

Background: The prion protein (PrPC) functions as a scaffold for cell surface signaling systems, and plays a role in neurodegenerative diseases that include clinical depression among their symptoms.
Results: PrPC-null mice showed depressive-like behavior concomitant with functional changes in monoaminergic systems.
Conclusion: PrPC regulates functions of monoaminergic synapses.
Significance: PrPC may be involved in major depression and related neuropsychiatric disorders.
Keywords: Alzheimer disease, depression, neurodegeneration, prion, prion disease, cerebral cortex, monoaminergic neurotransmission
Abstract
We sought to examine interactions of the prion protein (PrPC) with monoaminergic systems due to: the role of PrPC in both Prion and Alzheimer diseases, which include clinical depression among their symptoms, the implication of monoamines in depression, and the hypothesis that PrPC serves as a scaffold for signaling systems. To that effect we compared both behavior and monoaminergic markers in wild type (WT) and PrPC-null (PrP−/−) mice. PrP−/− mice performed poorly when compared with WT in forced swimming, tail suspension, and novelty suppressed feeding tests, typical of depressive-like behavior, but not in the control open field nor rotarod motor tests; cyclic AMP responses to stimulation of D1 receptors by dopamine was selectively impaired in PrP−/− mice, and responses to serotonin, but not to norepinephrine, also differed between genotypes. Contents of dopamine, tyrosine hydroxylase, and the 5-HT5A serotonin receptor were increased in the cerebral cortex of PrP−/−, as compared with WT mice. Microscopic colocalization, as well as binding in overlay assays were found of PrPC with both the 5HT5A and D1, but not D4 receptors. The data are consistent with the scaffolding of monoaminergic signaling modules by PrPC, and may help understand the pathogenesis of clinical depression and neurodegenerative disorders.
Introduction
The term prion, an abbreviation of proteinaceous infectious particle, underscores the prevailing concept that an infectious, self-propagating, protein-only particle is the cause of transmissible spongiform encephalopathies, a group of neurodegenerative disorders that include scrapie, Creutzfeldt-Jakob disease, fatal familial insomnia, bovine spongiform encephalopathy, and the chronic wasting disease of cervids (1, 2). The disease-related aggregates of abnormally folded protein, often referred to as prion scrapie (PrPSc), arise by structural conversion of the endogenous cellular protein known as the prion protein (PrPC),2 encoded by the human gene PRNP (Prnp in mouse) (3, 4). Although the abnormal conformers have historically received the most attention, there is growing interest in the physiological roles of the native, endogenous PrPC, due to its pleiotropic functions and more recently to the links with Alzheimer disease and cancer (see Refs. 5–7 for reviews).
The prion protein is mainly, albeit not exclusively, expressed at the surface of nerve and immune cells and, similar to other glycosylphosphatidylinositol-anchored proteins, PrPC associates with lipid rafts amid continuous trafficking around distinct plasma membrane domains and intracellular vesicles (8, 9). Experimental studies show that PrPC participates in events such as cell proliferation, differentiation, and survival through various signaling pathways (5, 6).
An increasing body of evidence supports the hypothesis that PrPC functions as a cell surface scaffold protein, that is, it serves as a hub for the assembly of diverse signaling modules involved in a variety of physiological functions (5, 10). Indeed, the scaffold hypothesis is consistent with the often controversial functional properties assigned to the prion protein, which extend far beyond the nervous system, including immune responses (11, 12), cancer biology (6, 13), heart oxidative stress (14), glucose homeostasis (15), and stem cell regulation (16, 17). Such widespread influence probably depends on the ability of PrPC to bind several partners, varied combinations of which populate the cell surface of distinct cell types (5, 10).
Functional consequences of the direct binding of PrPC to several neurotransmitter receptors were reported (18–21), and neurotransmission may be further affected by PrPC through co-regulation of the expression of neurotransmitter receptor subunits (22). In particular, antibody-mediated engagement of PrPC reduced signaling through adenylyl cyclase, phospholipases C and A2, from at least three subtypes of the G protein-coupled serotonin (5HT) receptor and modulated the cross-talk among these pathways in a cell line that expresses 5HT receptors as well as PrPC (23, 24).
Monoaminergic systems are disturbed as diseases progress in animal models of transmissible spongiform encephalopathies. Changes were reported in PrPSc-infected brains, of the levels of serotonin, dopamine, and norepinephrine (25–27), as well as in the activity of monoaminergic receptors (28) and related enzymes, such as monoaminooxidase B (29) and adenylyl cyclase (30). Moreover, serotonergic dysfunction was observed in human clinical cases of fatal familial insomnia (31), and dopaminergic depletion was reported in patients with Creutzfeldt-Jakob disease, associated with rapid cognitive decline and movement disorders (32, 33). Almost 40% of patients of sporadic Creutzfeldt-Jakob disease showed symptoms of major depression, a disorder linked to monoaminergic neurotransmission (34, 35). Notably, PrPC-null mice reportedly showed signs of depressive-like behavior, which were corrected by imipramine, a tricyclic antidepressant that acts primarily as a monoamine reuptake inhibitor (36).
On the other hand, the prion protein has also been identified as a ligand of oligomers of the β-amyloid peptide (37), which are closely tied with signal corruption in the brains of patients of Alzheimer disease (AD) (38, 39). Clinical depression has been associated with AD, both as a symptom and as a risk factor (40, 41), and despite current controversy as to the role of biogenic amines in AD-associated depression (42), both neuropathological and in vivo imaging studies demonstrated links between monoaminergic systems and AD (43–45).
A phage display screen conducted in our laboratory suggested that PrPC may bind both the 5HT5A serotonergic receptor and the serotonin transporter SERT.3 In light of the apparent connections among monoaminergic neurotransmission, cell surface PrPC, and behavioral output, we hypothesized that monoaminergic signaling might be affected in the absence of PrPC. The present study both extends behavioral analysis to Prnp knock-out mice of a well controlled genetic background, as well as discloses novel neurochemical changes that may underlie their depressive-like behavior.
Experimental Procedures
Animals
Experiments were done using male, 8–12 weeks old, C57BL/10 and PrPC knock-out mice (Npu Prnp−/−; 107), backcrossed to C57BL/10 mice for at least 10 generations. The founders of our colony were kindly provided by Drs. Bruce Chesebro and Richard Race (Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases, Hamilton, MT). Heterozygous mice were mated and homozygous F1 descendants were crossed to generate PrPC-null (Npu Prnp−/−) embryos and their respective wild-type controls (Npu Prnp+/+). Animals were housed in groups of five per cage, under a 12-h light/12-h dark cycle, with controlled room temperature and humidity. Food and water were provided ad libitum. All procedures followed institutional guidelines and were approved by the Animal Care and Use Committee of the Federal University of Rio de Janeiro.
Behavior
Forced Swim Test (FST)
The FST were conducted as described by Porsolt (46), with minor modifications (47). Mice were individually forced to swim for 6 min in an open cylinder (65 cm height, 30 cm diameter) filled with water (24 ± 1 °C) to a height of 45 cm. The testing sessions were preceded, 1 day earlier, by an habituation session in which each mouse was placed in the swimming cylinder for 15 min, as a means to reduce stress and/or anxiety on the next day. Water was changed between subjects, and after testing animals were dried under a warm light for 20 min. Immobility time was recorded along the 6 min of testing, by a trained researcher blind to group assignment. Mice were considered immobile when they ceased struggling and remained floating motionless in the water, making only movements necessary to keep their head above water.
Tail Suspension Test
Mice were suspended individually to 60 cm above the floor by adhesive tape placed ∼1 cm from the tip of the tail. Immobility time induced by tail suspension was recorded during 6 min according to the method described by Steru et al. (48) with minor modifications (49). Immobility was defined as the absence of any limb or body movements, except for those caused by breathing.
Novelty Suppress Feeding Test
Mice were deprived of food for 24 h before testing. At the time of testing, one food pellet was placed on a white filter paper located in the middle of a test arena (30 × 30 × 45 cm). One at a time, mice were placed in one of the corners of the arena and allowed to explore for a maximum of 10 min. Their latency to visit and begin chewing the food pellet was recorded. Immediately after the test, animals were transferred to their home cages and allowed to feed freely.
Locomotor Activity Assay (Open-field Test)
Mice were placed in the corner of a 30 × 30 × 45-cm box divided in 9 smaller squares. The number of squares crossed with all paws (crossings) and the raising the forepaws (rearings) were scored in 5-min sessions. The box was thoroughly cleaned with a solution of 30% alcohol and dried between subjects.
Accelerating Rotarod Test
Motor performance was tested using an accelerating Rotarod (Insight, Brazil). All mice were pre-trained in the apparatus to reach a stable performance and to minimize novelty stress. Training consisted of 3 sessions on 3 consecutive days, each session including 3 separate runs. In each training run, animals were placed on the rods at an initial speed of 6 rpm for 30 s. The speed was then increased gradually to 37 rpm over 5 min. If a mouse fell from the apparatus during a training session, it was immediately placed back on, until the end of the run. Between runs, mice rested for at least 2 min, to reduce stress and fatigue. On the day of testing, 5 consecutive trials were made with 15-min inter-trial intervals, and in each trial the speed was slowly increased from 6 to 37 rpm within 5 min. Results were expressed as latency of animals to fall from the apparatus.
Assay of Accumulation of cAMP
Mice were euthanized in a CO2 chamber and immediately decapitated. Brains were removed, and the cerebral cortex or striatum dissected out and cut into 400-μm slices using a tissue chopper. Slices were selected and distributed equally in 12-well plates filled with aCSF (126 mm NaCl, 12 mm MgCl2, 2.5 mm KCl, 21.4 mm NaHCO3, 11.1 mm glucose, 1.2 mm NaH2PO4, and 400 μm ascorbic acid), saturated with a 95% O2, 5% CO2 mixture, just before the experiment. A phosphodiesterase inhibitor (3-isobutyl-1-methylxanthine, 500 μm) was added for 15 min and then each well received a 40-min stimulus with forskolin, serotonin, dopamine, norepinephrine, SKF-38393, SCH-23390, SB-699551, or raclopride. Generation of cAMP was completely stopped by the addition of 10% trichloroacetic acid, and then assayed radiometrically, as previously described (50).
Western Blots
Mice cerebral cortex was dissected out in ice-cold PBS, and homogenized in a solution containing 2% SDS, 20 mm EDTA, and 62.5 Tris-HCl, pH 7.5. Samples were centrifuged at 10,000 × g for 10 min at 4 °C. Supernatants were collected and protein concentrations were determined using the BCA method. For each sample, 30 μg of protein were used for SDS-PAGE, transferred to PVDF membranes, and probed with: anti-D1 (1:400), anti-SERT (1:500), anti-5HT5A (1:400), anti-TH (1:1,000), anti-Phospho-TH Ser40 (Cell Signaling, number 2791, 1:1000), anti-tryptophan hydroxylase (TPH) (1:500), anti-DAT (1:500), anti-VMAT2 (1:500), and anti-5HT1A (Santa Cruz, sc-1459, 1:100). Anti-α-tubulin (Sigma, T6074, 1:50,000) and anti-GAPDH (Millipore, MAB374, 1:3000) antibodies were used as loading controls.
Extraction and Quantification of Monoamines
Brain monoamines and their immediate metabolites were measured by HPLC coupled with electrochemical detection (HPLC-ED), following a protocol adapted from Arita and co-workers (51). Briefly, animals were decapitated, brains were removed, and the cerebral cortex or striatum dissected out in ice-cold PBS. Perchloric acid was added to each sample to a final concentration of 0.1 m. Samples were sonicated (2 pulses of 5 s, 50 Hz) and then centrifuged (10,000 × g) for 10 min to remove precipitated proteins. Supernatants were used for HPLC. For normalization, pellets were resuspended and protein concentrations were quantified with a BCA kit (Thermo Fisher Scientific, Waltham, MA). Fast isocratic separation was obtained using a reverse phase LC-18 column (4.6 × 250 mm; Sigma) with the following mobile phase: 20 mm sodium phosphate dibasic; 20 mm citric acid, 10% methanol, 0.12 mm Na2EDTA, and 566 mg/liter of heptanesulfonic acid, pH 2.64.
Immunohistochemistry
Animals used for immunohistochemistry were anesthetized and transcardially perfused with saline followed by 4% paraformaldehyde in 0.1 m phosphate buffer. Brains were removed and placed in phosphate buffer containing increasing concentrations of sucrose (10, 15, and 30%), for cryoprotection. Frozen coronal sections (40 μm) were cut using a cryostat (Leica Microsystems, Wetzlar, Germany) and stored in TBS-AF (Tris-buffered saline, 0.05% sodium azide, 30% glycerol, and 15% sucrose) at 4 °C. For free-floating immunohistochemistry, sections were washed in PBS and blocked for 2 h with 0.25% Triton, 3% BSA, and 3% normal donkey serum. The following primary antibodies were then incubated overnight at 4 °C: anti-PrPC (Cayman, SAF83, 1:200), anti-D1 (Merck, US324390, 1:400), anti-SERT (Sigma, SAB4200039, 1:500), anti-5HT5A (Sigma, SAB2101110, 1:400), anti-TH (Millipore, MAB318, 1:400), and anti-TPH (Millipore, AB1541, 1:500). After washing with PBS, sections were incubated for 2 h at room temperature with Alexa Fluor secondary antibodies (Life Technologies), briefly stained with DAPI (1 μg/ml), and mounted on glass slides with n-propyl gallate.
Overlay Assay
Recombinant mouse PrPC was separated by SDS-PAGE in various lanes (5 μg/lane), and transferred to a nitrocellulose membrane. Protein migration was verified with Ponceau S staining. The membrane was incubated for 16 h at 4 °C in a solution containing 100 μg of a total protein extract from the cerebral cortex of wild type mice, in 50 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1 mm CaCl2, 1% BSA, and 0.5% Nonidet P-40. Then, individual lanes were cut, and each was subject to Western blotting with the appropriate antibody. In addition to D1, 5HT5A, and SERT, we also probed overlay membranes with an antibody to dopamine receptor D4 (Santa Cruz, SC-1439, 1:1000), as a control. Target proteins are detected only if binding to the previously identified PrPC band.
Statistical Analysis
Except where stated otherwise, data are presented as mean ± S.E. We used Student's t test, or analysis of variance followed by Bonferroni's post hoc test, to compare means among treatments. Differences between treated and control groups were considered significant when p < 0.05.
Results
We first tested for depressive-like traits in PrPC-null mice as compared with control littermates. In the “forced swim test,” PrPC-null mice were immobile for significantly longer periods than wild-type controls (Fig. 1A). Similar results were obtained with the “tail suspension test” (Fig. 1B). Such behavior, giving up struggling and remaining immobile in adverse situations, is commonly interpreted as a measure of “hopelessness,” and is susceptible to antidepressant treatments (52), thus likely indicative of a depressive-like state. A more complex procedure, known as “novelty suppressed feeding test,” also showed a difference between groups. Food-deprived PrPC-null mice had a higher latency to seek out a food pellet, when compared with wild-type animals (Fig. 1C). This increased latency may be interpreted as unwillingness to eat despite being hungry, a depressive-like behavior analogous to anhedonia (53).
FIGURE 1.

Mice depleted of PrPC display depressive-like behavior but no motor deficits. A–C, Prnp-null mice showed an increased immobility time when compared with wild-type controls in both FST (A, n = 8WT 8KO) and tail suspension test (B, n = 8WT, 8KO), and extended latency to eat in the novelty suppress feeding test (C, n = 8WT, 8KO). D–F, motor functions were not significantly different between genotypes in either the accelerating rotarod (D, n = 10WT, 10KO) nor in the open field (E, n = 8WT, 7KO). *, p < 0.05; **, p < 0.01.
To test whether motor deficits might explain the behavioral effects, we compared the performances of both wild-type and PrPC-knock-out mice in the “open field test” and in the more demanding “accelerating rotarod test.” No significant differences were found between genotypes either in the number of crossings/rearings measured in the open field, or in the latency to fall from the rotarod apparatus, thus indicating similar motor capacities in both groups (Fig. 1, D and E).
Changes in synaptic functions are likely to explain the behavioral differences between the mouse genotypes. Therefore, we assessed parameters of cortical monoaminergic transmission, through measurements of neurotransmitter-induced cAMP accumulation. As expected, serotonin, dopamine, and norepinephrine elicited accumulation of cAMP when administered to slices of the cerebral cortex of wild-type mice. In comparison, tissue from PrPC-null mice showed a significantly larger cAMP response when treated with relatively low concentrations of serotonin (1 nm; Fig. 2A) and, in contrast, showed a markedly reduced cAMP response to dopamine (100–300 μm; Fig. 2B). There were no significant differences in the cAMP response of slices treated with either norepinephrine or with higher concentrations of 5HT (Fig. 2, A and C). As a control, responses to forskolin, a direct activator of adenylyl cyclase, were the same in the cortex from both genotypes (Fig. 2, D and E). In sharp contrast with cerebral cortex, stimulation of striatum slices with dopamine, SKF-38393, or forskolin elicited similar responses in both genotypes (Fig. 2F).
FIGURE 2.

Monoamine-induced accumulation of cAMP in the cerebral cortex is genotype-related. A, serotonin induced an increase in cAMP only at 1 nm, at which point, Prnp-null cortical tissue showed a significantly larger response, compared with wild-type; B, responses to dopamine were significantly larger in wild-type at 100 mm or higher when compared with the unresponsive Prnp-null tissue. C, noradrenaline responses were similar in both genotypes. D and E, both concentration dependence (D) and kinetics (E) of adenylyl cyclase activation induced by forskolin in the cerebral cortex were similar in both genotypes, as were the responses to stimulation of striatum slices with dopamine (DA), SKF-38393 (SKF), or forskolin (FSK) (F). n = 3–6. *, p < 0.05; **, p < 0.01.
Interestingly, the D1-like receptor agonist SKF-38393 elicited an increase in cAMP similar to dopamine in the cerebral cortex of wild-type mice, whereas it had no effect on cAMP levels of PrPC-null tissue. Higher doses of SKF-38393 resulted in a reduced response in wild-type, although still failing to elicit any cAMP increase on knock-out tissue (Fig. 3A). The reason for the dose-dependent loss of wild-type response is unknown, but may be related to nonspecific activation of other G-protein-coupled receptors. Moreover, such high doses of SKF-38393 were shown to be potentially toxic and cause internalization of D1 receptors (54).
FIGURE 3.

Roles of D1-like and 5HT5A receptors in the differing cAMP responses of the cerebrocortical tissue from distinct genotypes. A, a D1-like receptor agonist elicited cAMP accumulation in wild-type, but not in Prnp-null tissue. B, a D1-like receptor antagonist completely blocked cAMP accumulation induced by 100 μm dopamine in wild-type tissue, with no effect in Prnp-null. C, a D2-like receptor antagonist had no effect on cAMP responses. D, a 5HT5A receptor antagonist partially prevented the larger increase in cAMP of Prnp-null tissue treated with 1 nm serotonin. n = 3–6. *, p < 0.05; **, p < 0.01.
The D1-like receptor antagonist SCH-23390 fully prevented dopamine-mediated accumulation of cAMP in wild-type slices, but it had no effect on PrPC knock-out tissue (Fig. 3B). To test whether an overactivation of D2-like receptors, rather than the apparent lack of D1-receptor response, is at play in the absence of PrPC, we examined the effect of the D2-like receptor antagonist raclopride. At 2 μm, raclopride had no effect on the net accumulation of cAMP induced by 100 μm dopamine in cortical slices of either genotype (Fig. 3C). We also tested the effects of a 5HT5A receptor antagonist. The relatively higher cAMP production stimulated by 1 nm serotonin in PrPC-null tissue when compared with wild-type (Figs. 2A and 3D) was partially prevented in the presence of SB-699551-A, upon which no significant difference was found between genotypes (Fig. 3D).
Western blots of monoamine-related proteins showed similar levels of SERT, TPH, 5-HT1A, and D1 in both genotypes. 5HT5A levels, however, were found increased by ∼30% in knock-out mice, and a similar increase was detected in the levels of TH (Fig. 4, B–D and F), albeit the proportion of TH phosphorylation in the Ser-40 residue was unchanged between groups (Fig. 4, A and E). In addition, we compared the total levels of 5HT, dopamine, noradrenaline, and their metabolites 5-hydroxyindoleacetic acid and 3,4-dihydroxyphenylacetic acid, in extracts from the cerebral cortex of wild-type and PrPC-null mice. A significant increase was found in total dopamine levels of PrPC-null tissue, whereas the other neurotransmitters and metabolites were similar in both genotypes (Fig. 5A). Again, there were no differences between the levels of either dopamine or 3,4-dihydroxyphenylacetic acid in the striatum of mice of both genotypes (Fig. 5B).
FIGURE 4.

Differing contents of monoamine-related proteins in wild-type and PrPC−/− mice. Western blots of protein extracts from the cerebral cortex show no significant differences between genotypes for TPH, SERT, 5HT1A, or D1 (B–D and F). Notice, however, that both 5HT5A and TH contents were significantly increased in Prnp-null mice (A and E). Relative level of phosphorylation of TH at the Ser-40 residue was similar in both groups (E). GAPDH was used as control in C, because the α-tubulin band overlapped with 5HT1A. n = 3–7; *, p < 0.05.
FIGURE 5.

Neurotransmitter and metabolite contents are selectively altered in cerebrocortical tissue. A, levels of dopamine were increased in Prnp-null cerebrocortical tissue when compared with wild-type, whereas neurotransmitter serotonin (5-HT) and noradrenaline (NA), as well as metabolites 5-hydroxyindoleacetic acid (5-HIAA, from serotonin) and 3,4-dihydroxyphenylacetic acid (DOPAC, from dopamine) were similar in both genotypes. B, total dopamine and DOPAC contents in the striatum were, however, similar in both genotypes. n = 6–12, *, p < 0.05.
Finally, we examined the pattern of immunostaining of proteins related to serotonergic and dopaminergic synapses, and compared those with the prion protein. In both genotypes, the general patterns of immunostaining were similar for tryptophan hydroxylase (TPH), the 5HT transporter (SERT), 5HT5A receptor, tyrosine hydroxylase (TH), as well as for the most abundant dopamine receptor in the cortex, D1R. Simultaneous PrPC immunostaining was strongly positive in the cerebral cortex of wild-type mice, as previously reported (55), and notably, it often co-localized with either D1R or 5HT5A (Fig. 6). These results were consistent with an overlay assay that resulted in binding of PrPC to D1R, but not to D4R, and to 5HT5A, as well as binding to SERT (Fig. 7).
FIGURE 6.
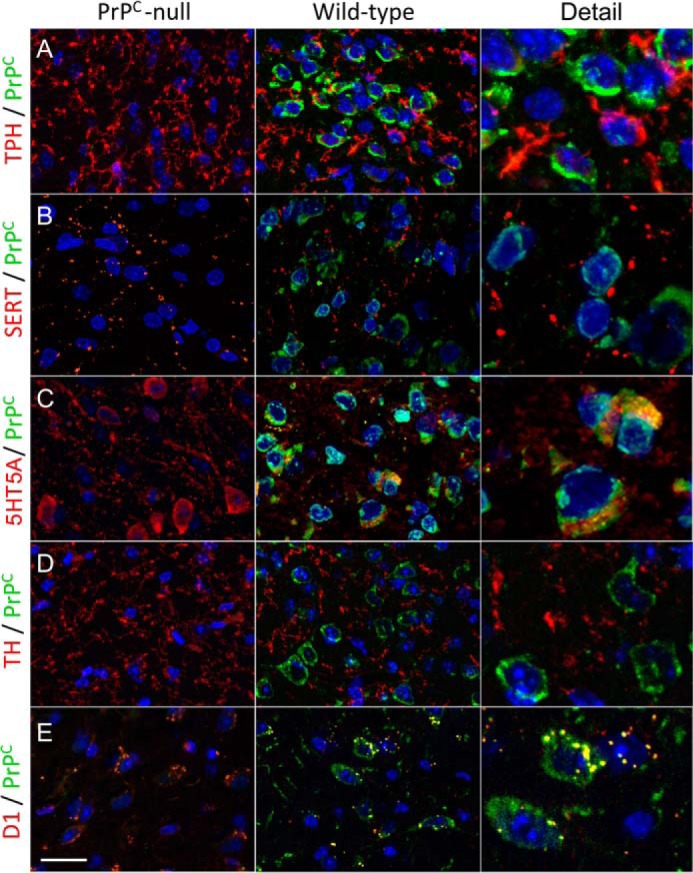
Distribution and colocalization of immunostaining to prion and monoamine-related proteins in the cerebral cortex. Labeling is shown for TPH, monoamine transporter (SERT), 5HT5A serotonin receptor, TH, or D1 dopamine receptor (red), and the prion protein (green) in confocal micrographs of coronal sections taken from the parietal cortex of wild-type (left) and Prnp-null (right) mice. Notice colocalization of PrPC with 5HT5A (C), and with D1 (E).
FIGURE 7.
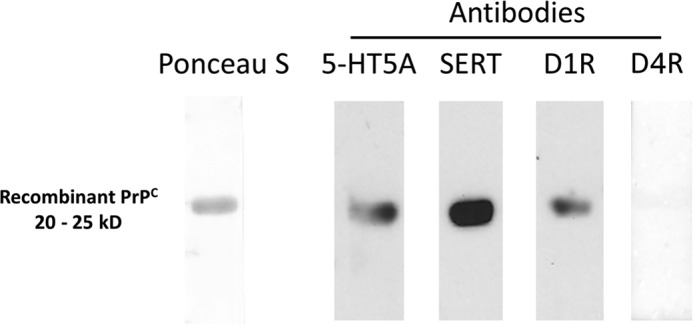
D1R, 5HT5A, and SERT bind to PrPC. Overlay assays of cerebrocortical protein extracts against recombinant PrPC were positive for phage display hits 5HT5A and SERT, and for D1 dopamine receptor, but not for D4 dopamine receptor.
Discussion
In the present study, we used 8–12-week-old wild-type and PrPC-null mice both to further explore a previous report of depressive-like traits in PrPC-deficient animals (36), and to examine monoaminergic signaling in the cerebral cortex. Among our results, the higher immobility in PrPC-null mice subject to the FST and tail suspension test, as well as their increased latency in novelty suppress feeding test, support the hypothesis that the lack of prion protein leads to depressive-like behavior in mice. Differing, however, from the study by Gadotti and co-workers (36), we did not find significant differences in motor function between genotypes.
Consistent with our results, no difference had been detected by other authors in the same motor tests when PrPC-null mice were compared with their wild-type littermates of a distinct genetic background, although an increase in locomotion and exploratory behavior was reported in mice that overexpress PrPC (56). An earlier study had, however, reported increased numbers of crossings in an open field by PrPC-null mice when compared with wild-type, but the data presented were not compelling (57). It should be noted that in both the latter studies, wild-type mice were descendants of the mating of C57/BL6 and 129/sv mice, whereas the PrPC-null mice were from an independent colony derived from a Prnp knock-out colony derived several years before (58). In turn, our colony derived from heterozygous mice has been inbred for several generations into the C57/BL10 background, thus leading to a higher degree of congenicity (59).
Our results are especially consistent with reports that motor deficits detectable by either rotarod or treadmill testing in PrPC-null mice are age-dependent. Thus, differing results in such tests were absent between wild-type and PrPC-null genotypes at 8–12 weeks of age, but apparent in older animals (60, 61). Other groups also reported age-dependent motor deficits restricted to aged PrPC-null mice (62, 63).
On the other hand, in a study of wild-type and PrPC-null mice inbred into the FVB genetic background, no difference between 8–12-week-old mice of either genotype were found in a forced swimming test (64), which differs from both our results (Fig. 1A), as well as of others (36). Notably, however, the FST in the Massimino study (64) was run for 3 min only, which differs greatly from the usual procedures, where the first 2 of a standard 6-min test are sometimes even discarded due to the hyperactivity of mice when introduced in the water pool (65). In addition, differing from our experiment, no pretraining/habituation was reported (64), whereas such pretraining helps prevent novelty stress and anxiety associated with the swimming test. Mice of the FVB strain, along with other Swiss-derived strains, are hyperactive, and show a rather low level of immobility even in standard FST (66, 67). Both differing genetic backgrounds and experimental protocols may, therefore, explain the distinct results found in our FST experiments, as compared with those of Massimino and co-workers (64). Overall, our data are consistent with the hypothesis that 8–12-week-old PrPC-null mice show a depressive-like behavior without motor defects, when compared with age-matched wild-type mice.
Notwithstanding that the monoamine hypothesis may be an oversimplification unlikely to fully explain the complexity of human depression (68–71), unbalance of monoaminergic systems is considered a key factor of the disease, and a prominent neurochemical feature of animal models (72, 73). Here we found several distinctions in monoaminergic function between wild-type and PrPC-null mice.
Two among six marker proteins of monoaminergic systems, the levels of which were estimated by Western blots, were found at distinct levels between PrPC-null and wild-type mice. The contents of both the 5HT5A serotonin receptor, and the limiting enzyme for dopamine synthesis, TH, were ∼30% higher in PrPC-null mice when compared with wild-type. Notably, the relative amounts of the main phosphorylated form of TH (pTHSer-40) were similar in both genotypes. Consistent with these findings, we also found a significant increase of total dopamine levels in PrPC-null cerebral cortex. Although phosphorylation of TH is the primary regulator of the production of dopamine in synapses (74), it is likely that the increased level of total TH, rather than a shift in phosphorylation, explains the chronically increased dopamine content.
Cyclic AMP is a common second messenger of monoaminergic signaling associated with depression (see Ref. 75 for review). Interestingly, when compared with the wild-type response, cerebrocortical tissue lacking PrPC responded with a significantly larger increase in cAMP to treatment with a low concentration of serotonin. In contrast, a drastically reduced cAMP response was observed in PrPC-null tissue stimulated with moderate concentrations of dopamine. The increases in cAMP induced either by norepinephrine or the potent adenylyl cyclase activator forskolin did not differ between knock-out and wild-type tissue, indicating that effects on monoaminergic transmission were neither generalized for metabotropic receptors, nor due to changes in the functional capacity of adenylyl cyclase. Differing from cerebrocortical tissue, slices of striatum from either PrPC-null or wild-type mice responded with similarly increased levels of cAMP, and had undistinguishable dopamine contents. These data are consistent with the lack of change in motor functions found in our behavioral experiments.
The D1-like receptors are the most abundant dopamine receptor in the cerebral cortex, and our experiments with selective agonists and antagonists showed that a lack of D1-like activity is mainly responsible for the unresponsiveness found in PrPC-null cortical slices. A plausible hypothesis is that the increased content of dopamine, due to the higher content of tyrosine hydroxylase in the brains of PrPC-null mice, may lead to sustained desensitization of D1 receptors (76). Alternatively, or perhaps in addition to desensitization, the presence of PrPC may be required for full responsiveness of D1-like receptors to dopamine, either due to direct regulation or to interfering with heterodimerization with D2-like receptors (77). The latter hypothesis is particularly interesting in view of the evidence that attributes the role of the dopaminergic system in clinical depression basically to the D2-like class of receptors (78).
In turn, there are numerous subtypes of serotonin receptor and their responses are difficult to isolate or reduce to a simple modulation of cAMP levels (79). Indeed, we found that the 5HT5A antagonist SB-699551-A appeared to diminish, albeit slightly, the serotonin-induced accumulation of cAMP in PrPC-null tissue, which fails to correlate with the reported negative coupling of this receptor type to adenylyl cyclase described upon transfection into cell lines (80–84). The 5HT5A receptor is relatively rare, apparently more abundant in astrocytes than in neurons (80, 85), and only recently a link began to be established between mood and cognition, and the presence of this receptor in cortical neurons (86).
The behavioral consequences of serotonin signaling through 5HT5A, relative to other receptor types, are in fact unclear. For example, SB-699551-A had no effect in the forced swim test, but induced sedation in rats (87), and, whereas increased cAMP has been linked to positive effects on memory consolidation (88), an impairment of this trait was reported by other investigators, in mice treated with the same 5HT5A antagonist (89). Furthermore, 5HT5A has been identified as an inhibitory autoreceptor that regulates the release of serotonin, if only in the absence of concurrent signaling through 5HT1A receptors (90). Therefore, the specific contribution of 5HT5A to serotonin-induced accumulation of cAMP in the cerebral cortex is difficult to assert. In this respect, it is particularly relevant to our results that the prion protein was also shown to modulate the activity of at least 3 other serotonin receptors, namely 5HT2B, 5HT1B/D, and 5HT2A, in an inducible serotonergic cell line (23, 24).
Although the changes reported here in monoaminergic control of intracellular cAMP in the cerebral cortex are insufficient to fully explain the depressive-like behavior of PrPC-null mice, they clearly demonstrate the involvement of monoaminergic systems in the behavioral effects of PrPC. The overlay assay showed that both the 5HT5A serotonin receptor and the SERT transporter, previously detected as potential ligands in a phage display screen, are indeed able to bind the prion protein, and confocal microscopy showed colocalization of PrPC with 5HT5A, albeit not with SERT. In addition, evidence for binding of PrPC to the D1 dopamine receptor was found in the overlay assay, and the two proteins colocalized in confocal micrographs. Both the 5HT5A and D1 receptors, therefore, add to a growing list of potential functional partners of PrPC consistent with the scaffold hypothesis, which already includes several neurotransmitter receptors, as well as other extracellular molecules (19, 20, 91–95). These findings further strengthen the hypothesis that PrPC provides a cell surface scaffold for multiple signaling modules (5, 10).
On the other hand, the changed contents of monoaminergc marker proteins are intriguing. Evidence has been reported of co-regulation of expression of the Prnp gene with both glutamate and γ-aminobutyric acid receptors (22), but protein content was not included in that study. Notably, whereas the content of both 5HT5A and TH were increased in PrPC-null tissue when compared with wild-type, no difference was detected for 5HT1A, D1 receptor, the SERT transporter, nor the TPH enzyme, all of which pertain to the same neurochemical pathways. Thus, the effect of PrPC upon proteostasis is selective, similar to other results of our laboratory that showed an effect of PrPC upon cellular content of a purinergic receptor but not of a metabotropic glutamate receptor, both of which bind to the prion protein.4 These data suggest that PrPC may interact and affect the functional properties of signaling systems not only at the cell surface, but also at the level of proteostasis.
Synaptic dysfunction and damage are prominent features of prion pathology, as seen both in post-mortem tissue of human patients and in animal models of the disease (96, 97). Several reports emerged in the last decade associating PrPC to synaptic function, structure, and plasticity (98) as well as memory and behavioral processes (99), often through the cAMP/PKA pathway. Notably, in elderly human subjects, variability in PRNP, the human gene encoding PrPC, was associated with a decline in cognitive performance (100, 101).
Human prion diseases, in particular the variant Creutzfeldt-Jakob, may present initial clinical symptoms involving psychiatric and behavioral disorders such as depression, anxiety, insomnia, and hallucinations (102). Much has been speculated that such deficits are related with the monoaminergic system (103, 104). It is possible that, as PrPSc begins to propagate, early loss of function of PrPC at the neuronal surface may interfere with monoaminergic signaling by modulating G-protein-coupled receptor responses. In addition, the evidence that implicates PrPC as a binding partner of Aβ peptide oligomers (37) extends the potential relevance of our data to neuropsychiatric manifestations of Alzheimer disease (105). Indeed, the behavioral deficits that indicate a depressive-like behavior of PrPC-null animals are reminiscent of those described in mice subject to injections of Aβ peptide oligomers (106). Further to the importance to specific neurodegenerative diseases, modulation of monoaminergic neurotransmission by PrPC may also be a potential target in major depression and related disorders.
Author Contributions
R. L. conceived and coordinated the study. D. B., L. E. S., F. G. M., and R. L. designed the experiments. D. B., L. E. S., T. A. A., and J. H. L. performed the experiments. D. B., L. E. S., F. G. M., and R. L. analyzed the data. D. B., L. E. S., and R. L. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We are indebted to Bernardo Stutz for coaching on chromatographic procedures.
This work was supported by grants and fellowships from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (to R. L.), Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (to R. L.), and National Institute of Translational Neuroscience (to F. G. M.). The authors declare that they have no conflicts of interest with the contents of this article.
T. A. Americo, M. H. Magdesian, and R. Linden, unpublished data.
M. V. Carneiro, T. A. Americo, M. F. Santiago, M. Z. Guimarães, and R. Linden, unpublished data.
- PrPC
- prion protein
- 5HT
- serotonin
- AD
- Alzheimer disease
- SERT
- serotonin transporter
- TPH
- tryptophan hydroxylase
- TH
- tyrosine hydroxylase
- FST
- forced swim test.
References
- 1. Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aguzzi A., Calella A. M. (2009) Prions: protein aggregation and infectious diseases. Physiol. Rev. 89, 1105–1152 [DOI] [PubMed] [Google Scholar]
- 3. Bolton D. C., McKinley M. P., Prusiner S. B. (1982) Identification of a protein that purifies with the scrapie prion. Science 218, 1309–1311 [DOI] [PubMed] [Google Scholar]
- 4. Smirnovas V., Baron G. S., Offerdahl D. K., Raymond G. J., Caughey B., Surewicz W. K. (2011) Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 18, 504–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Linden R., Martins V. R., Prado M. A., Cammarota M., Izquierdo I., Brentani R. R. (2008) Physiology of the prion protein. Physiol. Rev. 88, 673–728 [DOI] [PubMed] [Google Scholar]
- 6. Mehrpour M., Codogno P. (2010) Prion protein: from physiology to cancer biology. Cancer Lett. 290, 1–23 [DOI] [PubMed] [Google Scholar]
- 7. Laurén J. (2014) Cellular prion protein as a therapeutic target in Alzheimer's disease. J. Alzheimers Dis. 38, 227–244 [DOI] [PubMed] [Google Scholar]
- 8. Magalhães A. C., Silva J. A., Lee K. S., Martins V. R., Prado V. F., Ferguson S. S., Gomez M. V., Brentani R. R., Prado M. A. (2002) Endocytic intermediates involved with the intracellular trafficking of a fluorescent cellular prion protein. J. Biol. Chem. 277, 33311–33318 [DOI] [PubMed] [Google Scholar]
- 9. Morris R. J., Parkyn C. J., Jen A. (2006) Traffic of prion protein between different compartments on the neuronal surface, and the propagation of prion disease. FEBS Lett. 580, 5565–5571 [DOI] [PubMed] [Google Scholar]
- 10. Linden R., Cordeiro Y., Lima L. (2012) Allosteric function and dysfunction of the prion protein. Cell. Mol. Life Sci. 69, 1105–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Onodera T., Sakudo A., Tsubone H., Itohara S. (2014) Review of studies that have used knockout mice to assess normal function of prion protein under immunological or pathophysiological stress. Microbiol. Immunol. 58, 361–374 [DOI] [PubMed] [Google Scholar]
- 12. Mariante R. M., Nóbrega A., Martins R. A., Areal R. B., Bellio M., Linden R. (2012) Neuroimmunoendocrine regulation of the prion protein in neutrophils. J. Biol. Chem. 287, 35506–35515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martin-Lannerée S., Hirsch T. Z., Hernandez-Rapp J., Halliez S., Vilotte J. L., Launay J. M., Mouillet-Richard S. (2014) PrP(C) from stem cells to cancer. Front. Cell Dev. Biol. 2, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zanetti F., Carpi A., Menabò R., Giorgio M., Schulz R., Valen G., Baysa A., Massimino M. L., Sorgato M. C., Bertoli A., Di Lisa F. (2014) The cellular prion protein counteracts cardiac oxidative stress. Cardiovasc. Res. 104, 93–102 [DOI] [PubMed] [Google Scholar]
- 15. Strom A., Wang G. S., Scott F. W. (2011) Impaired glucose tolerance in mice lacking cellular prion protein. Pancreas 40, 229–232 [DOI] [PubMed] [Google Scholar]
- 16. Lopes M. H., Santos T. G. (2012) Prion potency in stem cells biology. Prion 6, 142–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miranda A., Ramos-Ibeas P., Pericuesta E., Ramirez M. A., Gutierrez-Adan A. (2013) The role of prion protein in stem cell regulation. Reproduction 146, R91–R99 [DOI] [PubMed] [Google Scholar]
- 18. Petrakis S., Irinopoulou T., Panagiotidis C. H., Engelstein R., Lindstrom J., Orr-Urtreger A., Gabizon R., Grigoriadis N., Sklaviadis T. (2008) Cellular prion protein co-localizes with nAChR β4 subunit in brain and gastrointestinal tract. Eur. J. Neurosci. 27, 612–620 [DOI] [PubMed] [Google Scholar]
- 19. Beraldo F. H., Arantes C. P., Santos T. G., Machado C. F., Roffe M., Hajj G. N., Lee K. S., Magalhães A. C., Caetano F. A., Mancini G. L., Lopes M. H., Américo T. A., Magdesian M. H., Ferguson S. S., Linden R., Prado M. A., Martins V. R. (2011) Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin γ1 chain. FASEB J. 25, 265–279 [DOI] [PubMed] [Google Scholar]
- 20. Beraldo F. H., Arantes C. P., Santos T. G., Queiroz N. G., Young K., Rylett R. J., Markus R. P., Prado M. A., Martins V. R. (2010) Role of α7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J. Biol. Chem. 285, 36542–36550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Black S. A., Stys P. K., Zamponi G. W., Tsutsui S. (2014) Cellular prion protein and NMDA receptor modulation: protecting against excitotoxicity. Front. Cell Dev. Biol. 2, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rangel A., Madroñal N., Gruart A., Gruart i Massó A., Gavín R., Llorens F., Sumoy L., Torres J. M., Delgado-García J. M., Del Río J. A. (2009) Regulation of GABA(A) and glutamate receptor expression, synaptic facilitation and long-term potentiation in the hippocampus of prion mutant mice. PLoS ONE 4, e7592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mouillet-Richard S., Pietri M., Schneider B., Vidal C., Mutel V., Launay J. M., Kellermann O. (2005) Modulation of serotonergic receptor signaling and cross-talk by prion protein. J. Biol. Chem. 280, 4592–4601 [DOI] [PubMed] [Google Scholar]
- 24. Mouillet-Richard S., Schneider B., Pradines E., Pietri M., Ermonval M., Grassi J., Richards J. G., Mutel V., Launay J. M., Kellermann O. (2007) Cellular prion protein signaling in serotonergic neuronal cells. Ann. N.Y. Acad. Sci. 1096, 106–119 [DOI] [PubMed] [Google Scholar]
- 25. Bassant M. H., Fage D., Dedek J., Cathala F., Court L., Scatton B. (1984) Monoamine abnormalities in the brain of scrapie-infected rats. Brain Res. 308, 182–185 [DOI] [PubMed] [Google Scholar]
- 26. Vidal C., Herzog C., Haeberle A. M., Bombarde C., Miquel M. C., Carimalo J., Launay J. M., Mouillet-Richard S., Lasmézas C., Dormont D., Kellermann O., Bailly Y. (2009) Early dysfunction of central 5-HT system in a murine model of bovine spongiform encephalopathy. Neuroscience 160, 731–743 [DOI] [PubMed] [Google Scholar]
- 27. Ledoux J. M. (2005) Effects on the serotoninergic system in sub-acute transmissible spongiform encephalopathies: current data, hypotheses, suggestions for experimentation. Med. Hypotheses 64, 910–918 [DOI] [PubMed] [Google Scholar]
- 28. Cross A. J., Kimberlin R. H., Crow T. J., Johnson J. A., Walker C. A. (1985) Neurotransmitter metabolites, enzymes and receptors in experimental scrapie. J. Neurol. Sci. 70, 231–241 [DOI] [PubMed] [Google Scholar]
- 29. Adjou K. T., Dilda P., Aumond P., Gueddari S., Deslys J. P., Dormont D., Seman M. (2008) Increase of monoamine oxidase-B activity in the brain of scrapie-infected hamsters. Neurochem. Int. 52, 1416–1421 [DOI] [PubMed] [Google Scholar]
- 30. Rasenick M. M., Valley S., Manuelidis E. E., Manuelidis L. (1986) Creutzfeldt-Jakob infection increases adenylate cyclase activity in specific regions of guinea pig brain. FEBS Lett. 198, 164–168 [DOI] [PubMed] [Google Scholar]
- 31. Wanschitz J., Klöppel S., Jarius C., Birner P., Flicker H., Hainfellner J. A., Gambetti P., Guentchev M., Budka H. (2000) Alteration of the serotonergic nervous system in fatal familial insomnia. Ann. Neurol. 48, 788–791 [PubMed] [Google Scholar]
- 32. Ragno M., Scarcella M. G., Cacchiò G., Capellari S., Di Marzio F., Parchi P., Trojano L. (2009) Striatal [123I] FP-CIT SPECT demonstrates dopaminergic deficit in a sporadic case of Creutzfeldt-Jakob disease. Acta Neurolog. Scand. 119, 131–134 [DOI] [PubMed] [Google Scholar]
- 33. Magnin E., Chopard G., Galmiche J., Brandel J. P., Rumbach L. (2011) Unusual dopaminergic depletion in variant Creutzfeldt-Jakob disease with early and rapid cognitive decline. Eur. Neurol. 65, 368. [DOI] [PubMed] [Google Scholar]
- 34. Wall C. A., Rummans T. A., Aksamit A. J., Krahn L. E., Pankratz V. S. (2005) Psychiatric manifestations of Creutzfeldt-Jakob disease: a 25-year analysis. J. Neuropsychiatry Clin. Neurosci. 17, 489–495 [DOI] [PubMed] [Google Scholar]
- 35. Cumbler E., Furfari K., Guerrasio J. (2009) Creutzfeldt-Jacob disease presenting as severe depression: a case report. CASES J. 2, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gadotti V. M., Bonfield S. P., Zamponi G. W. (2012) Depressive-like behaviour of mice lacking cellular prion protein. Behav. Brain Res. 227, 319–323 [DOI] [PubMed] [Google Scholar]
- 37. Laurén J., Gimbel D. A., Nygaard H. B., Gilbert J. W., Strittmatter S. M. (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 457, 1128–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Haass C., Selkoe D. J. (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112 [DOI] [PubMed] [Google Scholar]
- 39. Klein W. L. (2013) Synaptotoxic amyloid-beta oligomers: a molecular basis for the cause, diagnosis, and treatment of Alzheimer's disease? J. Alzheimers Dis. 33, S49–S65 [DOI] [PubMed] [Google Scholar]
- 40. Ownby R. L., Crocco E., Acevedo A., John V., Loewenstein D. (2006) Depression and risk for Alzheimer disease: systematic review, meta-analysis, and metaregression analysis. Arch. Gen. Psychiatry 63, 530–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Green R. C., Cupples L. A., Kurz A., Auerbach S., Go R., Sadovnick D., Duara R., Kukull W. A., Chui H., Edeki T., Griffith P. A., Friedland R. P., Bachman D., Farrer L. (2003) Depression as a risk factor for Alzheimer disease: the MIRAGE Study. Arch. Neurol. 60, 753–759 [DOI] [PubMed] [Google Scholar]
- 42. Khundakar A. A., Thomas A. J. (2015) Neuropathology of depression in Alzheimer's disease: current knowledge and the potential for new treatments. J. Alzheimers Dis. 44, 27–41 [DOI] [PubMed] [Google Scholar]
- 43. Meeks T. W., Ropacki S. A., Jeste D. V. (2006) The neurobiology of neuropsychiatric syndromes in dementia. Curr. Opin. Psychiatry 19, 581–586 [DOI] [PubMed] [Google Scholar]
- 44. Vermeiren Y., Van Dam D., Aerts T., Engelborghs S., De Deyn P. P. (2014) Monoaminergic neurotransmitter alterations in postmortem brain regions of depressed and aggressive patients with Alzheimer's disease. Neurobiol. Aging 35, 2691–2700 [DOI] [PubMed] [Google Scholar]
- 45. Hirao K., Pontone G. M., Smith G. S. (2014) Molecular imaging of neuropsychiatric symptoms in Alzheimer's and Parkinson's disease. Neurosci. Biobehav. Rev. 49C, 157–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Porsolt R. D., Bertin A., Jalfre M. (1977) Behavioral despair in mice: a primary screening test for antidepressants. Arch. Int. Pharmacodyn. Ther. 229, 327–336 [PubMed] [Google Scholar]
- 47. Autry A. E., Adachi M., Nosyreva E., Na E. S., Los M. F., Cheng P. F., Kavalali E. T., Monteggia L. M. (2011) NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475, 91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Steru L., Chermat R., Thierry B., Simon P. (1985) The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology 85, 367–370 [DOI] [PubMed] [Google Scholar]
- 49. Posser T., Kaster M. P., Baraúna S. C., Rocha J. B., Rodrigues A. L., Leal R. B. (2009) Antidepressant-like effect of the organoselenium compound ebselen in mice: evidence for the involvement of the monoaminergic system. Eur. J. Pharmacol. 602, 85–91 [DOI] [PubMed] [Google Scholar]
- 50. Gilman A. G. (1970) A protein binding assay for adenosine 3′:5′-cyclic monophosphate. Proc. Natl. Acad. Sci. U.S.A. 67, 305–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Arita D. Y., Di Marco G. S., Schor N., Casarini D. E. (2002) Purification and characterization of the active form of tyrosine hydroxylase from mesangial cells in culture. J. Cell Biochem. 87, 58–64 [DOI] [PubMed] [Google Scholar]
- 52. Yan H. C., Cao X., Das M., Zhu X. H., Gao T. M. (2010) Behavioral animal models of depression. Neurosci. Bull. 26, 327–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dulawa S. C., Hen R. (2005) Recent advances in animal models of chronic antidepressant effects: the novelty-induced hypophagia test. Neurosci. Biobehav. Rev. 29, 771–783 [DOI] [PubMed] [Google Scholar]
- 54. Gainetdinov R. R., Premont R. T., Bohn L. M., Lefkowitz R. J., Caron M. G. (2004) Desensitization of G protein-coupled receptors and neuronal functions. Annu. Rev. Neurosci. 27, 107–144 [DOI] [PubMed] [Google Scholar]
- 55. Salès N., Rodolfo K., Hässig R., Faucheux B., Di Giamberardino L., Moya K. L. (1998) Cellular prion protein localization in rodent and primate brain. Eur. J. Neurosci. 10, 2464–2471 [DOI] [PubMed] [Google Scholar]
- 56. Lobão-Soares B., Walz R., Prediger R. D., Freitas R. L., Calvo F., Bianchin M. M., Leite J. P., Landemberger M. C., Coimbra N. C. (2008) Cellular prion protein modulates defensive attention and innate fear-induced behaviour evoked in transgenic mice submitted to an agonistic encounter with the tropical coral snake Oxyrhopus guibei. Behav. Brain Res. 194, 129–137 [DOI] [PubMed] [Google Scholar]
- 57. Roesler R., Walz R., Quevedo J., de-Paris F., Zanata S. M., Graner E., Izquierdo I., Martins V. R., Brentani R. R. (1999) Normal inhibitory avoidance learning and anxiety, but increased locomotor activity in mice devoid of PrP(C). Brain Res. Mol. Brain Res. 71, 349–353 [DOI] [PubMed] [Google Scholar]
- 58. Büeler H., Fischer M., Lang Y., Bluethmann H., Lipp H. P., DeArmond S. J., Prusiner S. B., Aguet M., Weissmann C. (1992) Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356, 577–582 [DOI] [PubMed] [Google Scholar]
- 59. Cowin R. M., Bui N., Graham D., Green J. R., Grueninger S., Yuva-Paylor L. A., Syed A. U., Weiss A., Paylor R. (2011) Onset and progression of behavioral and molecular phenotypes in a novel congenic R6/2 line exhibiting intergenerational CAG repeat stability. PLoS ONE 6, e28409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nazor K. E., Seward T., Telling G. C. (2007) Motor behavioral and neuropathological deficits in mice deficient for normal prion protein expression. Biochim. Biophys. Acta 1772, 645–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Massimino M. L., Peggion C., Loro F., Stella R., Megighian A., Scorzeto M., Blaauw B., Toniolo L., Sorgato M. C., Reggiani C., Bertoli A. (2015) Age-dependent neuromuscular impairment in prion protein knock-out mice. Muscle Nerve 10.1002/mus.24708 [DOI] [PubMed] [Google Scholar]
- 62. Coitinho A. S., Roesler R., Martins V. R., Brentani R. R., Izquierdo I. (2003) Cellular prion protein ablation impairs behavior as a function of age. Neuroreport 14, 1375–1379 [DOI] [PubMed] [Google Scholar]
- 63. Gasperini L., Legname G. (2014) Prion protein and aging. Front. Cell Dev. Biol. 2, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Massimino M. L., Redaelli M., Bertoli A., Sorgato M. C., Mucignat-Caretta C. (2013) Altered behavioral aspects of aged mice lacking the cellular prion protein. Physiol. Behav. 119, 86–91 [DOI] [PubMed] [Google Scholar]
- 65. Can A., Dao D. T., Arad M., Terrillion C. E., Piantadosi S. C., Gould T. D. (2012) The mouse forced swim test. J. Visual Exp. 29, e3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Võikar V., Kõks S., Vasar E., Rauvala H. (2001) Strain and gender differences in the behavior of mouse lines commonly used in transgenic studies. Physiol. Behav. 72, 271–281 [DOI] [PubMed] [Google Scholar]
- 67. Can A., Blackwell R. A., Piantadosi S. C., Dao D. T., O'Donnell K. C., Gould T. D. (2011) Antidepressant-like responses to lithium in genetically diverse mouse strains. Genes Brain Behav. 10, 434–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wong M. L., Licinio J. (2001) Research and treatment approaches to depression. Nat. Rev. Neurosci. 2, 343–351 [DOI] [PubMed] [Google Scholar]
- 69. Nikolaus S., Antke C., Müller H. W. (2009) In vivo imaging of synaptic function in the central nervous system: II. mental and affective disorders. Behav. Brain Res. 204, 32–66 [DOI] [PubMed] [Google Scholar]
- 70. Zarate C., Duman R. S., Liu G., Sartori S., Quiroz J., Murck H. (2013) New paradigms for treatment-resistant depression. Ann. N.Y. Acad. Sci. 1292, 21–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Krishnadas R., Cavanagh J. (2012) Depression: an inflammatory illness? J. Neurol. Neurosurg. Psychiatry 83, 495–502 [DOI] [PubMed] [Google Scholar]
- 72. Berton O., Nestler E. J. (2006) New approaches to antidepressant drug discovery: beyond monoamines. Nat. Rev. Neurosci. 7, 137–151 [DOI] [PubMed] [Google Scholar]
- 73. Perona M. T., Waters S., Hall F. S., Sora I., Lesch K. P., Murphy D. L., Caron M., Uhl G. R. (2008) Animal models of depression in dopamine, serotonin, and norepinephrine transporter knockout mice: prominent effects of dopamine transporter deletions. Behav. Pharmacol. 19, 566–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lehmann I. T., Bobrovskaya L., Gordon S. L., Dunkley P. R., Dickson P. W. (2006) Differential regulation of the human tyrosine hydroxylase isoforms via hierarchical phosphorylation. J. Biol. Chem. 281, 17644–17651 [DOI] [PubMed] [Google Scholar]
- 75. Niciu M. J., Ionescu D. F., Mathews D. C., Richards E. M., Zarate C. A., Jr. (2013) Second messenger/signal transduction pathways in major mood disorders: moving from membrane to mechanism of action, part I: major depressive disorder. CNS Spectrums 18, 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Beaulieu J. M., Gainetdinov R. R. (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 63, 182–217 [DOI] [PubMed] [Google Scholar]
- 77. Pei L., Li S., Wang M., Diwan M., Anisman H., Fletcher P. J., Nobrega J. N., Liu F. (2010) Uncoupling the dopamine D1-D2 receptor complex exerts antidepressant-like effects. Nat. Med. 16, 1393–1395 [DOI] [PubMed] [Google Scholar]
- 78. Leggio G. M., Salomone S., Bucolo C., Platania C., Micale V., Caraci F., Drago F. (2013) Dopamine D3 receptor as a new pharmacological target for the treatment of depression. Eur. J. Pharmacol. 719, 25–33 [DOI] [PubMed] [Google Scholar]
- 79. Barnes N. M., Sharp T. (1999) A review of central 5-HT receptors and their function. Neuropharmacology 38, 1083–1152 [DOI] [PubMed] [Google Scholar]
- 80. Carson M. J., Thomas E. A., Danielson P. E., Sutcliffe J. G. (1996) The 5HT5A serotonin receptor is expressed predominantly by astrocytes in which it inhibits cAMP accumulation: a mechanism for neuronal suppression of reactive astrocytes. Glia 17, 317–326 [DOI] [PubMed] [Google Scholar]
- 81. Francken B. J., Jurzak M., Vanhauwe J. F., Luyten W. H., Leysen J. E. (1998) The human 5-HT5A receptor couples to Gi/Go proteins and inhibits adenylate cyclase in HEK 293 cells. Eur. J. Pharmacol. 361, 299–309 [DOI] [PubMed] [Google Scholar]
- 82. Hurley P. T., McMahon R. A., Fanning P., O'Boyle K. M., Rogers M., Martin F. (1998) Functional coupling of a recombinant human 5-HT5A receptor to G-proteins in HEK-293 cells. Br. J. Pharmacol. 124, 1238–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Thomas E. A., Matli J. R., Hu J. L., Carson M. J., Sutcliffe J. G. (2000) Pertussis toxin treatment prevents 5-HT(5a) receptor-mediated inhibition of cyclic AMP accumulation in rat C6 glioma cells. J. Neurosci. Res. 61, 75–81 [DOI] [PubMed] [Google Scholar]
- 84. Noda M., Yasuda S., Okada M., Higashida H., Shimada A., Iwata N., Ozaki N., Nishikawa K., Shirasawa S., Uchida M., Aoki S., Wada K. (2003) Recombinant human serotonin 5A receptors stably expressed in C6 glioma cells couple to multiple signal transduction pathways. J. Neurochem. 84, 222–232 [DOI] [PubMed] [Google Scholar]
- 85. Plassat J. L., Boschert U., Amlaiky N., Hen R. (1992) The mouse 5HT5 receptor reveals a remarkable heterogeneity within the 5HT1D receptor family. EMBO J. 11, 4779–4786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Goodfellow N. M., Bailey C. D., Lambe E. K. (2012) The native serotonin 5-HT(5A) receptor: electrophysiological characterization in rodent cortex and 5-HT(1A)-mediated compensatory plasticity in the knock-out mouse. J. Neurosci. 32, 5804–5809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kassai F., Schlumberger C., Kedves R., Pietraszek M., Jatzke C., Lendvai B., Gyertyán I., Danysz W. (2012) Effect of 5-HT5A antagonists in animal models of schizophrenia, anxiety and depression. Behav. Pharmacol. 23, 397–406 [DOI] [PubMed] [Google Scholar]
- 88. Giese K. P., Mizuno K. (2013) The roles of protein kinases in learning and memory. Learn. Mem. 20, 540–552 [DOI] [PubMed] [Google Scholar]
- 89. Gonzalez R., Chávez-Pascacio K., Meneses A. (2013) Role of 5-HT5A receptors in the consolidation of memory. Behav. Brain Res. 252, 246–251 [DOI] [PubMed] [Google Scholar]
- 90. Thomas D. R., Soffin E. M., Roberts C., Kew J. N., de la Flor R. M., Dawson L. A., Fry V. A., Coggon S. A., Faedo S., Hayes P. D., Corbett D. F., Davies C. H., Hagan J. J. (2006) SB-699551-A (3-cyclopentyl-N-[2-(dimethylamino)ethyl]-N-[(4′-{[(2-phenylethyl)amino]methyl}-4-biphenylyl)methyl]propanamide dihydrochloride), a novel 5-ht5A receptor-selective antagonist, enhances 5-HT neuronal function: evidence for an autoreceptor role for the 5-ht5A receptor in guinea pig brain. Neuropharmacology 51, 566–577 [DOI] [PubMed] [Google Scholar]
- 91. Rieger R., Edenhofer F., Lasmézas C. I., Weiss S. (1997) The human 37-kDa laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nat. Med. 3, 1383–1388 [DOI] [PubMed] [Google Scholar]
- 92. Graner E., Mercadante A. F., Zanata S. M., Forlenza O. V., Cabral A. L., Veiga S. S., Juliano M. A., Roesler R., Walz R., Minetti A., Izquierdo I., Martins V. R., Brentani R. R. (2000) Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res. Mol. Brain Res. 76, 85–92 [DOI] [PubMed] [Google Scholar]
- 93. Zanata S. M., Lopes M. H., Mercadante A. F., Hajj G. N., Chiarini L. B., Nomizo R., Freitas A. R., Cabral A. L., Lee K. S., Juliano M. A., de Oliveira E., Jachieri S. G., Burlingame A., Huang L., Linden R., Brentani R. R., Martins V. R. (2002) Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 21, 3307–3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Schmitt-Ulms G., Legname G., Baldwin M. A., Ball H. L., Bradon N., Bosque P. J., Crossin K. L., Edelman G. M., DeArmond S. J., Cohen F. E., Prusiner S. B. (2001) Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 314, 1209–1225 [DOI] [PubMed] [Google Scholar]
- 95. Um J. W., Kaufman A. C., Kostylev M., Heiss J. K., Stagi M., Takahashi H., Kerrisk M. E., Vortmeyer A., Wisniewski T., Koleske A. J., Gunther E. C., Nygaard H. B., Strittmatter S. M. (2013) Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79, 887–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Clinton J., Forsyth C., Royston M. C., Roberts G. W. (1993) Synaptic degeneration is the primary neuropathological feature in prion disease: a preliminary study. Neuroreport 4, 65–68 [DOI] [PubMed] [Google Scholar]
- 97. Senatore A., Restelli E., Chiesa R. (2013) Synaptic dysfunction in prion diseases: a trafficking problem? Int. J. Cell Biol. 2013, 543803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Caiati M. D., Safiulina V. F., Fattorini G., Sivakumaran S., Legname G., Cherubini E. (2013) PrPC controls via protein kinase A the direction of synaptic plasticity in the immature hippocampus. J. Neurosci. 33, 2973–2983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Coitinho A. S., Freitas A. R., Lopes M. H., Hajj G. N., Roesler R., Walz R., Rossato J. I., Cammarota M., Izquierdo I., Martins V. R., Brentani R. R. (2006) The interaction between prion protein and laminin modulates memory consolidation. Eur. J. Neurosci. 24, 3255–3264 [DOI] [PubMed] [Google Scholar]
- 100. Berr C., Helbecque N., Sazdovitch V., Mohr M., Amant C., Amouyel P., Alpérovitch A., Hauw J. J. (2003) Polymorphism of the codon 129 of the prion protein (PrP) gene and neuropathology of cerebral ageing. Acta Neuropathol. 106, 71–74 [DOI] [PubMed] [Google Scholar]
- 101. Croes E. A., Dermaut B., Houwing-Duistermaat J. J., Van den Broeck M., Cruts M., Breteler M. M., Hofman A., van Broeckhoven C., van Duijn C. M. (2003) Early cognitive decline is associated with prion protein codon 129 polymorphism. Ann. Neurol. 54, 275–276 [DOI] [PubMed] [Google Scholar]
- 102. Thompson A., MacKay A., Rudge P., Lukic A., Porter M. C., Lowe J., Collinge J., Mead S. (2014) Behavioral and psychiatric symptoms in prion disease. Am. J. Psychiatry 171, 265–274 [DOI] [PubMed] [Google Scholar]
- 103. Baumgarten H. G., Lachenmayer L. (2004) Serotonin neurotoxins: past and present. Neurotox. Res. 6, 589–614 [DOI] [PubMed] [Google Scholar]
- 104. Cools R., Roberts A. C., Robbins T. W. (2008) Serotoninergic regulation of emotional and behavioural control processes. Trends Cogn. Sci. 12, 31–40 [DOI] [PubMed] [Google Scholar]
- 105. Gimbel D. A., Nygaard H. B., Coffey E. E., Gunther E. C., Laurén J., Gimbel Z. A., Strittmatter S. M. (2010) Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 30, 6367–6374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ledo J. H., Azevedo E. P., Clarke J. R., Ribeiro F. C., Figueiredo C. P., Foguel D., De Felice F. G., Ferreira S. T. (2013) Amyloid-β oligomers link depressive-like behavior and cognitive deficits in mice. Mol. Psychiatry 18, 1053–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Manson J. C., Clarke A. R., Hooper M. L., Aitchison L., McConnell I., Hope J. (1994) 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol. Neurobiol. 8, 121–127 [DOI] [PubMed] [Google Scholar]