

Background: Hepatic steatosis is an increasing health concern associated with metabolic syndrome.
Results: RGC-32 deficiency protects mice from HFD- and ethanol-induced hepatic steatosis by decreasing hepatic lipogenic gene expression.
Conclusion: RGC-32 is a novel regulator for hepatic lipogenesis.
Significance: RGC-32 may be a novel target for treating hepatic steatosis.
Keywords: gene expression, lipogenesis, liver, metabolism, transcription, response gene to complement 32, hepatic steatosis, stearoyl-CoA desaturase 1
Abstract
Hepatic steatosis is associated with insulin resistance and metabolic syndrome because of increased hepatic triglyceride content. We have reported previously that deficiency of response gene to complement 32 (RGC-32) prevents high-fat diet (HFD)-induced obesity and insulin resistance in mice. This study was conducted to determine the role of RGC-32 in the regulation of hepatic steatosis. We observed that hepatic RGC-32 was induced dramatically by both HFD challenge and ethanol administration. RGC-32 knockout (RGC32−/−) mice were resistant to HFD- and ethanol-induced hepatic steatosis. The hepatic triglyceride content of RGC32−/− mice was decreased significantly compared with WT controls even under normal chow conditions. Moreover, RGC-32 deficiency decreased the expression of lipogenesis-related genes, sterol regulatory element binding protein 1c (SREBP-1c), fatty acid synthase, and stearoyl-CoA desaturase 1 (SCD1). RGC-32 deficiency also decreased SCD1 activity, as indicated by decreased desaturase indices of the liver and serum. Mechanistically, insulin and ethanol induced RGC-32 expression through the NF-κB signaling pathway, which, in turn, increased SCD1 expression in a SREBP-1c-dependent manner. RGC-32 also promoted SREBP-1c expression through activating liver X receptor. These results demonstrate that RGC-32 contributes to the development of hepatic steatosis by facilitating de novo lipogenesis through activating liver X receptor, leading to the induction of SREBP-1c and its target genes. Therefore, RGC-32 may be a potential novel drug target for the treatment of hepatic steatosis and its related diseases.
Introduction
Hepatic steatosis, also known as fatty liver, is characterized by the accumulation of triglycerides in the liver (1) and is a uniform response of the liver to excessive energy intake or alcohol consumption (2–4). It is the most common liver disease in the United States, with a high prevalence in the obese and type 2 diabetic population, but it is probably underestimated as a cause for cirrhosis (5). Therefore, there is an increasing interest in understanding the pathogenesis of the fatty liver so that effective therapies may be developed by targeting factors that sustain steatosis.
Although the molecular mechanism regulating the development of hepatic steatosis is complex, recent studies have shown that targeting enzymes important for lipogenesis in the liver may be an effective approach to treat hepatic steatosis (6). It is well established that sterol regulatory element (SRE)3 binding protein (SREBP) 1c (SREBP-1c), a transcriptional factor primarily expressed in the liver, is a critical mediator for the transcription of lipogenic enzymes (7). Stearoyl-CoA desaturase 1 (SCD1), a lipogenic gene regulated by SREBP-1c, plays a central role in the de novo lipogenic pathway (8). SCD1 catalyzes the rate-limiting step in the biosynthesis of monounsaturated fatty acids from saturated fatty acids. It introduces a single double bond into its substrates, palmitic acid (16:0) and stearic acid (18:0), to generate palmitoleic (16:1n-7) and oleic acid (18:1n-9). These products are the most abundant fatty acids found in triglycerides, cholesterol esters, and phospholipids. Mice with a global or liver-specific knockout of SCD1 are protected from diet-induced obesity, hepatic steatosis, and insulin resistance (9–11).
Response gene to complement 32 (RGC-32) is expressed in numerous organs and tissues, including the liver (12). Functionally, RGC-32 plays an important role in cell proliferation, differentiation (13–15), fibrosis (16), and cancer (17–19). We found recently that RGC-32 deficiency (RGC32−/−) protected mice from high-fat diet (HFD)-induced obesity and insulin resistance through alleviating inflammation and increasing energy expenditure of adipose tissue (20). In this study, we found that RGC32−/− prevented HFD- and ethanol-induced hepatic steatosis in mice through suppressing de novo lipogenesis. Our results indicate that RGC-32 is potentially a novel regulator of hepatic lipid metabolism.
Experimental Procedures
Animals and Diets
RGC32−/− mice on the C57BL/6 background were generated and genotyped as described previously (21). Parallel line WT C57BL/6 mice were purchased from The Jackson Laboratory. Age-matched WT and RGC32−/− male mice were maintained on a regular chow for 8 weeks, after which they were fed with either normal chow (13% calories from fat, 3.07 kcal/g; catalog no. 5053, LabDiet) or an HFD (40% calories from fat, 4.5 kcal/g; catalog no. D12108C, Research Diets) for an additional 12 weeks. Mice were fasted overnight and anesthetized (2.0% isoflurane), and blood was collected by direct cardiac puncture. The liver was removed carefully and weighed. For generating ethanol-induced fatty liver, 8-week-old WT and RGC32−/− male mice received three acute doses of ethanol (4.7 g/kg bodyweight, 40% diluted in water, v/v) with a 12-h interval by oral gavage. Mice were sacrificed 12 h after the final administration, and whole livers were collected. A portion of the liver was fixed in 4% paraformaldehyde for histological analysis, whereas the other portions were stored at −80 °C for other assays. All animals were housed under conventional conditions in the animal care facilities and received humane care in compliance with the Principles of Laboratory Animal Care formulated by the National Society for Medical Research and the Guide for the Care and Use of Laboratory Animals. All experimental procedures were approved by Institutional Animal Care and Use Committee of the University of Georgia.
Isolation and Culture of Primary Hepatocytes
Hepatocytes were isolated from 8- to 12-week-old male C57BL/6 mice as described previously (22). Briefly, mice were anesthetized with 2.0% isoflurane, portal veins were cannulated, and livers were perfused with buffer A (10 mmol/liter HEPES, 143 mmol/liter NaCl, 7 mmol/liter KCl, and 0.2 mmol/liter EDTA (pH 7.4)), followed by buffer B (50 mmol/liter HEPES, 100 mmol/liter NaCl, 7 mmol/liter KCl, and 5 mmol/liter CaCl2 (pH 7.6)) containing type IV collagenase (0.5 g/liter, Sigma) at 37 °C for 10 min. After perfusion, the livers were excised, transferred to DMEM, and cut into small pieces. The cell suspension was passed through 70-μm nylon mesh filters, centrifuged at 50 × g, and resuspended in DMEM supplemented with 10% FBS to inhibit residual collagenase activity. The hepatocytes were then plated onto gelatin-coated plates at a density of 105 cells/cm2 and cultured in DMEM supplemented with 10% FBS. The viability of the cells was tested with 0.4% trypan blue.
RNA Extraction and Real-time Quantitative RT-PCR (qPCR) Analysis
qPCR was performed as described previously (23). Briefly, total RNA from tissues or cultured cells was extracted using TRIzol reagent (Invitrogen). 1 μg of total RNA was reverse-transcribed using the iScript cDNA synthesis kit (Bio-Rad). qPCR was performed in a Mx3005P qPCR machine using SYBR Green Master Mix (Agilent Technologies). Each sample was amplified in triplicate. The expression of each gene was normalized with cyclophilin.
Western Blot Analysis
Western blotting was performed as described previously (24). Antibodies against SREBP-1c, FAS, LXR-α (Abcam), SCD1, p-Akt, Akt, p-NF-κB, NF-κB, α-tubulin (Cell Signaling Technology), and RGC-32 were used. Protein expressions were detected with an enhanced chemiluminescence kit (Millipore).
Histological Analysis
Liver sections (5 μm) were deparaffinized and stained with H&E. Oil Red O staining of the frozen liver sections (12 μm) and quantitative analysis were performed as described previously (25). Images were captured with a Nikon microscope.
Measurement of Desaturase Indices
Total lipids were extracted from serum or liver samples, methylated, and analyzed by gas chromatography as described previously (26). Briefly, total fatty acid composition was determined using a Shimadzu GC-2010 analyzer. The column was a DB-23 (catalog no. 123-2332, J&W Scientific): length, 30 m; inner diameter, 0.32-mm-wide bore; film thickness, 0.25 μm. The carrier gas is helium with a column flow rate of 2.5 ml/min. The analysis of fatty acid methyl esters was made on the basis of the area under the curve, calculated with Shimadzu Class VP 7.4 software. Fatty acid identification was made on the basis of retention times of authenticated fatty acid methyl ester standards (Matreya LLC Inc.). Our primary analysis focused on the principle substrates (16:0, 18:0) and products of SCD1 (16:1n-7, 18:1n-9). The desaturase indices were calculated as the ratios of 16:1/16:0 and 18:1/18:0.
SCD1 Promoter Luciferase Reporter Constructs
The mouse SCD1 promoter DNA fragments from −500 or −250 to +300 bp relative to the mapped transcription start sites were obtained by PCR amplification from mouse genomic DNA using primers containing restriction enzyme sites of KpnI or XhoI. The PCR fragments were digested with restriction enzymes and cloned into the pGL4 vector. The SRE mutation was generated by replacing AGAG to CGCG as described previously (27, 28) using a site-directed mutagenesis kit (Agilent). The promoter regions of PCR products were verified by sequencing.
Transient Transfection and Luciferase Assay
Transient transfection and the luciferase assay were performed as described previously (29). Briefly, mouse primary hepatocytes were cultured for 24 h and then transduced with an adenovirus expressing GFP (Ad-GFP), RGC-32 shRNA (Ad-shRGC32), or RGC-32 (Ad-RGC32). 24 h later, different SCD1 promoter constructs were transfected into the cells with Lipofectamine LTX (Invitrogen) for 48 h, followed by a luciferase assay using the Dual-Luciferase reporter assay system (Promega). For each sample, a Renilla luciferase vector was cotransfected as an internal control, and promoter activities were normalized accordingly.
ChIP
A ChIP assay was performed as described previously (29). Briefly, mouse primary hepatocytes were transduced with Ad-GFP, Ad-shRGC32, or Ad-RGC32 for 48 h. Chromatin complexes were immunoprecipitated with SREBP-1c antibody, normal rabbit IgG (negative control), LXR-α antibody (Abcam), or normal mouse IgG (negative control). Semiquantitative PCR and qPCR were performed to amplify the SCD1 promoter region containing functional SRE using the primer set 5′-ATC AGC CCA CAG CAA AGA GG-3′ (forward) and 5′-AGC CCG TCT TGT CAT TGG CC-3′ (reverse), and, to amplify the SREBP-1c promoter region containing the functional LXR element, the primer set 5′-ATG GTT GCC TGT GCG GCA GGG GTT-3′ (forward) and 5′-GGT TTC TCC CGG TGC TCT GAA T-3′ (reverse) was used.
Statistical Analysis
Data are presented as means ± S.D., and the number of independent experiments is indicated for each data set. For statistical analysis, two groups were compared using two-tailed Student's t tests, and three groups were evaluated by one-way analysis of variance followed by Tukey's multiple comparisons. Four groups were evaluated by two-way analysis of variance followed by Bonferroni post hoc tests for multiple comparisons using GraphPad Prism 5.0 software. p < 0.05 was considered statistically significant.
Results
RGC-32 Deficiency Prevented HFD-induced Hepatic Steatosis
Our previous studies have shown that RGC-32 expression in adipose tissue was induced by HFD challenge, and RGC-32 deficiency prevented HFD-induced obesity and insulin resistance (20). It is well known that obesity is often accompanied by hepatic steatosis (30). To test whether RGC-32 affects liver during HFD-induced obesity, we first detected RGC-32 expression in liver tissues from WT mice fed normal chow or a 12-week HFD. We found that both mRNA and protein expression of RGC-32 was dramatically up-regulated by the HFD challenge (Fig. 1, A and B). Then we examined whether RGC-32 plays a role in the regulation of HFD-induced hepatic steatosis. Both H&E and Oil Red O staining showed that HFD-fed WT mice had severe hepatic steatosis with numerous lipid droplets (Fig. 1, C–E). However, RGC32−/− dramatically alleviated HFD-induced hepatic steatosis, as evidenced by significantly fewer lipid droplets (Fig. 1, C–E). The hepatic triglyceride content of RGC32−/− mice was also significantly lower than that of WT mice fed both normal chow and an HFD (Fig. 1F). Consistently, an HFD significantly increased the liver weight of WT but not RGC32−/− mice (Fig. 1G). These data indicate that RGC32−/− protects mice from developing HFD-induced hepatic steatosis.
FIGURE 1.

RGC-32 deficiency prevented HFD-induced hepatic steatosis. A and B, RGC-32 mRNA (A) and protein (B) expression in liver tissues of WT mice fed with normal chow or a 12-week HFD (n = 6). C and D, representative images of H&E- (C) and Oil Red O-stained (D) sections of livers from WT and RGC32−/− mice fed normal chow and an HFD as indicated. Microsteatosis and macrosteatosis are indicated by short and long arrows, respectively. E, quantification of Oil Red O staining. ORO, Oil Red O; a.u., arbitrary units. F and G, liver triglyceride (TG) content (F) and liver weights (G) of WT and RGC32−/− mice fed normal chow and an HFD (n = 6). *, p < 0.05 and **, p < 0.01 compared with WT chow groups; ##, p < 0.01 compared with WT HFD groups.
RGC-32 Deficiency Reduced de Novo Hepatic Lipogenesis
Hepatic steatosis can occur as a result of increased lipogenesis and/or reduced β-oxidation (6). To determine how RGC-32 affects HFD-induced hepatic steatosis, we detected a variety of genes involved in lipogenesis and β-oxidation of the liver. Interestingly, SREBP-1c, FAS, and SCD1 mRNA expression was decreased dramatically in livers of HFD-fed RGC32−/− mice compared with HFD-fed WT control mice (Fig. 2A), whereas no significant change of β-oxidation-related genes was observed (Fig. 2A). Decreased expression of SREBP-1c, FAS, and SCD1 proteins was also evident in RGC32−/− mice fed both normal chow and an HFD (Fig. 2, B–E), consistent with RGC32−/− mice having a lower hepatic triglyceride content when fed both normal chow and an HFD (Fig. 1, C–E). SCD1 is the rate-limiting enzyme in lipogenesis. It catalyzes the desaturation of long-chain saturated fatty acids into monounsaturated fatty acids (11), the major components of membrane phospholipids, triglycerides, and cholesterol esters. The desaturase indices were calculated as the product-to-substrate ratio of the fatty acids metabolized by SCD1 (16:1n-7/16:0 and 18:1n-9/18:0) and were used as a proxy for the activity of the enzyme (31, 32). Consistent with the decreased expression of SCD1, significantly reduced desaturase indices of C16 and C18 fatty acids were observed in both the liver and serum of RGC32−/− mice fed both normal chow and an HFD (Fig. 3, A–D). These data suggest that RGC-32 deficiency may ameliorate hepatic steatosis through inhibiting de novo hepatic lipogenesis.
FIGURE 2.
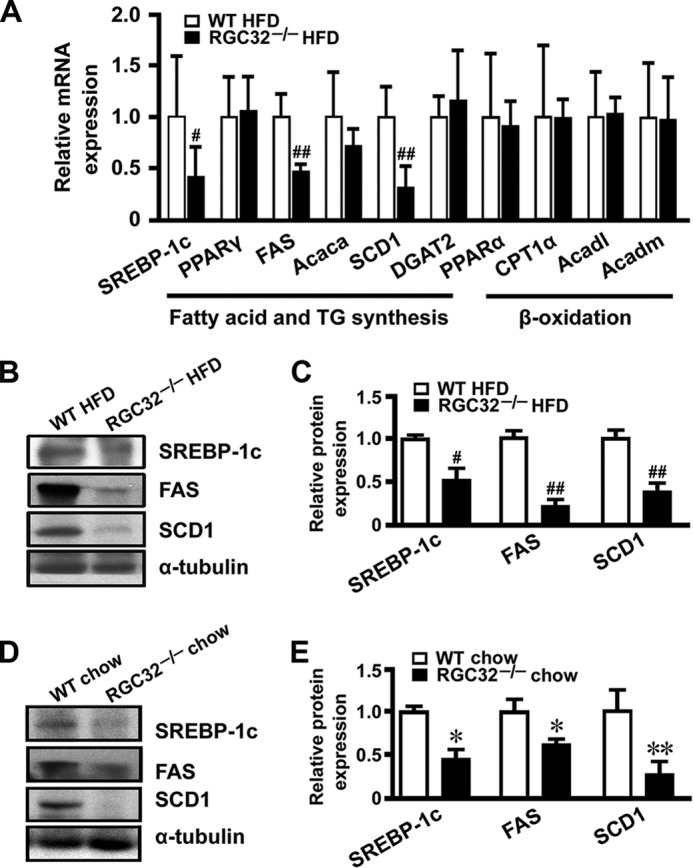
RGC-32 deficiency reduced the expression of lipogenic genes. A, mRNA expression of fatty acid-, triglyceride synthesis-, and β-oxidation-related genes in the livers of HFD-fed WT and RGC32−/− mice (n = 6). B–E, protein expression of SREBP-1c, FAS, and SCD1 in livers from WT and RGC32−/− mice fed an HFD (B and C) or normal chow (D and E) were detected by Western blot analysis and normalized to α-tubulin (n = 6). *, p < 0.05 and **, p < 0.01 compared with WT chow groups; #, p < 0.05 and ##, p < 0.01 compared with WT HFD groups.
FIGURE 3.

RGC-32 deficiency reduced SCD1 activity. A–D, ratios of monounsaturated fatty acids (16:1n-7 and 18:1n-9) to saturated fatty acids (16:0 and 18:0) in livers (A and B) and sera (C and D) of WT and RGC32−/− mice fed normal chow and an HFD (n = 6). *, p < 0.05 and **, p < 0.01 compared with WT chow groups; #, p < 0.05 and ##, p < 0.01 compared with WT HFD groups.
RGC-32 Deficiency Protected Mice from Ethanol-induced Hepatic Steatosis
In addition to HFD-induced hepatic steatosis, increased lipogenesis was also observed in ethanol-induced fatty liver (4, 33, 34). To determine whether RGC-32 plays a broader role in de novo hepatic lipogenesis, we examined the effect of RGC-32 on the development of hepatic steatosis in response to ethanol administration. Ethanol administration dramatically increased RGC-32 expression at both mRNA and protein levels in the liver of WT mice (Fig. 4, A–C). Although H&E staining did not show an alteration in morphology (Fig. 4D), Oil Red O staining revealed that RGC32−/− mouse liver had significantly fewer lipid droplets compared with the WT control both with and without ethanol administration (Fig. 4, E and F). Importantly, ethanol significantly induced the expression of SREBP-1c, FAS, and SCD1 in the livers of WT mice both at the mRNA and protein levels, whereas this effect was dramatically inhibited in RGC32−/− mice (Fig. 5, A–C). These results demonstrate that RGC-32 deficiency also protects mice from ethanol-induced hepatic steatosis through inhibiting de novo lipogenesis.
FIGURE 4.

RGC-32 deficiency prevented ethanol-induced hepatic steatosis. A, RGC-32 mRNA expression in liver tissues of WT mice with or without ethanol administration (n = 6). B and C, RGC-32 protein expression in liver tissues of WT mice with or without ethanol administration was detected by Western blot analysis and normalized to α-tubulin (n = 6). D and E, representative images of H&E- (D) and Oil Red O-stained (E) sections of livers from WT and RGC32−/− mice with or without ethanol administration as indicated. Ctrl, control. F, quantification of Oil Red O staining. ORO, Oil Red O; a.u., arbitrary units. **, p < 0.01 compared with WT groups; ##, p < 0.01 compared with WT ethanol groups.
FIGURE 5.

RGC-32 deficiency ameliorated ethanol-induced lipogenesis. A, SREBP-1c, FAS, and SCD1 mRNA expression in liver tissues of WT mice with or without ethanol administration (n = 6). B and C, SREBP-1c, FAS, and SCD1 protein expression in liver tissues of WT and RGC32−/− mice with or without ethanol administration was detected by Western blot analysis and normalized to α-tubulin (n = 6). *, p < 0.05 and **, p < 0.01 compared with WT groups; #, p < 0.05 and ##, p < 0.01 compared with WT ethanol groups.
Insulin and Ethanol Increased Hepatic RGC-32 Expression through the NF-κB Signaling Pathway
To determine the mechanism by which an HFD induced hepatic RGC-32 expression, mouse primary hepatocytes were isolated and treated with glucose and insulin, and then RGC-32 expression was detected. As shown in Fig. 6A, glucose slightly increased RGC-32 expression, whereas insulin dramatically induced RGC-32 expression, suggesting that insulin is the major contributor to increased RGC-32 expression during HFD challenge. To further determine how insulin regulated RGC-32 expression, we pretreated mouse primary hepatocytes with LY294002 or PDTC to block the PI3K-Akt and NF-κB signaling pathways, respectively, before insulin treatment. PDTC, but not LY294002, blocked insulin-induced RGC-32 expression (Fig. 6, B–E), suggesting that insulin increased RGC-32 expression through the NF-κB signaling pathway. To determine whether ethanol induces RGC-32 expression in vitro and whether it shares a common mechanism with insulin, we treated mouse primary hepatocytes with ethanol with or without PDTC pretreatment. As shown in Fig. 6F, ethanol increased RGC-32 expression, and PDTC blocked this effect. These data suggest that both insulin and ethanol induce hepatic RGC-32 expression through the NF-κB signaling pathway.
FIGURE 6.

Insulin and ethanol increased hepatic RGC-32 expression through the NF-κB signaling pathway. A, mouse primary hepatocytes were treated with 25 mm glucose, 100 nm insulin, or vehicle (control, Ctrl) for 24 h. RGC-32 protein expression was detected by Western blot analysis and normalized to α-tubulin. B and C, mouse primary hepatocytes were preincubated with 10 μm LY294002 (PI3K inhibitor), 25 μm PDTC (NF-κB inhibitor), or vehicle for 1 h and then treated with 100 nm insulin or vehicle for 24 h as indicated. RGC-32 protein expression was detected by Western blot analysis and normalized to α-tubulin. D and E, p-Akt and p-NF-κB were detected by Western blot analysis and normalized to total Akt and NF-κB, respectively. F, mouse primary hepatocytes were preincubated with 25 μm PDTC or vehicle for 1 h and then treated with 50 mm ethanol for 24 h. RGC-32 protein expression was detected by Western blot analysis and normalized to α-tubulin. All results are representative of at least three independent experiments. *, p < 0.05 and **, p < 0.01 compared with control groups; #, p < 0.05 and ##, p < 0.01 compared with insulin or ethanol treatment groups.
RGC-32 Regulated Hepatic SCD1 Expression in a SREBP-1c-dependent Manner
Previous studies have shown that SCD1 is regulated at the transcriptional level via an SREBP-1c-dependent mechanism (28, 35). Our results showed that RGC-32 deficiency prevented HFD- and ethanol-induced SREBP-1c and SCD1 expression in the liver (Figs. 2 and 5). To determine the mechanism by which RGC-32 regulates hepatic SCD1 gene expression, mouse primary hepatocytes were transduced with Ad-GFP, Ad-shRGC32, or Ad-RGC32. Consistent with the in vivo findings, RGC-32 knockdown down-regulated, whereas RGC-32 overexpression up-regulated, the expression of SREBP-1c and SCD1 at both the mRNA and protein levels (Fig. 7, A–C), suggesting that RGC-32 directly regulates SREBP-1c and SCD1 expression in hepatocytes. Because RGC-32 does not have a DNA binding domain, we hypothesized that RGC-32 may regulate SCD1 expression through SREBP-1c. An SCD1 promoter analysis showed that RGC-32 knockdown decreased, whereas RGC-32 overexpression increased, SCD1 promoter activities (Fig. 8A). Importantly, deletion or mutation of SRE in the SCD1 promoter abolished the function of RGC-32 (Fig. 8A), suggesting that RGC-32 regulates SCD1 in an SRE-dependent manner. Furthermore, RGC-32 knockdown decreased, whereas RGC-32 overexpression increased, SREBP-1c binding to the SCD1 promoter (Fig. 8, B and C). Because RGC-32 regulates SREBP-1c expression directly (Fig. 7, A–C), the change of SREBP-1c binding to the SCD1 promoter may result from the change in SREBP-1c expression level. To further verify that RGC-32 regulation of SCD1 is dependent on SREBP-1c, we knocked down SREBP-1c in RGC-32-overexpressed cells and found that blockade of SREBP-1c significantly inhibited RGC-32-induced SCD1 expression (Fig. 8, D and E), demonstrating that RGC-32 increases hepatic SCD1 expression in an SREBP-1c-dependent manner.
FIGURE 7.

RGC-32 directly regulated hepatic SREBP-1c and SCD1 expression. Mouse primary hepatocytes were transduced with Ad-GFP, Ad-shRGC32, or Ad-RGC32 for 48 h. A, qPCR was performed to detect mRNA expression of RGC-32, SREBP-1c, and SCD1 as indicated. B and C, RGC-32, SREBP-1c, and SCD1 protein expression was detected by Western blot analysis and normalized to α-tubulin. All results are representative of at least three independent experiments. *, p < 0.05 and **, p < 0.01 compared with Ad-GFP groups.
FIGURE 8.

RGC-32 regulated hepatic SCD1 expression in an SREBP-1c-dependent manner. A, mouse primary hepatocytes were transduced with Ad-GFP, Ad-shRGC32, or Ad-RGC32 for 24, followed by transfection of different SCD1 promoter constructs, as indicated, for 48 h. A luciferase assay was performed. Luciferase activity was normalized to Renilla activity. B and C, mouse primary hepatocytes were transduced with Ad-GFP, Ad-shRGC32, or Ad-RGC32 for 48 h, and a ChIP assay was performed. SREBP-1c binding enrichment was detected by RT-PCR (B) and qPCR (C). D and E, mouse primary hepatocytes were transfected with scramble (siCtrl) or SREBP-1c siRNA (siSREBP-1c) for 24 h and then transduced with Ad-GFP or Ad-RGC32 for 48 h as indicated. SCD1, RGC-32, and SREBP-1c were detected by Western blot analysis and normalized to α-tubulin. All results are representative of at least three independent experiments. *, p < 0.05 and **, p < 0.01 compared with Ad-GFP groups; ##, p < 0.01 compared with siCtrl + AdRGC32 groups.
RGC-32 Regulated Hepatic SREBP-1c Expression through Activating LXR
Previous studies have shown that the binding of LXR to the functional LXR element in the SREBP-1c promoter is critical for SREBP-1c transcription (36–38). Neither RGC-32 knockdown nor RGC-32 overexpression had any effect on LXR protein expression (Fig. 9, A and B). However, arachidonic acid, an inhibitor of LXR activation, blocked RGC-32-induced SREBP-1c expression (Fig. 9, C and D), suggesting that RGC-32 may be involved in LXR activation. Therefore, we tested LXR binding to the SREBP-1c promoter using a ChIP assay. As shown in Fig. 9, E and F, RGC-32 knockdown decreased, whereas RGC-32 overexpression increased, LXR binding to the SREBP-1c promoter, suggesting that RGC-32 regulates hepatic SREBP-1c expression through activating LXR. Taken together, these data demonstrate that RGC-32 increases hepatic SREBP-1c expression through LXR activation, followed by enhancing SREBP-1c binding to the SCD1 promoter, leading to activation of SCD1 gene expression and lipogenesis.
FIGURE 9.

RGC-32 regulated hepatic SREBP-1c expression through activating LXR. A and B, mouse primary hepatocytes were transduced with Ad-GFP, Ad-shRGC32, or Ad-RGC32 for 48 h. LXR-α and RGC-32 were detected by Western blot analysis and normalized to α-tubulin. C and D, mouse primary hepatocytes were preincubated with 100 μm arachidonic acid (AA) or vehicle for 1 h and then transduced with Ad-GFP or Ad-RGC32 for 48 h. SREBP-1c and RGC-32 were detected by Western blot analysis and normalized to α-tubulin. E and F, mouse primary hepatocytes were transduced with Ad-GFP, Ad-shRGC32, or Ad-RGC32 for 48 h, and a ChIP assay was performed. LXR-α binding enrichment was detected by RT-PCR (E) and qPCR (F). All results are representative of at least three independent experiments. *, p < 0.05 and **, p < 0.01 compared with Ad-GFP groups; ##, p < 0.01 compared with Ad-RGC32 groups.
Discussion
Nonalcoholic fatty liver disease is defined as a fatty liver (liver fat > 5–10% of liver weight) not due to excess alcohol consumption or other causes of steatosis (39). It is the most common cause of liver dysfunction and associated with obesity, metabolic syndrome, dyslipidemia, insulin resistance, and type 2 diabetes (6, 39). Our study demonstrates that RGC-32 plays a critical role in the development of hepatic steatosis. RGC-32 is strongly induced in the liver by HFD challenge, which is consistent with the finding of Guo et al. (40). RGC-32 deficiency prevents HFD-induced hepatic steatosis, which may explain the improved insulin sensitivity in HFD-fed RGC32−/− mice. More importantly, the liver triglyceride content of RGC32−/− mice is much lower than that of WT mice, even under normal chow conditions, suggesting that RGC-32 is essential for regulating hepatic triglyceride content under both physiological and pathological conditions.
Excessive accumulation of hepatic triglycerides can occur as a result of increased fat synthesis or reduced fat oxidation (41, 42). Studies in humans and rodents have demonstrated that it is mainly linked to enhanced de novo lipid synthesis via the lipogenic pathway in the liver, whereas lipid disposal via β-oxidation is less important (6, 43, 44). It is now well accepted that the transcription factor SREBP-1c, induced by insulin through LXR activation (38), mediates the transcriptional effect on the enzymes involved in fatty acid synthesis and triglyceride synthesis, including FAS and SCD1 (8, 45, 46). Although lack of FAS reduces the rate of lipogenesis, liver-specific FAS knockout fails to protect mice against the development of fatty liver but rather exacerbates it because of a reduction in β-oxidation (47). Mice with a global or liver-specific knockout of SCD1 show decreased lipogenic gene expression and increased β-oxidation and are protected from diet-induced obesity and hepatic steatosis (9–11). Our results show that RGC-32 affects hepatic lipid accumulation by regulating the expression of lipogenic genes because SREBP-1c, FAS, and SCD1 are decreased significantly in the livers of RGC32−/− mice. However, RGC-32 appears not to be involved in the regulation of fatty acid β-oxidation because the expression of peroxisome proliferator-activated receptor α and its downstream target carnitine palmitoyltransferase 1α are not affected in HFD-fed RGC32−/− mice compared with the WT controls. Peroxisome proliferator-activated receptor α is a key transcription factor regulating several genes important for fatty acid β-oxidation. These observations are consistent with previous findings showing that SCD1 function in hepatic steatosis is independent of peroxisome proliferator-activated receptor α (48). Remarkably, RGC-32 regulates SCD1 gene transcription, although RGC-32 does not directly bind to the SCD1 promoter. It appears that RGC-32 increases SREBP-1c expression through LXR activation and increases SREBP-1c binding to the SRE in the SCD1 promoter, which is critical for SCD1 transcription. Therefore, the beneficial effects of RGC-32 deficiency are at least partially due to the suppression of hepatic de novo lipogenesis.
In addition to nonalcoholic fatty liver disease, alcoholic liver disease is also a serious health problem. Alcoholic fatty liver is the earliest and most common response of the liver to alcohol and may be a precursor of more severe forms of liver injury, like steatohepatitis, progressive fibrosis, cirrhosis, and hepatocellular carcinoma (4, 49). Although the pathogenesis and diagnosis are different between nonalcoholic and alcoholic fatty liver disease, excessive triglyceride accumulation in the liver induced by activation of lipogenesis is their common feature. Our data demonstrate that RGC-32 is a common regulator for the hepatic lipogenesis observed in both alcoholic and nonalcoholic hepatic steatosis. RGC-32 deficiency prevents both HFD- and ethanol-induced hepatic steatosis through suppressing hepatic de novo lipogenesis by reducing the lipogenic genes SREBP-1c, FAS, and SCD1.
Our results also demonstrate that insulin instead of glucose is the major contributor to increased hepatic RGC-32 expression under HFD challenge. The study of Guo et al. (40) shows that glucose increases RGC-32 expression more dramatically than insulin in vascular endothelial cells. This discrepancy is likely due to cell type-specific effects. In vascular endothelial cells, RGC-32 is involved in glucose homeostasis (40), whereas, in hepatocytes, RGC-32 participates in lipogenesis. Taken together, our study identifies RGC-32 as a novel regulator for both nonalcoholic and alcoholic hepatic steatosis, which makes RGC-32 a potential drug target in preventing or reversing the process of hepatic steatosis.
Author Contributions
X. B. C. and S. Y. C. conceived the study, designed and analyzed the experiments, and wrote the paper. X. B. C. performed the majority of experiments. J. N. L. performed the Western blot analysis. All authors reviewed the results and approved the final version of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants HL119053 and HL123302. This work was also supported by National Natural Science Foundation of China Grant 81328002. The authors declare that they have no conflicts of interest with the contents of this article.
- SRE
- sterol regulatory element
- SREBP
- sterol regulatory element binding protein
- HFD
- high-fat diet
- qPCR
- quantitative PCR
- FAS
- fatty acid synthase
- LXR
- liver X receptor
- PDTC
- pyrrolidine dithiocarbamate.
References
- 1. Abdelmalek M. F., Diehl A. M. (2007) Nonalcoholic fatty liver disease as a complication of insulin resistance. Med. Clin. North Am. 91, 1125–1149, ix [DOI] [PubMed] [Google Scholar]
- 2. Lettner A., Roden M. (2008) Ectopic fat and insulin resistance. Curr. Diab. Rep. 8, 185–191 [DOI] [PubMed] [Google Scholar]
- 3. Naveau S., Giraud V., Borotto E., Aubert A., Capron F., Chaput J. C. (1997) Excess weight risk factor for alcoholic liver disease. Hepatology 25, 108–111 [DOI] [PubMed] [Google Scholar]
- 4. You M., Fischer M., Deeg M. A., Crabb D. W. (2002) Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J. Biol. Chem. 277, 29342–29347 [DOI] [PubMed] [Google Scholar]
- 5. Cheung O., Sanyal A. J. (2010) Recent advances in nonalcoholic fatty liver disease. Curr. Opin. Gastroenterol. 26, 202–208 [DOI] [PubMed] [Google Scholar]
- 6. Postic C., Girard J. (2008) Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J. Clin. Invest. 118, 829–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ferré P., Foufelle F. (2007) SREBP-1c transcription factor and lipid homeostasis: clinical perspective. Horm. Res. 68, 72–82 [DOI] [PubMed] [Google Scholar]
- 8. Dobrzyn A., Ntambi J. M. (2005) Stearoyl-CoA desaturase as a new drug target for obesity treatment. Obes. Rev. 6, 169–174 [DOI] [PubMed] [Google Scholar]
- 9. Cohen P., Miyazaki M., Socci N. D., Hagge-Greenberg A., Liedtke W., Soukas A. A., Sharma R., Hudgins L. C., Ntambi J. M., Friedman J. M. (2002) Role for stearoyl-CoA desaturase-1 in leptin-mediated weight loss. Science 297, 240–243 [DOI] [PubMed] [Google Scholar]
- 10. Ntambi J. M., Miyazaki M., Stoehr J. P., Lan H., Kendziorski C. M., Yandell B. S., Song Y., Cohen P., Friedman J. M., Attie A. D. (2002) Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc. Natl. Acad. Sci. U.S.A. 99, 11482–11486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miyazaki M., Flowers M. T., Sampath H., Chu K., Otzelberger C., Liu X., Ntambi J. M. (2007) Hepatic stearoyl-CoA desaturase-1 deficiency protects mice from carbohydrate-induced adiposity and hepatic steatosis. Cell Metab. 6, 484–496 [DOI] [PubMed] [Google Scholar]
- 12. Badea T. C., Niculescu F. I., Soane L., Shin M. L., Rus H. (1998) Molecular cloning and characterization of RGC-32, a novel gene induced by complement activation in oligodendrocytes. J. Biol. Chem. 273, 26977–26981 [DOI] [PubMed] [Google Scholar]
- 13. Li F., Luo Z., Huang W., Lu Q., Wilcox C. S., Jose P. A., Chen S. (2007) Response gene to complement 32, a novel regulator for transforming growth factor-β-induced smooth muscle differentiation of neural crest cells. J. Biol. Chem. 282, 10133–10137 [DOI] [PubMed] [Google Scholar]
- 14. Fosbrink M., Cudrici C., Tegla C. A., Soloviova K., Ito T., Vlaicu S., Rus V., Niculescu F., Rus H. (2009) Response gene to complement 32 is required for C5b-9 induced cell cycle activation in endothelial cells. Exp. Mol. Pathol. 86, 87–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang J. N., Shi N., Xie W. B., Guo X., Chen S. Y. (2011) Response gene to complement 32 promotes vascular lesion formation through stimulation of smooth muscle cell proliferation and migration. Arterioscler. Thromb. Vasc. Biol. 31, e19–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li Z., Xie W. B., Escano C. S., Asico L. D., Xie Q., Jose P. A., Chen S. Y. (2011) Response gene to complement 32 is essential for fibroblast activation in renal fibrosis. J. Biol. Chem. 286, 41323–41330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fosbrink M., Cudrici C., Niculescu F., Badea T. C., David S., Shamsuddin A., Shin M. L., Rus H. (2005) Overexpression of RGC-32 in colon cancer and other tumors. Exp. Mol. Pathol. 78, 116–122 [DOI] [PubMed] [Google Scholar]
- 18. Vlaicu S. I., Tegla C. A., Cudrici C. D., Fosbrink M., Nguyen V., Azimzadeh P., Rus V., Chen H., Mircea P. A., Shamsuddin A., Rus H. (2010) Epigenetic modifications induced by RGC-32 in colon cancer. Exp. Mol. Pathol. 88, 67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim D. S., Lee J. Y., Lee S. M., Choi J. E., Cho S., Park J. Y. (2011) Promoter methylation of the RGC32 gene in nonsmall cell lung cancer. Cancer 117, 590–596 [DOI] [PubMed] [Google Scholar]
- 20. Cui X. B., Luan J. N., Ye J., Chen S. Y. (2015) RGC32 deficiency protects against high-fat diet-induced obesity and insulin resistance in mice. J. Endocrinol. 224, 127–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cui X. B., Guo X., Chen S. Y. (2013) Response gene to complement 32 deficiency causes impaired placental angiogenesis in mice. Cardiovasc Res. 99, 632–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boileau P., Wolfrum C., Shih D. Q., Yang T. A., Wolkoff A. W., Stoffel M. (2002) Decreased glibenclamide uptake in hepatocytes of hepatocyte nuclear factor-1α-deficient mice: a mechanism for hypersensitivity to sulfonylurea therapy in patients with maturity-onset diabetes of the young, type 3 (MODY3). Diabetes 51, S343–348 [DOI] [PubMed] [Google Scholar]
- 23. Li L., Fan D., Wang C., Wang J. Y., Cui X. B., Wu D., Zhou Y., Wu L. L. (2011) Angiotensin II increases periostin expression via Ras/p38 MAPK/CREB and ERK1/2/TGF-β1 pathways in cardiac fibroblasts. Cardiovasc. Res. 91, 80–89 [DOI] [PubMed] [Google Scholar]
- 24. Cui X. B., Wang C., Li L., Fan D., Zhou Y., Wu D., Cui Q. H., Fu F. Y., Wu L. L. (2012) Insulin decreases myocardial adiponectin receptor 1 expression via PI3K/Akt and FoxO1 pathway. Cardiovasc Res. 93, 69–78 [DOI] [PubMed] [Google Scholar]
- 25. Mehlem A., Hagberg C. E., Muhl L., Eriksson U., Falkevall A. (2013) Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat. Protoc. 8, 1149–1154 [DOI] [PubMed] [Google Scholar]
- 26. Hofacer R., Magrisso I. J., Jandacek R., Rider T., Tso P., Benoit S. C., McNamara R. K. (2012) Omega-3 fatty acid deficiency increases stearoyl-CoA desaturase expression and activity indices in rat liver: positive association with non-fasting plasma triglyceride levels. Prostaglandins Leukot. Essent. Fatty Acids 86, 71–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tabor D. E., Kim J. B., Spiegelman B. M., Edwards P. A. (1999) Identification of conserved cis-elements and transcription factors required for sterol-regulated transcription of stearoyl-CoA desaturase 1 and 2. J. Biol. Chem. 274, 20603–20610 [DOI] [PubMed] [Google Scholar]
- 28. Kim E., Liu N. C., Yu I. C., Lin H. Y., Lee Y. F., Sparks J. D., Chen L. M., Chang C. (2011) Metformin inhibits nuclear receptor TR4-mediated hepatic stearoyl-CoA desaturase 1 gene expression with altered insulin sensitivity. Diabetes 60, 1493–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xie W. B., Li Z., Shi N., Guo X., Tang J., Ju W., Han J., Liu T., Bottinger E. P., Chai Y., Jose P. A., Chen S. Y. (2013) Smad2 and myocardin-related transcription factor B cooperatively regulate vascular smooth muscle differentiation from neural crest cells. Circ. Res. 113, e76–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Unger R. H. (2002) Lipotoxic diseases. Annu. Rev. Med. 53, 319–336 [DOI] [PubMed] [Google Scholar]
- 31. Hulver M. W., Berggren J. R., Carper M. J., Miyazaki M., Ntambi J. M., Hoffman E. P., Thyfault J. P., Stevens R., Dohm G. L., Houmard J. A., Muoio D. M. (2005) Elevated stearoyl-CoA desaturase-1 expression in skeletal muscle contributes to abnormal fatty acid partitioning in obese humans. Cell Metab. 2, 251–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Warensjö E., Ohrvall M., Vessby B. (2006) Fatty acid composition and estimated desaturase activities are associated with obesity and lifestyle variables in men and women. Nutr. Metab. Cardiovasc. Dis. 16, 128–136 [DOI] [PubMed] [Google Scholar]
- 33. Yin H. Q., Kim M., Kim J. H., Kong G., Kang K. S., Kim H. L., Yoon B. I., Lee M. O., Lee B. H. (2007) Differential gene expression and lipid metabolism in fatty liver induced by acute ethanol treatment in mice. Toxicol. Appl. Pharmacol. 223, 225–233 [DOI] [PubMed] [Google Scholar]
- 34. Wada S., Yamazaki T., Kawano Y., Miura S., Ezaki O. (2008) Fish oil fed prior to ethanol administration prevents acute ethanol-induced fatty liver in mice. J. Hepatol. 49, 441–450 [DOI] [PubMed] [Google Scholar]
- 35. Miyazaki M., Dobrzyn A., Man W. C., Chu K., Sampath H., Kim H. J., Ntambi J. M. (2004) Stearoyl-CoA desaturase 1 gene expression is necessary for fructose-mediated induction of lipogenic gene expression by sterol regulatory element-binding protein-1c-dependent and -independent mechanisms. J. Biol. Chem. 279, 25164–25171 [DOI] [PubMed] [Google Scholar]
- 36. Ou J., Tu H., Shan B., Luk A., DeBose-Boyd R. A., Bashmakov Y., Goldstein J. L., Brown M. S. (2001) Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc. Natl. Acad. Sci. U.S.A. 98, 6027–6032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yoshikawa T., Shimano H., Yahagi N., Ide T., Amemiya-Kudo M., Matsuzaka T., Nakakuki M., Tomita S., Okazaki H., Tamura Y., Iizuka Y., Ohashi K., Takahashi A., Sone H., Osuga Ji J., Gotoda T., Ishibashi S., Yamada N. (2002) Polyunsaturated fatty acids suppress sterol regulatory element-binding protein 1c promoter activity by inhibition of liver X receptor (LXR) binding to LXR response elements. J. Biol. Chem. 277, 1705–1711 [DOI] [PubMed] [Google Scholar]
- 38. Chen G., Liang G., Ou J., Goldstein J. L., Brown M. S. (2004) Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl. Acad. Sci. U.S.A. 101, 11245–11250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Neuschwander-Tetri B. A., Caldwell S. H. (2003) Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology 37, 1202–1219 [DOI] [PubMed] [Google Scholar]
- 40. Guo S., Philbrick M. J., An X., Xu M., Wu J. (2014) Response gene to complement 32 (RGC-32) in endothelial cells is induced by glucose and helpful to maintain glucose homeostasis. Int. J. Clin. Exp. Med. 7, 2541–2549 [PMC free article] [PubMed] [Google Scholar]
- 41. Tateya S., Rizzo-De Leon N., Handa P., Cheng A. M., Morgan-Stevenson V., Ogimoto K., Kanter J. E., Bornfeldt K. E., Daum G., Clowes A. W., Chait A., Kim F. (2013) VASP increases hepatic fatty acid oxidation by activating AMPK in mice. Diabetes 62, 1913–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sheng L., Cho K. W., Zhou Y., Shen H., Rui L. (2011) Lipocalin 13 protein protects against hepatic steatosis by both inhibiting lipogenesis and stimulating fatty acid β-oxidation. J. Biol. Chem. 286, 38128–38135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lewis G. F., Carpentier A., Adeli K., Giacca A. (2002) Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr. Rev. 23, 201–229 [DOI] [PubMed] [Google Scholar]
- 44. Ferré P., Foufelle F. (2010) Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes. Metab. 12, 83–92 [DOI] [PubMed] [Google Scholar]
- 45. Shimano H., Yahagi N., Amemiya-Kudo M., Hasty A. H., Osuga J., Tamura Y., Shionoiri F., Iizuka Y., Ohashi K., Harada K., Gotoda T., Ishibashi S., Yamada N. (1999) Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J. Biol. Chem. 274, 35832–35839 [DOI] [PubMed] [Google Scholar]
- 46. Moon Y. A., Shah N. A., Mohapatra S., Warrington J. A., Horton J. D. (2001) Identification of a mammalian long chain fatty acyl elongase regulated by sterol regulatory element-binding proteins. J. Biol. Chem. 276, 45358–45366 [DOI] [PubMed] [Google Scholar]
- 47. Chakravarthy M. V., Pan Z., Zhu Y., Tordjman K., Schneider J. G., Coleman T., Turk J., Semenkovich C. F. (2005) “New” hepatic fat activates PPARα to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 1, 309–322 [DOI] [PubMed] [Google Scholar]
- 48. Miyazaki M., Dobrzyn A., Sampath H., Lee S. H., Man W. C., Chu K., Peters J. M., Gonzalez F. J., Ntambi J. M. (2004) Reduced adiposity and liver steatosis by stearoyl-CoA desaturase deficiency are independent of peroxisome proliferator-activated receptor-α. J. Biol. Chem. 279, 35017–35024 [DOI] [PubMed] [Google Scholar]
- 49. Ge X., Antoine D. J., Lu Y., Arriazu E., Leung T. M., Klepper A. L., Branch A. D., Fiel M. I., Nieto N. (2014) High mobility group box-1 (HMGB1) participates in the pathogenesis of alcoholic liver disease (ALD). J. Biol. Chem. 289, 22672–22691 [DOI] [PMC free article] [PubMed] [Google Scholar]