

Background: GRK4 mediates GPCR phosphorylation and is implicated in hypertension.
Results: The crystal structure of human GRK4α A486V is presented. Phosphorylation assays highlight kinetic differences between wild-type GRK4α and GRK4α A486V.
Conclusion: GRK4α has unusual features that help explain the effects of GRK4 mutations and GRK4 biology.
Significance: This work provides a structural basis for GRK4 function.
Keywords: G-protein-coupled receptor (GPCR), hypertension, kinetics, phosphorylation, serine/threonine protein kinase, x-ray crystallography, dopamine D1 receptor, GRK4
Abstract
G-protein-coupled receptor (GPCR) kinases (GRKs) bind to and phosphorylate GPCRs, initiating the process of GPCR desensitization and internalization. GRK4 is implicated in the regulation of blood pressure, and three GRK4 polymorphisms (R65L, A142V, and A486V) are associated with hypertension. Here, we describe the 2.6 Å structure of human GRK4α A486V crystallized in the presence of 5′-adenylyl β,γ-imidodiphosphate. The structure of GRK4α is similar to other GRKs, although slight differences exist within the RGS homology (RH) bundle subdomain, substrate-binding site, and kinase C-tail. The RH bundle subdomain and kinase C-terminal lobe form a strikingly acidic surface, whereas the kinase N-terminal lobe and RH terminal subdomain surfaces are much more basic. In this respect, GRK4α is more similar to GRK2 than GRK6. A fully ordered kinase C-tail reveals interactions linking the C-tail with important determinants of kinase activity, including the αB helix, αD helix, and the P-loop. Autophosphorylation of wild-type GRK4α is required for full kinase activity, as indicated by a lag in phosphorylation of a peptide from the dopamine D1 receptor without ATP preincubation. In contrast, this lag is not observed in GRK4α A486V. Phosphopeptide mapping by mass spectrometry indicates an increased rate of autophosphorylation of a number of residues in GRK4α A486V relative to wild-type GRK4α, including Ser-485 in the kinase C-tail.
Introduction
The G-protein-coupled receptor (GPCR)2 family of membrane-bound receptors constitutes an important component of cellular signaling in a multitude of cellular processes. GPCR activity is modulated in part by phosphorylation of Ser and Thr residues in the intracellular loops and/or C-terminal tail. Phosphorylation is accomplished by a family of G-protein-coupled receptor kinases (GRK) that are members of the protein kinase A, G, and C (AGC) family of Ser/Thr kinases (1, 2). GPCR phosphorylation promotes high affinity binding of arrestin to the cytoplasmic surface of the receptor. This hinders further G-protein binding and activation and leads to receptor desensitization and internalization. GRK activity is essential to down-regulate GPCR activity, and GRK dysregulation is implicated in a wide range of disorders, including cardiac disease, night blindness, and Parkinson disease (2).
In contrast to the large diversity within the GPCR family, there are only seven members of the GRK family. GRK1–7 are multidomain proteins, consisting of a short N-terminal region followed by a regulator of G-protein signaling (RGS) homology domain (RH domain) and a highly conserved Ser/Thr kinase domain (1). The GRK C termini are highly divergent, but all contain features to facilitate membrane localization (3). GRK2 and GRK3 also have a C-terminal pleckstrin homology domain that binds G-protein βγ (Gβγ) subunits (2). The RH domain contains binding sites for many proteins, including calmodulin, actin, caveolin, and RKIP and plays regulatory and membrane localization roles (4, 5).
There are three GRK subfamilies based upon sequence identity. The GRK1 subfamily consists of GRK1 (rhodopsin kinase) and GRK7, which are expressed almost exclusively in the retina, whereas the GRK2 subfamily contains the widely expressed GRK2 and GRK3 (also called β-adrenergic receptor kinase-1 and -2). The GRK4 family contains GRK4, GRK5, and GRK6. GRK5 and GRK6 are widely expressed, but GRK4 expression is mainly restricted to the testis, cerebellum, and proximal tubules of the kidney. GRK4 is unusual as it is expressed as four splice variants (GRK4α, -β, -γ, and -δ) that differ in their cellular localization, receptor specificity, and ability to interact with other proteins (Fig. 1) (6–8).
FIGURE 1.

Sequence alignment of GRK4 splice variants α, β, γ, and δ with representative members of the GRK family. All sequences are human. Every 10th residue in GRK4α is denoted by a black asterisk above the alignment. The four GRK4 splice variants result from alternative splicing of exons 2 and 15 in the RH domain, with GRK4β missing exon 2 (residues 18–49), GRK4γ missing exon 15 (residues 516–561), and GRK4δ missing exons 2 and 15 (residues 18–49 and 516–561) relative to full-length GRK4 (GRK4α). The sequence of the kinase domain is identical for all four GRK4 splice variants. Conserved residues are shown in cyan; the DFG motif at the start of the activation loop (DLG in the GRK family) is shown in red, and GRK4 hypertension-related mutations are in magenta. The SA/VV motif (residues 485–487 in human GRK4α) is in blue. This motif is STV in human GRK5, human GRK6, human GRK1, dog GRK4, and bovine GRK4; SVI in rabbit GRK4; and SVV in rat, chimp, and mouse GRK4. The secondary structural elements of GRK4α are shown above the alignment, with cylinders representing α-helices, arrows representing β-strands, and lines representing unstructured residues. Secondary structural elements are colored as follows: RH terminal subdomain in orange; RH bundle subdomain in blue; kinase domain β-strands in yellow; and kinase domain α-helices in green. Disordered residues at the N and C termini in the structure of GRK4α are in gray. Additional helices at the N and C termini of GRK6 bound to sangivamycin (αNT and αCT) (41) are shown in purple.
The generally accepted model of GRK-mediated GPCR desensitization is that the GRK only phosphorylates the agonist-bound GPCR and that phosphorylation is required for desensitization (2). However, a number of studies suggest that the GRK4 subfamily is atypical, as GRK4–6 constitutively phosphorylate a number of GPCRs and can recruit arrestin binding in the absence of ligand (9, 10). Moreover, GRK4 desensitizes the GABA receptor in the absence of phosphorylation, indicating that a direct physical interaction may be sufficient to hinder G-protein-GPCR coupling (11, 12).
GRK4 is implicated in the regulation of blood pressure via effects upon dopamine signaling in the kidney (13), with high basal levels of phosphorylation of the dopamine D1 receptor (D1R) associated with hypertension (14). The link between D1R and GRK4 is supported by three single nucleotide polymorphisms in GRK4γ (R65L, A142V, and A486V) (15). These variants show increased GRK4 activity in renal proximal tubule and CHO cells and caused phosphorylation and agonist-independent uncoupling of D1R from its G-protein·effector enzyme complex. Transgenic mice overexpressing GRK4γ A142V or A486V also exhibit hypertension, when placed on regular (A142V) or high salt (A486V) diets (16).
Although similar studies for GRK4 hypertension variants have not been performed for the other isoforms, genetic studies in humans that do not distinguish between the four isoforms also link GRK4 single nucleotide polymorphisms, including A486V, with essential and/or salt-sensitive hypertension in some but not all populations (2, 8, 17–21). All four GRK4 isoforms are expressed in renal proximal tubule cells where GRK4α and GRK4γ constitutively phosphorylate D1R and D3R (15, 22) thus implying these two isoforms could play a role in salt reabsorption and hypertension in vivo. The role of GRK4 in hypertension may not be confined to dopamine receptor signaling, as GRK4 single nucleotide polymorphisms are also associated with increased expression of the angiotensin II type 1 receptor, which is another important determinant of blood pressure (8, 13). GRK4 variants have also been linked with decreased efficacy of beta blocker therapy and increased risk of adverse cardiovascular events in hypertensive patients (23).
All four GRK4 isoforms contain an intact kinase domain, but alternate splicing in GRK4β and -δ would result in loss of α0 and α1 helices of the RH domain, and GRK4γ and -δ would lose the C-terminal end of α10 and all of the α11 helix (Fig. 1). This loss of secondary structure brings the folding and stability of GRK4β, -γ, and -δ into question. Because of this observation, and given that the A486V polymorphism is genetically linked to hypertension in humans (20), the A486V variant in the context of GRK4α is the subject of this study.
X-ray structures of GRK1, GRK2, and GRK6 have been published (24–29). Three recent structures of GRK5 bound to AMPPNP, sangivamycin, and an inhibitor (CCG215022) are also in press (30, 31). The lack of a GRK4 structure hinders understanding of GRK4 function in particular and of the GRK family in general. Moreover, structures of each GRK would aid in the structure-guided design of selective GRK modulators for multiple disease indications. To address this gap, we present the 2.6 Å resolution structure of the hypertension-associated A486V polymorphic variant of GRK4α crystallized in the presence of the nonhydrolyzable ATP analog AMPPNP. We also present enhanced autophosphorylation by A486V as a possible mechanism for increased activity of the hypertension-related A486V mutation. These data unmask unique features of GRK4 within the GRK family and provide insights into the origins of substrate specificity and promiscuity in this important family of kinases.
Experimental Procedures
Cloning and Expression of GRK4α
DNA corresponding to full-length human GRK4α (NCBI accession number, NM_182982) was cloned into pVL1393 vector (Abvector, San Diego). The wild-type and A486V constructs were generated with the two modifications to the reference sequence. First, the putative palmitoylation sites (Cys-563 and Cys-578) were mutated to Ser to ensure a homogeneous preparation and eliminate the need for detergent during purification (26). Second, a tobacco etch virus protease-cleavable His8 affinity tag was incorporated at the C terminus. Sequences were confirmed with Sanger sequencing. Baculoviruses harboring GRK4α DNA were used to infect Sf21 insect cells at a multiplicity of infection of 0.1. Cells were grown at 27 °C and harvested 72 h post-infection by centrifugation (5000 × g, 20 min, 4 °C). The expression conditions were identified from a screen that varied multiplicity of infection (0.1–2), expression time (48–72 h), and cell type (Sf21 and Trichoplusia ni) using the Piccolo robot (TAP Biosystems, UK). Cell pellets were flash-frozen and stored at −80 °C.
Purification of GRK4α
Cell pellets were resuspended on ice (4.5 ml/g of cell weight) in lysis buffer (50 mm HEPES, pH 7.4, 0.5 m NaCl, 20 mm imidazole, 2 mm DTT, 10% (v/v) glycerol, 50 units/g benzonase, 2 mm MgCl2). EDTA-free protease inhibitor tablets (1 tablet/100 ml of lysate) (Roche Applied Science) were added, and cells were lysed by homogenization using a Dounce homogenizer, followed by two passes through a microfluidizer (Avestin, Ottawa, Canada). The lysate was clarified by centrifugation (45,000 × g, 45 min, 4 °C) and loaded onto a 10-ml HisTrap FF crude column (GE Healthcare), equilibrated with buffer A (50 mm HEPES, pH 7.4, 0.5 m NaCl, 40 mm imidazole, 2 mm DTT, 10% (v/v) glycerol). The column was washed with 10 column volumes of buffer A, and GRK4α was eluted from the column with a linear gradient of imidazole (40–500 mm). Fractions containing GRK4α were pooled and diluted 4-fold into buffer B (50 mm MES, pH 6.5, 1 mm EDTA, 2 mm DTT, 10% (v/v) glycerol) and loaded onto a 5-ml HiTrap SP HP column (GE Healthcare). After washing with buffer B containing 200 mm NaCl, a 5-ml HiTrap Heparin HP column (GE Healthcare) was attached to the output of the SP column, and GRK4α was eluted using a linear gradient of 200 mm to 1 m NaCl.
Fractions containing GRK4α were pooled, and the pH was adjusted to 7.5 for overnight tobacco etch virus protease cleavage at 4 °C, using a molar ratio of 30:1 GRK4α/TEV protease. The cleaved protein was purified using a 1-ml HisTrap FF column, followed by a HiLoad 16/60 Superdex 75 column (GE Healthcare), pre-equilibrated in buffer C (50 mm MES, pH 6.5, 0.5 m NaCl, 0.5 mm EDTA, 2 mm DTT or TCEP, 10% (v/v) glycerol). Samples were concentrated to 0.7 mg/ml using 10,000 molecular weight cutoff spin concentrators (EMD Millipore, Billerica, MA) and were either flash-frozen and stored at −80 °C or buffer-exchanged into buffer D (50 mm MES, pH 6.5, 0.25 m NaCl, 0.5 mm EDTA, 2 mm TCEP, 5% (v/v) glycerol) and concentrated to 10 mg/ml for crystallization. The final protein concentration was determined by amino acid analysis (AAA service Laboratory, Boring, OR) and molecular mass confirmed with whole-protein mass spectrometry. Approximate final yields were 0.2 mg/liter.
Analytical Ultracentrifugation
Sedimentation equilibrium experiments were performed with a Beckman XL-I centrifuge. Prior to loading, GRK4α A486V was dialyzed against buffer D at 4 °C for 24 h. Data were collected at 4 °C in a 12-mm path length 6-sector cell using loading concentrations spanning 3–12 μm and at rotor speeds ranging from 10,000 to 40,000 rpm. Data were analyzed using Origin (OriginLab, Northampton, MA) and fit to ideal single species and monomer-dimer models. Solvent densities of 1.029 g ml−1 and partial molar volumes of 0.726 ml g−1 were calculated using Sednterp (University of New Hampshire). Molecular mass and baseline offset were allowed to float during fits to the ideal single-species model. The log of the dissociation constant and the base-line offset were allowed to float while fitting using the self-associating model.
Crystallization
GRK4α (10 mg/ml) was mixed with AMPPNP/MgCl2 (final concentrations of 8 mm AMPPNP and 10 mm MgCl2). Initial crystals were grown using sitting-drop vapor diffusion at 4 °C in 0.1 m sodium citrate, pH 5–6.5, 15–27.5% (w/v) polyethylene glycol 3350 (PEG3350). Microseeding into 0.1 m sodium citrate, pH 5.5, 10% (w/v) PEG3350 improved crystal quality.
Data Collection and Structure Determination
The crystals were transferred to cryoprotectant buffer containing 20% (w/v) ethylene glycol in 12% (w/v) PEG3350 and vitrified by plunging into liquid nitrogen. The x-ray diffraction data were collected at Industrial Macromolecular Crystallography Association beamline 17-ID at the Advanced Photon Source. Data were reduced and scaled using HKL2000 (HKL Research, Charlottesville, VA). The structure was built and refined in iterative cycles using Coot (32) and autoBUSTER (Global Phasing). The AMPPN ligand (identifier AN2) was parameterized using Grade Version 1.2.8 with MOGUL and semi-empirical (RM1) QM restraints. Simulated annealing omit maps for the C-tail (residues Ala-471–Ala-493) and AMPPN ligands were calculated using the PHENIX software suite (34) with standard defaults. The dimerization interface was calculated with areaimol (CCP4) using a 1.5-Å probe. Figures were made using PyMOL Version 1.7.4 (Schrödinger, LLC). Atomic coordinates and structure factors were deposited in the PDB (code 4YHJ).
Structure Analysis and Peptide Docking
Cα kinase structure overlays were performed in PyMOL (Schrödinger, LLC). The large lobe angle was calculated by superimposing all structures over the hinge domain and then measuring the degree of rotation required to align the large lobes using the Cα of Met-267 in the middle of the hinge as the pivot point (26). This method gives an angle of rotation between PKB1 (1O6K) and GRK2 (3CIK) of 21°, which is comparable with the value of 20° reported previously (26). The GSK3β peptide from PKB (PDB code 1O6K) was spliced directly into the GRK4α structure using Maestro Version 9.7 (Schrödinger, LLC). AMPPN was remodeled to ATP in complex with two magnesium ions, and the structure was energy-minimized using the Polak-Ribiere conjugate gradient method, OPLS 2005 force field, convergence threshold of 0.05 kJ/mol, default water solvation, and fixed protein heavy atoms.
Peptide Phosphorylation Mobility Shift Assay
Wild-type and mutant D1R peptides containing 5-carboxyfluorescein at the N terminus and an amidated C terminus were purchased from CPC Scientific (Sunnyvale, CA). Peptide sequences are shown in Fig. 9. For ATP Km determination, GRK4α, ATP, and D1R-L1 peptide were diluted in assay buffer (50 mm HEPES, pH 7.5, 0.1 m NaCl, 10 mm MgCl2, 2 mm TCEP, 0.01% Tween 20, 0.1% (w/v) bovine serum albumin) and mixed to final concentrations of 10 nm GRK4, 2.4 μm to 5 mm ATP, 2 μm peptide, 0.13% (v/v) DMSO. Phosphorylated and unphosphorylated peptides were separated with a LabChip EZ Reader 12-Sipper Chip (catalog no. 760404) (PerkinElmer Life Sciences) and monitored in real time for 45 min using a Caliper LabChip EZ ReaderII instrument (PerkinElmer Life Sciences). The following instrument settings were used for separation: pressure = −1.2 p.s.i.; upstream voltage = −400 V; downstream voltage = −1500 V; post sample buffer sip time = 80 s; and lamp intensity = 100%. Product formation was determined as a ratio of product to substrate fluorescence, converted to moles, and plotted as a function of time. The initial rate at each ATP concentration was calculated from the slope using SigmaPlot (Systat, San Jose, CA). Only conditions under which product formation was linear with time were included for analysis, and the number of replicates was ≥3 for all experiments. Experiments were performed in 1, 2.5, 5, or 10% (v/v) DMSO to establish assay tolerance to DMSO.
FIGURE 9.
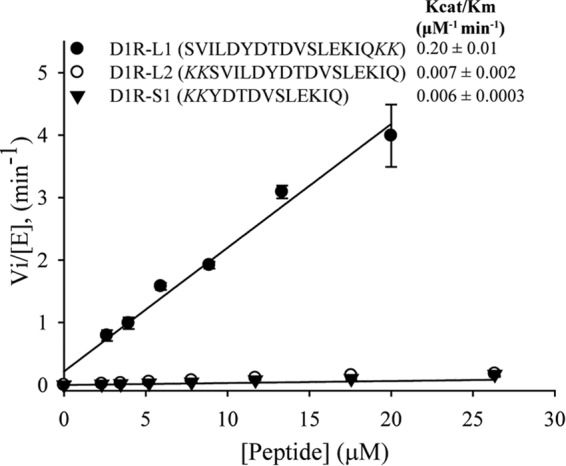
Initial rates of D1R peptide phosphorylation by GRK4α. kcat/Km values for D1R-L1, D1R-L2, and D1R-S1 are displayed on the figure. The data are derived from three independent experiments and the means ± S.D. are shown. Initial rate of D1R-S2 (YDTDVSLEKIQ) was very similar to D1R-S1 (kcat/Km of D1R-S2 = 0.0076 ± 0.0013 μm−1 min−1).
For autophosphorylation experiments, wild-type GRK4α and GRK4α A486V were diluted to 4 μm using assay buffer and mixed 1:1 with 400 μm ATP (+ATP preincubation) or assay buffer (−ATP preincubation). Samples were incubated at 22 °C for 45 min and then diluted 50-fold using assay buffer. Diluted samples were mixed 1:1 with 80 μm ATP and 4 μm D1R-L1 peptide (+ATP preincubation) or 84 μm ATP and 4 μm D1R-L1 peptide (−ATP preincubation) (final assay conditions: 20 nm GRK4α, 42 μm ATP, 2 μm peptide, 0.13% DMSO). Peptide phosphorylation was monitored for 45 min on the Caliper LabChip instrument as above.
To compare catalytic efficiency of different peptides, GRK4α was preincubated with ATP in assay buffer for 45 min at 22 °C and then mixed with increasing concentrations of peptide. Final conditions were 10 nm GRK4, 200 μm ATP, 1.8–30 μm peptide, 2% (v/v) DMSO. Instrument conditions were the same as D1R-L1 for all peptides except D1R-S2, where the conditions needed to be modified to see separation of substrate and product (pressure = −1.5 p.s.i.; upstream voltage e = −400 V; downstream voltage = −2200 V; and post sample buffer sip time = 160 s). Lamp intensity was also varied from 10 to 100% to ensure the fluorescence product signal was not saturated at higher peptide concentrations. Because of saturating substrate fluorescence under some conditions, product formation was measured directly rather than as a ratio of product to substrate. Channel-to-channel variability in signal intensity was normalized by monitoring the fluorescence of a 2 μm peptide solution run under the same conditions in each of the 12 channels.
Mass Spectrometry Analysis of Phosphorylation
Whole-protein mass spectrometry was performed with a ThermoFisher LTQ mass spectrometer. Peptide phosphomapping experiments were undertaken at the Southwest Michigan Innovation Center. GRK4α and GRK4α A486V were mixed with ATP in phosphorylation buffer (50 mm HEPES, pH 7.5, 0.1 m NaCl, 10 mm MgCl2) at final concentrations of 20 nm GRK4α and 40 μm ATP to mimic the conditions used in the −ATP preincubation peptide phosphorylation mobility shift assays. Samples were incubated for 0–30 min at 22 °C prior to quenching with 20 mm EDTA. Samples were either treated with trypsin in solution using filter-aided sample preparation or subjected to SDS-PAGE followed by in-gel trypsin digestion. Resulting peptides were analyzed by LC-MS/MS using a Waters Q-Tof Premier mass spectrometer on-line with a Waters NanoAcquity UPLC. Data were processed using PLGS (Waters). Output files were used to implicate phosphopeptides based on calculated mass using PepCompare (Southwest Michigan Innovation Center) and mass deviation of ± 0.03 Da. Confirmation of site-specific phosphorylation was performed manually using Waters MassLynx software. Phosphopeptide intensities were compared with the sum of the top three most intense nonphosphorylated peptides at each time point (35). Peptide sequence coverage averaged 89% for wild-type GRK4α and GRK4α A486V.
Results
Overall Structure of GRK4α A486V
The structure of GRK4α A486V crystallized in the presence of AMPPNP was solved at 2.6 Å resolution (Fig. 2). Data collection statistics are summarized in Table 1. Two molecules of GRK4α A486V are present in the asymmetric unit, and the final model contains residues 25–525 in chain A and 25–475 and 489–526 in chain B (Fig. 2A). Although the crystals were grown in the presence of AMPPNP, the γ-phosphate is not present in unbiased (Fig. 2C) and final refined electron density maps. Based on the electron density and previous reports of hydrolysis of AMPPNP to AMPPN in other kinase structures under mildly acidic conditions (36), AMPPN was modeled into both GRK4 molecules in the asymmetric unit.
FIGURE 2.
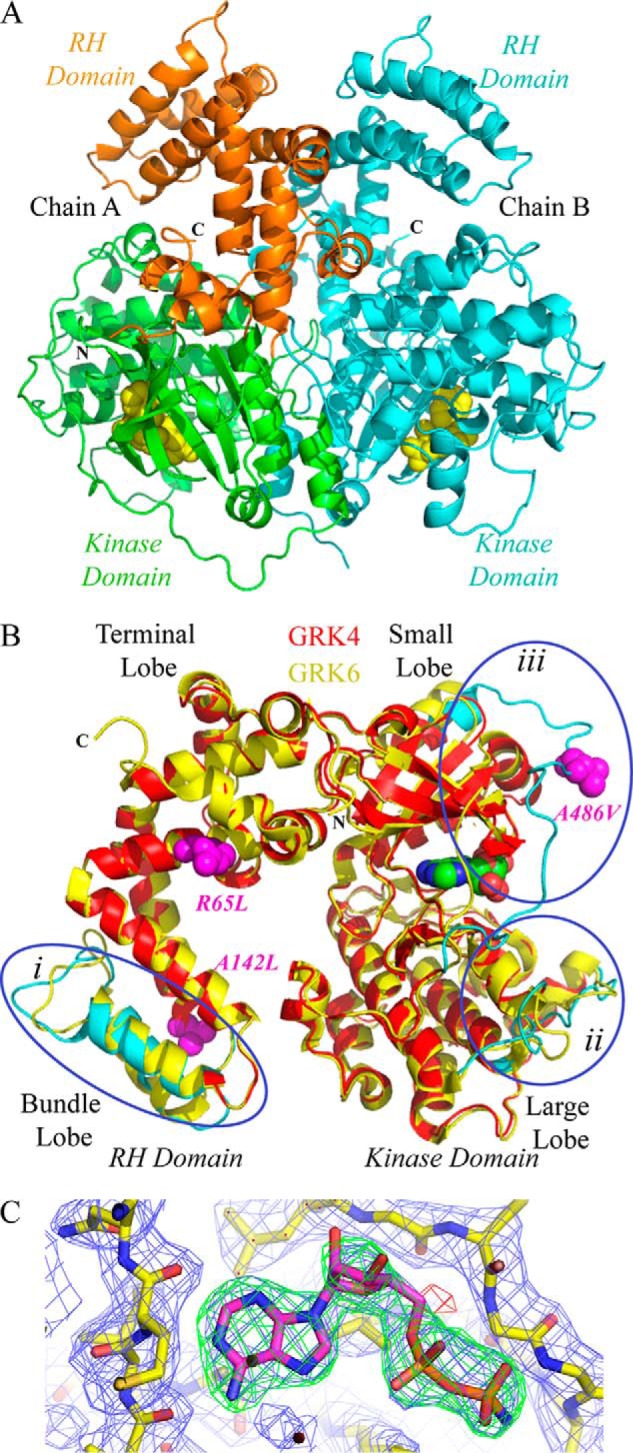
Crystal structure of human GRK4α A486V. A, crystal structure contains two molecules in the asymmetric unit. The N terminus (N) and C terminus (C) of chain A are labeled, and the C terminus is labeled for chain B. The N terminus of chain B is on the back of the structure as shown and is not visible. The RH domain of chain A is shown in orange, and the kinase domain of chain A is in green. Chain B is shown in cyan. AMPPN is in space-fill. B, GRK4α chain A (shown in red) overlaid with chain A of GRK6 bound to AMPPNP (26) (shown in yellow). Regions of the proteins that vary between the two structures are highlighted on the GRK4α structure in cyan. These are as follows: (i) the bundle subdomain of the RH domain; (ii) the substrate-binding channel of the kinase domain large lobe (e.g. αD–αE and αF-αG loops), and (iii) the region around the C-tail of the kinase domain. Hypertension-related mutations of GRK4 (R65L, A142L, and A486V) are shown in magenta. C, simulated annealing omit maps for AMPPN. 2m|Fo| − D|Fc|map (blue mesh) contoured at 1σ. The m|Fo| − D|Fc| maps are contoured at ±3σ and shown as green (positive density) and red (negative density) meshes.
TABLE 1.
Crystallographic data and refinement statistics
Values in parentheses are for the highest resolution bin.
| Crystallographic data | |
| Wavelength (Å) | 1.0 |
| Space group | P3121 |
| Unit cell parameters | a=b=104.8Å, c=221.8Å |
| α=β=90°, γ=120° | |
| Resolution (Å) | 2.60 |
| Observed reflections | 438,426 |
| Unique reflections | 44,032 |
| Completeness (%) | 99.5 (99.5) |
| Rsym (%)a | 9.1 (53) |
| 〈I/σ(I)〉 | 10.6 (2) |
| Refinement statistics | |
| Resolution limit (Å) | 47.33–2.60 (2.67–2.60) |
| Reflections used | 44,032 (3035) |
| R factor (%)b | 19.7 (24.8) |
| Rfree (%) | 24.6 (33.9) |
| No. of solvent molecules | 134 |
| r.m.s.d. bond length (Å)c | 0.010 |
| r.m.s.d. bond angle (°) | 1.2 |
| Ramachandran plot statistics (%) | |
| Residues in most favored region | 95.33 |
| Residues in additional allowed region | 3.25 |
| Residues in disallowed regions | 1.42 |
a Rsym (%) = Σ|Ii − 〈I〉|/ΣIi.
b R factor (%) = Σ‖Fo|−| Fc‖/Σ Fo|, where |Fo| and |Fc| are observed and calculated structure factor amplitudes, respectively; Rfree was calculated from a randomly chosen 10% of reflections excluded from refinement, and R factor was calculated for the remaining 90% of reflections.
c r.m.s.d. is the root mean square deviation from ideal geometry.
The overall GRK4α A486V monomer structure is most similar to that of GRK6 bound to AMPPNP (PDB code, 2ACX) (Fig. 2B) (26). The Cα r.m.s.d. between the kinase domains of these two structures is 0.6 Å. The Cα root mean square deviations with GRK1 (PDB code, 3C4Z), GRK2 (PDB code, 3CIK), and sangivamycin-bound GRK6 (PDB code, 3NYN) are larger at 1.1, 2.2, and 1.7 Å, respectively. The kinase domain of GRK4α also overlays well with PKB bound to GSK3β peptide (PDB code, 1O6K), with Cα r.m.s.d. of 1.6 Å.
Kinase Domain
GRK4α exhibits the typical AGC bilobal kinase fold that is also seen for GRK1, GRK2, and GRK6 (24–29). The protein contains some but not all elements necessary for full AGC kinase activation. In comparison with the closed conformation of active PKB (37), the N- and C-terminal lobes of GRK4α are in a more open conformation and would require a rotation of 12° to resemble the fully closed PKB-GSK3β peptide structure. The kinase domain of GRK4α is also more open than the sangivamycin-bound GRK6 structure by 8°, but it is similar to or slightly more closed than GRK1 (PDB code, 3C4Z), GRK6-AMPPNP (PDB code, 2ACX), and GRK2 (PDB code 3CIK), with rotational differences of 2, 5, and 8°, respectively.
Although the kinase domain of GRK4α is more open than for a fully active AGC kinase, features of the active state of AGC kinases are present. The DFG motif (DLG in the GRK family) is in the active DFG in conformation, and the catalytic Lys-216 is appropriately positioned to make interactions with Glu-235 in the fully ordered αC helix, and with the β-phosphate of the bound nucleotide (Fig. 3A). The P-loop is ordered and anchored by interactions between backbone carbonyls of Gly-196 and Gly-197 of the P-loop and the side chain of Arg-222 of helix αB (Fig. 3A).
FIGURE 3.

Key interactions in the GRK4α kinase domain. A, C-tail of GRK4α is shown in cyan, and the rest of the kinase domain is in yellow. Key residues and secondary structure elements in the C-tail and small lobe are labeled, and notable salt bridges and hydrogen bonds are shown. Conserved residues associated with GPCR and peptide phosphorylation in GRK1, GRK2, or GRK6 are highlighted in magenta. Ser-485 (autophosphorylation site) is in green, and the A486V hypertension-related polymorphism is shown in orange. AMPPN is shown in space-fill. B, simulated annealing omit map for the C-tail of GRK4α. The 2m|Fo| − D|Fc| map (blue mesh) contoured at 1σ. The m|Fo| − D|Fc| maps are contoured at ±3σ and shown as green (positive density) and red (negative density) meshes. Residues are colored as in A. C, structural overlay of the AST loop of GRK4α with two previous structures of GRK6. GRK4α is shown in cyan; GRK6 bound to AMPPNP (26) is in green, and GRK6 bound to sangivamycin (41) is in orange. Distances (Å) between specific atoms are shown. A hydrogen bond between the backbone amide of Asp-468 and the side chain hydroxyl of Tyr-473 previously observed in the structure of GRK6-sangivamycin (41) is conserved in GRK4α (corresponding residues for GRK4α are Asp-469 and Tyr-474). This is in contrast to GRK6-AMPPNP, where the distance between these atoms is over 14 Å. D, C-tail of GRK4α A486V (shown in cyan) overlaid with GRK6-sangivamycin (41) (shown in orange). The hydrogen bond between Lys-475 and Arg-190 in GRK6-sangivamycin is shown with black dashed lines and the Asp-476–Asp-479 turn in GRK6-sangivamycin is shown in gray. The SA/VV motif in GRK4α (STV in GRK6) is highlighted in italics.
The C-tail of the kinase domain (residues 451–503 in GRK4α) is fully ordered in chain A and partially ordered in chain B, with electron density missing for residues 476–488 (Fig. 3A). As with all AGC kinases, the C-tail of GRK4α begins with the C-lobe tether and active-site tether (AST) loop. The AST loop is formed by a hydrogen bond between the backbone amide of Asp-469 and the side chain hydroxyl of Tyr-474 (Fig. 3C), and it is preserved in both chains of GRK4, even though the whole C-tail is only visible in chain A. In contrast, the two equivalent residues in the GRK6-AMPPNP structure are over 13 Å apart and could not support formation of this hydrogen bond (26). The remainder of the C-tail forms a belt over the P-loop and αB helix of the N-lobe, with a combination of polar and nonpolar interactions stabilizing this conformation(Fig. 3A).
The backbone density in the C-tail belt region of GRK4α is continuous (Fig. 3B), although density is missing for the side chains of Glu-482, Gln-483, and Val-486. The side chains of Leu-479, Asp-480, Ile-481, and Phe-484, however, are well defined and act as anchors. Key residues in this region include Val-478 that packs against the P-loop. Also, the well resolved side chain carboxylate of Asp-480 is 3.4 Å from the backbone amide of Val-192 in the P-loop indicating a possible polar interaction. Three hydrophobic residues (Val-487, Ile-490, and Leu-492) pack against the side of the αB helix, with the backbone carbonyl groups of Val-487 and Ile-490 stabilizing the side chain of Lys-225 in the αB helix. Parts of the C-tail belt also pack against a symmetry related molecule (Fig. 3B). The C-terminal part of the C-tail is anchored in place by ionic interactions between Tyr-491, Asp-493, Asp-496, and Lys-224 in the αB helix. The start and end of the C-tail overlay well with the structure of GRK6-sangivamycin (Fig. 3D). However, the path taken by the C-tail belt is shifted significantly in GRK4α compared with GRK6-sangivamycin, with the residues around the STV motif flipped almost 180° in GRK4α when compared with GRK6-sangivamycin.
RH Domain of GRK4α
Two of the three GRK4 hypertension-associated polymorphisms (R65L and A142V) are located in the RH domain (Fig. 2B, magenta). The core of the RH domain in GRK4α contains 12 α-helices (α0–α11) that form bundle (α4–α7) and terminal (α0–α3 and α8–α11) subdomains. Residues in the RH bundle subdomain and kinase C-terminal lobe of GRK4α form a large, continuous acidic region similar to GRK2 (Fig. 4A). However, in GRK6 this region is much more basic. The structural differences between GRK4α and related proteins map specifically to helices α5 and α6 in the four-helix bundle subdomain of the RH domain. This region displays relatively low sequence conservation within the GRK family (25% identity and 59% similarity between GRK4α and GRK6 and 3% identity and 34% similarity between GRK4α and GRK2) and is the Gαq-binding site in GRK2 (38).
FIGURE 4.

Electrostatic surface of GRK4α compared with GRK2 and GRK6. The electrostatic surface is contoured to ±6 kT, with negative surface charge shown in red and positive surface charge in blue. The ATP binding pocket is highlighted by arrows. A, GRK2 (28) and GRK4α and GRK6 (26) are oriented as in Fig. 2B, with the RH domain on the left and kinase domain on the right. B, rotation by 180° around the vertical relative to A.
In contrast to the RH bundle subdomain and kinase C-terminal lobe described above, the RH terminal subdomain and N-terminal lobe are much more conserved with respect to surface charge distribution (Fig. 4A). This is not surprising as a number of regulatory proteins are reported to interact with this GRK face, including caveolin that binds to a conserved region straddling the acidic and basic patches (amino acids 63–71 in GRK2) (39). In addition, the basic patch contains binding sites for calmodulin, actin (residues 20–39), and phosphatidylinositol 4,5-bisphosphate (residues 22–28) (4, 5, 40) and is a likely membrane-binding surface (41).
GRK4α Displays Unusual Dimer Architecture in the Crystal Structure
The crystallographic dimer interface of GRK4α includes 4945 Å2 and is on the opposite side of the molecule from the acidic face described above (Figs. 4B and 5A). The activation loop of one monomer tucks into the dimer interface and packs against the RH and kinase domains of the opposing monomer, with a mixture of polar and nonpolar interactions (Fig. 5B). Also present is a favorable stacking interaction between Arg-237 in the αC helix of each monomer (Fig. 5C). The α7 helices of the RH domain form the edge of the GRK4α dimer interface and are arranged in a partially offset back-to-back manner, with key interactions between the side chain of Arg-148 in one monomer and the backbone carbonyl of Asn-152 in the other monomer (Fig. 5D). The GRK4α dimerization interface contrasts to the GRK1 and GRK6 dimer structures (Fig. 5A) (26, 27, 41), including GRK6/AMPPNP, in which the interface (3321 Å2) is mediated almost exclusively by α0–α1, α9, and α11 helices of the RH terminal subdomain (Fig. 5E) and is on a distinct face of the protein when compared with GRK4α (Fig. 5A).
FIGURE 5.

Comparison of GRK4α and GRK6. A, chains A of GRK4α (kinase domain shown in yellow and RH domain in red) and GRK6 (26) (shown in green) are overlaid. The corresponding B chains are shown in light blue for GRK4α and magenta for GRK6. The ligand is shown as space-fill. B, activation loop of one monomer (highlighted in blue and the rest of kinase domain in yellow) interacts with residues in the RH domain, kinase N-lobe, and kinase C-lobe of the other monomer (shown in cyan). C, Arg-237 of each αC helix monomer stacked on top of each other. D, interactions between residues in the α7 helix of the RH domain in monomers A (cyan) and B (red). E, alignment of residues in the RH domain (α0-α1 helix, α9 helix, and C-terminal end of α11 helix) that form a hydrophobic domain-swap interface in GRK6 and GRK1 crystal structures (26, 27, 41) or a C-terminal/RH domain packing interface within monomeric GRK5 structures (30, 31). Conserved residues in the domain-swap interface of GRK6-AMPPNP (26) are highlighted in yellow, and those also interacting in the GRK5 monomer structures (30, 31) are in green. The LXXDL motif (residues 534–538 of human GRK5) is conserved in GRK4α as described previously (30) and is underlined.
In contrast to the GRK4α crystal structure, sedimentation equilibrium analysis shows that GRK4α is a monomer in solution with a Kd above 12 μm (the highest concentration analyzed). The data are accounted for by an ideal single species model with a molecular mass of 63.6 ± 3 kDa (data not shown), similar to that expected for the monomer (67.4 kDa). Fits do not exhibit a systematic deviation of residuals nor a systematic change in apparent molecular mass with loading concentration or rotor speed (data not shown).
Enhanced Autophosphorylation of GRK4α A486V
Mass spectrometry was used to interrogate GRK4α autophosphorylation before and after incubation with ATP/MgCl2. Purified wild-type GRK4α and GRK4α A486V showed a mixture of unphosphorylated and singly phosphorylated species by whole-protein mass spectrometry (data not shown). Phosphopeptide-mapping analysis confirmed that this was due to low levels of phosphorylation at a number of residues, rather than at one particular residue (Fig. 6A). Treatment with 40 μm ATP/MgCl2 for 30 min resulted in an average of 3–6 phosphate groups per GRK4α by whole-protein mass spectrometry (data not shown). Phosphomapping analysis showed an initial increase in the abundance of one phosphopeptide relative to the basal state (Fig. 6B), although another did not change (Fig. 6C). Two new phosphorylation sites were also observed (Ser-139 and Ser-485). Phosphorylation of Ser-485 was much more efficient in GRK4α A486V compared with wild-type GRK4α (Fig. 6D). Autophosphorylation of a peptide containing Ser-244, Ser-249, and Thr-256 was also different between wild-type GRK4 and GRK4 A486V (Fig. 6E). Unlike Ser-485, this peptide was phosphorylated prior to ATP incubation. However, the abundance of this peptide in GRK4α A486V increased markedly after ATP treatment but did not change in wild-type GRK4α. With the exception of Ser-244 and Ser-249 that are buried in the structure, all of the autophosphorylation sites in GRK4α are solvent-exposed (Fig. 6F).
FIGURE 6.

Phosphopeptide mapping of wild-type GRK4α and GRK4α A486V with LC-MS/MS. A, phosphorylated peptides are highlighted in yellow, and phosphorylated residues in the basal state are underlined. Residues that show an increase in phosphorylation after incubation with ATP in GRK4α A486V (but not wild-type GRK4) are highlighted in green, and those that increase in both GRK4α variants are in magenta. Non-native serine residues C563S and C578S are shown in red. Residues that can be unambiguously assigned as phosphorylation sites are shown in boldface. For peptides where unambiguous assignment is not possible, all potential phosphorylation sites are highlighted. Vertical lines indicate instances where multiple versions of the same peptide are observed due to incomplete cleavage. Residues that were not observed in zero time point in-gel trypsin digest samples are highlighted in gray. B–E, effects of ATP incubation upon intensities of four representative phosphopeptides. Phosphopeptide intensities were normalized using the sum of the three most intense nonphosphorylated peptides. F, phosphorylation sites mapped on the crystal structure of GRK4α. Phosphorylated residues are shown as space-fill and colored as in A. Hypertension-related mutations are shown in yellow.
Autophosphorylation Affects Activity of Wild-type GRK4α but Not GRK4α A486V
A peptide based on D1R corresponding to residues 421–434 (D1R-L1) was used for substrate phosphorylation studies following autophosphorylation. Phosphorylation of D1R-L1 by wild-type GRK4α preincubated with ATP was linear with time (Fig. 7A). In contrast, a significant lag and a 2-fold decrease in rate of product formation were seen without ATP preincubation that persisted for the first 5–10 min (Fig. 7A). This result suggests that autophosphorylation plays a role in the activation of wild-type GRK4α. In contrast, preincubation with ATP had little effect on the initial rate of D1R-L1 phosphorylation by the A486V variant (Fig. 7A).
FIGURE 7.
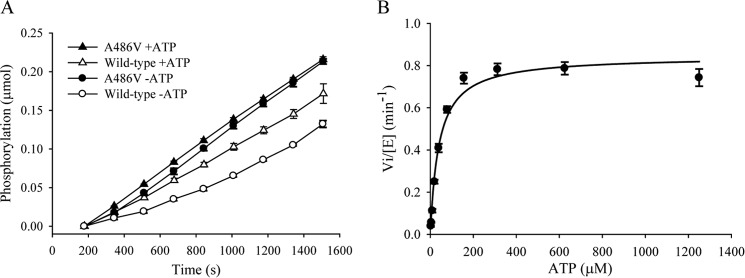
Kinetic studies of wild-type GRK4α and GRK4α A486V upon incubation with ATP. A, rate of D1R peptide phosphorylation is affected by preincubation with ATP for wild-type GRK4α (open circles versus open triangles) but not for GRK4α A486V (black circles versus black triangles). B, Km for ATP is 40 μm.
Substrate Binding and Recognition by GRK4
The substrate-binding channel of GRK4α is lined by residues of the kinase activation loop and the αD–αE and αF–αG loops of the C-lobe. Overall, the substrate-binding channel is neutral to basic, especially when compared with the acidic channel of PKB. The basic nature of the GRK4α-binding pocket is due in large part to a patch of Lys and Arg residues in the αF–αG loop. The αD–αE and αF–αG loops are poorly conserved across the AGC and GRK families, and the full αF–αG loop is not always visible in crystal structures of these kinases. However, this region was fully resolved in both monomers of GRK4α and is visibly different to related structures of GRK2 and GRK6 (Fig. 8).
FIGURE 8.
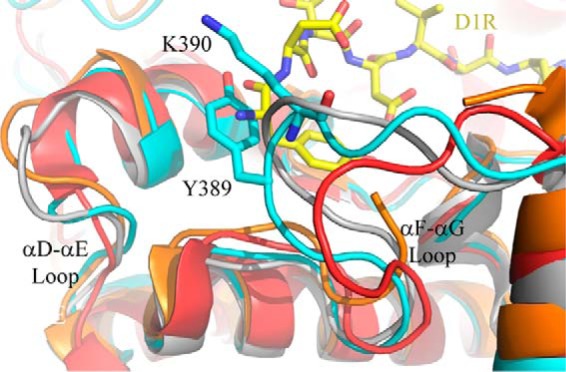
Overlay of the αD–αE and αF–αG loops of GRK kinase large lobes. GRK4α A486V is shown in cyan; GRK6-AMPPMP (26) is in gray; GRK6-sangivamycin (41) is in orange, and GRK2 (28) is in red. The D1R peptide (shown in yellow) was docked into GRK4α by structural alignment to the GSK3β peptide-bound structure of PKB (37). Docking based on this structure results in multiple clashes at the N-terminal side of the peptide with Tyr-389 and Lys-390 in the αF–αG loop.
The conformation of the D1R-L1 peptide bound to GRK4α was modeled by docking D1R-L1 into GRK4α using the PKB-GSK3β peptide structure as an initial model. Ser-431 was confirmed with mass spectrometry as the relevant phosphorylation site in the context of the peptide D1R-L1 by replacing Thr-428 or Ser-431 with Ala and observing that T428A remains a substrate but S431A does not (data not shown). A number of favorable interactions were formed with minimal side chain rearrangement. However, Tyr-389 and Lys-390 in the αF–αG loop of GRK4α clashed with the N terminus of the peptide (Fig. 8).
To understand substrate requirements for GRK4, wild-type and mutant D1R peptides of two different lengths (D1R-L and D1R-S) were tested for phosphorylation by GRK4α. Peptide solubility precluded direct determination of Km and Vmax values, so catalytic efficiencies between peptides were compared in the linear range of product conversion. The kcat/Km value for D1R-L1 was 0.20 ± 0.01 μm−1 min−1. However, removal of the first five residues of the peptide caused an ∼30-fold decrease in catalytic efficiency of GRK4α (Fig. 9, D1R-S1). This decrease in the kcat/Km value was not affected by the presence of two non-native C-terminal Lys residues that were necessary to enable sufficient capillary electrophoresis separation of substrate and product (data not shown). However, transferring these non-native Lys residues to the N terminus of D1R-L1 caused a significant decrease in catalytic efficiency (Fig. 9, D1R-L2).
Discussion
Here, we present the structure of a hypertension-associated A486V variant of GRK4α bound to an ATP analog. GRK4α A486V adopts the AGC kinase fold in a partially active conformation, with some but not all elements necessary for full activation. The additional N-terminal helix and C-terminal region observed in the GRK6-sangivamycin structure (41) are not visible in GRK4α A486V and are presumably disordered. Order or disorder of these regions may simply be ligand-dependent or the result of different crystallization conditions. Alternatively, the differences may be due to diversity in the activation mechanism of GRK4α and GRK6. It may be that, for GRK6, nucleotide binding promotes full ordering of the N-terminal helix, although in GRK4α, ordering of this region is not necessary for full activation, or ordering is promoted instead by GPCR binding.
GRK4α A486V crystallized with two molecules in the asymmetric unit that appear to associate closely (Fig. 2). The 4945 Å2 crystallographic dimer interface of GRK4α is larger than typical protein-protein interfaces (1000–2000 Å2 per molecule) (42), suggesting functional relevance to this apparent dimer. However, sedimentation equilibrium experiments suggest that GRK4α is a monomer in solution, as seen also for GRK6 and GRK5 (26, 31). Of the residues that form the core of a hydrophobic domain-swap dimer interface in the crystal structures of GRK6 and GRK1 (Fig. 5A), only two are conserved in human GRK4α (Fig. 5E), and it is unlikely that human GRK4 would form the same dimer. In GRK1-L166K a mutation in the domain-swap dimer interface caused the protein to crystallize in a monomeric form with the overall structure unchanged (43). The equivalent residue in GRK4 is Ser-166, and the structure of GRK4α is reminiscent of GRK1-L166K at this position. In GRK1-L166K, the C terminus packs against the RH domain within the same monomer. The GRK4α structure ends three residues earlier than GRK1-L166K but is also consistent with a similar packing interaction. Although it is not visible in the structure, GRK4α contains the LXXDL motif (Fig. 5E) that forms part of the same interface in two very recent crystal structures of GRK5 (30, 31).
It is possible to prevent dimerization and membrane localization of GRK5 by substitution of residues 165–169 with the equivalent residues in GRK4 (44). Notably, residues 165–169 are involved in the domain-swap interface in GRK6 and the RH/C-tail interface in GRK5 and GRK1-L166K (Fig. 5E). According to Xu et al. (44), GRK4 is primarily cytoplasmic, whereas GRK5 and GRK6 are predominantly localized to the cell membrane, and it is possible that cytoplasmic localization of GRK4 may be caused by an inability to dimerize. This would indicate divergence of GRK4 from other GRKs in cellular localization and regulation. Mechanisms of GRK4 regulation may also be species-specific, as there is significant sequence diversity between GRK4 orthologs (Fig. 5E). The domain-swap dimer interface residues in GRK6 are well conserved in rat GRK4 but not in human or chimp GRK4 (Fig. 5E). In contrast, the LXXDL motif that forms part of the interface between the RH domain and C-terminal region in GRK5 structures (30, 31) is conserved in human GRK4 but not in rat or mouse GRK4 (Fig. 5E). Human GRK4 may dimerize in vivo through a mechanism that may or may not be similar to the GRK4α crystal structure. It is also possible that the GRK4α crystallization dimer interface reflects a binding site for other proteins, as GRKs interact with a variety of proteins in addition to GPCRs (1). The mechanisms governing GRK4 activity, cellular localization, and in vivo relevance (or not) of GRK4 dimerization are important avenues for further exploration.
The kinase C-tail contains a number of conserved features and has been identified as an important regulatory module across the AGC kinase family (45). Peptide and GPCR phosphorylation studies suggest that GPCRs bind to GRKs at multiple sites, with high affinity binding to activate the GRK and lower affinity binding to the kinase active site (46). The C-tail is thought to play a role in kinase domain closure and activation (47–49). Full ordering of the C-tail is seen in chain A of the GRK4α structure (Fig. 3). C-tail ordering has not been observed in any other GRK structure with AMPPNP, ATP, or ADP bound (26, 27) with the very recent exception of GRK5-AMPPNP (31). This ordering allowed visualization of key residues known to affect phosphorylation of peptides and GPCRs in related GRKs (Fig. 3A, magenta). The C-tail in AMPPNP-bound GRK4α is similar, but not identical, to that in the GRK6-sangivamycin structure, in which the two lobes of the kinase adopt a more closed conformation than for GRK4α-AMPPNP (Fig. 3D) (41). The start and end of the C-tail overlay well, with features such as the AST loop hydrogen bond conserved between the two structures (Fig. 3C). The central part of the C-tail (residues 470–490) is less well defined but is shifted in GRK4α compared with GRK6-sangivamycin (Fig. 3D). A very recent structure of GRK5-AMPPNP (31) also shows the C-tail displaced compared with GRK6-sangivamycin, indicating likely conformational flexibility. Consistent with this, the small lobe of GRK4–6 contains a number of positively charged residues adjacent to the C-tail capable of making many different interactions, and the weak side chain electron density in the central part of the belt in GRK4α indicates flexibility. There are also symmetry-related molecules adjacent to the C-tail in the GRK4α chain A (Fig. 3B) and GRK5-AMPPNP (31). The different conformations observed may therefore be a result of crystal packing, conformational flexibility, and/or ordering of the N-terminal helix. However, the interactions linking the C-tail to important determinants of kinase activity, including the P-loop, αB helix, and αC helix, suggest that the C-tail conformation of GRK4α seen here may be relevant in vivo.
The C-tail contains the hypertension-related A486V mutation. Because of the poor density at this position in the structure, it is difficult to speculate on the functional consequences of the polymorphism. However, it is possible that the presence of Val at this position compared with Ala may alter the conformational preference of the C-tail and favor GPCR binding. Ala-486 is contained in a partially conserved SXV motif of the kinase C-tail, where X is residue 486 in human GRK4α (Fig. 1). The SXV motif varies across species (Fig. 1), and it is possible that the A486V polymorphism may result in a gain-of-function for human GRK4. However, the story may be more complicated than this, as wild-type GRK4α shows constitutive activity in HEK293 cells for D1R, but wild-type GRK4γ or GRK4γ A486V do not (9). This suggests that both the splice variant and mutation may play a role in GRK4 function (9).
Autophosphorylation of STV in the GRK5 kinase C-tail has been previously reported to increase the enzymatic activity of GRK5 by 15–20-fold for GPCRs and 2-fold for peptides (50). GRK4 is the most related protein to GRK5, although purified GRK4 has not previously been shown to autophosphorylate (46). This study demonstrates that GRK4α autophosphorylates Ser-485 in vitro and that phosphorylation of Ser-485 is much more efficient in GRK4α A486V than in wild-type GRK4α. GRK4α also autophosphorylates at a number of other residues that differ in basal phosphorylation or kinetics between wild-type GRK4α and GRK4 A486Vα (Fig. 6). All phosphorylation sites are on the surface of GRK4α except Ser-244 and Ser-249 that are buried in the structure and are much less likely to be phosphorylated than Thr-256 of the same peptide (Fig. 6E). In the context of a model peptide there is a lag in activity for wild-type GRK4α in the absence of preincubation with ATP that is not seen in GRK4α A486V (Fig. 7). These observations suggest that autophosphorylation may play a role in activation of GRK4α and in differences between wild-type GRK4 and GRK4 A486V in vivo. Alternatively, GRK4α A486V may not require autophosphorylation for activity, although wild-type GRK4α does. Further studies using full-length dopamine receptors are necessary to confirm the differences in behavior between wild-type GRK4α and GRK4α A486V seen using D1R peptides.
D1R and D3R are thought to be two of the most physiologically relevant receptors for water and electrolyte transport in the kidney (14, 15, 22) and are associated with hypertension. A peptide based on D1R residues 421–434 (D1R-L1) was chosen for phosphorylation studies, as D1R is a physiological substrate of GRK4. GRK4α exhibits a lag in D1R-L1 phosphorylation compared with GRK4α A486V. The difference between wild-type and A486V can be explained by enhanced autophosphorylation kinetics for GRK4α A486V relative to wild-type GRK4. Alternatively, it may be that autophosphorylation is not required for full activity of the A486V variant. Removal of five residues (SVILD) at the N terminus of the peptide, or addition of two non-native Lys residues at the N terminus, significantly decreased rates of phosphorylation. The N terminus of the D1R-L1 peptide may pack against residues in GRK4α to form interactions that are diminished or lost by their removal. Depending on the exact path taken by D1R, N-terminal non-native Lys residues could interact unfavorably with a number of Lys and Arg in the active-site region. The αF–αG loop would need to rearrange if D1R takes a similar path to GSK3β (Fig. 8). However, D1R is likely very flexible around the phosphorylation site, and it is possible that it may take a different path across GRK4α. Conformational changes of GRK4α upon GPCR binding may also alleviate this clash.
The surface charge distribution of the RH bundle subdomain of GRK4α resembles that of GRK2 and not GRK6 (Fig. 4A). GRK2 contains a Gα-binding site in this region (29), and although G-protein binding to GRK4α has not been shown directly, GRK4γ binds to Gαs and Gα13 in transfected HEK293T cells (33). The sequence of GRK4α in the RH bundle subdomain is identical to GRK4γ (Fig. 1), and based on the surface charge similarity between GRK2 and GRK4α, it is tempting to speculate that the RH domain of GRK4 may also interact with Gα. Although this is an interesting concept, sequence conservation across the GRK family is poor in this region, and further studies are needed to determine whether differences in surface charge between GRK2, GRK4α, and GRK6 are relevant in vivo. In summary, we present the structure of the human GRK4α Α486V complex with AMPNN. GRK4α-AMPPN is similar to GRK6-AMPPNP, although features of an active kinase C-tail that are missing in GRK6-AMPPNP, such as the AST loop hydrogen bond, are present in GRK4α. This indicates that GRK4α A486V may be in a more active conformation than GRK6 crystallized with AMPPNP. Consistent with this, the kinase domain of GRK4α is slightly more closed (5°) than GRK6-AMPPNP, although a further 12° rotation would be required to resemble the fully active structure of PKB bound to GSK3β peptide. Finally, phosphomapping studies suggest a mechanistic basis for differences between GRK4α and GRK4α A486V.
Author Contributions
S. J. A. purified GRK4, performed peptide phosphorylation and sedimentation equilibrium experiments, assisted with structure determination, and wrote the paper. G. P. and J. C. R. crystallized GRK4. S. K. M. and S. M. S. determined and analyzed the crystal structure. R. E. D., R. E. F., and J. M. S. designed and performed cloning, virus generation, expression testing, and large scale expression. D. L. H. and P. Z. assisted with phosphorylation experiments. S. A. J. designed and performed peptide docking and structural comparisons. K. W. R. assisted with GRK4 purification and performed autophosphorylation studies. S. J. A., P. L. D., and K. J. L. designed the study. K. J. L. also provided technical assistance with sedimentation equilibrium experiments and wrote the paper. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Gregory Cavey and Chris Hendrickson at the Southwest Michigan Innovation Center for mass spectrometry peptide phosphopeptide mapping experiments.
The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 4YHJ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- GPCR
- G-protein-coupled receptor
- AMPPNP
- 5′-adenylyl β,γ-imidodiphosphate
- AST
- active-site tether
- D1R
- dopamine D1 receptor
- D3R
- dopamine D3 receptor
- GRK
- GPCR kinase
- PKB
- protein kinase B
- RGS
- regulator of G-protein signaling
- RH
- RGS homology
- r.m.s.d.
- root mean-square deviation
- AGC
- protein kinase A, G, and C
- TCEP
- tris(2-carboxyethyl)phosphine
- PDB
- Protein Data Bank
- AMPPN
- adenosine 5′-diphosphoramidate.
References
- 1. Ribas C., Penela P., Murga C., Salcedo A., García-Hoz C., Jurado-Pueyo M., Aymerich I., Mayor F., Jr. (2007) The G protein-coupled receptor kinase (GRK) interactome: role of GRKs in GPCR regulation and signaling. Biochim. Biophys. Acta 1768, 913–922 [DOI] [PubMed] [Google Scholar]
- 2. Gurevich E. V., Tesmer J. J., Mushegian A., Gurevich V. V. (2012) G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol. Ther. 133, 40–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Homan K. T., Glukhova A., Tesmer J. J. (2013) Regulation of G protein-coupled receptor kinases by phospholipids. Curr. Med. Chem. 20, 39–46 [PubMed] [Google Scholar]
- 4. Pitcher J. A., Fredericks Z. L., Stone W. C., Premont R. T., Stoffel R. H., Koch W. J., Lefkowitz R. J. (1996) Phosphatidylinositol 4,5-bisphosphate (PIP2)-enhanced G protein-coupled receptor kinase (GRK) activity. Location, structure, and regulation of the PIP2 binding site distinguishes the GRK subfamilies. J. Biol. Chem. 271, 24907–24913 [DOI] [PubMed] [Google Scholar]
- 5. Pronin A. N., Satpaev D. K., Slepak V. Z., Benovic J. L. (1997) Regulation of G protein-coupled receptor kinases by calmodulin and localization of the calmodulin binding domain. J. Biol. Chem. 272, 18273–18280 [DOI] [PubMed] [Google Scholar]
- 6. Sallese M., Mariggiò S., Collodel G., Moretti E., Piomboni P., Baccetti B., De Blasi A. (1997) G protein-coupled receptor kinase GRK4. Molecular analysis of the four isoforms and ultrastructural localization in spermatozoa and germinal cells. J. Biol. Chem. 272, 10188–10195 [DOI] [PubMed] [Google Scholar]
- 7. Premont R. T., Macrae A. D., Stoffel R. H., Chung N., Pitcher J. A., Ambrose C., Inglese J., MacDonald M. E., Lefkowitz R. J. (1996) Characterization of the G protein-coupled receptor kinase GRK4. Identification of four splice variants. J. Biol. Chem. 271, 6403–6410 [DOI] [PubMed] [Google Scholar]
- 8. Jose P. A., Soares-da-Silva P., Eisner G. M., Felder R. A. (2010) Dopamine and G protein-coupled receptor kinase 4 in the kidney: role in blood pressure regulation. Biochim. Biophys. Acta 1802, 1259–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rankin M. L., Marinec P. S., Cabrera D. M., Wang Z., Jose P. A., Sibley D. R. (2006) The D1 dopamine receptor is constitutively phosphorylated by G protein-coupled receptor kinase 4. Mol. Pharmacol. 69, 759–769 [DOI] [PubMed] [Google Scholar]
- 10. Li L., Homan K. T., Vishnivetskiy S. A., Manglik A., Tesmer J. J., Gurevich V. V., Gurevich E. V. (2015) G protein-coupled receptor kinases of the GRK4 protein subfamily phosphorylate inactive G protein-coupled receptors (GPCRs). J. Biol. Chem. 290, 10775–10790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Perroy J., Adam L., Qanbar R., Chénier S., Bouvier M. (2003) Phosphorylation-independent desensitization of GABA(B) receptor by GRK4. EMBO J. 22, 3816–3824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kanaide M., Uezono Y., Matsumoto M., Hojo M., Ando Y., Sudo Y., Sumikawa K., Taniyama K. (2007) Desensitization of GABA(B) receptor signaling by formation of protein complexes of GABA(B2) subunit with GRK4 or GRK5. J. Cell. Physiol. 210, 237–245 [DOI] [PubMed] [Google Scholar]
- 13. Harris R. C., Zhang M. Z. (2012) Dopamine, the kidney, and hypertension. Curr. Hypertens. Rep. 14, 138–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sanada H., Jose P. A., Hazen-Martin D., Yu P. Y., Xu J., Bruns D. E., Phipps J., Carey R. M., Felder R. A. (1999) Dopamine-1 receptor coupling defect in renal proximal tubule cells in hypertension. Hypertension 33, 1036–1042 [DOI] [PubMed] [Google Scholar]
- 15. Felder R. A., Sanada H., Xu J., Yu P. Y., Wang Z., Watanabe H., Asico L. D., Wang W., Zheng S., Yamaguchi I., Williams S. M., Gainer J., Brown N. J., Hazen-Martin D., Wong L. J., et al. (2002) G protein-coupled receptor kinase 4 gene variants in human essential hypertension. Proc. Natl. Acad. Sci. U.S.A. 99, 3872–3877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Felder R. A., Jose P. A. (2006) Mechanisms of disease: the role of GRK4 in the etiology of essential hypertension and salt sensitivity. Nat. Clin. Pract. Nephrol. 2, 637–650 [DOI] [PubMed] [Google Scholar]
- 17. Liu C., Xi B. (2012) Pooled analyses of the associations of polymorphisms in the GRK4 and EMILIN1 genes with hypertension risk. Int. J. Med. Sci. 9, 274–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sanada H., Yatabe J., Midorikawa S., Hashimoto S., Watanabe T., Moore J. H., Ritchie M. D., Williams S. M., Pezzullo J. C., Sasaki M., Eisner G. M., Jose P. A., Felder R. A. (2006) Single-nucleotide polymorphisms for diagnosis of salt-sensitive hypertension. Clin. Chem. 52, 352–360 [DOI] [PubMed] [Google Scholar]
- 19. Speirs H. J., Katyk K., Kumar N. N., Benjafield A. V., Wang W. Y., Morris B. J. (2004) Association of G-protein-coupled receptor kinase 4 haplotypes, but not HSD3B1 or PTP1B polymorphisms, with essential hypertension. J. Hypertens. 22, 931–936 [DOI] [PubMed] [Google Scholar]
- 20. Rayner B., Ramesar R. (2015) The importance of G protein-coupled receptor kinase 4 (GRK4) in pathogenesis of salt sensitivity, salt sensitive hypertension and response to antihypertensive treatment. Int. J. Mol. Sci. 16, 5741–5749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sanada H., Yoneda M., Yatabe J., Williams S. M., Bartlett J., White M. J., Gordon L. N., Felder R. A., Eisner G. M., Armando I., Jose P. A. (2015) Common variants of the G protein-coupled receptor type 4 are associated with human essential hypertension and predict the blood pressure response to angiotensin receptor blockade. Pharmacogenomics J. 10.1038/tpj.2015.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Villar V. A., Jones J. E., Armando I., Palmes-Saloma C., Yu P., Pascua A. M., Keever L., Arnaldo F. B., Wang Z., Luo Y., Felder R. A., Jose P. A. (2009) G protein-coupled receptor kinase 4 (GRK4) regulates the phosphorylation and function of the dopamine D3 receptor. J. Biol. Chem. 284, 21425–21434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vandell A. G., Lobmeyer M. T., Gawronski B. E., Langaee T. Y., Gong Y., Gums J. G., Beitelshees A. L., Turner S. T., Chapman A. B., Cooper-DeHoff R. M., Bailey K. R., Boerwinkle E., Pepine C. J., Liggett S. B., Johnson J. A. (2012) G protein receptor kinase 4 polymorphisms: β-blocker pharmacogenetics and treatment-related outcomes in hypertension. Hypertension 60, 957–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lodowski D. T., Barnhill J. F., Pyskadlo R. M., Ghirlando R., Sterne-Marr R., Tesmer J. J. (2005) The role of Gβγ and domain interfaces in the activation of G protein-coupled receptor kinase 2. Biochemistry 44, 6958–6970 [DOI] [PubMed] [Google Scholar]
- 25. Lodowski D. T., Pitcher J. A., Capel W. D., Lefkowitz R. J., Tesmer J. J. (2003) Keeping G proteins at bay: a complex between G protein-coupled receptor kinase 2 and Gβγ. Science 300, 1256–1262 [DOI] [PubMed] [Google Scholar]
- 26. Lodowski D. T., Tesmer V. M., Benovic J. L., Tesmer J. J. (2006) The structure of G protein-coupled receptor kinase (GRK)-6 defines a second lineage of GRKs. J. Biol. Chem. 281, 16785–16793 [DOI] [PubMed] [Google Scholar]
- 27. Singh P., Wang B., Maeda T., Palczewski K., Tesmer J. J. (2008) Structures of rhodopsin kinase in different ligand states reveal key elements involved in G protein-coupled receptor kinase activation. J. Biol. Chem. 283, 14053–14062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tesmer J. J., Tesmer V. M., Lodowski D. T., Steinhagen H., Huber J. (2010) Structure of human G protein-coupled receptor kinase 2 in complex with the kinase inhibitor balanol. J. Med. Chem. 53, 1867–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tesmer V. M., Kawano T., Shankaranarayanan A., Kozasa T., Tesmer J. J. (2005) Snapshot of activated G proteins at the membrane: the Gαaq-GRK2-Gβγ complex. Science 310, 1686–1690 [DOI] [PubMed] [Google Scholar]
- 30. Homan K. T., Waldschmidt H. V., Glukhova A., Cannavo A., Song J., Cheung J. Y., Koch W. J., Larsen S. D., Tesmer J. J. (June 1, 2015) Crystal structure of G protein-coupled receptor kinase 5 in complex with a rationally designed inhibitor. J. Biol. Chem. 290, 10.1074/jbc.M115.647370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Komolov K. E., Bhardwaj A., Benovic J. L. (June 1, 2015) Atomic structure of G protein-coupled receptor kinase 5 (GRK5) reveals distinct structural features novel for GRKs. J. Biol. Chem. 290, 10.1074/jbc.M115.647297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Keever L. B., Jones J. E., Andresen B. T. (2008) G protein-coupled receptor kinase 4γ interacts with inactive Gα(s) and Gα13. Biochem. Biophys. Res. Commun. 367, 649–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Adams P. D., Afonine P. V., Bunkoczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Silva J. C., Gorenstein M. V., Li G. Z., Vissers J. P., Geromanos S. J. (2006) Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol. Cell. Proteomics 5, 144–156 [DOI] [PubMed] [Google Scholar]
- 36. Ferguson A. D., Sheth P. R., Basso A. D., Paliwal S., Gray K., Fischmann T. O., Le H. V. (2011) Structural basis of CX-4945 binding to human protein kinase CK2. FEBS Lett. 585, 104–110 [DOI] [PubMed] [Google Scholar]
- 37. Yang J., Cron P., Good V. M., Thompson V., Hemmings B. A., Barford D. (2002) Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat. Struct. Biol. 9, 940–944 [DOI] [PubMed] [Google Scholar]
- 38. Day P. W., Tesmer J. J., Sterne-Marr R., Freeman L. C., Benovic J. L., Wedegaertner P. B. (2004) Characterization of the GRK2 binding site of Gαq. J. Biol. Chem. 279, 53643–53652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Carman C. V., Lisanti M. P., Benovic J. L. (1999) Regulation of G protein-coupled receptor kinases by caveolin. J. Biol. Chem. 274, 8858–8864 [DOI] [PubMed] [Google Scholar]
- 40. Freeman J. L., De La Cruz E. M., Pollard T. D., Lefkowitz R. J., Pitcher J. A. (1998) Regulation of G protein-coupled receptor kinase 5 (GRK5) by actin. J. Biol. Chem. 273, 20653–20657 [DOI] [PubMed] [Google Scholar]
- 41. Boguth C. A., Singh P., Huang C. C., Tesmer J. J. (2010) Molecular basis for activation of G protein-coupled receptor kinases. EMBO J. 29, 3249–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Arkin M. R., Tang Y., Wells J. A. (2014) Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem. Biol. 21, 1102–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tesmer J. J., Nance M. R., Singh P., Lee H. (2012) Structure of a monomeric variant of rhodopsin kinase at 2.5 A resolution. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 622–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xu H., Jiang X., Shen K., Fischer C. C., Wedegaertner P. B. (2014) The regulator of G protein signaling (RGS) domain of G protein-coupled receptor kinase 5 (GRK5) regulates plasma membrane localization and function. Mol. Biol. Cell 25, 2105–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kannan N., Haste N., Taylor S. S., Neuwald A. F. (2007) The hallmark of AGC kinase functional divergence is its C-terminal tail, a cis-acting regulatory module. Proc. Natl. Acad. Sci. U.S.A. 104, 1272–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Palczewski K. (1997) GTP-binding-protein-coupled receptor kinases-two mechanistic models. Eur. J. Biochem. 248, 261–269 [DOI] [PubMed] [Google Scholar]
- 47. Huang C. C., Yoshino-Koh K., Tesmer J. J. (2009) A surface of the kinase domain critical for the allosteric activation of G protein-coupled receptor kinases. J. Biol. Chem. 284, 17206–17215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sterne-Marr R., Leahey P. A., Bresee J. E., Dickson H. M., Ho W., Ragusa M. J., Donnelly R. M., Amie S. M., Krywy J. A., Brookins-Danz E. D., Orakwue S. C., Carr M. J., Yoshino-Koh K., Li Q., Tesmer J. J. (2009) GRK2 activation by receptors: role of the kinase large lobe and carboxyl-terminal tail. Biochemistry 48, 4285–4293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Beautrait A., Michalski K. R., Lopez T. S., Mannix K. M., McDonald D. J., Cutter A. R., Medina C. B., Hebert A. M., Francis C. J., Bouvier M., Tesmer J. J., Sterne-Marr R. (2014) Mapping the putative G protein-coupled receptor (GPCR) docking site on GPCR kinase 2: insights from intact cell phosphorylation and recruitment assays. J. Biol. Chem. 289, 25262–25275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kunapuli P., Gurevich V. V., Benovic J. L. (1994) Phospholipid-stimulated autophosphorylation activates the G protein-coupled receptor kinase GRK5. J. Biol. Chem. 269, 10209–10212 [PubMed] [Google Scholar]