

Background: Components of GABA catabolism feed into sleep and potential energy pathways.
Results: We identified a metabolic phenotype in Drosophila mutants of GABA turnover and traced it to a limit in glutamate, which is not relevant for sleep.
Conclusion: GABA regulates metabolic and sleep homeostasis through independent mechanisms.
Significance: Neurological disorders involving GABA disruption may be associated with metabolic problems.
Keywords: amino acid, Drosophila metabolism, glutamate, mitochondria, tricarboxylic acid cycle (TCA cycle) (Krebs cycle)
Abstract
Breakdown of the major sleep-promoting neurotransmitter, γ-aminobutyric acid (GABA), in the GABA shunt generates catabolites that may enter the tricarboxylic acid cycle, but it is unknown whether catabolic by-products of the GABA shunt actually support metabolic homeostasis. In Drosophila, the loss of the specific enzyme that degrades GABA, GABA transaminase (GABAT), increases sleep, and we show here that it also affects metabolism such that flies lacking GABAT fail to survive on carbohydrate media. Expression of GABAT in neurons or glia rescues this phenotype, indicating a general metabolic function for this enzyme in the brain. As GABA degradation produces two catabolic products, glutamate and succinic semialdehyde, we sought to determine which was responsible for the metabolic phenotype. Through genetic and pharmacological experiments, we determined that glutamate, rather than succinic semialdehyde, accounts for the metabolic phenotype of gabat mutants. This is supported by biochemical measurements of catabolites in wild-type and mutant animals. Using in vitro labeling assays, we found that inhibition of GABAT affects energetic pathways. Interestingly, we also observed that gaba mutants display a general disruption in bioenergetics as measured by altered levels of tricarboxylic acid cycle intermediates, NAD+/NADH, and ATP levels. Finally, we report that the effects of GABAT on sleep do not depend upon glutamate, indicating that GABAT regulates metabolic and sleep homeostasis through independent mechanisms. These data indicate a role of the GABA shunt in the development of metabolic risk and suggest that neurological disorders caused by altered glutamate or GABA may be associated with metabolic disruption.
Introduction
The GABA shunt is the biochemical pathway responsible for the catabolism of GABA, the main inhibitory neurotransmitter in eukaryotic organisms (1). This reaction is catalyzed through the activity of the catabolic enzyme, GABA transaminase (GABAT),3 which breaks down GABA into the products succinic semialdehyde (SSA) and glutamate using a keto acid as an amino acceptor (Fig. 1). Studies have confirmed that both SSA and glutamate can enter the TCA cycle through independent pathways; SSA may do so if broken down into SUCC by the enzyme succinic semialdehyde dehydrogenase (SSADH) (2); glutamate can be incorporated into the TCA cycle if broken down into α-ketoglutarate (α-KG) by glutamate dehydrogenase (GDH) (Fig. 1) (3). However, despite over 40 years of study, a role of the GABA shunt pathway in regulating cellular metabolism has not been demonstrated. Thus, it is unknown whether glutamate or succinate derived from GABAT is required to support the metabolic demands of an organism.
FIGURE 1.
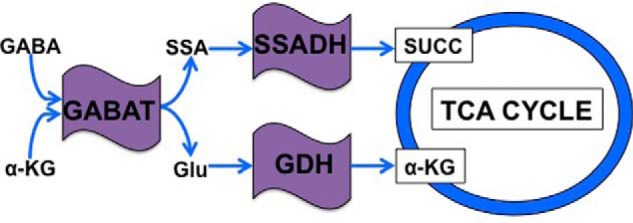
Schema of the metabolic connections between GABAT and the TCA cycle. Once transported into mitochondria, GABA is converted by GABAT into SSA and glutamate (Glu) using α-KG as an amino acceptor. SSA is then reduced by SSADH to form SUCC, which then enters the TCA cycle. If retained in the mitochondria, GABAT-derived glutamate is catabolized by GDH into α-KG, which also enters the TCA cycle.
We recently demonstrated that GABAT plays a role in sleep homeostasis in Drosophila (4). Specifically, fly strains with a null (gabatPL00338) or hypomorphic mutation (gabatf01602) in gabat sleep a total of 100–200 min more than wild-type controls. Although the excess sleep is likely due to the high GABA levels in these mutants, it may also be attributable to altered glutamate levels. Glutamate is a wake-promoting neurotransmitter in eukaryotes and is directly metabolized by the enzyme glutamate decarboxylase into GABA, the main sleep-promoting molecule in mammals and flies (5–8).
In this study, we report that in addition to a sleep phenotype, GABAT mutants exhibit severe metabolic stress because they fail to thrive on a sucrose diet. We investigated how GABAT regulates metabolism by determining whether the following by-products of GABAT, glutamate or SSA, are functionally relevant to the metabolic phenotype. Because glutamate and SSA are components of bioenergetic pathways, we also assessed whether the mutants exhibit abnormal function of brain energy homeostasis. Finally, we determined whether GABAT regulates metabolic and sleep homeostasis through the same mechanism.
Experimental Procedures
Drosophila Strains
All fruit fly strains (Drosophila melanogaster) were maintained at room temperature on standard cornmeal/molasses medium. An isogenic line (w1118) was used as the wild-type strain in this study (9). This strain, as well as cg1640EPgy2, got1wHy, ELAV-GAL4, gabatPL00338, gdhEY07150, ssadhP37186 (Bloomington stock center); gabatf01602 (Exelixis collection at Harvard Medical School); Repo-GAL4 (gift from Gero Miesenböck, Julie Simpson, and Vanessa Auld); and the UAS-GABATvh transgene (4), were used in this study. All strains were outcrossed to our isogenized wild-type strain for 5–7 generations prior to behavioral analysis.
Food Preparation and Behavioral Analysis
For experiments analyzing the metabolic and sleep phenotype of fly strains, we placed adult male fruit flies (age, 1–5 days) into glass tubes containing either sucrose media (5% sucrose and 1.5% agar), regular food (standard cornmeal/molasses medium with 2.5% yeast), or sucrose + amino acid supplement (0.05 m amino acid in 5% sucrose with 1.5% agar). The following amino acids used in this study were purchased from Sigma: l-alanine (A7627); l-cysteine hydrochloride (C1276); l-glutamic acid (G12510); l-glutamine (G3126); l-isoleucine (I2752); l-leucine (L8912); l-phenylalanine (P2126); l-tryptophan (T0254); l-tyrosine (T3754); and l-valine (V0500). For the amino acids that are soluble in water, we neutralized the solution to pH 7.2 to 0.4 before dissolving in 5% sucrose with 1.5% agar. Flies were then placed into Trikinetics Activity Monitors (Trikinetics, Waltham, MA) housed in a temperature-regulated Precision incubator model 818 (Thermo Electron Corp., Marietta, OH) under 12 h of light followed by 12 h of dark.
To analyze the metabolic phenotype of a strain, we assessed whether flies could survive in locomotor tubes containing sucrose food, regular food, or sucrose food + amino acid supplement for 7 days. For the sleep experiment, we monitored the locomotor activity patterns of individual flies on sucrose supplemented with an amino acid in 1-min bins using the DAM File Scan (version 1.1.06, Trikinetics). To analyze both phenotypes, we used pySolo (10). To compare sleep time between treatments, we took the average total sleep time (in minutes) of each fly from day 2 to day 4. Group means of a treatment were then calculated from the average sleep time of the individual flies within the treatment.
Methods for High Performance Liquid Chromatography (HPLC), Brain Extraction
gabatf and wild-type strains were entrained to 12 h of light followed by 12 h of dark at 25 °C for 5 days. On the 5th day, four replicates of 25 male gabatf and four replicates of wild-type males were placed in locomotor tubes containing sucrose media. At ZT0 of the following day, we removed males from the locomotor tubes and placed them on ice until their brains were extracted. We then anesthetized 4–5 males on CO2, placed the anesthetized flies in 75% ethanol, and then transferred them to a dissecting dish with chilled 1× phosphate-buffered saline (PBS) solution. Dissected brains were transferred to an Eppendorf tube with chilled 1× PBS. This process was repeated for 20 brains per genotype. We then spun the samples at 10,000 × g for 10 s, pipetted out the 1× PBS, and stored samples at −80 °C until they were shipped to the Neurochemistry Core at Vanderbilt University for tissue extraction and HPLC analysis.
Tissue Extraction
At the Neurochemistry Core at Vanderbilt University, brain samples were homogenized using a tissue dismembrator in 100–750 μl of 0.1 m TCA, which contained 10−2 m sodium acetate, 10−4 m EDTA, 5 ng/ml isoproterenol (as internal standard), and 10.5% methanol (pH 3.8). Samples were spun in a microcentrifuge at 10,000 × g for 20 min. The supernatant was removed and stored at −80 °C. The pellet was saved for protein analysis using the BCA protein assay kit purchased from Pierce. The supernatant was then thawed and spun for 20 min. Samples of the supernatant were then analyzed for glutamate and GABA.
HPLC Methods
Glutamate and GABA levels were determined by the Waters AccQ-Tag system utilizing a Waters 2475 fluorescence detector (11). The AccQ-Tag method is based on derivatizing reagent developed specifically for amino acid analysis (6-aminoquinolyl-N-hydroxysuccinimidyl carbamate). Briefly, 10-μl samples of the supernatant were diluted with 70 μl of borate buffer to which 20-μl aliquots of 6-aminoquinol-N-hydroxysuccinimidyl carbamate and 10 μl of 250 pmol/μl α-aminobutyric acid (as internal standard) were added to form the fluorescent derivatives. After heating the mixture for 10 min at 37 °C, 10 μl of derivatized samples were injected into the HPLC system, consisting of a Waters 2707 autosampler, two 515 HPLC pumps, column heater (37 °C), and the fluorescence detector. Separation of the amino acids was accomplished by means of a Waters AccQ-Tag chemistry package (Waters) and supplied buffers. Mobile phase A is Waters AccQTag chemistry eluent A and contains 19% sodium acetate, 7% phosphoric acid, 2% triethylamine, and 72% water. Eluent B is 60% acetonitrile in water. The gradient table for HPLC is shown in Table 1. HPLC control and data acquisition were managed by Empower 2 software.
TABLE 1.
Gradient table for HPLC analysis
| Time | Eluent A | Eluent B |
|---|---|---|
| min | ||
| 0 | 100 | 0 |
| 0.5 | 98 | 2 |
| 15 | 93 | 7 |
| 19 | 90 | 10 |
| 32 | 67 | 33 |
| 33 | 67 | 33 |
| 34 | 0 | 100 |
| 37 | 0 | 100 |
| 38 | 100 | 0 |
| 50 | 100 | 0 |
Methods for Liquid Chromatography-Mass Spectrometry (LC-MS), Metabolite Extraction of Whole Flies and Fly Heads
The quantity of tissue required for the LC-MS analysis prevented us from evaluating metabolite concentrations in fly brains; therefore, we performed metabolite extraction on fly bodies (SUCC) or heads (α-KG and NAD+/NADH) in separate experiments. To measure SUCC in fly bodies, three replicate samples of 30 male ssadhHP, gabatf, and wild-type flies were placed on sucrose food for 4 days. To measure α-KG and NAD+/NADH levels in fly heads, three replicate samples of 120 male gabatf and wild-type flies were placed on sucrose food for 4 days. On ZT0 of the 4th day, males from each replicate were anesthetized on CO2, and each sample was frozen in dry ice for 10 min. Metabolites from either fly bodies (SUCC) or fly heads (α-KG and NAD+/NADH) were then extracted by first adding 40 μl of 1× PBS directly to the Eppendorf tube, and then the sample was homogenized. After homogenization, 200 μl of methanol and 100 μl of chloroform were added, and samples were then sonicated for 15 min. 100 μl of chloroform and distilled H2O were then added, and samples were subsequently centrifuged for 7 min at 13,000 rpm at 4 °C. 200 μl was pipetted from the upper aqueous layer of each sample into a new Eppendorf tube. Samples were evaporated for 2–24 h in a SpeedVac and stored at −80 °C until LC-MS analysis.
LC-MS Protocol
LC-MS for the TCA intermediates was performed on a Waters Acquity UPLC coupled to a Waters TQD mass spectrometer. Liquid chromatography conditions and mass spectrometer parameters were based on methods described by Yuan et al. (12), with chromatographic separation of metabolites performed using a Waters BEH Amide (100 mm × 2.1 mm × 2.5 μm) column. Data processing was accomplished using Waters TargetLynx software (version 4.1).
Cell Culture Methods
Drosophila central nervous system (CNS) cells ML-DmBG2 (Drosophila Genomics Resource Center, Bloomington, IN) were grown in Shields and Sang M3 insect media, with 0.5 g/liter potassium bicarbonate, 1 g/liter select yeast extract (Sigma), 2.5 g/liter bactopeptone (BD Biosciences), 10 μg/ml insulin, 10% FBS (Thermo Scientific), and 1× penicillin/streptomycin. For the labeling experiment, conditions were adapted from Qu et al. (19). Briefly, cells in a confluent T75 flask were switched to media without FBS, and 6 mm glucose and 0.5 mm 13C5 glutamate (Cambridge Isotope Laboratories, Tewksbury, MA) were added for 2 h, with or without 1 mm ethanolamine O-sulfate (Sigma) (19). Cells were extracted by washing with cold PBS, followed by scraping into 1 ml of cold PBS. After cell counting, cells were spun down; 600 μl of 2:1 methanol/chloroform were added to the cells, and TCA intermediaries were extracted as described above.
NMR Spectroscopy and Fractional Enrichment Analysis
Extracted cell samples were dissolved in phosphate buffer with 10% D2O and 0.255 mm 2,2-dimethyl-2-silapentane-5-sulfonic acid (Cambridge Isotope, Ltd.) as internal standard. The samples (180 μl) were transferred into 3-mm NMR tubes for 1H NMR spectroscopy (Bruker Biospin, Billerica, MA). All the spectra were acquired in a 700 MHz Bruker Avance III HD NMR spectrometer (Bruker Biospin) fitted with a 3-mm TXI probe and Bruker sample jet system. The spectra were acquired using a pulse program of the form relaxation delay-90-t-90-τm-90-acquire (NOESYPR1d in Bruker pulse program) with saturation on water signal during relaxation delay and mixing time (τm). For each spectrum, 512 scans were performed with acquisition of 76,000 data points. The FIDs were zero-filled to 128,000 and Fourier-transformed with a line broadening of 0.1 Hz. The spectra were imported into Chenomx Profiler version 8.0 (Edmonton, Canada), and the lactate doublet peak at (1.32 ppm) was profiled along with the satellites (13C-1H coupling) around it. The fractional enrichment was calculated as concentration of the [13C]lactate divided by the total pool calculated from [12C]- and [13C]lactate.
Methods for Measuring ATP in Fly Heads
To measure ATP in fly heads, 12 replicate samples of five male gabatf and wild-type flies were placed on sucrose food for 4 days. On ZT0 of the 4th day, males from each replicate were anesthetized by CO2, and each sample was frozen in dry ice for 10 min. Heads were then extracted and homogenized in 6 m guanidine HCl. The ATP concentrations of fly head samples were determined by the luciferin-luciferase reaction. ATP assay mix (Sigma, catalog no. FLAAM) was prepared by adding 5 ml of ATP assay mix dilution buffer (Sigma, catalog no. FLAAB) and 5 ml of H2O. Samples were diluted 1:100 in ATP assay mix dilution buffer, and 80 μl of sample was added to wells in a 96-well plate. 20 μl of ATP assay mix was added using a multichannel pipette, and luminescence was read on a luminometer plate reader (Luminoskan Ascent, Thermo Scientific, Franklin, MA). Luminescence values were converted to an ATP concentration by preparing an ATP standard curve.
Statistical Analysis
All statistical analyses were performed using JMP PRO version 10 (SAS Institute, Cary, NC). To compare the differences among the proportion of individuals that were alive at the end of an experiment (proportion alive), we used a Fisher's exact test. To compare differences in the proportion of individuals alive at the end of an experiment among groups that exceeded two, we used a one-way ANOVA between subjects followed by a Tukey post hoc test. To compare the differences in means between two genotypes, we used an independent sample t test. To compare differences in means among groups that exceeded two, we used a one-way ANOVA between subjects followed by a Tukey post hoc test. Principal component analysis for TCA intermediates was performed using SIMCA version 13 (Umetrics, Umeå, Sweden).
Results
Fly Mutants of Gabat Exhibit Overt Signals of Metabolic Stress
The GABA shunt is the biochemical pathway responsible for the catabolism of GABA, the main inhibitory neurotransmitter in eukaryotes. Because components of this shunt are also implicated in energetic pathways (Fig. 1) (2, 3), we were interested to know whether mutants of GABAT exhibit overt signs of metabolic stress. To address this, we evaluated the survival of gabat mutants in the absence of nutrients (cornmeal, dry yeast, agar, and molasses), which might compensate for biochemical pathways affected by GABAT. Therefore, we placed males of wild-type, null (gabatPL), and hypomorphic (gabatf) gabat strains on a sucrose diet for 7 days and recorded the proportion of individuals that survived the treatment. We found that both gabat mutant strains show reduced survival (3–4%) on sucrose food (Fig. 2a). Furthermore, it appears that the increased mortality is specifically due to a dysfunction in metabolism because transfer of gabatf males to regular food results in survival levels equivalent to those of wild-type flies (Fig. 2b). For the remainder of the study, we show data for the hypomorphic gabatf strain and will refer to the reduced survival of gabatf on sucrose food as a “metabolic phenotype.”
FIGURE 2.
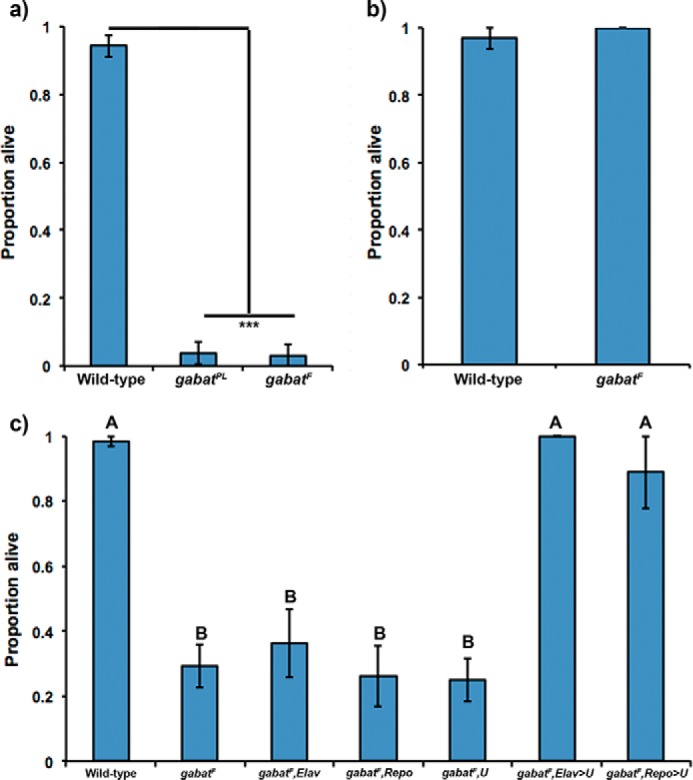
Fly mutants of gabat exhibit overt signals of metabolic stress. a, mutants of gabat cannot survive on sucrose food. When fed a sucrose-only diet, males of our gabatPL and gabatf strains die after 7 days. Fisher's exact test, ***, p < 0.0001. b, GABAT metabolic phenotype is rescued on regular food. When males of the gabatf strain are transferred to a regular food diet, they no longer exhibit overt signals of metabolic stress. Fisher's exact test, p = 0.24. c, metabolic phenotype of gabatf is rescued by driving gabat expression in either neurons or glia. A one-way ANOVA between subjects was conducted to compare the effect of genotype on lethality in gabatf flies carrying UAS-GABATvH (U) driven by GAL4 drivers in glial cells using Repo-GAL4 (Repo) or neurons using ELAV-GAL4 (Elav). The following lines were used as a negative controls: gabatf; gabatf,Elav; gabatf,Repo; and gabatf,U. There was a significant effect of genotype on lethality at the p < 0.05 level for the four groups (F(6) = 30.8, p < 0.0001). Lines with different letter values (A and B) are statistically different as determined by the Tukey post hoc HSD test.
Microarray expression data (13) indicate that gabat is expressed in neurons and non-GABAergic glial cells (4). To determine whether neuronal or glial GABAT regulates the metabolic requirements of the brain, we drove the expression of a gabat construct in either neurons or glia of the gabatf mutant background using the responder UAS-GABATvh and a cell-specific driver (neurons, ELAV-GAL4; glia, Repo-GAL4). We found that we could rescue the metabolic phenotype of gabatf in either neurons or glia (Fig. 2c), indicating that it is caused by a general disruption in brain metabolism.
Biochemical Pathway Linking GABAT to Succinate Is Normal in gabatf Flies
Within mitochondria, GABAT deaminates GABA to form SSA and glutamate (Fig. 1). The aldehyde of SSA is then oxidized by SSADH to SUCC, which may enter the TCA cycle (2, 14). To test whether the metabolic phenotype of gabatf is caused by a decrease of GABAT-derived succinate, we assessed mutants of ssadh on sucrose food. Interestingly, we found that ssadhHP mutants can survive on a sucrose food diet (Fig. 3a), indicating that GABAT regulates metabolic stress independently of succinate. In fact, analysis of SUCC levels of wild-type, gabatf, and ssadhHP flies using liquid-chromatography mass spectrometry (LC-MS) did not reveal a difference among the groups (Fig. 3b), suggesting that alternative enzymes (such as succinyl-CoA ligase, EC 6.2.1.5) compensate for any GABAT-derived reduction in SUCC. Overall, these data suggest that the metabolic phenotype of gabatf is not caused by a disruption in GABAT-derived SUCC.
FIGURE 3.
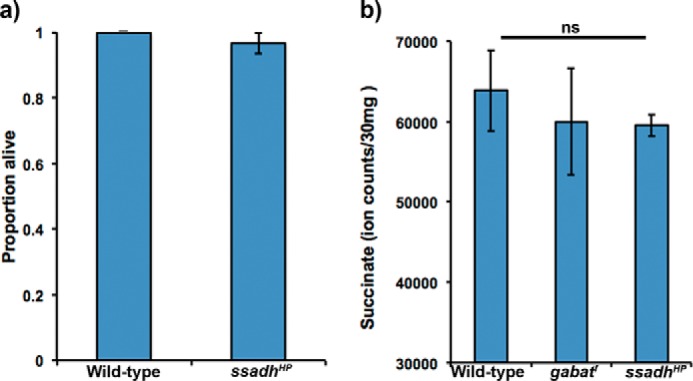
Biochemical pathway linking GABAT-derived SSA to the TCA cycle is normal in gabatf flies. a, mutants of SSADH do not exhibit a metabolic phenotype on sucrose food. Fisher's exact test, p = 1.0. b, succinate concentrations are equivalent among wild-type, gabatf, and ssadhHP strains. A one-way ANOVA between subjects was conducted to compare the effect of genotype on succinate concentrations among wild-type, gabatf, ssadhHP strains. There was no significant effect of genotype on succinate concentrations for the three groups (F(3) = 0.19, p = 0.84). ns, not significant.
Biochemical Pathway Linking GABAT to Glutamate and α-KG Is Impaired in gabatf Flies
As our data suggested that metabolic stress of gabatf is not caused by a reduction in SUCC (Fig. 3), we next assessed whether glutamate metabolism is disrupted. To address this, we evaluated whether gabatf mutants exhibit abnormal concentrations of brain GABA and glutamate after 1 day of sucrose food treatment. Using HPLC, we found that brain glutamate levels are half (Fig. 4a) and GABA levels are 6 times as high (Fig. 4b) in gabatf flies, as compared with controls also maintained on sucrose. We reported previously that brain GABA levels are twice as high as wild-type on regular food (4), and so it appears that the increase in GABA levels of gabatf is exacerbated on sucrose medium.
FIGURE 4.
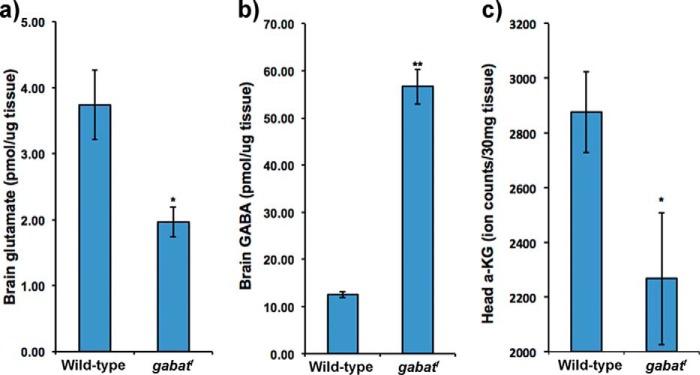
Biochemical pathway linking GABAT-derived glutamate to the TCA cycle is impaired in gabatf flies. a, brain glutamate levels are reduced in gabatf mutants. Using HPLC, we compared brain glutamate levels between gabatf and wild-type strains after 1 day of sucrose food treatment. Independent sample t tests, *, p < 0.05. b, brain GABA levels are increased in gabatf mutants. Using HPLC, we compared brain GABA levels between gabatf and wild-type after 1 day of sucrose food treatment. Independent sample t tests, **, p < 0.005. c, head α-KG levels are lower in gabatf. Using liquid chromatography-mass spectrometry, we compared head α-KG levels between wild-type and gabatf flies after a 4-day treatment of sucrose food. Independent sample t tests, *, p < 0.05.
Once formed by GABAT, glutamate could have several metabolic fates, which include its direct utilization for glutamine formation, participation in the purine nucleotide cycle, incorporation into proteins or the tripeptide glutathione, and breakdown by GDH into α-KG that can enter the TCA cycle (Fig. 1) (15). We predicted that if GABAT-derived glutamate was an important source of α-KG, then α-KG levels would be low in gabatf mutants. In support of this prediction, we found, using LC-MS, that α-KG levels are significantly lower in heads of gabatf than in wild type (Fig. 4c). Next, we evaluated whether mutants of GDH have a metabolic phenotype on sucrose food. If glutamate-derived α-KG is important for energy homeostasis, we predicted that GDH mutants would also exhibit a metabolic phenotype. Similarly to gabatf, the survival of gdhEY07150 flies on sucrose was significantly reduced (p < 0.03): Mwild type ±S.D. = 1.0 ± 0.0; Mgdh ± S.D. = 0.81 ± 0.40. We suspect that the metabolic phenotype of homozygous gdhEY07150 mutants is not as robust as that of the gabatf mutants because they are likely hypomorphs; the insertion site of the P element of gdhEY07150 is in the last intron of the gene, depending on the isoform. We were unable to generate viable homozygotes from other, perhaps null, mutant lines available at the Bloomington Drosophila Stock Center. Overall, these data suggest that the biochemical pathway linking GABAT-derived glutamate to the TCA substrate, α-KG, is impaired in gabatf mutant flies and that this impairment may contribute to metabolic stress.
Deficits in Glutamate Cause the Overt Metabolic Phenotype of gabatf
As our data suggested that gabatf mutants have impaired glutamate metabolism (Fig. 4), we next sought to experimentally verify that this disruption causes the gabatf metabolic phenotype on sucrose food. The gabatf mutants have low brain glutamate levels (Fig. 4a), so we first asked whether adding glutamate to the sucrose food would rescue the metabolic phenotype. We found that this was the case (Fig. 5a).
FIGURE 5.

Deficiency in glutamate causes the metabolic phenotype of gabatf. a, glutamate rescues the metabolic phenotype of gabatf. When supplementing sucrose food with glutamate (final concentration = 0.05 m), the metabolic phenotype of gabatf mutant males is rescued. Fisher's exact test, ***, p < 0.0005. b, specific amino acids rescue the metabolic phenotype of gabatf males. When we supplement the sucrose diet with the nine amino acids that can be metabolized into glutamate by specific aminotransferases, we found that the following amino acids rescue the metabolic phenotype: alanine, glutamine, leucine, phenylalanine, tyrosine, and valine. Fisher's exact test, *, p < 0.05; **, p < 0.005; ***, p < 0.0005. c, survival curves of gabatf flies on specific diets. Using data from b, we plotted the proportion of live gabatf mutants as a function of the number of days the mutants were given a specific diet. Rescue data from b were compared on day 7. d, GOT1 mediates the metabolic rescue of gabatf by alanine. We placed males of our wild-type, gabatf, ;got1wHy;, and got1wHy;;gabatf strain on alanine, aspartate, and sucrose food. A one-way ANOVA between subjects was conducted to compare the effect of a food treatment on survival among wild-type, gabatf, ;got1wHy;, and ;got1wHy;gabatf strains. There was a significant effect of the food treatment on survival for the 12 groups (F(11) = 156.1, p < 0.0001). When combined with ;got1wHy;, the metabolic phenotype of gabatf cannot be rescued by alanine or aspartate. Different levels (A–C) indicate statistical significance according to Tukey post hoc tests.
To establish specificity for disrupted glutamate metabolism in the lethality phenotype of gabatf mutants on sucrose, we next evaluated whether the amino acids that can be directly metabolized to glutamate through specific aminotransferases can also rescue the gabatf metabolic phenotype. Previous work has shown that aminotransferases play a prominent role in regulating the synthesis and degradation of glutamate by transferring the amino group of specific amino acids to α-KG to produce glutamate (16). Of the nine amino acids that can be directly metabolized into glutamate, we found that six can rescue lethality (Fig. 5, b and c; Table 2).
TABLE 2.
Amino acids that can be metabolized to glutamate by specific aminotransferases in D. melanogaster
Results of Fig. 5b are listed in the Proportion alive column. Data on the aminotransferase and gene were compiled from Kanehisa et al. (18). NA means not applicable.
| Treatment | Aminotransferase | Gene (Dmel) | Proportion alive ± S.E. |
|---|---|---|---|
| Alanine | EC 2.6.1.2 | cg1640 | 0.75 ± 0.08a |
| Cysteine | EC 2.6.1.1 | got1, got2 | 0.17 ± 0.07 |
| Glutamine | EC 2.6.1.16 | gfat1, gfat2 | 0.61 ± 0.09a |
| Isoleucine | EC 2.6.1.42 | cg1673 | 0.00 ± 0.00 |
| Leucine | EC 2.6.1.42 | cg1673 | 1.00 ± 0.00a |
| Phenylalanine | EC 2.6.1.1 | got1, got2 | 0.38 ± 0.09b |
| EC 2.6.1.5 | cg1461 | ||
| Tryptophan | EC 2.6.1.1 | got1, got2 | 0.06 ± 0.04 |
| Tyrosine | EC 2.6.1.1 | got1, got2 | 0.24 ± 0.08c |
| EC 2.6.1.5 | cg1461 | ||
| Valine | EC 2.6.1.42 | cg1673 | 0.81 ± 0.08a |
| Sucrose | NA | 0.04 ± 0.04 |
a p < 0.0005.
b p < 0.005.
c p < 0.05.
Next, we sought to determine experimentally whether the amino acids that rescue the gabatf metabolic phenotype (Fig. 5, b and c; Table 2) do so through the action of specific aminotransferases. We were particularly interested in whether alanine degradation into glutamate by alanine aminotransferase (ALT) or indirectly by glutamic-oxaloacetic transaminase (GOT) accounts for the alanine rescue of the gabatf mutants. Numerous epidemiological studies in humans have reported that abnormal ALT or GOT regulation is associated with metabolic disease (17), and although the mechanism by which it does so is unknown, a hypothesis proposed by Sookoian and Pirola (17) postulates that metabolic pathology is caused by a disruption in glutamate metabolism as ALT and GOT are major regulators of steady-state glutamate levels. Biochemically, ALT degrades alanine into glutamate using α-KG as a substrate (R00258 (18)): l-alanine + α-KG → pyruvate + l-glutamate. However, if alanine is broken down into aspartate by aspartate 4-decarboxylase ((R00397 (18)) l-alanine + CO2 → l-aspartate), GOT may use aspartate to form glutamate (R00355 (18)): l-aspartate + α-KG → oxaloacetate + l-glutamate. To address the relevant pathway, we created the double mutant strains of ALT, GABAT (cg1640EPgy2;;gabatf) as well as GOT, GABAT (;got1wHy;gabatf).
In creating the double mutant lines, we found that the cg1640EPgy2;;gabatf combination was lethal, so we could not determine whether ALT mediates rescue of the gabatf metabolic phenotype on alanine food. However, we found that GOT1 is required for the rescue of gabatf by alanine treatment because the double mutant strain (;got1wHy;gabatf) exhibited the gabatf metabolic phenotype on alanine food (Fig. 5d). These and the glutamate food rescue (Fig. 5a) data together support the idea that impaired glutamate metabolism causes the metabolic phenotype of gabatf.
Mutants of Gabat Show Disrupted Energy Homeostasis
Disrupted metabolism in gabatf mutants (Figs. 4 and 5) suggested that the GABA shunt is connected to bioenergetics pathways. Although catabolites of the GABA shunt generate substrates (such as glutamate (1)) that can be used within the TCA cycle (19), to our knowledge, the GABA shunt has not been linked to cellular metabolism. To determine whether this connection exists, we tested whether inhibiting the shunt in vitro using ethanolamine O-sulfate (EOS), a selective, irreversible inhibitor of GABAT (20), would increase the utilization of exogenous glutamate to compensate for glutamate normally generated by the enzyme. Thus, we added [13C5]glutamate to a Drosophila CNS cell line (ML-DmBG2) with or without EOS for 2 h. To assess incorporation of the exogenous glutamate into the cellular energy pathways, we measured labeled carbon incorporation into lactate. We found labeled lactate increased with EOS inhibition (Fig. 6a), suggesting a functional link between GABAT, glutamate, and energy metabolism. Furthermore, we measured TCA intermediates in gabatf, ssadhHP, and wild-type fly heads and observed differences in gabatf compared with wild-type and ssadhHP (Fig. 6b). The principal components analysis of loading plots among these genotypes revealed a disruption in the TCA metabolite profile (Fig. 6c).
FIGURE 6.

gabat mutants display deficiencies in bioenergetics. a, Drosophila CNS cells were fed 0.5 mm [13C5]glutamate with or without 1 mm of the GABAT inhibitor EOS. Labeling of lactate from glutamate via the TCA cycle was measured in cell extracts using 1H NMR. Fractional enrichment of labeled lactate was calculated from lactate satellite peaks (centered around 1.32 ppm, d peak of lactate CH3) with respect to the total lactate pool (all in micromolar). A two-tailed Student's t test demonstrated a significantly higher fractional enrichment for lactate in the presence of GABAT inhibition (*, p < 0.05), suggesting increased exogenous glutamate uptake and incorporation into the TCA cycle. b, principal components analysis of TCA metabolite profiles on gabatf, ssadhHP, and wild-type fly heads using LC-MS demonstrates divergence in the metabolite profiles of the gabatf flies compared with wild-type and ssadhHP. Each point represents a fly head sample, with labels indicating group (Gaba = gabatf; Ssadh = ssadhHP; WT = wild-type; numbers indicate biological and analytical replicates, respectively). The overlap in WT and SsadhHP suggests TCA metabolites are highly similar, and divergence with gabatf indicates a unique TCA metabotype. c, principal components analysis loadings plot depicting the disruption in the TCA metabolite profile among the biological groups, suggesting energetic imbalance in flies with a mutation in GABAT. From this plot, fumarate, malate, lactate, citrate/isocitrate, and aconitate can be interpreted to be relatively high in gabatf compared with the other biological groups. The difference between gabatf and ssadhHP/WT is explained by 60% of the variance in the data (R2X = 0.599). d, NAD+/NADH ratios; e, ATP concentrations in heads of wild-type and gabatf flies. Independent sample t test (*, p < 0.05).
These data suggested the existence of an energetic imbalance in gabatf flies. To further test whether this was the case, we evaluated the ratio of NAD+/NADH, which is related to the redox status of mitochondria and can control carbon entry into the TCA cycle, whereby reduced NAD+/NADH levels slow carbon entry into the TCA cycle (21). Interestingly, we observed a significantly higher NAD+/NADH ratio in gabatf than in wild type (Fig. 6d), implying that the mutants have reduced energy levels. Measurement of ATP levels also supported the idea of an energetic imbalance in gabatf flies as these were lower than wild type (Fig. 6e). Overall, these data indicate that loss of gabat impairs energy homeostasis.
GABAT Regulates Sleep Independently of Metabolic Homeostasis
Our data suggest that a limit in glutamate causes the gabatf metabolic phenotype (Fig. 5d). Previously, we observed that gabatf mutants have a sleep phenotype, sleeping on average a total of 100–200 min more than wild-type controls (4). Because glutamate is one of the main wake-promoting neurotransmitters, we tested whether low glutamate also accounts for the sleep phenotype of gabatf. Thus, we placed male flies (age, 1–5 days) on a sucrose diet supplemented with each of the amino acids, including glutamate, which rescues the metabolic phenotype on sucrose (Fig. 5, b and c; Table 2). We predicted that if a limit in brain glutamate accounts for the gabatf sleep phenotype, then the amino acids that rescue metabolism should also rescue sleep. We did not find support for this hypothesis, because none of the amino acids reduced the total sleep time of gabatf relative to that of the negative controls (Fig. 7). This suggests that GABAT regulates sleep independently of metabolic homeostasis.
FIGURE 7.
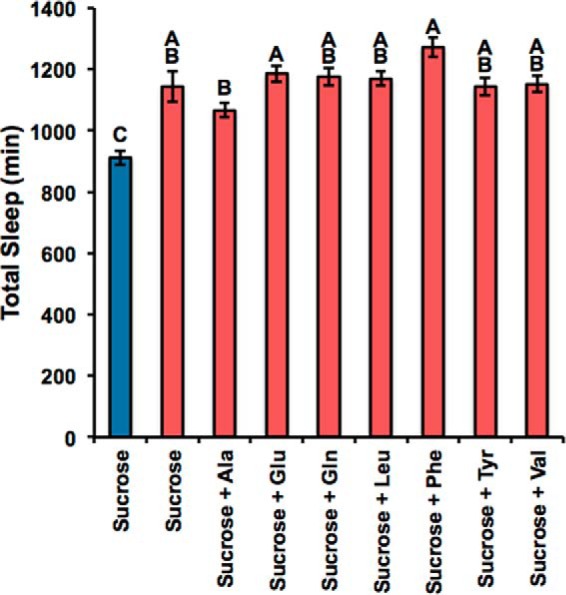
GABAT regulates sleep independently of glutamate. When we fed gabatf mutant (red) and wild-type (blue) flies a sucrose diet enhanced with the amino acids that can be metabolized into glutamate and can rescue the metabolic phenotype (Fig. 5, b and c; Table 2), we found that the sleep phenotype of gabatf cannot be rescued, suggesting that GABAT regulates sleep independently of metabolic homeostasis. We used a one-way ANOVA between subjects to compare total sleep time (minutes) of gabatf mutant and wild-type flies fed sucrose, alanine, glutamate, glutamine, leucine, phenylalanine, tyrosine, and valine. There was a significant effect of the food treatment on total sleep at the p < 0.05 level for the 13 groups, F(8) = 9.68, p < 0.0001. We used Tukey post hoc comparisons to compute the groups that were statistically significant: different levels (A–C) indicate statistical significance. None of the amino acids reduced sleep relative to the negative controls.
Discussion
We show here that the GABA shunt is functionally relevant for regulating metabolic homeostasis in vivo. Mutants of gabat cannot survive on sucrose food (Fig. 2a), an effect that is reversed when they are placed on regular food (Fig. 2b). Furthermore, we show that GABAT regulates metabolism generally in the brain because we can rescue the metabolic phenotype of gabatf in either neurons or glia (Fig. 2c). GABAT is a mitochondrial enzyme, which breaks down GABA into the by-products SSA and glutamate using α-KG as an amino acceptor (Fig. 1). Interestingly, our analysis of the mechanism by which GABAT regulates metabolic homeostasis on sucrose food indicates that it is not through SSA (Fig. 3), but rather glutamate. This is based upon the following: (a) correlative data suggesting that the biochemical pathway linking GABAT-derived glutamate to a TCA cycle substrate is disrupted (Fig. 4), and (b) experimental evidence showing that the correlation is functionally significant (Fig. 5). These data provide new perspectives on the pathogenesis of brain-associated metabolic disorders by introducing a role of glutamate metabolism via the GABA shunt in the development of metabolic risk (17).
How does GABAT regulate metabolism via glutamate in vivo? One possibility is that it regulates energy homeostasis through a link to the TCA cycle (Fig. 1). It is well established that glutamate can be incorporated into the TCA cycle (2, 22), although it is unknown whether this represents glutamate derived through GABAT action. We show that GABAT impacts the conversion of glutamate into lactate, a process that normally involves the TCA cycle (Fig. 6a). However, we do not know whether the GABA/glutamate carbon skeleton is utilized in the TCA cycle nor whether changes to the TCA cycle cause gabat mutants to die on sucrose food. Although we found evidence that energy homeostasis is disrupted in GABAT mutants (Fig. 6, b–e), it is possible that effects on the TCA cycle are secondary to other effects of gabat loss.
Given that a deficiency in glutamate causes the metabolic stress of gabatf on sucrose food (Fig. 5d), it is surprising that cysteine, isoleucine, and tryptophan, amino acids that can be metabolized into glutamate (Table 2), could not rescue the metabolic phenotype of gabatf (Fig. 5, b and c, and Table 2). One explanation is that our mutant strain could not properly ingest cysteine, isoleucine, and tryptophan. Cysteine is a particularly odorous amino acid due to its thiol side chain, and so we suspect that the flies were repelled by its obnoxious smell. We found support for this explanation because most wild-type flies did not survive on cysteine food either (proportion alive ± S.D., 0.03 ± 0.0004). We suggest that our mutants could not properly catabolize tryptophan or isoleucine into glutamate. Tryptophan is highly hydrophobic and so may have had limited solubility in sucrose solution. The branched chain amino acids (isoleucine, leucine, and valine) are also hydrophobic, but our data indicate leucine and valine were ingested in sufficient quantity because these rescued the gabatf metabolic phenotype (Fig. 5, b and c; Table 2). We therefore suspect the branched-chain aminotransferase (EC 2.6.1.2) does not work as effectively using isoleucine as a substrate. However, we acknowledge also that reasons other than conversion to glutamate could contribute to the rescue by specific amino acids.
Although this study establishes that GABAT is functionally important for regulating metabolic homeostasis through glutamate, we found that glutamate had no effect on the long sleeping phenotype observed previously in gabatf mutants (4). This is interesting given that glutamate signaling tends to be wake-promoting in eukaryotes. It is possible that the metabolic phenotype of gabatf is so severe that most exogenously provided glutamate is incorporated into metabolic processes (15). In this scenario, the remaining glutamate levels are not sufficiently high to counteract the effects of high GABA, and in fact, we found by using HPLC that GABA is 6× higher in the brains of gabatf mutants maintained on sucrose (Fig. 4b). As reported previously (5–8), GABA is the major sleep-promoting neurotransmitter in flies and in mammals.
To our knowledge, this is the first study to report the dual role of GABAT in regulating two fundamental processes, metabolic and sleep homeostasis. We show here that mutants of gabat exhibit overt signals of metabolic stress (Fig. 2), and we previously described a long sleeping phenotype of these mutants (4). Our data support the idea that GABAT regulates metabolic homeostasis by increasing the availability of glutamate (Fig. 5), and it likely promotes wakefulness by decreasing GABA (4). Together these data provide a mechanistic account of the role GABAT in metabolism and sleep. Previous studies have identified correlations between an individual's behavioral state and cellular function, particularly with respect to metabolism. For example, using genome-wide expression profiling, Cirelli et al. (23) showed that transcripts associated with energy metabolism, including those involved in glutamate synthesis, are up-regulated during wake, a phenomenon that likely supports the elevated brain metabolic requirements during this state (24).
In humans, GABAT deficiency is a rare but fatal disease. Neuropathic conditions co-morbid with the disorder include psychomotor retardation, convulsions, and hyper-reflexia (25, 26), but metabolic analysis of energy substrates has not been conducted. Our data suggest that a disruption in GABA and glutamate metabolism could be one mechanism by which a deficiency in GABAT produces neuropathic effects.
Author Contributions
S. E. M., S. R., W. F. C., A. M. W., and A. Sehgal, conceived and designed the study. S. E. M. and A. Sehgal wrote the manuscript. S. E. M., S. R., A. Sengupta, Z. Y., J. C. L., and C. H. M. designed and performed experiments and analyzed data. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
HPLC analysis was performed by Raymond Johnson (Neurochemistry Core at Vanderbilt University).
This work was supported, in whole or in part, by National Institutes of Health Predoctoral Training Grant HL07953 (to S. E. M.), Pharmacology T32 Training Grant T32 GM008076 (to S. R.), Grant EY015537 (to C. H. M.), and Grant 2P40OD010949-10A1 (to The Drosophila Genomics Resource Center). The authors declare that they have no conflicts of interest with the contents of this article.
- GABAT
- GABA transaminase
- SSA
- succinic semialdehyde
- SSADH
- succinic semialdehyde dehydrogenase
- α-KG
- α-ketoglutarate
- GDH
- glutamate dehydrogenase
- ANOVA
- analysis of variance
- TCA
- tricarboxylic acid
- SUCC
- succinate
- GOT
- glutamic-oxaloacetic transaminase
- ALT
- alanine aminotransferase
- EOS
- ethanolamine O-sulfate.
References
- 1. Balázs R., Machiyama Y., Hammond B. J., Julian T., Richter D. (1970) The operation of the γ-aminobutyrate bypath of the tricarboxylic acid cycle in brain tissue in vitro. Biochem. J. 116, 445–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Feehily C., Karatzas K. (2013) Role of glutamate metabolism in bacterial responses towards acid and other stresses. J. Appl. Microbiol. 114, 11–24 [DOI] [PubMed] [Google Scholar]
- 3. Berg J. M., Tymoczko J. L., Stryer L. (2007) Biochemistry, 6th Ed., Friedman & Co., New York [Google Scholar]
- 4. Chen W. F., Maguire S., Sowcik M., Luo W., Koh K., Sehgal A. (2015) A neuron-glia interaction involving GABA transaminase contributes to sleep loss in sleepless mutants. Mol. Psychiatry 20, 240–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Agosto J., Choi J. C., Parisky K. M., Stilwell G., Rosbash M., Griffith L. C. (2008) Modulation of GABAA receptor desensitization uncouples sleep onset and maintenance in Drosophila. Nat. Neurosci. 11, 354–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chung B. Y., Kilman V. L., Keath J. R., Pitman J. L., Allada R. (2009) The GABA(A) receptor RDL acts in peptidergic PDF neurons to promote sleep in Drosophila. Curr. Biol. 19, 386–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Parisky K. M., Agosto J., Pulver S. R., Shang Y., Kuklin E., Hodge J. J., Kang K., Kang K., Liu X., Garrity P. A., Rosbash M., Griffith L. C. (2008) PDF cells are a GABA-responsive wake-promoting component of the Drosophila sleep circuit. Neuron 60, 672–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saper C. B., Fuller P. M., Pedersen N. P., Lu J., Scammell T. E. (2010) Sleep state switching. Neuron 68, 1023–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koh K., Joiner W. J., Wu M. N., Yue Z., Smith C. J., Sehgal A. (2008) Identification of SLEEPLESS, a sleep-promoting factor. Science 321, 372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gilestro G. F., Cirelli C. (2009) pySolo: a complete suite for sleep analysis in Drosophila. Bioinformatics 25, 1466–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cohen S. A., Michaud D. P. (1993) Synthesis of a fluorescent derivatizing reagent, 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate, and its application for the analysis of hydrolysate amino acids via high-performance liquid chromatography. Anal. Biochem. 211, 279–287 [DOI] [PubMed] [Google Scholar]
- 12. Yuan M., Breitkopf S. B., Yang X., Asara J. M. (2012) A positive/negative ion–switching, targeted mass spectrometry based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc. 7, 872–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. St Pierre S. E., Ponting L., Stefancsik R., McQuilton P., and FlyBase Consortium. (2014) Flybase 102–advanced approaches to interrogating FlyBase. Nucleic Acids Res. 42, D780–D788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Studart-Guimarães C., Fait A., Nunes-Nesi A., Carrari F., Usadel B., Fernie A. (2007) Reduced expression of succinyl-coenzyme A ligase can be compensated for by up-regulation of the γ-aminobutyrate shunt in illuminated tomato leaves. Plant Physiol. 145, 626–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McKenna M. (2007) The glutamate-glutamine cycle is not stoichiometric: fates of glutamate in brain. J. Neurosci. Res. 85, 3347–3358 [DOI] [PubMed] [Google Scholar]
- 16. Brosnan M. E., Brosnan J. T. (2009) Hepatic glutamate metabolism: a tale of 2 hepatocytes. Am. J. Clin. Nutr. 90, 857S–861S [DOI] [PubMed] [Google Scholar]
- 17. Sookoian S., Pirola C. (2012) Alanine and aspartate aminotransferase and glutamine-cycling pathway: their roles in pathogenesis of metabolic syndrome. World J. Gastroenterol. 18, 3775–3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kanehisa M., Goto S., Sato Y., Kawashima M., Furumichi M., Tanabe M. (2014) Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qu H., Konradsen J. R., van Hengel M., Wolt S., Sonnewald U. (2001) Effect of glutamine and GABA on [U-13C]glutamate metabolism in cerebellar astrocytes and granule neurons. J. Neurosci. Res. 66, 885–890 [DOI] [PubMed] [Google Scholar]
- 20. Fowler L. J., John R. A. (1972) Active-site-directed irreversible inhibition of rat brain 4-aminobutyrate aminotransferase by ethanolamine o-sulphate in vitro and in vivo. Biochem. J. 130, 569–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fendt S.-M., Bell E. L., Keibler M. A., Olenchock B. A., Mayers J. R., Wasylenko T. M., Vokes N. I., Guarente L., Vander Heiden M. G., Stephanopoulos G. (2013) Reductive glutamine metabolism is a function of the α-ketoglutarate to citrate ratio in cells. Nat. Commun. 4, 2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bouché N., Fromm H. (2004) GABA in plants: just a metabolite? Trends Plant Sci. 9, 110–115 [DOI] [PubMed] [Google Scholar]
- 23. Cirelli C., Gutierrez C. M., Tononi G. (2004) Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron 41, 35–43 [DOI] [PubMed] [Google Scholar]
- 24. Steriade M., Timofeev I. (2003) Neuronal plasticity in thalamocortical networks during sleep and waking oscillations. Neuron 37, 563–576 [DOI] [PubMed] [Google Scholar]
- 25. Medina-Kauwe L. K., Tobin A. J., De Meirleir L., Jaeken J., Jakobs C., Nyhan W. L., Gibson K. M. (1999) 4-Aminobutyrate aminotransferase (GABA-transaminase) deficiency. J. Inherit. Metab. Dis. 22, 414–427 [DOI] [PubMed] [Google Scholar]
- 26. Jaeken J., Casaer P., de Cock P., Corbeel L., Eeckels R., Eggermont E., Schechter P. J., Brucher J. M. (1984) γ-Aminobutyric acid-transaminase deficiency: a newly recognized inborn error of neurotransmitter metabolism. Neuropediatrics 15, 165–169 [DOI] [PubMed] [Google Scholar]