different modes by which these different type II topoisomerase inhibitors may act on the malaria enzyme with selectivity over the human enzyme, a schematic model was generated based on the crystal structures of inhibitor-protein-DNA tertiary complexes of HsTopoIIβ (40) and bacterial DNA gyrase (14). From an end view (Fig. 6B), the DNA substrate (gray) is wrapped by TopoII polypeptides (dark gray surface). Ciprofloxacin (cyan) makes contact with the enzyme (dark gray surface) and the DNA (gray) (Fig. 6B). The quinolone group of ciprofloxacin (cyan) rests between the DNA base pairs (gray), whereas the piperazine and carboxyl groups of the quinolone contact the topoisomerase (14). In contrast, the new bacterial DNA gyrase inhibitor, GSK299423 (green) inhibits topoisomerases by a different mode (Fig. 6B). After inserting the quinoline-carbonitrile group into the DNA double-helix base pairs, the oxothiolo-pyridine group of GSK299423 sits on the topoisomerase at a noncatalytic dimeric interface of the cleavage reunion domains (14). Finally, the polycyclic core of etoposide (purple) sits between DNA base pairs, whereas the diacetophenone end extends into the protein topoisomerase (Fig. 6B) (40). Superimposing the PfTopoII (PF3d7_1433500) sequence on the existing crystal structures from other species points to a number of malarial amino acid residues that are different in the parasite enzyme compared with the human counterpart, in addition to other distant protein variations that likely contribute to selectivity. In a reported crystal structure showing the binding mode of ciprofloxacin with topoisomerase, the carboxyl group of the quinolone contacted Ser-84 of gyrase A (13). The corresponding residue in PfTopoII is Ser-786 (Fig. 1D). Thus, selective inhibition of PfTopoII by GSK299423 over the human enzyme may be due to the shared residues between parasite and the bacterial enzymes at the interface of the cleavage reunion domains that are responsible for bacterial gyrase inhibition. There are also differences; the crystal structure of Staphylococcus aureus gyrase-bound GSK299423 (14) displayed Met-121 as one of five important residues for selective inhibition of enzyme. The corresponding residue in PfTopoII is Ala-827 and in HsTopoIIα and HsTopoIIβ, it is Pro-803 and Pro-819, respectively. Lessons from malarial dihydrofolate reductase-thymidylate synthase teach us that, in addition to active site host-parasite differences in drug binding, other host-parasite differences such as expression levels and nature of gene regulation can also contribute to drug selectivity in unexpected ways (30).
In summary, we have expressed functional P. falciparum topoisomerase II and developed fast reliable methods to purify and assay the enzyme. With existing TopoII inhibitors, we have also demonstrated that this platform is ready for screening new molecules, for crystallization of this high value target, and for informative lead optimization campaigns that should lead to new classes of potent, lasting antimalarials.
Author Contributions
D. G. M. and P. K. R. conceived the biochemistry experiments, analyzed the data, and wrote the manuscript. S. Ku. analyzed the structural data and assisted in writing of the manuscript. S. Ko. synthesized key inhibitors. J. W. conducted cell-based experiments and helped write the manuscript.
Acknowledgments
We thank Kalyan Krishnamoorthy for the protein mass spectrometry protocol and Laura Chery for critically reading the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants AI093380, AI099280, and AI089688. The authors declare that they have no conflicts of interest with the contents of this article.
- PfTopoII
- P. falciparum topoisomerase II
- kDNA
- kinetoplast catenated DNA
- NAD
- N-terminal ATPase domain
- CRD
- cleavage reunion domain
- CTD
- C-terminal domain
- TAE buffer
- 40 mm Tris acetate with 1 mm EDTA at pH 8.3.
References
- 1. Murray C. J., Rosenfeld L. C., Lim S. S., Andrews K. G., Foreman K. J., Haring D., Fullman N., Naghavi M., Lozano R., Lopez A. D. (2012) Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet 379, 413–431 [DOI] [PubMed] [Google Scholar]
- 2. WHO (2014) World Malaria Report, World Health Organization, Geneva, Swtzerland [Google Scholar]
- 3. Rathod P. K., McErlean T., Lee P. C. (1997) Variations in frequencies of drug resistance in Plasmodium falciparum. Proc. Natl. Acad. Sci. U.S.A. 94, 9389–9393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guler J. L., Freeman D. L., Ahyong V., Patrapuvich R., White J., Gujjar R., Phillips M. A., DeRisi J., Rathod P. K. (2013) Asexual populations of the human malaria parasite, Plasmodium falciparum, use a two-step genomic strategy to acquire accurate, beneficial DNA amplifications. PLoS Pathog. 9, e1003375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dondorp A. M., Nosten F., Yi P., Das D., Phyo A. P., Tarning J., Lwin K. M., Ariey F., Hanpithakpong W., Lee S. J., Ringwald P., Silamut K., Imwong M., Chotivanich K., Lim P., Herdman T., An S. S., Yeung S., Singhasivanon P., Day N. P., Lindegardh N., Socheat D., White N. J. (2009) Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 361, 455–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miller L. H., Ackerman H. C., Su X. Z., Wellems T. E. (2013) Malaria biology and disease pathogenesis: insights for new treatments. Nat. Med. 19, 156–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ariey F., Witkowski B., Amaratunga C., Beghain J., Langlois A. C., Khim N., Kim S., Duru V., Bouchier C., Ma L., Lim P., Leang R., Duong S., Sreng S., Suon S., Chuor C. M., Bout D. M., Ménard S., Rogers W. O., Genton B., Fandeur T., Miotto O., Ringwald P., Le Bras J., Berry A., Barale J. C., Fairhurst R. M., Benoit-Vical F., Mercereau-Puijalon O., Ménard D. (2014) A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505, 50–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kappe S. H., Vaughan A. M., Boddey J. A., Cowman A. F. (2010) That was then but this is now: malaria research in the time of an eradication agenda. Science 328, 862–866 [DOI] [PubMed] [Google Scholar]
- 9. Wang J. C. (1996) DNA topoisomerases. Annu. Rev. Biochem. 65, 635–692 [DOI] [PubMed] [Google Scholar]
- 10. Mitscher L. A. (2005) Bacterial topoisomerase inhibitors: quinolone and pyridone antibacterial agents. Chem. Rev. 105, 559–592 [DOI] [PubMed] [Google Scholar]
- 11. Widdowson K., Hennessy A. (2010) Advances in structure-based drug design of novel bacterial topoisomerase inhibitors. Future Med. Chem. 2, 1619–1622 [DOI] [PubMed] [Google Scholar]
- 12. Pommier Y., Leo E., Zhang H., Marchand C. (2010) DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 17, 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nitiss J. L. (2009) Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 9, 338–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bax B. D., Chan P. F., Eggleston D. S., Fosberry A., Gentry D. R., Gorrec F., Giordano I., Hann M. M., Hennessy A., Hibbs M., Huang J., Jones E., Jones J., Brown K. K., Lewis C. J., May E. W., Saunders M. R., Singh O., Spitzfaden C. E., Shen C., Shillings A., Theobald A. J., Wohlkonig A., Pearson N. D., Gwynn M. N. (2010) Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466, 935–940 [DOI] [PubMed] [Google Scholar]
- 15. Berger J. M., Gamblin S. J., Harrison S. C., Wang J. C. (1996) Structure and mechanism of DNA topoisomerase II. Nature 379, 225–232 [DOI] [PubMed] [Google Scholar]
- 16. Cortázar T. M., Coombs G. H., Walker J. (2007) Leishmania panamensis: comparative inhibition of nuclear DNA topoisomerase II enzymes from promastigotes and human macrophages reveals anti-parasite selectivity of fluoroquinolones, flavonoids and pentamidine. Exp. Parasitol. 116, 475–482 [DOI] [PubMed] [Google Scholar]
- 17. Gardner M. J., Hall N., Fung E., White O., Berriman M., Hyman R. W., Carlton J. M., Pain A., Nelson K. E., Bowman S., Paulsen I. T., James K., Eisen J. A., Rutherford K., Salzberg S. L., Craig A., Kyes S., Chan M. S., Nene V., Shallom S. J., Suh B., Peterson J., Angiuoli S., Pertea M., Allen J., Selengut J., Haft D., Mather M. W., Vaidya A. B., Martin D. M., Fairlamb A. H., Fraunholz M. J., Roos D. S., Ralph S. A., McFadden G. I., Cummings L. M., Subramanian G. M., Mungall C., Venter J. C., Carucci D. J., Hoffman S. L., Newbold C., Davis R. W., Fraser C. M., Barrell B. (2002) Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419, 498–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mehlin C., Boni E., Buckner F. S., Engel L., Feist T., Gelb M. H., Haji L., Kim D., Liu C., Mueller N., Myler P. J., Reddy J. T., Sampson J. N., Subramanian E., Van Voorhis W. C., Worthey E., Zucker F., Hol W. G. (2006) Heterologous expression of proteins from Plasmodium falciparum: results from 1000 genes. Mol. Biochem. Parasitol. 148, 144–160 [DOI] [PubMed] [Google Scholar]
- 19. Vedadi M., Lew J., Artz J., Amani M., Zhao Y., Dong A., Wasney G. A., Gao M., Hills T., Brokx S., Qiu W., Sharma S., Diassiti A., Alam Z., Melone M., Mulichak A., Wernimont A., Bray J., Loppnau P., Plotnikova O., Newberry K., Sundararajan E., Houston S., Walker J., Tempel W., Bochkarev A., Kozieradzki I., Edwards A., Arrowsmith C., Roos D., Kain K., Hui R. (2007) Genome-scale protein expression and structural biology of Plasmodium falciparum and related Apicomplexan organisms. Mol. Biochem. Parasitol. 151, 100–110 [DOI] [PubMed] [Google Scholar]
- 20. Raghu Ram E. V., Kumar A., Biswas S., Chaubey S., Siddiqi M. I., Habib S. (2007) Nuclear gyrB encodes a functional subunit of the Plasmodium falciparum gyrase that is involved in apicoplast DNA replication. Mol. Biochem. Parasitol. 154, 30–39 [DOI] [PubMed] [Google Scholar]
- 21. Roy A., D'Annessa I., Nielsen C. J., Tordrup D., Laursen R. R., Knudsen B. R., Desideri A., Andersen F. F. (2011) Peptide Inhibition of Topoisomerase IB from Plasmodium falciparum. Mol. Biol. Int. 2011, 854626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Phillips M. A., Gujjar R., Malmquist N. A., White J., El Mazouni F., Baldwin J., Rathod P. K. (2008) Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite Plasmodium falciparum. J. Med. Chem. 51, 3649–3653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Coteron J. M., Marco M., Esquivias J., Deng X., White K. L., White J., Koltun M., El Mazouni F., Kokkonda S., Katneni K., Bhamidipati R., Shackleford D. M., Angulo-Barturen I., Ferrer S. B., Jiménez-Díaz M. B., Gamo F. J., Goldsmith E. J., Charman W. N., Bathurst I., Floyd D., Matthews D., Burrows J. N., Rathod P. K., Charman S. A., Phillips M. A. (2011) Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J. Med. Chem. 54, 5540–5561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mudeppa D. G., Pang C. K., Tsuboi T., Endo Y., Buckner F. S., Varani G., Rathod P. K. (2007) Cell-free production of functional Plasmodium falciparum dihydrofolate reductase-thymidylate synthase. Mol. Biochem. Parasitol. 151, 216–219 [DOI] [PubMed] [Google Scholar]
- 25. Mudeppa D. G., Rathod P. K. (2013) Expression of functional Plasmodium falciparum enzymes using a wheat germ cell-free system. Eukaryot. Cell 12, 1653–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leaver-Fay A., Tyka M., Lewis S. M., Lange O. F., Thompson J., Jacak R., Kaufman K., Renfrew P. D., Smith C. A., Sheffler W., Davis I. W., Cooper S., Treuille A., Mandell D. J., Richter F., Ban Y. E., Fleishman S. J., Corn J. E., Kim D. E., Lyskov S., Berrondo M., Mentzer S., Popović Z., Havranek J. J., Karanicolas J., Das R., Meiler J., Kortemme T., Gray J. J., Kuhlman B., Baker D., Bradley P. (2011) ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 28. Caron P. R., Watt P., Wang J. C. (1994) The C-terminal domain of Saccharomyces cerevisiae DNA topoisomerase II. Mol. Cell Biol. 14, 3197–3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 30. Zhang K., Rathod P. K. (2002) Divergent regulation of dihydrofolate reductase between malaria parasite and human host. Science 296, 545–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schmidt B. H., Osheroff N., Berger J. M. (2012) Structure of a topoisomerase II-DNA-nucleotide complex reveals a new control mechanism for ATPase activity. Nat. Struct. Mol. Biol. 19, 1147–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meczes E. L., Gilroy K. L., West K. L., Austin C. A. (2008) The impact of the human DNA topoisomerase II C-terminal domain on activity. PLoS One 3, e1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cheesman S. J. (2000) The topoisomerases of protozoan parasites. Parasitol. Today 16, 277–281 [DOI] [PubMed] [Google Scholar]
- 34. Singh P. K., Chan P. F., Hibbs M. J., Vazquez M. J., Segura D. C., Thomas D. A., Theobald A. J., Gallagher K. T., Hassan N. J. (2011) High-yield production and characterization of biologically active GST-tagged human topoisomerase IIα protein in insect cells for the development of a high-throughput assay. Protein Expr. Purif 76, 165–172 [DOI] [PubMed] [Google Scholar]
- 35. Taylor J. A., Burton N. P., Maxwell A. (2012) High-throughput microtitre plate-based assay for DNA topoisomerases. Methods Mol. Biol. 815, 229–239 [DOI] [PubMed] [Google Scholar]
- 36. Barnabé N., Hasinoff B. B. (2001) High-throughput fluorescence flow-injection topoisomerase II inhibition assay. J. Chromatogr. B Biomed Sci. Appl. 760, 263–269 [DOI] [PubMed] [Google Scholar]
- 37. Jahnz M., Medina M. A., Schwille P. (2005) A novel homogenous assay for topoisomerase II action and inhibition. Chembiochem 6, 920–926 [DOI] [PubMed] [Google Scholar]
- 38. Sahai B. M., Kaplan J. G. (1986) A quantitative decatenation assay for type II topoisomerases. Anal. Biochem. 156, 364–379 [DOI] [PubMed] [Google Scholar]
- 39. Kelly J. M., McRobert L., Baker D. A. (2006) Evidence on the chromosomal location of centromeric DNA in Plasmodium falciparum from etoposide-mediated topoisomerase-II cleavage. Proc. Natl. Acad. Sci. U.S.A. 103, 6706–6711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu C. C., Li T. K., Farh L., Lin L. Y., Lin T. S., Yu Y. J., Yen T. J., Chiang C. W., Chan N. L. (2011) Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 333, 459–462 [DOI] [PubMed] [Google Scholar]
- 41. Cheesman S., McAleese S., Goman M., Johnson D., Horrocks P., Ridley R. G., Kilbey B. J. (1994) The gene encoding topoisomerase II from Plasmodium falciparum. Nucleic Acids Res. 22, 2547–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cheesman S., Horrocks P., Tosh K., Kilbey B. (1998) Intraerythrocytic expression of topoisomerase II from Plasmodium falciparum is developmentally regulated. Mol. Biochem. Parasitol. 92, 39–46 [DOI] [PubMed] [Google Scholar]
- 43. Oehring S. C., Woodcroft B. J., Moes S., Wetzel J., Dietz O., Pulfer A., Dekiwadia C., Maeser P., Flueck C., Witmer K., Brancucci N. M., Niederwieser I., Jenoe P., Ralph S. A., Voss T. S. (2012) Organellar proteomics reveals hundreds of novel nuclear proteins in the malaria parasite Plasmodium falciparum. Genome Biol. 13, R108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Florens L., Liu X., Wang Y., Yang S., Schwartz O., Peglar M., Carucci D. J., Yates J. R., 3rd, Wu Y. (2004) Proteomics approach reveals novel proteins on the surface of malaria-infected erythrocytes. Mol. Biochem. Parasitol. 135, 1–11 [DOI] [PubMed] [Google Scholar]
- 45. Lasonder E., Janse C. J., van Gemert G. J., Mair G. R., Vermunt A. M., Douradinha B. G., van Noort V., Huynen M. A., Luty A. J., Kroeze H., Khan S. M., Sauerwein R. W., Waters A. P., Mann M., Stunnenberg H. G. (2008) Proteomic profiling of Plasmodium sporozoite maturation identifies new proteins essential for parasite development and infectivity. PLoS Pathog. 4, e1000195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Silvestrini F., Lasonder E., Olivieri A., Camarda G., van Schaijk B., Sanchez M., Younis Younis S., Sauerwein R., Alano P. (2010) Protein export marks the early phase of gametocytogenesis of the human malaria parasite Plasmodium falciparum. Mol. Cell. Proteomics 9, 1437–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lindner S. E., Swearingen K. E., Harupa A., Vaughan A. M., Sinnis P., Moritz R. L., Kappe S. H. (2013) Total and putative surface proteomics of malaria parasite salivary gland sporozoites. Mol. Cell. Proteomics 12, 1127–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Auparakkitanon S., Wilairat P. (2000) Cleavage of DNA induced by 9-anilinoacridine inhibitors of topoisomerase II in the malaria parasite Plasmodium falciparum. Biochem. Biophys. Res. Commun. 269, 406–409 [DOI] [PubMed] [Google Scholar]
- 49. Chavalitshewinkoon P., Leelaphiwat S., Wilairat P. (1994) Partial purification and characterization of DNA topoisomerase II from Plasmodium falciparum. Southeast Asian J. Trop. Med. Public Health 25, 32–36 [PubMed] [Google Scholar]
- 50. Pang C. K., Hunter J. H., Gujjar R., Podutoori R., Bowman J., Mudeppa D. G., Rathod P. K. (2009) Catalytic and ligand-binding characteristics of Plasmodium falciparum serine hydroxymethyltransferase. Mol. Biochem. Parasitol. 168, 74–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hekmat-Nejad M., Rathod P. K. (1996) Kinetics of Plasmodium falciparum thymidylate synthase: interactions with high-affinity metabolites of 5-fluoroorotate and D1694. Antimicrob. Agents Chemother. 40, 1628–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ganesan K., Ponmee N., Jiang L., Fowble J. W., White J., Kamchonwongpaisan S., Yuthavong Y., Wilairat P., Rathod P. K. (2008) A genetically hard-wired metabolic transcriptome in Plasmodium falciparum fails to mount protective responses to lethal antifolates. PLoS Pathog. 4, e1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weissig V., Vetro-Widenhouse T. S., Rowe T. C. (1997) Topoisomerase II inhibitors induce cleavage of nuclear and 35-kb plastid DNAs in the malarial parasite Plasmodium falciparum. DNA Cell Biol. 16, 1483–1492 [DOI] [PubMed] [Google Scholar]
- 54. Dahl E. L., Rosenthal P. J. (2007) Multiple antibiotics exert delayed effects against the Plasmodium falciparum apicoplast. Antimicrob Agents Chemother. 51, 3485–3490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Goodman C. D., Su V., McFadden G. I. (2007) The effects of anti-bacterials on the malaria parasite Plasmodium falciparum. Mol. Biochem. Parasitol. 152, 181–191 [DOI] [PubMed] [Google Scholar]
- 56. Mahmoudi N., Ciceron L., Franetich J. F., Farhati K., Silvie O., Eling W., Sauerwein R., Danis M., Mazier D., Derouin F. (2003) In vitro activities of 25 quinolones and fluoroquinolones against liver and blood stage Plasmodium spp. Antimicrob. Agents Chemother. 47, 2636–2639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nenortas E., Kulikowicz T., Burri C., Shapiro T. A. (2003) Antitrypanosomal activities of fluoroquinolones with pyrrolidinyl substitutions. Antimicrob. Agents Chemother. 47, 3015–3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Romero I. C., Saravia N. G., Walker J. (2005) Selective action of fluoroquinolones against intracellular amastigotes of Leishmania (Viannia) panamensis in vitro. J. Parasitol. 91, 1474–1479 [DOI] [PubMed] [Google Scholar]