

Background: IcmF exhibits low isovaleryl-CoA/pivalyl-CoA mutase (PCM) activity.
Results: IcmF mutants designed to enhance PCM activity were susceptible to inactivation prompting a bioinformatics search for a “bona fide” PCM.
Conclusion: A B12-dependent PCM was identified, cloned, and expressed and exhibited PCM activity.
Significance: The newly discovered PCM could be useful in bioremediation and biosynthetic reactions.
Keywords: adenosylcobalamin (AdoCbl), enzyme, enzyme catalysis, enzyme kinetics, enzyme mutation, cofactor, isomerase
Abstract
Adenosylcobalamin-dependent isomerases catalyze carbon skeleton rearrangements using radical chemistry. We have recently demonstrated that an isobutyryl-CoA mutase variant, IcmF, a member of this enzyme family that catalyzes the interconversion of isobutyryl-CoA and n-butyryl-CoA also catalyzes the interconversion between isovaleryl-CoA and pivalyl-CoA, albeit with low efficiency and high susceptibility to inactivation. Given the biotechnological potential of the isovaleryl-CoA/pivalyl-CoA mutase (PCM) reaction, we initially attempted to engineer IcmF to be a more proficient PCM by targeting two active site residues predicted based on sequence alignments and crystal structures, to be key to substrate selectivity. Of the eight mutants tested, the F598A mutation was the most robust, resulting in an ∼17-fold increase in the catalytic efficiency of the PCM activity and a concomitant ∼240-fold decrease in the isobutyryl-CoA mutase activity compared with wild-type IcmF. Hence, mutation of a single residue in IcmF tuned substrate specificity yielding an ∼4000-fold increase in the specificity for an unnatural substrate. However, the F598A mutant was even more susceptible to inactivation than wild-type IcmF. To circumvent this limitation, we used bioinformatics analysis to identify an authentic PCM in genomic databases. Cloning and expression of the putative AdoCbl-dependent PCM with an α2β2 heterotetrameric organization similar to that of isobutyryl-CoA mutase and a recently characterized archaeal methylmalonyl-CoA mutase, allowed demonstration of its robust PCM activity. To simplify kinetic analysis and handling, a variant PCM-F was generated in which the αβ subunits were fused into a single polypeptide via a short 11-amino acid linker. The fusion protein, PCM-F, retained high PCM activity and like PCM, was resistant to inactivation. Neither PCM nor PCM-F displayed detectable isobutyryl-CoA mutase activity, demonstrating that PCM represents a novel 5′-deoxyadenosylcobalamin-dependent acyl-CoA mutase. The newly discovered PCM and the derivative PCM-F, have potential applications in bioremediation of pivalic acid found in sludge, in stereospecific synthesis of C5 carboxylic acids and alcohols, and in the production of potential commodity and specialty chemicals.
Introduction
5′-Deoxyadenosylcobalamin (AdoCbl),3 a derivative of cobalamin, is used as a cofactor by enzymes that catalyze 1,2 rearrangement reactions using radical chemistry (1–5). The distinguishing feature of the cofactor is its cobalt-carbon bond, which has a bond dissociation energy of ∼31 kcal/mol (6) and holds the key to its utility as a transient source of radicals for initiating chemically challenging isomerization reactions (7). Within the family of AdoCbl-dependent isomerases, the acyl-CoA mutases are the most rapidly growing subclass and several new members have been described in recent years (Fig. 1A) (3). Of these, methylmalonyl-CoA mutase (MCM), which catalyzes the interconversion of methylmalonyl-CoA to succinyl-CoA, is the best studied (1, 5, 8). MCM is the most widely distributed of the AdoCbl-dependent isomerases and is found in organisms ranging from bacteria to man. Relatives of MCM, which catalyze carbon skeleton rearrangements on structurally similar substrates, include isobutyryl-CoA mutase (ICM) (9), ethylmalonyl-CoA mutase (ECM) (10), and 2-hydroxyisobutyryl-CoA mutase (HCM) (11) (Fig. 1A).
FIGURE 1.
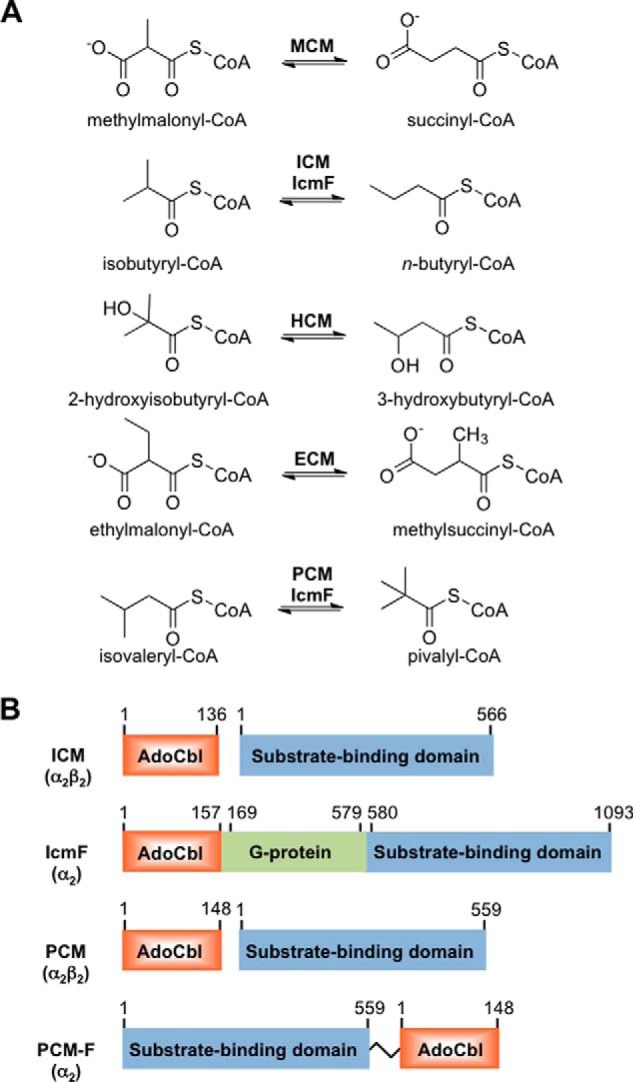
Comparison of reactions catalyzed and organization of acyl-CoA mutases. A, reactions catalyzed by acyl-CoA mutases. IcmF can also catalyze the PCM reaction, albeit inefficiently. B, organization of ICM and PCM variants. ICM and PCM are heterotetramers in which two small B12-binding subunits interact with two large substrate-binding subunits. IcmF is a natural variant in which the small and large subunits found in ICM are fused via a middle G-protein chaperone domain. PCM-F is an artificially engineered variant of PCM (this study) in which the large and small subunits have been fused via an 11-amino acid linker as described under “Experimental Procedures.”
Our laboratory has described a variant of ICM, IcmF (for isobutyryl-CoA mutase fused), which is encoded by many bacterial genomes (12). In IcmF, the separate B12- and substrate-binding subunits found in ICM are fused in a single polypeptide chain together with a G-protein chaperone (Fig. 1B). Thus, IcmF is composed of the three domains, an N-terminal AdoCbl-binding domain, a middle G-protein domain, and a C-terminal substrate-binding domain and displays both GTPase activity and AdoCbl-dependent isomerase activity. Recently, we demonstrated that the Cupriavidus metallidurans (Cm) IcmF catalyzes an additional reaction, i.e. isomerization of isovaleryl-CoA and pivalyl-CoA, designated as pivalyl-CoA mutase (PCM) activity (Fig. 1A) (13). The structural difference between isobutyryl-CoA and the substrate analog, isovaleryl-CoA, is but one carbon unit. From a biotechnological viewpoint, IcmF could be potentially useful as a reagent for bioremediation of pivalic acid found in pharmaceutical wastewaters (pivalic acid esters are used as pro-drugs) and/or for the biosynthesis of a simple β-nonfunctional α-quaternary carboxylic acid (3, 14). Another potential application of IcmF is in metabolic engineering of pathways to produce branched C4 and C5 building blocks that can subsequently be converted to useful derivatives, for example, the corresponding isobutyl and pivalyl alcohols. The potential utility of CmIcmF for isomerizing isovaleryl-CoA/pivalyl-CoA is, however, limited by its inefficiency; it displays an ∼1000-fold lower catalytic efficiency for the isomerization of isovaleryl-CoA versus isobutyryl-CoA and a high susceptibility to inactivation (13).
Sequence alignment of the active site residues in acyl-CoA mutases indicate substitutions at a few key positions that are predicted to be important for substrate specificity (Fig. 2A) (11–13, 15), which can be located in the crystal structures of MCM (16) and CmIcmF (17) (Fig. 2B). The residues corresponding to Phe-598 and Gln-742 in CmIcmF are Tyr-89 and Arg-207 in the Propionibacterium shermanii MCM, where they interact with the carboxylate moiety of the substrate (Fig. 2B). In HCM, the corresponding amino acids are Ile and Gln. Although the substitution of Arg in MCM by Gln in IcmF, ICM, and HCM likely reflects the absence of an anionic carboxylate substituent in their respective substrates, the substitution of Tyr-89 (in MCM) by Phe (in IcmF/ICM) and Ile in (HCM) could be important for accommodating differences in substrate bulk (12, 13). Tyr-89 in MCM is also proposed to play a role in labializing the cobalt-carbon bond of AdoCbl following substrate binding (18). Although the residues corresponding to Tyr-89 and Arg-207 are also found in ECM, substitutions at two other active site residues are predicted to be important for its substrate specificity. ECM catalyzes the interconversion of ethylmalonyl-CoA and methylsuccinyl-CoA, which are one-carbon longer than the corresponding substrates for MCM (10). In fact, MCM can also catalyze the successive isomerization of glutaryl-CoA to methylsuccinyl-CoA and ethylmalonyl-CoA, albeit with very low efficiency (19). Substitutions of His-328 and Asn-366 in MCM by Gly and Pro are proposed to accommodate the longer substrate in ECM. The MCM residues Tyr-89 and Arg-207 are conserved in ECM and presumably make similar contacts with the carboxylate substituent of the substrate as they do in MCM (Fig. 2).
FIGURE 2.

Substitutions at key active site residues underlie differences in substrate specificities in acyl-CoA mutases. A, multiple sequence alignment of substrate-binding domains of different AdoCbl-dependent acyl-CoA mutases. IcmF from C. metallidurans (YP_582365), PCM from X. autotrophicus strain Py2 (ABS70241), ICM from Streptomyces cinnamonensis (AAC08713), MCM from P. shermanii (YP_003687736), ECM from Rhodobacter sphaeroides (YP_354045), and HCM from Aquincola tertiaricarbonis strain L108 (AFK77668) are shown. Four residues (Phe-598, Gln-742, His-863, and Asn-901 in IcmF) predicted to be important for substrate binding, are highlighted in black. Identical and similar residues are indicated with asterisks and dots, respectively. Two residues, Phe and Gln, found in ICM and IcmF are substituted by Tyr and Arg in MCM and ECM or Ile and Gln in HCM, respectively. ECM differs from other acyl-CoA mutases by the substitution of His-863 and Asn-901 in IcmF to Gly and Pro, respectively. All accession numbers are from the NCBI Protein database. B, comparison of active site structures of IcmF (left) and MCM (right). The CmIcmF and P. shermanii MCM structures were generated using Protein Data Bank files 4XC6 and 4REQ, respectively. In the IcmF structure, n-butyryl-CoA is modeled in from Protein Data Bank file 4XC8. Two striking differences in the active site residues are the substitutions of Phe-598 and Gln-742 in IcmF (left) by Leu-87 and Asn-204 in PCM and Tyr-89 and Arg-207 in MCM (right), which are predicted to be important for substrate specificity. The histidine residues on the lower face of the corrin ring are provided by the protein and coordinate to the cobalt. In PCM, Leu-87 and Asn-204 are from the large subunit, whereas His-21 is from the small subunit.
In this study, we engineered CmIcmF to enhance its isovaleryl-CoA mutase activity at the expense of its isobutyryl-CoA mutase activity by introducing mutations at Phe-598 and Gln-742 (Fig. 2B). The F598A mutation switched the substrate specificity of IcmF and enhanced the catalytic efficiency of the isovaleryl-CoA mutase over the native isobutyryl-CoA mutase activity ∼4000-fold. However, the mutation also enhanced the propensity of the enzyme to inactivation during turnover. This prompted us to use bioinformatics analysis to identify a PCM from Xanthobacter autotrophicus and herein, we report its initial characterization.
Experimental Procedures
Materials
AdoCbl, GTP, GDP, isobutyryl-CoA, isovaleryl-CoA, and dl-3-hydroxybutyryl-CoA were purchased from Sigma. Valeric acid was purchased from Fluka. Dithiothreitol and tris(2-carboxyethyl)phosphine hydrochloride were from Gold Biotechnology (St. Louis, MO). All other chemicals were purchased from Sigma or Fisher Scientific, and were used without further purification.
Site-directed Mutagenesis of CmIcmF
Mutagenesis was performed using the QuikChange XL site-directed mutagenesis kit (Agilent Technologies) using the CmIcmF cloned in the pMCSG7 plasmid as a template. The following set of forward primers in which the mutagenic codon is underlined were synthesized by Integrated DNA Technologies (Coralville, IA) and used to introduce missense mutations: F598A, 5′-CCC ACG CGC ATG GCC GCG GGC GAG GG-3′; F598G, 5′-CCC ACG CGC ATG GGC GCG GGC GAG GG-3′; F598I, 5′-CC ACG CGC ATG ATC GCG GGC GAG-3′; F598L, 5′-CCC ACG CGC ATG TTA GCG GGC GAG GGC GAT G-3′; F598V, 5′-CC ACG CGC ATG GTC GCG GGC GAG-3′; Q742A, 5′-CTG AAG GAA GAC CAG GGC GCG AAT ACG TGC ATC TTC TC-3′; Q742L, 5′-AAG GAA GAC CAG GGC CTG AAT ACG TGC ATC TTC TC-3′; Q742N, 5′-CTG AAG GAA GAC CAG GGC AAT AAT ACG TGC ATC TTC TCG-3′.
The sequences of the reverse primers were complementary to those of the forward primers. The mutations were confirmed by nucleotide sequence determination at the University of Michigan DNA Sequencing Core.
Expression and Purification of CmIcmF
Recombinant wild-type and mutant IcmFs were expressed and purified as described previously (13).
Cloning of PCM Subunits
The genomic DNA from X. autotrophicus strain Py2 (ATCC BAA-1158) was purchased from ATCC (Manassas, VA). The large and small subunits of PCM cDNAs were PCR-amplified by PfuTurbo High-Fidelity DNA polymerase (Agilent Technologies) using the following primers containing NdeI and HindIII restriction sites (underlined): large subunit (forward): 5′-AGT ATA CAT ATG AAC CAG GCC GCG GTG-3′, and large subunit (reverse), 5′-T AGT AAG CTT TCA GGC GAT GAT TAG GGG ATG-3′; small subunit (forward), 5′-CTA GTA CAT ATG ATC CAT GCC GGC ACG-3′, and small subunit (reverse), 5′-A GAT AAG CTT TCA TGG GTT TGC CTC CG-3′. The PCR products of the large and small subunits were digested with NdeI and HindIII and subcloned into the pET-28 vector (Novagen).
Subsequently, the resulting vector was used as a template for ligation-independent cloning (LIC) for the large subunit with the following primers, 5′-TAC TTC CAA TCC AAT GCA ATG AAC CAG GCC GCG GTG-3′ (forward) and 5′-T TAT CCA CTT CCA ATG TTA TTA TCA GGC GAT GAT TAG GGG ATG-3′ (reverse), and subcloned into a LIC vector, which introduces a His6-MBP tag and tobacco etch virus (TEV) protease cleavage site at the N terminus of the desired protein. Three additional residues, Ser-Asn-Ala, remained at the N terminus of the large subunit of PCM after removal of the His6-MBP tag.
Construction of an Expression Plasmid for Fusion Protein between Large and Small PCM Subunits
To generate a fusion of the large and small subunits of PCM (PCM-F), a co-expression system was initially constructed using pETDuet-1 vector (Novagen), in which the large subunit was inserted into multicloning site 1 (MCS1) using XbaI and HindIII restriction sites, and the small subunit was inserted into MCS2 using NdeI and XhoI restriction sites. Subsequently, the second translation initiation site between MCS1 and MCS2 was deleted and an 11-amino acid linker sequence, (Gly-Gln)5-Gly, was inserted between the large and small subunits simultaneously by the inverse PCR method using the following 5′-phosphorylated primers: 5′-GGA CAA GGA CAA GGA CAA GGA CAA GGA CAA GGA ATG ATC CAT GCC GGC ACG-3′ (forward) and 5′-GGC GAT GAT TAG GGG ATG GC-3′ (reverse). The resulting fragment was ligated and transformed, and subsequently used as a template for LIC. The insert was amplified with the following primers for LIC cloning, 5′-TAC TTC CAA TCC AAT GCA ATG AAC CAG GCC GCG GTG-3′ (forward) and 5′-T TAT CCA CTT CCA ATG TTA TTA TCA TGG GTT TGC CTC CGA-3′ (reverse), and inserted into a LIC vector, which introduces a His6-SUMO tag and TEV protease cleavage site at the N terminus of the desired protein. Three residues, Ser-Asn-Ala, were added to the N terminus of the expressed protein following removal of the His6-SUMO tag.
Expression and Purification of PCM and PCM-F
Escherichia coli BL21(DE3) was transformed with the vector expressing the large subunit of PCM and grown overnight at 37 °C in 5 ml of Luria-Bertani medium containing ampicillin (100 μg/ml). Then, 6 liters of the same medium containing ampicillin was inoculated with the starter culture and grown at 37 °C. After 4 h, when the A600 had reached 0.6–0.8, the temperature was reduced to 15 °C. The cultures were induced with 0.1 mm isopropyl β-d-1-thiogalactopyranoside, and the cells were harvested 20 h later. The cells pellets were stored at −80 °C until use. The cell pellets were suspended in 150 ml of buffer containing 50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 20 mm imidazole, 0.2 mg/ml of lysozyme, 1 mm PMSF and one protease inhibitor mixture tablet (Roche Applied Science). The cell suspension was stirred at 4 °C for 40 min and then sonicated (power setting = 6) on ice for 10 min at 30-s intervals separated by 60-s cooling periods. The sonicate was centrifuged at 35,000 × g for 30 min, and the supernatant was loaded onto a Ni-Sepharose 6 Fast Flow column (2.5 × 8 cm, GE Healthcare) pre-equilibrated with Buffer A (50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 20 mm imidazole). The column was washed with 300 ml of Buffer A containing 20 mm imidazole and eluted with 300 ml of a linear gradient ranging from 20 to 300 mm imidazole in Buffer A. The fractions of interest were identified by SDS-PAGE analysis, pooled, and concentrated to 5 ml, and dialyzed overnight against 1 liter of dialysis buffer containing 100 mm Tris-HCl, pH 8.0, 100 mm NaCl, and 1 mm 2-mercaptoethanol and His6-MBP tag was cleaved with TEV protease (1 mg of TEV protease/100 mg of protein). The dialyzed protein was loaded on to amylose resin column (New England Biolabs) to remove His6-MBP tag, and the flow-through fraction was collected and loaded on to a Superdex 200 column (120 ml, GE Healthcare) pre-equilibrated with 50 mm HEPES-NaOH, pH 7.5, 100 mm NaCl. The fractions of interest were pooled, concentrated, frozen in liquid nitrogen, and stored at −80 °C until further use. The His6-tagged small subunit of PCM was expressed as described for the large subunit with the exception that kanamycin (50 μg/ml) was used as the antibiotic and purified using a Ni-Sepharose 6 Fast Flow column and followed by gel filtration using a Superdex 200 column. PCM-F was expressed as a fusion with a His6-tagged SUMO protein and expressed and purified using the same procedures as described above for the large subunit with the exception of the amylose resin column, which was excluded.
Enzyme Assays
Enzyme activity was monitored in 50 mm HEPES-NaOH, pH 7.5, containing 100 mm NaCl and 10 mm MgCl2 in a total volume of 0.6 ml. The reaction mixture contained 0.5–20 μm enzyme, 100 μm AdoCbl, and 0.1–5 mm isovaleryl-CoA or isobutyryl-CoA ± 1 mm nucleotides (GTP or GDP) at 37 °C. The enzymes were preincubated with AdoCbl (± 1 mm nucleotides in the case of IcmF), and the reaction was started by addition of substrate. At various time points (0.5–60 min), 100-μl aliquots were removed and quenched with 50 μl of 2 n KOH containing 0.2 mm valeric acid used as an internal standard. Following addition of 50 μl of H2SO4 (15%, v/v), the reaction mixture was saturated with solid NaCl and extracted with ethyl acetate (125 μl). The extract was analyzed directly by gas chromatography (GC, Agilent Technologies) using a 0.25-μm DB-FFAP (30-m × 0.25 mm inner diameter) capillary column (Agilent Technologies). A 5-μl sample was injected in the pulsed splitless mode. The oven temperature was initially at 80 °C. Following the sample injection, the temperature was raised to 150 °C at a rate of 10 °C/min and maintained at 150 °C for 2 min. Retention times for the compounds of interest were as follows: isobutyric acid, 5.85 min; pivalic acid, 5.99 min; n-butyric acid, 6.5 min; isovaleric acid, 6.96 min; and valeric acid, 7.78 min.
To determine the kinetic parameters for the isovaleryl-CoA mutase and isobutyryl-CoA mutase activities of IcmF, the initial rate of the reaction was plotted against substrate concentration and fitted to the Michaelis-Menten equation, where v = Vmax × [S]/(Km + [S]).
To determine the kinetic parameters of isovaleryl-CoA mutase activity of PCM, the initial rate of the reaction was plotted against substrate concentration and fitted to the Hill equation, where v = Vmax × [S]n/(Km + [S]n).
Enzyme-monitored Turnover
Spectral changes in AdoCbl bound to IcmF and PCM were monitored by UV-visible spectroscopy at 25 °C in 50 mm HEPES-NaOH, pH 7.5, containing 100 mm NaCl and 10 mm MgCl2. Substrates (1 mm final concentration of isovaleryl-CoA or isobutyryl-CoA) were added to 30 μm AdoCbl-loaded enzyme. With IcmF, the influence of the G-protein domain on catalytic turnover was checked by addition of 5 mm GTP or GDP to the reaction mixture. The amount of cob(II)alamin formed under steady-state turnover conditions was calculated from the decrease in absorbance at 525 nm upon substrate addition using a value of Δϵ525 nm of −4.8 mm−1 cm−1 (20). During the course of the reaction, AdoCbl was gradually converted to enzyme-bound aquocobalamin (OH2Cbl) as indicated by the appearance of the 351-nm absorption peak. The increase in absorption at 351 nm (A) was fitted to a single exponential function, A = A0 + A1(1 − e−kt) where k is the observed rate constant for inactivation, A0 is the initial absorbance at 351 nm, t is time in minutes, and A1 is the amplitude.
Isothermal Titration Calorimetry
Isothermal titration calorimetry experiments were performed in triplicate as described previously (12) and the data were analyzed using MicrocalORIGIN software. Briefly, binding of AdoCbl to PCM was monitored as follows. Enzyme (10 μm small subunit of PCM or PCM-F) in 50 mm HEPES-NaOH buffer, pH 7.5, containing 100 mm NaCl was titrated with 30 × 10-μl aliquots of a 0.4 mm solution of AdoCbl at 20.0 °C. The calorimetric signals were integrated and the data were well fitted to a single-site binding model to estimate the equilibrium association constant, KA, and the binding enthalpy, ΔH0. The Gibbs free energy of binding, ΔG0, and the entropic contribution to the binding free energy, −TΔS0, were calculated using Equations 1 and 2.
Bioinformatics Analysis
The integrated microbial genomes database web-based tools were used for BLAST search and operon analysis (21). A multiple sequence alignments were constructed using a stand-alone version of ClustalX version 2.1.
Results
Structure-based Redesign of IcmF
Our efforts at enhancing the catalytic activity of IcmF with isovaleryl-CoA as substrate were guided by sequence comparisons of IcmF with other acyl-CoA mutases (10, 11) and by the crystal structures of IcmF (17) and MCM (16, 22). Sequence comparisons revealed the presence of a putative acyl-CoA mutase annotated as MCM-like in which Leu and Asn substitute Phe-598 and Gln-742, respectively, in IcmF (Fig. 2A). Hence, these two residues were targeted by multiple missense mutations (Table 1). To better accommodate the longer isovaleryl-CoA substrate, we introduced residues with smaller side chains at Phe-598 (Ala, Gly, Ile, Leu, and Val) and Gln-742 (Ala, Leu, and Asn). The yield of each of the purified mutants (∼5.0 mg/liter of culture) was similar to that of wild-type IcmF and the purity of each mutant protein was judged to be >95% by SDS-PAGE analysis (data not shown). The mutants, like wild-type IcmF, eluted as a single peak by gel filtration chromatography with a molecular mass of 279 kDa, consistent with being homodimers.
TABLE 1.
Comparison of the kinetic parameters for the isovaleryl-CoA mutase and isobutyryl-CoA mutase activities of wild-type and mutant IcmFs, PCM, and PCM-F
Enzyme activities were determined at 37 °C and represent the average of at least three independent experiments ± S.D. The activity of PCM was measured in the presence of a 3:1 ratio of small:large subunit. Wild-type and the mutants in the first column refer to IcmF.
| Isovaleryl-CoA |
Isobutyryl-CoA |
|||||
|---|---|---|---|---|---|---|
| kcata | Km | kcat/Km | kcata | Km | kcat/Km | |
| min−1 | mm | mm−1 min−1 | min−1 | mm | mm−1 min−1 | |
| Wild-type | 3.7 ± 0.1 | 1.4 ± 0.1 | 2.6 | 2900 ± 400 | 1.1 ± 0.3 | 2600 |
| F598A | 22 ± 2 | 0.51 ± 0.15 | 43 | 4.8 ± 0.4 | 0.45 ± 0.11 | 11 |
| F598G | NDb | ND | ND | ND | ND | ND |
| F598I | 3.6 ± 0.2 | 0.58 ± 0.12 | 6.2 | 8.4 ± 0.3 | 0.36 ± 0.05 | 23 |
| F598L | 8.4 ± 0.8 | 0.56 ± 0.16 | 15 | 76 ± 4 | 0.89 ± 0.10 | 85 |
| F598V | 15 ± 1 | 0.62 ± 0.16 | 24 | 12 ± 2 | 0.67 ± 0.23 | 18 |
| Q742A | ND | ND | ND | 18 ± 2 | 0.79 ± 0.23 | 23 |
| Q742L | ND | ND | ND | ND | ND | ND |
| Q742N | ND | ND | ND | 110 ± 10 | 2.4 ± 0.3 | 46 |
| PCM | 14 ± 3 | 0.37 ± 0.13 | 38 | ND | ND | ND |
| PCM-F | 23 ± 1 | 0.20 ± 0.01 | 115 | ND | ND | ND |
a The kcat values for IcmF, PCM, and PCM-F were calculated per α, αβ, and α unit, respectively of each enzyme.
b ND is not detected. The detection limit of the GC assay is 5 pmol of product.
Isovaleryl-CoA Mutase and Isobutyryl-CoA Mutase Activities of IcmF
The isovaleryl-CoA mutase and isobutyryl-CoA mutase activities of the mutants were compared with wild-type IcmF (Table 1). The F598A mutation simultaneously enhanced isovaleryl-CoA mutase activity 6-fold (0.18 ± 0.02 μmol min−1 mg−1) and diminished isobutyryl-CoA activity >600-fold (0.039 ± 0.003 μmol min−1 mg−1), with respect to wild-type IcmF (0.030 ± 0.001 μmol min−1 mg−1 and 24 ± 3 μmol min−1 mg−1, respectively). Substitution of Phe-598 with the nonaromatic hydrophobic residues isoleucine, leucine, and valine, either maintained or increased isovaleryl-CoA mutase activity, whereas isobutyryl-CoA mutase activity was diminished albeit with less dramatic effects than the F598A mutation. In addition, mutations at Phe-598 decreased the Km for isovaleryl-CoA and therefore increased the catalytic efficiency (kcat/Km) of the isovaleryl-CoA mutase reaction compared with wild-type IcmF. The activity of the F598G mutant was not detectable with either substrate. Mutation of Gln-742 to alanine, leucine, or asparagine, resulted in undetectable isovaleryl-CoA mutase activity and significantly diminished isobutyryl-CoA mutase activity (Table 1).
Inactivation Kinetics of IcmF Mutants
The active sites in the CmIcmF dimer exhibit identical affinity for AdoCbl (Kd = 0.27 ± 0.01 μm, n = 2.1 ± 0.1) in contrast to the different affinities reported for the Geobacillus kaustophilus IcmF (Kd1 = 0.081 ± 0.014 μm and Kd2 = 1.98 ± 0.42 μm) (12). To assess whether the low or undetectable activity of several of the CmIcmF mutants in Table 1 was due to their enhanced propensity for inactivation, the spectrum of AdoCbl bound to IcmF was monitored following addition of isovaleryl-CoA (Fig. 3). For these experiments, IcmF was reconstituted with 2 eq of AdoCbl and the absence of free AdoCbl was confirmed by analyzing the spectrum of the filtrate obtained after concentrating the sample mixture using an Amicon concentrator (50-kDa cutoff).
FIGURE 3.

Enzyme-monitored turnover by wild-type and F598A IcmF. Spectra of (A) wild-type and (B) F598A IcmF (with 2 eq or 30 μm bound AdoCbl) in 50 mm HEPES-NaOH, pH 7.5, containing 100 mm NaCl and 10 mm MgCl2 at 25 °C (solid lines) and after addition of 1 mm isovaleryl-CoA (dotted lines), which resulted in the formation of cob(II)alamin with an absorption maximum at 471 nm. C, rapid oxidation of cob(II)alamin to OH2Cbl was observed during reaction of F598A IcmF (with 2 eq or 30 μm AdoCbl bound) with 1 mm isovaleryl-CoA in 50 mm HEPES-NaOH, pH 7.5, containing 100 mm NaCl and 10 mm MgCl2 at 25 °C in the dark. D, the time-dependent increase at 351 nm (denoting OH2Cbl formation) was plotted in the presence of oxygen. The data presented here are representative of at least three experiments and were fit to a single exponential equation described under “Experimental Procedures.”
Addition of isovaleryl-CoA to wild-type holo-IcmF resulted in formation of the intermediate, cob(II)alamin, as evidenced by the decrease in absorbance at 530 nm and increase at 471 nm (Fig. 3A). Using a Δϵ525 nm of −4.8 mm−1 cm−1 (20), cob(II)alamin was estimated to represent ∼20% of the cofactor under steady-state turnover conditions (Table 2). In contrast, AdoCbl was quantitatively converted to cob(II)alamin when isovaleryl-CoA was added to F598A (Fig. 3B) or to the F598I/L or Q742A mutants (Table 2). Accumulation of all the cofactor as cob(II)alamin indicates a change in the reaction coordinate such that the barrier to one or more steps following cob(II)alamin formation and preceding reformation of AdoCbl, has increased.
TABLE 2.
Comparison of inactivation kinetics for wild-type and mutant IcmFs, PCM, and PCM-F
Inactivation rates for OH2Cbl formation were determined in the presence of isovaleryl-CoA at 25 °C and represent the average of at least three independent experiments ± S.D.
| Enzyme | kobs | Cob(II)alamina |
|---|---|---|
| min−1 | % | |
| Wild-type IcmF | 0.035 ± 0.001 | 20 |
| F598A IcmF | 0.12 ± 0.02 | 100 |
| F598G IcmF | NDb | ND |
| F598I IcmF | 0.080 ± 0.007 | 100 |
| F598L IcmF | 0.14 ± 0.02 | 100 |
| F598V IcmF | 0.077 ± 0.007 | 90 |
| Q742A IcmF | 0.10 ± 0.01 | 100 |
| Q742L IcmF | 0.0090 ± 0.0002 | <10 |
| Q742N IcmF | ND | ND |
| PCMc | 0.014 ± 0.002 | 5 |
| PCM-F | 0.016 ± 0.002 | 5 |
a The concentration of the catalytic intermediate, cob(II)alamin was calculated 2–5 min after adding isovaleryl-CoA under aerobic conditions. Errors were <10%.
b ND, not determined due to rapid formation of OH2Cbl following addition of isovaleryl-CoA.
c The inactivation rate of PCM was measured in the presence of a 1:2 or 1:3 ratio of small:large subunit and were found to be indistinguishable.
Because cob(II)alamin can be oxidized to OH2Cbl, its increased accumulation in the mutants could lead to an increased propensity for OH2Cbl formation. OH2Cbl is a hallmark of inactivation in AdoCbl-dependent enzymes (23) and is characterized by an increase in absorption at 351 and 530 nm (Fig. 3C). The F598A mutant inactivates 3-fold more rapidly than wild-type IcmF as monitored by the increase in absorbance at 351 nm (Fig. 3D, Table 2).
The F598G and Q742N mutants showed rapid conversion of AdoCbl to OH2Cbl under aerobic conditions without detectable accumulation of the cob(II)alamin intermediate (Table 2). Hence, enzyme-monitored turnover indicates that the IcmF mutants engineered to have enhanced isovaleryl-CoA mutase activity are simultaneously more susceptible to inactivation. The only exception is Q742L IcmF, which showed a lower proportion of the cofactor in the cob(II)alamin state under steady-state turnover conditions and a lower rate of OH2Cbl formation (Table 2). However, Q742L IcmF did not exhibit isovaleryl-CoA mutase activity (Table 1).
Effects of Nucleotides and Reductants on the Isovaleryl-CoA Mutase Activity of IcmF
Next, the ability of nucleotides that bind to the G-protein domain of IcmF, on isovaleryl-CoA mutase activity was assessed with the most active mutant, F598A. The G-protein domain interacts with both the B12 and substrate-binding domains in IcmF (17). The presence of GTP or GDP increased the maximal isovaleryl-CoA mutase activity of wild-type IcmF by ∼4-fold (Fig. 4A). However, the same conditions had virtually no effect on the isovaleryl-CoA mutase activity of the F598A mutant (Fig. 4B). Furthermore, whereas the nucleotides stabilized wild-type IcmF against inactivation, they had little or no effect on the F598A mutant (Fig. 4, C and D).
FIGURE 4.

Effects of nucleotides on the isovaleryl-CoA mutase activity of IcmF. A and B, comparison of the time courses for the reaction catalyzed by (A) wild-type and (B) F598A IcmF at 37 °C in the absence or presence of nucleotides. The reaction conditions were 2 μm IcmF dimer, 100 μm AdoCbl, 50 mm HEPES-NaOH buffer, pH 7.5, 100 mm NaCl, 10 mm MgCl2 ± 1 mm nucleotides, and 1 mm isovaleryl-CoA at 37 °C. The data represent the mean ± S.D. of three independent experiments. C and D, time dependence of the inactivation of wild-type (C) and F598A (D) IcmF in the absence or presence of nucleotides. The increase in absorbance at 351 nm corresponding to formation of OH2Cbl via oxidation of the initially formed cob(II)alamin intermediate during turnover was monitored as described under “Experimental Procedures.” The spectra were recorded between 0 and 60 min. The data presented here are representative of at least three experiments and were fit to a single exponential equation described under “Experimental Procedures.”
In a further attempt to stabilize IcmF against inactivation, the effect of several reductants was tested. Both DTT and 2-mercaptoethanol enhanced the maximal isovaleryl-CoA mutase activity of wild-type IcmF ∼5-fold and F598A IcmF ∼2-fold, respectively, whereas GSH and tris(2-carboxyethyl)phosphine hydrochloride were without effect (data not shown).
Identification of a Putative Pivalyl-CoA/Isovaleryl-CoA Mutase
Given the high susceptibility of the most active IcmF mutant to inactivation, we used bioinformatics analysis to identify a “bona fide” isovaleryl-CoA/pivalyl-CoA mutase. We examined sequences that are annotated in the databases as MCM-like proteins but differ in the two active site residues that are predicted to be important for substrate specificity especially in organisms with multiple copies of acyl-CoA mutases (24, 25). Specifically, we identified a group of genes that encode a substrate-binding subunit where Leu and Asn substitute Phe-598 and Gln-742, respectively, in IcmF. We concentrated on X. autotrophicus strain Py2 where Xaut_5043 and Xaut_5044 encode the large and small subunits, respectively, of an AdoCbl-dependent mutase, tentatively identified as a PCM. In the large subunit of the putative PCM, Leu-87 and Asn-204 correspond to Phe-598 and Gln-742 in IcmF, respectively (Fig. 2). In addition, a gene encoding a putative G-protein chaperone (Xaut_5042) is present in the same operon. The bacterial MeaB and human CblA are G-protein chaperones that gate docking of AdoCbl to bacterial and human MCM, respectively, whereas the corresponding G-protein in IcmF is fused between the small and large subunits (Fig. 1B). The putative PCM is similar in organization to ICM, which exists as an α2β2 heterotetramer with the small and large subunits binding AdoCbl and substrate, respectively (9).
Purification and Kinetic Properties of PCM
The recombinant large subunit of PCM was purified as a His6-MBP-tagged protein. Following cleavage of the tag, the large subunit was obtained with a yield of ∼4 mg of protein/liter of culture. The band observed by SDS-PAGE analysis corresponds to the predicted mass of 62.3 kDa for the large subunit (Fig. 5A). Size exclusion chromatography on a calibrated Superdex 200 column yielded an estimated molecular mass of 135 kDa, consistent with the large subunit being a homodimer (not shown).
FIGURE 5.

Characterization of PCM and PCM-F. A, the purity of the large and small subunits of PCM and PCM-F were judged by SDS-PAGE analysis. B, dependence of the isovaleryl-CoA mutase activity of PCM on the ratio of the small:large subunit. The reaction mixture contained 1 mm isovaleryl-CoA in 50 mm HEPES-NaOH buffer, pH 7.5, 100 mm NaCl, 10 mm MgCl2 at 37 °C and varying ratios of the small:large subunit as described under “Experimental Procedures.” C, dependence of isovaleryl-CoA mutase activity of PCM (open circles) and PCM-F (closed circles) on the concentration of isovaleryl-CoA (0.1–1.0 mm) in 50 mm HEPES-NaOH buffer, pH 7.5, 100 mm NaCl, 10 mm MgCl2 at 37 °C. The ratio of the small to large subunit of PCM was fixed at 3:1. The kinetic parameters derived from this analysis are reported in Table 1. D, time dependence of the inactivation of PCM (closed circles) and PCM-F (closed triangles). The inactivation of wild-type and F598A IcmF (Fig. 3D) are also included for comparison.
The recombinant small subunit of PCM was purified as an N-terminal His6-tagged protein and ∼6 mg of highly pure protein were obtained per liter of culture (Fig. 5A). The small subunit eluted with an apparent mass of 20 kDa by gel filtration chromatography, suggesting that it exists as a monomer based on the predicted mass of the polypeptide of 17.7 kDa. Gel filtration of a 1:1 mixture of the large and small subunits of PCM in the presence of AdoCbl, showed no evidence of complex formation, indicating weak interaction between the subunits under these conditions.
Next, the dependence of the isovaleryl-CoA mutase activity of PCM was assessed at increasing small:large subunit ratios (Fig. 5B). Maximal activity was observed at an ∼1:1 ratio. The specific activity (0.18 ± 0.04 μmol min−1 mg−1 of protein) and steady-state kinetic parameters for PCM were determined at a 3:1 molar ratio of the small:large subunit. The activity of PCM showed a sigmoidal dependence on the concentration of isolvaleryl-CoA (Fig. 5C). From a Hill plot analysis of the data, the following values were estimated: Km for isovaleryl-CoA = 0.37 ± 0.13 mm, kcat = 14 ± 3 min−1, and n (Hill coefficient) = 1.4 ± 0.4, indicating positive cooperativity. Unlike IcmF, the activity of PCM was resistant to inactivation (Table 2 and Fig. 5D).
Purification and Kinetic Characteristics of PCM-F
Because the weak interaction between the large and small subunits of PCM complicates kinetic analysis, we created a variant in which the large and small subunits were linked in a single polypeptide by an 11-amino acid long (Gly-Gln)5-Gly sequence to give the fusion protein, PCM-F (Fig. 1B). This strategy has been used previously to study an AdoCbl-dependent isomerase, glutamate mutase, which like PCM, exhibits an α2β2, stoichiometry (26). Highly pure PCM-F was obtained at a yield of ∼2 mg of protein/liter of culture (Fig. 5A). Gel filtration chromatography yielded an apparent mass of 168 kDa indicating that PCM-F with a subunit molecular mass of 78.8 kDa, exists as an α2 homodimer.
The activity of PCM-F showed a sigmoidal dependence on the concentration of isovaleryl-CoA (Fig. 5C). From a Hill plot analysis of the data, the following values were estimated: Km for isovaleryl-CoA = 0.20 ± 0.01 mm, kcat = 23 ± 1 min−1, and n (Hill coefficient) = 2.0 ± 0.3. The catalytic efficiency (kcat/Km) of PCM-F is 3-fold higher than for PCM.
Binding of AdoCbl to PCM and PCM-F
The energetics of AdoCbl binding to the PCM and PCM-F were monitored by isothermal titration calorimetry (Table 3). Binding of AdoCbl to the small subunit of PCM (Kd = 12.5 ± 1.9 μm) exhibits a ΔG of −6.6 ± 0.1 kcal/mol and is enthalpically favored. The Kd for AdoCbl is lower for PCM-F (4.9 ± 0.3 μm) than for PCM and exhibits a ΔG of −7.3 ± 0.2 kcal/mol, which is also enthalpically driven. Hence, the presence of the “built-in” large subunit in PCM-F enhances the affinity for AdoCbl ∼2.6-fold.
TABLE 3.
Thermodynamic parameters for the binding of AdoCbl to PCM
The experiments were performed in 50 mm HEPES-NaOH, pH 7.5, 100 mm NaCl at 20 °C as described under “Experimental Procedures.” The data represent the mean ± S.D. of three independent experiments.
| n | Kd | ΔH | TΔS | ΔG | |
|---|---|---|---|---|---|
| μm | kcal/mol | kcal/mol | kcal/mol | ||
| PCM-small subunit | 0.9 ± 0.1 | 12.5 ± 1.9 | −14.4 ± 2.3 | −7.8 ± 2.4 | −6.6 ± 0.1 |
| PCM-F | 0.9 ± 0.1 | 4.9 ± 0.3 | −18.2 ± 10.1 | −11.1 ± 10.1 | −7.3 ± 0.2 |
Discussion
Our understanding of pivalic acid metabolism in nature is very limited. Some bacteria are known to mineralize pivalic acid to carbon dioxide (14). Some bacteria use pivalic acid as a starter unit and incorporate it into tertiary-butyl fatty acids, iso-even or anteiso-fatty acids and in Streptomyces avermitilis, into the antibiotic avermectin (27). In addition, pivalic acid (or pivalyl-CoA) might be formed during catabolism of marine sponge-derived polytheonamides in which the tert-butyl modification of threonine is introduced post-translationally (28). Isovaleryl-CoA is produced during leucine catabolism via the branched chain keto acid dehydrogenase or in myxobateria, via a mevalonate-dependent isoprenoid biosynthesis pathway involving 3-hydroxy-3-methylglutaryl-CoA synthase (29). In several bacteria, the gene encoding IcmF is coregulated with those involved in branched-chain amino acid degradation (30) or fatty acid catabolism (13). The potential for utilizing a catalyst with isovaleryl-CoA/pivalyl-CoA isomerase activity for metabolic engineering to produce branched C4 and C5 building blocks or the corresponding alcohol derivatives, is untapped.
Until this study, IcmF was the only known albeit inefficient, catalyst with isovaleryl-CoA/pivalyl-CoA isomerase activity, and with high susceptibility to inactivation (13). The PCM described in this study is derived from X. autotrophicus strain Py2, a facultative aerobe that was first isolated from black pool sludge, has versatile metabolic capabilities, and is able to utilize 2-hydroxyisobutyric acid, propene, and trichloroethylene, 1-butene among others, as carbon sources (31). The large subunit of PCM has Leu and Asn residues that are also seen in other genes annotated as being MCM-like in the database (Fig. 6A). At this point, it is not clear whether or not these AdoCbl-dependent acyl-CoA mutases, which are distinct from all previously characterized ones, are indeed bona fide PCMs. In X. autotrophicus strain Py2, the PCM genes are located in an operon that also encodes a homolog of the G-protein chaperone described for MCM, and two other genes annotated as phenylacetic acid degradation protein-like (paaD) and phenylacetic acid-CoA ligase-like (paaK) (Fig. 6B). Although the ligase is presumably important for converting the acid to a CoA ester needed for the AdoCbl-dependent isomerase activity, the role of PCM in X. autotrophicus remains to be established. In E. coli, 14 genes are involved in the phenylacetic acid degradation pathway (32).
FIGURE 6.

PCM sequence comparison and gene organization. A, multiple sequence alignment of substrate-binding domains of PCMs. PCMs from X. autotrophicus (Xaut_5043), Burkholderia phenoliruptrix (locus tag BPACDRAFT_03564), Cycloclasticus sp. (locus tag Ga0055576_00391), and Nocardioides sp. CF8 (locus tag CF8_0950) are shown. The two conserved residues (Leu-87 and Asn-204, X. autotrophicus numbering) predicted to be important for substrate binding, are highlighted in black. Identical and similar residues are indicated with asterisks and dots, respectively. B, organization of the PCM-encoding operon from X. autotrophicus. The genes involved in the operon are annotated as paaK (phenylacetate-CoA ligase-like, Xaut_5040), paaD (phenylacetic acid degradation protein-like, Xaut_5041), a meaB homolog encoding a G-protein chaperone (Xaut_5042), pcmA (large subunit of PCM, Xaut_5043), pcmB (small subunit of PCM, Xaut_5044), and tetR (a transcriptional regulator, Xaut_5045).
Most acyl-CoA mutases exhibit fairly stringent substrate specificities, which appear to be dictated by a limited number of differences in key active site residues. In an effort to exploit the measurable but low isovaleryl/pivalyl-CoA mutase activity reported for wild-type IcmF (13), we rationally targeted two active site residues, Phe-598 and Gln-742, making a total of eight substitutions. Of the individual missense mutations, F598A IcmF was the most active and led to an ∼4000-fold enhancement of the isovaleryl-CoA mutase relative to the isobutyryl-CoA mutase activity compared with wild-type enzyme (Table 1). However, F598A IcmF and the other mutants were even more susceptible than wild-type IcmF to inactivation during turnover (Table 2), a problem that was only marginally improved by the presence of reductants, DTT and 2-mercaptoethanol.
A similar switch in substrate selectivity has been previously reported for MCM where the corresponding active site residues, Tyr-89 and Arg-207, were targeted by mutagenesis (33). The double mutant, Y89F/R207Q, mimicked the corresponding active site residues in ICM (and the then unknown IcmF). In contrast to wild-type MCM, the double mutant bound the ICM substrates, n-butyryl-CoA and isobutyryl-CoA, but instead of catalyzing their isomerization, promoted inactivation via a suicidal internal electron transfer mechanism. Among the mutants characterized in this study, the F598I IcmF resembles HCM with Ile-598 and Gln-742 corresponding to Ile-90 and Gln-208 in HCM (11). Therefore, we examined the possibility that F598I IcmF exhibits HCM activity. However, dl-3-hydroxybutyryl-CoA was not converted to product as judged by an HPLC-based assay (data not shown) although cob(II)alamin formation was seen indicating that the first step, cobalt-carbon bond homolysis, had occurred. These results indicate that residues that dictate the narrow substrate specificity of acyl-CoA mutases are distinct from those that promote the 1,2 rearrangement reactions. Alternatively, mutations that permit accommodation of non-native substrates also lead to a misalignment in the active site residues so that unwanted side reactions such as oxidation of cob(II)alamin, are suppressed. Loss of reaction fidelity is seen in mutants in other AdoCbl-dependent enzymes, e.g. diol dehydratase, which catalyzes the conversion of 1,2-propanediol and 1,2-ethanediol to the corresponding aldehydes, and undergoes mechanism-based inactivation in the presence of glycerol. Interestingly, the S301A and Q336A mutations in the catalytic subunit of diol dehydratase confer resistance to glycerol-dependent inactivation relative to the wild-type enzyme indicating that these residues are involved in inactivation in the native enzyme (34).
The susceptibility of AdoCbl-dependent radical enzymes to irreversible inactivation during catalysis necessitates their reliance on chaperones for their reactivation. In the subclass of AdoCbl-dependent enzymes to which diol dehydratase belongs, reactivating factors mediate an ATP-dependent exchange of enzyme-bound OH2Cbl with free AdoCbl (23, 35). These reactivases have sequence similarity to DnaK and other members of the Hsp70 family of molecular chaperones and lower sequence similarity to the large subunits of corresponding mutases (23). In contrast, the G-protein chaperones associated with AdoCbl-dependent acyl-CoA mutases belong to the G3E family of P-loop metallochaperones (36). In MCM where the role of the G-protein chaperone MeaB is best characterized (37), the chaperone uses GTP hydrolysis to power expulsion of cob(II)alamin when 5′-deoxyadenosine is lost from the active site (37). MeaB diminishes the inactivation rate of the mutase ∼15- and ∼3-fold in the presence of GTP and GDP, respectively (20). The human ortholog of MeaB is CblA and it is reported to prevent inactivation and increase the enzymatic activity of human MCM (38, 39). In IcmF, which has a built-in G-protein chaperone, GTP and GDP protect against inactivation in wild-type but not in F598A IcmF (Fig. 4, Table 2) indicating that this residue might be important for communication between the mutase and G-protein domains. The GTPase activity of IcmF is not affected in the Phe-598 mutants (kcat is 30–40 min−1), which is comparable with that of wild-type IcmF (34 ± 2 min−1 at 37 °C).
In summary, our efforts to rationally redesign the IcmF active site to improve its isovaleryl-CoA mutase activity yielded a protein that was a better catalyst but also more susceptible to inactivation. They also led to characterization of the first AdoCbl-dependent PCM exhibiting isovaleryl-CoA mutase activity, which was resistant to inactivation. We engineered a variant in which the two weakly interacting subunits of PCM were fused into a single polypeptide to obtain PCM-F, which was also highly active as an isovaleryl-CoA mutase. PCM-F could be useful in applications ranging from bioremediation to stereospecific synthesis of C5 carboxylic acids and alcohols.
Author Contributions
K. K. designed, performed, and analyzed the experiments and wrote the manuscript. V. C. helped conceive the experiments, conducted bioinformatics analysis, and edited the manuscript. R. B. helped conceive the experiments, analyzed the data and co-wrote the manuscript. All authors approved the final version of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant DK45776 (to R. B.) and a postdoctoral fellowship for research abroad from the Japan Society for the Promotion of Science (to K. K). The authors declare that they have no conflicts of interest with the contents of this article.
- AdoCbl
- 5′-deoxyadenosylcobalamin
- ECM
- ethylmalonyl-CoA mutase
- HCM
- 2-hydroxyisobutyryl-CoA mutase
- ICM
- isobutyryl-CoA mutase
- IcmF
- isobutyryl-CoA mutase fused with its G-protein chaperone
- MCM
- methylmalonyl-CoA mutase
- OH2Cbl
- aquocobalamin
- PCM
- pivalyl-CoA mutase
- PCM-F
- engineered fusion construct of PCM
- LIC
- ligation-independent cloning
- TEV
- tobacco etch virus
- MBP
- maltose-binding protein.
References
- 1. Banerjee R. (2003) Radical carbon skeleton rearrangements: catalysis by coenzyme B12-dependent mutases. Chem. Rev. 103, 2083–2094 [DOI] [PubMed] [Google Scholar]
- 2. Banerjee R. (2001) Radical peregrinations catalyzed by coenzyme B12-dependent enzymes. Biochemistry 40, 6191–6198 [DOI] [PubMed] [Google Scholar]
- 3. Cracan V., Banerjee R. (2012) Novel B12-dependent acyl-CoA mutases and their biotechnological potential. Biochemistry 51, 6039–6046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Frey P. A. (2014) Travels with carbon-centered radicals: 5′-deoxyadenosine and 5′-deoxyadenosine-5′-yl in radical enzymology. Acc. Chem. Res. 47, 540–549 [DOI] [PubMed] [Google Scholar]
- 5. Dowling D. P., Croft A. K., Drennan C. L. (2012) Radical use of Rossmann and TIM barrel architectures for controlling coenzyme B12 chemistry. Annu. Rev. Biophys. 41, 403–427 [DOI] [PubMed] [Google Scholar]
- 6. Hay B. P., Finke R. G. (1987) Thermolysis of the Co-C bond in adenosylcorrins: 3. quantifiation of the axial base effect in adenosylcobalamin by the synthesis and thermolysis of axial base-free adenosylcobinamide: insights into the energetics of enzyme-assisted cobalt-carbon bond homolysis. J. Am. Chem. Soc. 109, 8012–8018 [Google Scholar]
- 7. Halpern J. (1985) Mechanisms of coenzyme B12-dependent rearrangements. Science 227, 869–875 [DOI] [PubMed] [Google Scholar]
- 8. Banerjee R., Chowdhury S. (1999) Methylmalonyl-CoA mutase. in Chemistry and Biochemistry of B12 (Banerjee R., ed) pp. 707–730, John Wiley and Sons, New York [Google Scholar]
- 9. Ratnatilleke A., Vrijbloed J. W., Robinson J. A. (1999) Cloning and sequencing of the coenzyme B12-binding domain of isobutyryl-CoA mutase from Streptomyces cinnamonensis, reconstitution of mutase activity, and characterization of the recombinant enzyme produced in Escherichia coli. J. Biol. Chem. 274, 31679–31685 [DOI] [PubMed] [Google Scholar]
- 10. Erb T. J., Rétey J., Fuchs G., Alber B. E. (2008) Ethylmalonyl-CoA mutase from Rhodobacter sphaeroides defines a new subclade of coenzyme B12-dependent acyl-CoA mutases. J. Biol. Chem. 283, 32283–32293 [DOI] [PubMed] [Google Scholar]
- 11. Yaneva N., Schuster J., Schäfer F., Lede V., Przybylski D., Paproth T., Harms H., Müller R. H., Rohwerder T. (2012) Bacterial acyl-CoA mutase specifically catalyzes coenzyme B12-dependent isomerization of 2-hydroxyisobutyryl-CoA and (S)-3-hydroxybutyryl-CoA. J. Biol. Chem. 287, 15502–15511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cracan V., Padovani D., Banerjee R. (2010) IcmF is a fusion between the radical B12 enzyme isobutyryl-CoA mutase and its G-protein chaperone. J. Biol. Chem. 285, 655–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cracan V., Banerjee R. (2012) Novel coenzyme B12-dependent interconversion of isovaleryl-CoA and pivalyl-CoA. J. Biol. Chem. 287, 3723–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Probian C., Wülfing A., Harder J. (2003) Anaerobic mineralization of quaternary carbon atoms: isolation of denitrifying bacteria on pivalic acid (2,2-dimethylpropionic acid). Appl. Environ. Microbiol. 69, 1866–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rohwerder, Müller H. (2007) New bacterial cobalamin-dependent CoA-carbonyl mutases involved in degradation pathways. in Vitamin B: New Research (Elliot C. M., ed) pp. 81–98, Nova Science Publishers, Hauppauge, NY [Google Scholar]
- 16. Mancia F., Evans P. R. (1998) Conformational changes on substrate binding to methylmalonyl CoA mutase and new insights into the free radical mechanism. Structure 6, 711–720 [DOI] [PubMed] [Google Scholar]
- 17. Jost M., Cracan V., Hubbard P. A., Banerjee R., Drennan C. L. (2015) Visualization of a radical B12 enzyme with its G-protein chaperone. Proc. Natl. Acad. Sci. U.S.A. 112, 2419–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vlasie M. D., Banerjee R. (2003) Tyrosine 89 accelerates Co-carbon bond homolysis in methylmalonyl-CoA mutase. J. Am. Chem. Soc. 125, 5431–5435 [DOI] [PubMed] [Google Scholar]
- 19. Padmakumar R., Banerjee R. (1995) A carbon skeleton walk: a novel double rearrangement of glutaryl-CoA catalyzed by the human methylmalonyl-CoA mutase. Biofactors 5, 83–86 [PubMed] [Google Scholar]
- 20. Padovani D., Banerjee R. (2006) Assembly and protection of the radical enzyme, methylmalonyl-CoA mutase, by its chaperone. Biochemistry 45, 9300–9306 [DOI] [PubMed] [Google Scholar]
- 21. Markowitz V. M., Chen I. M., Palaniappan K., Chu K., Szeto E., Grechkin Y., Ratner A., Jacob B., Huang J., Williams P., Huntemann M., Anderson I., Mavromatis K., Ivanova N. N., Kyrpides N. C. (2012) IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res. 40, D115–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mancia F., Keep N. H., Nakagawa A., Leadlay P. F., McSweeney S., Rasmussen B., Bösecke P., Diat O., Evans P. R. (1996) How coenzyme B12 radicals are generated: the crystal structure of methylmalonyl-coenzyme A mutase at 2-Å resolution. Structure 4, 339–350 [DOI] [PubMed] [Google Scholar]
- 23. Toraya T. (2014) Cobalamin-dependent dehydratases and a deaminase: radical catalysis and reactivating chaperones. Arch. Biochem. Biophys. 544, 40–57 [DOI] [PubMed] [Google Scholar]
- 24. Rezanka T., Kolouchová I., Cejková A., Sigler K. (2012) Biosynthesis and metabolic pathways of pivalic acid. Appl. Microbiol. Biotechnol. 95, 1371–1376 [DOI] [PubMed] [Google Scholar]
- 25. Rohwerder T., Breuer U., Benndorf D., Lechner U., Müller R. H. (2006) The alkyl tert-butyl ether intermediate 2-hydroxyisobutyrate is degraded via a novel cobalamin-dependent mutase pathway. Appl. Environ. Microbiol. 72, 4128–4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen H. P., Marsh E. N. (1997) Adenosylcobalamin-dependent glutamate mutase: examination of substrate and coenzyme binding in an engineered fusion protein possessing simplified subunit structure and kinetic properties. Biochemistry 36, 14939–14945 [DOI] [PubMed] [Google Scholar]
- 27. Rezanka T., Siristova L., Schreiberová O., Rezanka M., Masák J., Melzoch K., Sigler K. (2011) Pivalic acid acts as a starter unit in a fatty acid and antibiotic biosynthetic pathway in Alicyclobacillus, Rhodococcus, and Streptomyces. Environ. Microbiol. 13, 1577–1589 [DOI] [PubMed] [Google Scholar]
- 28. Freeman M. F., Gurgui C., Helf M. J., Morinaka B. I., Uria A. R., Oldham N. J., Sahl H. G., Matsunaga S., Piel J. (2012) Metagenome mining reveals polytheonamides as posttranslationally modified ribosomal peptides. Science 338, 387–390 [DOI] [PubMed] [Google Scholar]
- 29. Bode H. B., Ring M. W., Schwär G., Altmeyer M. O., Kegler C., Jose I. R., Singer M., Müller R. (2009) Identification of additional players in the alternative biosynthesis pathway to isovaleryl-CoA in the myxobacterium Myxococcus xanthus. ChemBioChem 10, 128–140 [DOI] [PubMed] [Google Scholar]
- 30. Kazakov A. E., Rodionov D. A., Alm E., Arkin A. P., Dubchak I., Gelfand M. S. (2009) Comparative genomics of regulation of fatty acid and branched-chain amino acid utilization in proteobacteria. J. Bacteriol. 191, 52–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ensign S. A., Small F. J., Allen J. R., Sluis M. K. (1998) New roles for CO2 in the microbial metabolism of aliphatic epoxides and ketones. Arch. Microbiol. 169, 179–187 [DOI] [PubMed] [Google Scholar]
- 32. Ferrández A., Miñambres B., García B., Olivera E. R., Luengo J. M., García J. L., Díaz E. (1998) Catabolism of phenylacetic acid in Escherichia coli: characterization of a new aerobic hybrid pathway. J. Biol. Chem. 273, 25974–25986 [DOI] [PubMed] [Google Scholar]
- 33. Vlasie M. D., Banerjee R. (2004) When a spectator turns killer: suicidal electron transfer from cobalamin in methylmalonyl-CoA mutase. Biochemistry 43, 8410–8417 [DOI] [PubMed] [Google Scholar]
- 34. Yamanishi M., Kinoshita K., Fukuoka M., Saito T., Tanokuchi A., Ikeda Y., Obayashi H., Mori K., Shibata N., Tobimatsu T., Toraya T. (2012) Redesign of coenzyme B12 dependent diol dehydratase to be resistant to the mechanism-based inactivation by glycerol and act on longer chain 1,2-diols. FEBS J. 279, 793–804 [DOI] [PubMed] [Google Scholar]
- 35. Toraya T., Mori K. (1999) A reactivating factor for coenzyme B12-dependent diol dehydratase. J. Biol. Chem. 274, 3372–3377 [DOI] [PubMed] [Google Scholar]
- 36. Leipe D. D., Wolf Y. I., Koonin E. V., Aravind L. (2002) Classification and evolution of P-loop GTPases and related ATPases. J. Mol. Biol. 317, 41–72 [DOI] [PubMed] [Google Scholar]
- 37. Padovani D., Banerjee R. (2009) A G-protein editor gates coenzyme B12 loading and is corrupted in methylmalonic aciduria. Proc. Natl. Acad. Sci. U.S.A. 106, 21567–21572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Froese D. S., Kochan G., Muniz J. R., Wu X., Gileadi C., Ugochukwu E., Krysztofinska E., Gravel R. A., Oppermann U., Yue W. W. (2010) Structures of the human GTPase MMAA and vitamin B12-dependent methylmalonyl-CoA mutase and insight into their complex formation. J. Biol. Chem. 285, 38204–38213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Takahashi-Íñiguez T., García-Arellano H., Trujillo-Roldán M. A., Flores M. E. (2011) Protection and reactivation of human methylmalonyl-CoA mutase by MMAA protein. Biochem. Biophys. Res. Commun. 404, 443–447 [DOI] [PubMed] [Google Scholar]