

Background: The pathophysiological importance of autophagy in renal tubules during the development of diabetic nephropathy (DN) has been implicated.
Results: Autophagy was inactivated because lysosomal membrane permeabilization and lysosomal dysfunction were triggered by advanced glycation end products.
Conclusion: The autophagy-lysosome pathway is disrupted in renal tubules in DN.
Significance: The findings open a new field for studying the mechanisms of DN.
Keywords: autophagy, diabetic nephropathy, lysosome, reactive oxygen species (ROS), receptor for advanced glycation end products (RAGE)
Abstract
It has been suggested that autophagy protects renal tubular epithelial cells (TECs) from injury in diabetic nephropathy (DN). However, the manner in which the autophagy-lysosome pathway is changed in this state remains unclear. In this study of DN, we investigated the autophagic activity and lysosomal alterations in vivo and in vitro. We found that autophagic vacuoles and SQSTM1-positive proteins accumulated in TECs from patients with DN and in human renal tubular epithelial cell line (HK-2 cells) treated with advanced glycation end products (AGEs), the important factors that involved in the pathogenesis of DN. In HK-2 cells, exposure to AGEs caused a significant increase in autophagosomes but a marked decrease in autolysosomes, and the lysosomal turnover of LC3-II was not observed, although LC3-II puncta were co-localized with the irregular lysosomal-associated membrane protein1 granules after AGEs treatment. Furthermore, lysosomal membrane permeabilization was triggered by AGEs, which likely resulted in a decrease in the enzymatic activities of cathepsin B and cathepsin L, the defective acidification of lysosomes, and suppression of the lysosomal degradation of DQ-ovalbumin. Oxidative stress evoked by AGEs-receptor for AGE interaction likely played an important role in the lysosomal dysfunction. Additionally, ubiquitinated proteins were co-localized with SQSTM1-positive puncta and accumulated in HK-2 cells after exposure to AGEs, indicating blocked degradation of SQSTM1-positive and ubiquitinated aggregates. Taken together, the results show that lysosomal membrane permeabilization and lysosomal dysfunction are triggered by AGEs, which induce autophagic inactivation in TECs from patients with DN. Disruption of the autophagy-lysosome pathway should be focused when studying the mechanisms underlying DN.
Introduction
In recent years, diabetic nephropathy (DN),3 a common consequence of type 1 and 2 diabetes mellitus, has garnered much attention because of its high prevalence, poor prognosis, and heavy economic burden. In renal intrinsic cells, diabetes mellitus-mediated alterations of extra- and intracellular metabolism and hemodynamics influence DN (1), and advanced glycation end products (AGEs) play a key role during this process (2). Although some potential targets for renoprotective therapies, including AGEs inhibitors, renin-angiotensin-aldosterone system blockers, and peroxisome proliferator activated receptor-γ agonists, have been studied and tested in experimental and clinical models, the development of end stage kidney disease remains a major concern (3). Therefore, more specific pathophysiologic mechanisms underlying DN should be identified to develop new treatment modalities and strategies for this devastating disease.
Autophagy activation is an important mechanism that degrades damaged organelles and abnormal and misfolded proteins to sustain cellular metabolism (4). Autophagy is initiated by the formation of a double-membraned autophagosome, which then fuses with the lysosome to form an autophagolysosome, in which the enclosed cargo is degraded by lysosomal acid hydrolases (5). Therefore, autophagy cannot function when any part of this process is interrupted. Lysosomal dysfunction is a major cause of autophagy inactivation that leads to the accumulation and aggregation of proteins, including defective organelles and autophagy substrates, resulting in cellular toxicity (6, 7).
Renal tubular epithelial cells (TECs) are key players in orchestrating events in DN (8). TECs exhibit a certain level of autophagy under physiological conditions. Autophagy activation is an important mechanism that protects TECs against various injuries induced by aristolochic acid, cisplatin, cyclosporine A, or endotoxemia (9–12). We also found that autophagy activation may reduce TEC injury induced by urinary proteins (13). Notably, several studies have suggested a role for the autophagy of TECs in DN (14). However, the manner in which the autophagic pathway changes during DN remains unclear. In our study, the changes of autophagy-lysosome pathway are investigated that occur in TECs after exposure to AGEs.
Experimental Procedures
Patients
The Institutional Review Board of the Affiliated Hospital of Guangdong Medical College approved this study. All of the clinical data from 17 patients (35–80 years) at the Affiliated Hospital of Guangdong Medical College were deidentified. Kidney tissue specimens were obtained from biopsy-proven DN patients (n = 11) with mild, diffuse, or nodular glomerulosclerosis. The kidney specimens (n = 6) obtained from patients with mild urinary protein excretion or only hematuria and characterized with a minimal change in histology were used as controls.
Cell Culture and Treatments
Human proximal tubular HK-2 cells (ATCC, Manassas, VA) were maintained in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) under standard conditions. The cells were exposed to 100 μg/ml nonglycated control bovine serum albumin (Co-BSA) or AGE-BSA (2221–10; BioVision, Mountain View, CA) for 0, 6, 12, and 24 h. Then the protein levels of microtubule-associated protein 1 light chain 3B (LC3B), lysosomal-associated membrane protein1 (LAMP1), cathepsin B (CB), SQSTM1, and ubiquitin, as well as the activities of CB, CD, and CL were measured. Also, ovalbumin dequenching and Lyso-Tracker Red (LTR) uptake were tested. Subsequently, after exposure to Co-BSA and AGE-BSA, the cells were incubated with 100 nm bafilomycin A1 (ab120497; Abcam, Cambridge, MA), 200 μg/ml leupeptin (L9783; Sigma) or 10 μm chloroquine (C6628; Sigma) for LC3-II turnover assay at the 12-h time point. After pretreatment with 10 μg/ml anti-receptor for AGE (RAGE; MAB11451; R&D Systems, Minneapolis, MN), 1 mm N-acetyl cysteine (NAC; A7250; Sigma), 2000 units/ml catalase (219261-100KU; Millipore, Billerica, MA), 100 μm dansylcadaverine (D4008; Sigma), or 100 μg/ml genistein (G6649; Sigma), HK-2 cells were exposure to Co-BSA and AGE-BSA for quantifying the production of reactive oxygen species (ROS) at 45-min time point, for ovalbumin dequenching assay and LTR uptake test at 12-h time point.
Enzymatic Assay
To measure the activity of CB, CD, or CL, fluorescence-based assay kits (K140–100, K143–100, and K142–100; BioVision) were used. After cleavage of the synthetic substrate by the cell lysate, the released fluorescence was quantified using a fluorescence plate reader according to the manufacturer's instructions.
Immunofluorescence Study
An immunostaining analysis of the tissues and cells was performed as described previously (15). Rabbit anti-LC3B, rabbit anti-LAMP1, mouse anti-CB, mouse anti-LAMP1 (ab51520, ab24170, ab58802, and ab25630; Abcam), mouse anti-SQSTM1, mouse anti-ubiquitin (sc28359 and sc8017; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-SQSTM1 (PM045; MBL, Nagoya, Japan), and Alexa Fluor® 488 donkey anti-rabbit and Alexa Fluor® 594 donkey anti-mouse IgG antibodies (A21206 and A21203; Invitrogen) were used for staining. The images were taken with a TCS SP5 II confocal microscope (Leica Microsystems, Wetzlar, Germany). The LC3-II- or SQSTM1-positive puncta/cell were counted in at least 30 HK-2 cells for each group. The puncta were assessed blindly by two independent investigators, and the slides were mixed with a set of normal slides to ensure that the examiner was blinded to its information. The expression level of LC3-II or SQSTM1 in each proximal renal tubule was first graded on a scale of 0–4, and the average of these scores was subsequently computed as described previously (13).
Reactive Oxygen Species Assay
After incubated by 10 μm 2′-7′-dichlorodihydrofluorescein diacetate (S0033; Beyotime Institute of Biotechnology, Jiangsu, China) for 20 min, HK-2 cells were trypsinized and resuspended in PBS. The fluorescence was determined by FACSCalibur flow cytometer (FACSCanto II, San Jose, CA), with excitation at 488 nm and emission at 525 nm.
Ovalbumin Dequenching Assay and LTR Uptake Test
After exposure to AGE-BSA for 12 h, the HK-2 cells were incubated with 10 μg/ml DQ-ovalbumin (D12053; Invitrogen) for an additional 2 h or with 50 nm LTR (L-7528; Invitrogen) for an additional 15 min in complete DMEM at 37 °C. For the ovalbumin dequenching assay, the cells were washed with PBS and fixed with 4% paraformaldehyde. Green fluorescent DQ-ovalbumin puncta in individual HK-2 cells were counted, and the average numbers of dots in at least 30 cells are presented in the figures. For the LTR uptake test, the cultures were washed with PBS to remove any excess lysosomal marker, and the images were taken with a TCS SP5 II confocal microscope. The mean fluorescent intensity value of the LTR was measured, and the data are expressed as fold changes relative to the control. In addition, HK-2 cells were trypsinized and resuspended in PBS for FACS analysis to measure the mean fluorescent intensities of fluorescent DQ-ovalbumin and LTR. To determine whether the dequenched DQ-ovalbumin vesicles were co-localized with lysosomes, after DQ-ovalbumin incubation, the cells were fixed with 4% paraformaldehyde at room temperature and permeabilized with ice-cold methanol at −20 °C for 10 min, respectively. Following washed in PBS, the cells were labeled with mouse anti-LAMP1 and visualized by second antibody.
Plasmid Transfection
HK-2 cells were transfected with the mRFP-GFP tandem fluorescent-tagged LC3 (tfLC3) plasmid (Addgene, Cambridge, MA) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After transfection, the cells were treated with Co-BSA, AGE-BSA, 10 μm rapamycin (553210; Calbiochem, La Jolla, CA), or AGE-BSA plus 10 μm chloroquine for an additional 12 h to assess autophagosome and autolysosome formation as described previously (16).
Western Blot Analysis
Western blot analysis was performed as described previously (15). The primary antibodies against LC3B (L7543; Sigma), SQSTM1 protein (Santa Cruz), ubiquitin (Santa Cruz), and tubulin (ab59680; Abcam) and HRP-conjugated secondary antibodies (A0208 and A0216; Beyotime Institute of Biotechnology) were used.
Statistical Analysis
All of the statistical tests were performed using SPSS 16.0. All of the data are expressed as the means ± S.E., counts, or percentage. Two-group comparisons were performed using an independent sample t test unless otherwise indicated. Multiple group comparisons were performed using analysis of variance followed by Bonferroni or Dunnett post hoc tests. Differences with a p value less than 0.05 were considered statistically significant.
Results
Autophagic Vacuoles Are Accumulated in TECs during the Progression of DN
The clinical characteristics of the 17 enrolled patients were presented in Table 1. There was no statistical significance between the DN group and the control group with regard to sex or average age. Compared with the controls, the hemoglobin level decreased, whereas the serum creatinine, blood urea nitrogen, and incidence of diabetic retinopathy and neuropathy increased in DN patients. The HbA1c, blood fat, serum uric acid, and urinary protein levels and the incidence of hypertension were higher, but the serum albumin was lower in the DN patients than in controls.
TABLE 1.
Clinical characteristics of the enrolled patients
| Groups | Control | DN | p value |
|---|---|---|---|
| Sample size | 6 | 11 | |
| Female sex (%)a | 2 (33.3%) | 4 (36.4%) | 0.67 |
| Age (years)b | 57.3 ± 6.0 | 61.2 ± 3.5 | 0.60 |
| HbA1c (%)b | 5.0 ± 0.4 | 7.2 ± 0.6 | <0.05 |
| Hemoglobin (g/liter)b | 118.2 ± 4.1 | 100.2 ± 6.3 | 0.07 |
| Total cholesterol (mmol/liter)b | 4.4 ± 0.5 | 7.3 ± 0.4 | <0.01 |
| Triglyceride (mmol/liter)b | 1.3 ± 0.1 | 2.2 ± 0.3 | <0.05 |
| Serum albumin (g/liter)b | 43.2 ± 2.4 | 28.0 ± 2.4 | <0.001 |
| Serum creatinine (μmol/liter)b | 88.6 ± 10.1 | 124.6 ± 13.0 | 0.08 |
| Blood urea nitrogen (mmol/liter)b | 5.6 ± 0.5 | 7.7 ± 0.7 | 0.07 |
| Serum uric acid (μmol/liter)b | 324.0 ± 20.8 | 376.6 ± 12.7 | <0.05 |
| 24-h urinary protein (g)b | 0.5 ± 0.1 | 4.6 ± 0.9 | <0.01 |
| Number of hypertension cases (%)a | 1 (16.7%) | 8 (72.7%) | <0.05 |
| Number of diabetic retinopathy/neuropathy cases (%)a | 0 | 5 (45.5%) | 0.06 |
a The data are expressed as counts (%), and the p values for the between group differences were calculated using the Wilcoxon rank sum test.
b The data are expressed as the means ± S.E., and the p values for the two-group comparisons were calculated using an independent sample t test.
In our study, we utilized immunofluorescent technology to examine the expression of the LC3-II, a key marker of autophagy, in TECs both in vivo and in vitro. As shown in Fig. 1 (A and C) compared with the control, there were more LC3-II-positive puncta in the renal TECs from patients with DN. To mimic the pathological process of kidney disease induced by diabetes mellitus, the human renal tubular epithelial cell line HK-2 was exposed to AGE-BSA in vitro. The AGE-BSA concentration at 100 μg/ml was utilized, because this dose was reported to represent AGEs level in the serum of diabetic patients (17). Before incubation with AGE-BSA, only a few LC3-II puncta were detected in the HK-2 cells through immunofluorescence staining (Fig. 1, B and D). After exposure to 100 μg/ml AGE-BSA, the number of LC3-II-positive puncta tended to increase at 3 h (data not shown) and significantly increased at 6, 12, and 24 h (Fig. 1, B and D). A similar pattern of LC3-II expression was observed by Western blotting (Fig. 1E).
FIGURE 1.

Quantitative changes in autophagic vacuoles and autophagy substrate in TECs during the development of DN. A and C, immunofluorescent staining of LC3 and SQSTM1 and quantitative changes in autophagic vacuoles and SQSTM1-positive puncta in renal tubules from DN patients or controls. Scale bar, 20 μm. B and D, immunofluorescent staining of LC3 and SQSTM1 and quantitative changes in autophagic vacuoles and SQSTM1-positive puncta in HK-2 cells after exposure to AGE-BSA for 0, 6, 12, and 24 h. Scale bar, 20 μm. E, Western blot analysis of LC3 or SQSTM1 after exposure to AGE-BSA for 0, 6, 12, and 24 h. Densitometry was performed for the quantification, and the ratio of LC3-II or SQSTM1 to tubulin is expressed as the fold change compared with the level in the control. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Autophagy Is Inhibited by AGEs in TECs during DN
To explore whether the accumulation of autophagic vacuoles was a sign of autophagic activation or a hint of impaired autophagic degradation, the autophagy substrate SQSTM1/p62 was next examined. As shown in Fig. 1 (A and C), more SQSTM1-positive dots were observed in the tubular cells from DN patients than the controls. The in vitro immunofluorescence and Western blot assays also confirmed that the SQSTM1 protein was increased in HK-2 cells after exposure to AGE-BSA for 6, 12 and 24 h (Fig. 1, B, D, and E), which is most likely an indicator of autophagy inhibition.
To separately evaluate the extent of autophagosome and autolysosome accumulation, tfLC3 was transfected into HK-2 cells. In HK-2 cells overexpressing tfLC3, green and red puncta were clearly visible. The numbers of both GFP (green) and mRFP (red) dots were significantly increased after exposure to rapamycin (an autophagy inducer) or AGE-BSA for 12 h (Fig. 2, A and B). There were more yellow and red dots in cells after rapamycin exposure than the controls (Fig. 2, A and C), indicating an increase in the formation of both autophagosomes and autolysosomes. However, only yellow, but not red dots were found increased in AGE-BSA-treated cells, suggesting that a large number of autophagosomes were not degraded by lysosomes (Fig. 2, A and C). We then treated cells with the lysosomal inhibitor chloroquine and found that the numbers of yellow and free red puncta were not significantly changed in AGE-BSA- and chloroquine-treated cells compared with the cells treated with AGE-BSA only (Fig. 2, A and C). It further indicates that AGE-BSA results in the inhibition of autophagosome degradation.
FIGURE 2.

Quantitative changes in autophagosomes and autolysosomes in HK-2 cells after exposure to AGE-BSA, rapamycin, or AGE-BSA plus chloroquine. A, fluorescent microscopic analysis of HK-2 cells transfected with plasmid constructs harboring LC3 fused with a tandem mRFP-GFP tag (tfLC3) and treated with Co-BSA, AGE-BSA (100 μg/ml), rapamycin (RAP, 10 μm), or AGE-BSA plus chloroquine (CQ, 10 μm) for 12 h. The yellow puncta indicate autophagosomes (arrowheads). The free red puncta indicate autolysosomes (arrows). Scale bar, 10 μm. B, quantitative data for green or red puncta per cell. C, quantitative data for yellow puncta or free red puncta per cell. *, p < 0.05 (for green or red puncta); ***, p < 0.001 (for green or red puncta); ^^^, p < 0.001 (for free red puncta); #, p < 0.05 (for yellow puncta); ###, p < 0.001 (for yellow puncta); ns, no significance.
It is well known that bafilomycin A1, leupeptin, and chloroquine block the fusion of autophagosomes and lysosomes, inhibit protease activity, and increase intralysosomal pH, respectively (18). Therefore, LC3-II turnover was also examined in the presence and absence of these lysosomal inhibitors (19). We found that the protein levels of LC3-II were significantly increased after exposure to AGE-BSA for 12 h. However, LC3-II expression was not further elevated by the addition of bafilomycin A1, leupeptin, or chloroquine by Western blot assay (Fig. 3A). Consistent with the Western blot results, an additive increase in LC3-II accumulation was not observed by immunostaining in the bafilomycin A1-, leupeptin- or chloroquine- and AGE-BSA-treated group compared with the group treated with AGE-BSA only (Fig. 3, B and C). These data indicate that LC3-II is not degraded by lysosomes and that autophagic activity is inhibited upon the exposure of HK-2 cells to AGE-BSA.
FIGURE 3.
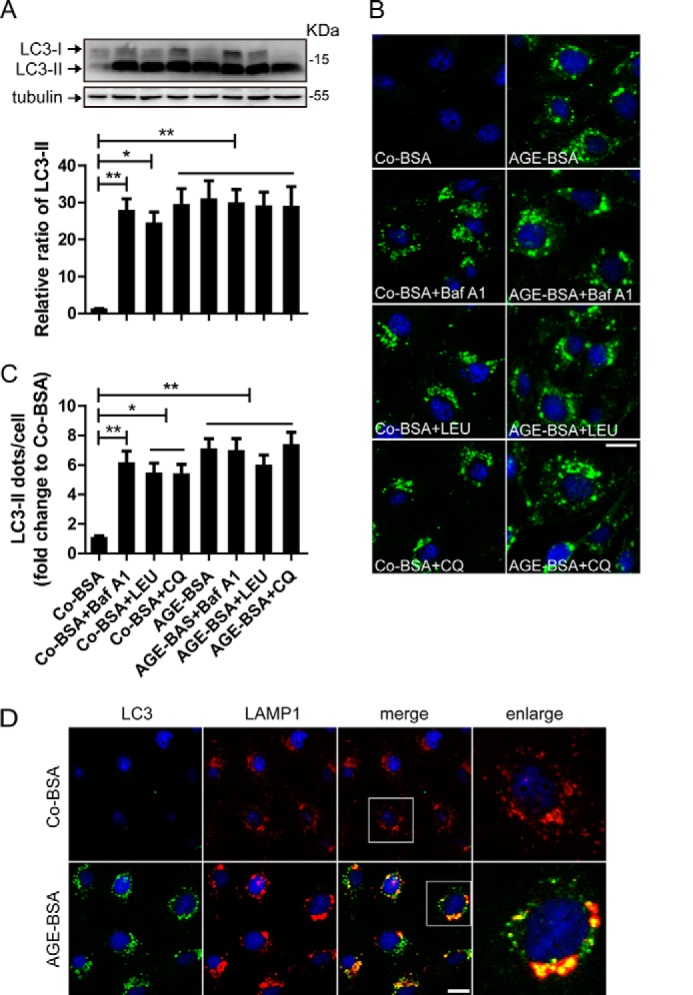
Autophagosome and lysosome fusion analysis and LC3 turnover assay in vitro. A, Western blot analysis of LC3 and the difference in the LC3-II levels between samples with and without bafilomycin A1 (Baf A1), leupeptin (LEU), and chloroquine (CQ) was compared after exposure to AGE-BSA or Co-BSA. B and C, immunofluorescence analysis of LC3 and the difference in LC3-II puncta between samples with and without bafilomycin A1, LEU, and CQ was compared after exposure to AGE-BSA or Co-BSA. LC3-II turnover (comparing columns 5–8 with column 4) was not observed after exposure to AGE-BSA. D, immunofluorescent signals doubled labeled by LC3 (green) and LAMP1 (red). Scale bar, 20 μm. **, p < 0.01; ***, p < 0.001.
To determine whether AGE-BSA blocked the fusion of autophagosomes and lysosomes, LC3 and LAMP1 were double-stained. We found that LAMP1 granules had a uniform distribution in Co-BSA-treated cells. After treatment with AGE-BSA, cytoplasmic LAMP1 granules became irregular and larger, and most of them were co-localized with the LC3-II-positive puncta (Fig. 3D). It seems that the increased LC3-II dots can fuse with the irregular LAMP1 granules.
Autophagy Inhibition Is Attributed to Impaired Lysosomal Activity after Exposure to AGEs
We next examined the lysosomal activity because the lysosome-mediated degradation system plays a key role in autophagy activation. For enzyme activity in lysosomes, AGE-BSA but not the nonglycated Co-BSA induced a significant decrease in CB activity at 6, 12, and 24 h (Fig. 4A). Similar results were obtained for CL activity, although CD activity was not changed significantly at any time points (just showed a decreasing tendency) (Fig. 4A). Different from the activities of CB and CL, CD is reported to remain active at neutral pH (20), which might explain the unchanged activity of CD. We subsequently used LTR to label and track the acidic intracellular compartments (lysosomes) in live cells. Co-BSA treatment did not change the fluorescence at any time points (data not shown). However, AGE-BSA abolished the LTR staining from 6 to 24 h when examined by fluorescence microscopy (Fig. 4, B and C). LTR signal evidenced by mean fluorescent intensity also declined after exposure to AGE-BSA for 6, 12, or 24 h by FACS analysis (Fig. 4D). These data indicate a reduction in the intracellular acidic components and lysosomal impairment after AGE treatment. To further evaluate the efficiency of lysosome-mediated proteolytic degradation, a self-quenched substrate for proteases, DQ-ovalbumin, was used. A large number of dequenched DQ-ovalbumin dots were co-localized with LAMP1-positive puncta in Co-BSA-treated HK-2 cells, indicating the lysosomal degradation of DQ-ovalbumin. However, accompanying an occurrence of irregular and larger LAMP1 granules, few DQ-ovalbumin vesicles were detected in AGE-BSA-treated cells (Fig. 5A). After exposure to AGE-BSA, the mean DQ-ovalbumin puncta per cell were significantly decreased at 6, 12, or 24 h when compared with 0 h (Fig. 5, B and C). Also, the mean fluorescent intensity of DQ-ovalbumin was lower in AGE-BSA-treated cells than nontreated control, as assessed by flow cytometry (Fig. 5D). All of these data suggest a decrease in the degradative potential of the lysosome after treatment with AGE-BSA.
FIGURE 4.

Effects of AGE-BSA on enzymatic activity and LTR fluorescence in HK-2 cells. A, proteolytic activity of CB, CL, and CD in HK-2 cells after exposure to Co-BSA or AGE-BSA for different times. B and C, LTR staining and mean fluorescent intensity in HK-2 cells after exposure to 100 μg/ml AGE-BSA for 0, 6, 12, or 24 h, as assessed by fluorescence microscopy. D, LTR signals and mean fluorescent intensity in HK-2 cells analyzed by flow cytometry. Scale bar, 20 μm. ns, no significance. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FIGURE 5.

Effect of AGE-BSA on lysosomal degradation of DQ-ovalbumin in HK-2 cells. A, fluorescent signals indicating the LAMP1 labeled lysosomes (red) and the dequenched DQ-ovalbumin vesicles (green) after exposure to 100 μg/ml AGE-BSA for 12 h. B, cleaved fluorescent DQ-ovalbumin (green) in HK-2 cells after exposure to AGE-BSA for 0, 6, 12, or 24 h. C, quantitative change in the number of green DQ-ovalbumin puncta per cell after exposure to AGE-BSA for different times. D, the mean fluorescent intensity of DQ-ovalbumin signal after exposure to AGE-BSA for different times, as analyzed by flow cytometry. Scale bar, 20 μm. *, p < 0.05; **, p < 0.01.
RAGE-mediated Oxidative Stress Contributes to the Lysosomal Impairment after Exposure to AGEs
It is well known that AGEs can mediate multiple effects via a specific receptor RAGE (21), so the possible action of RAGE on lysosomes was subsequently tested. AGE-BSA could significantly abolish LTR staining and DQ-ovalbumin fluorescence. However, these actions were inhibited partially by anti-RAGE antibody pretreatment under the microscope (Figs. 6A and 7A). Similar results were obtained when the mean fluorescent intensities of LTR and DQ-ovalbumin were assessed by flow cytometer (Figs. 6B and 7B), indicating that AGEs-RAGE interaction is an important mechanism underlying lysosomal dysfunction. Furthermore, we found that the production of ROS was significantly enhanced after exposure of HK-2 cells to AGE-BSA for 12 h. However, similar to the effect of antioxidant NAC or catalase, anti-RAGE antibody notably suppressed the ROS production mediated by AGE-BSA (Fig. 7C). Interestingly, pretreatment with antioxidant NAC or catalase also improved the lysosomal dysfunction in AGE-BSA-treated cells, characterized by an increase in LTR and DQ-ovalbumin fluorescence signals (Figs. 6, A and B, and 7, A and B). These data suggest that AGEs-RAGE axis evokes oxidative stress generation, which plays an important role in lysosomal impairment. Finally, the endocytosis inhibitors genistein and dansylcadaverine (22, 23) were also used in our study to test the possible effect of endocytosed AGE-BSA on lysosomes. Lysosomal dysfunction was not significantly affected by pretreatment with the endocytosis inhibitors (data not shown), indicating that the effect of AGEs is endocytosis-independent.
FIGURE 6.
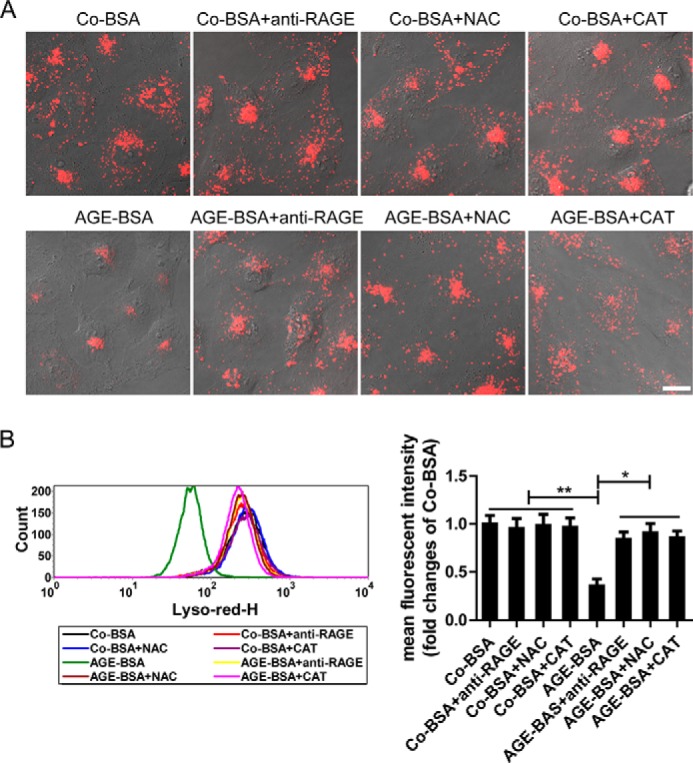
Effects of pretreatment with an anti-RAGE antibody or antioxidants on LTR fluorescence in HK-2 cells. A, LTR fluorescence assessed by fluorescence microscopy in HK-2 cells that were pretreated with an anti-RAGE antibody or NAC or catalase (CAT). Scale bar, 20 μm. B, LTR signals and mean fluorescent intensity analyzed by flow cytometry in HK-2 cells that were pretreated as described in A. *, p < 0.05; **, p < 0.01.
FIGURE 7.
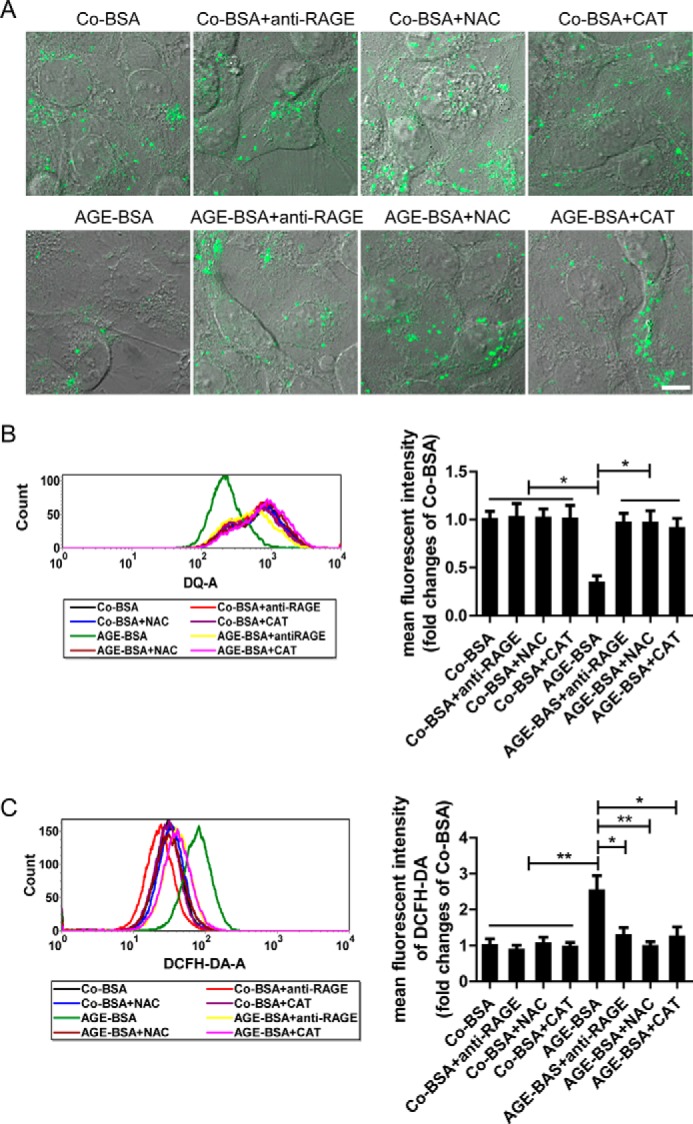
Effect of pretreatment with an anti-RAGE antibody or antioxidants on the degradation of DQ-ovalbumin and ROS production in HK-2 cells. A, dequenched DQ-ovalbumin after exposure to Co-BSA or AGE-BSA in HK-2 cells that were pretreated with an anti-RAGE antibody or NAC or catalase (CAT), as assessed by fluorescence microscopy. Scale bar, 10 μm. B, dequenched fluorescent DQ-ovalbumin and mean fluorescent intensity analyzed by flow cytometry in HK-2 cells that were pretreated as described in A. C, ROS levels after exposure to Co-BSA or AGE-BSA in HK-2 cells that were pretreated as described in A, as assessed by flow cytometry. *, p < 0.05; **, p < 0.01.
Lysosomal Membrane Permeabilization (LMP) Is Triggered by AGEs in TECs during DN
LMP causes the release of cathepsins from the lysosomal lumen to the cytosol. Therefore, we ascertained the cathepsin immunoreactivity to determine whether LMP occurs in TECs. After exposure to Co-BSA, HK-2 cells displayed fine, granular, perinuclear CB immunoreactivity at all time points. However, in the AGE-BSA-treated cells, the CB granules were irregular and dispersed, suggesting the release of CB from the lysosomes into the cytosol (Fig. 8, A and B). To further clarify the relationship between CB leakage and LMP, CB and LAMP1 were double-stained. In Co-BSA-treated HK-2 cells, LAMP1-immunoreactive granules were regularly distributed in the cytosol, and most of those were co-localized with CB. Accompanying the dispersed CB expression, irregular and larger cytoplasmic LAMP1 granules were detected in many cells after exposure to AGE-BSA (Fig. 8, A and C). In kidney preparations from the controls, the majority of TECs presented regular CB granules, which were co-localized with LAMP1 puncta. However, diffuse CB immunostaining, accompanied by dispersed LAMP1 immunofluorescence, was observed in many TECs and even in whole tubules (Fig. 8D). This finding indicates that LMP occurs in the TECs of DN patients, which may contribute to lysosomal dysfunction.
FIGURE 8.
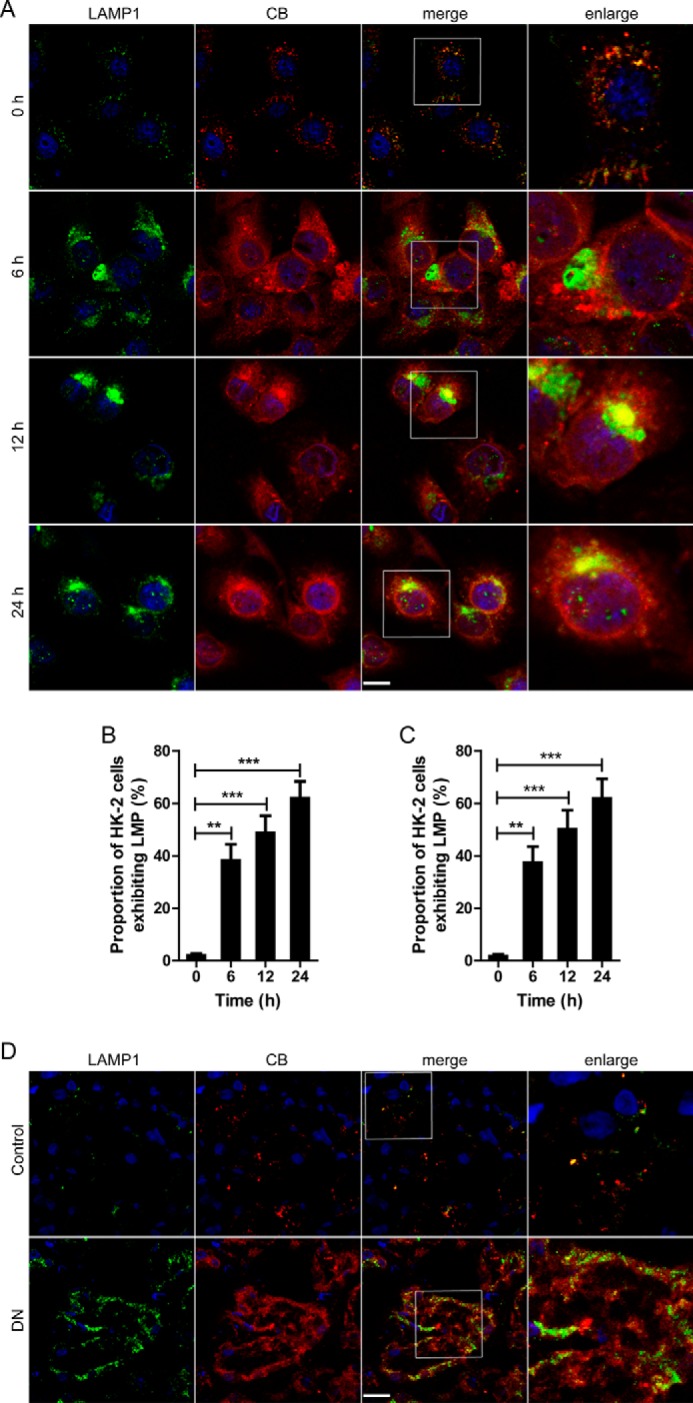
Changes of the distribution of CB and LAMP1 in TECs during the development of DN. A, immunofluorescent staining of LAMP1 and CB in HK-2 cells after exposure to 100 μg/ml AGE-BSA for 0, 6, 12, or 24 h. The red immunofluorescence in the upper panel shows the leakage of CB from lysosomes into the cytoplasm that is accompanied by dispersed LAMP1 (green fluorescence). Scale bar, 20 μm. B, proportion of HK-2 cells exhibiting LMP after exposure to AGE-BSA for 0, 6, 12, and 24 h, assessed by CB immunostaining. C, proportion of HK-2 cells exhibiting LMP after exposure to AGE-BSA for 0, 6, 12, and 24 h, assessed by LAMP1 immunostaining. D, immunofluorescent staining of LAMP1 and CB in the TECs from patients with DN or controls. **, p < 0.01; ***, p < 0.001.
Ubiquitinated Proteins Accumulate after Exposure to AGEs
To determine the functional importance of autophagy inhibition and lysosomal impairment in TECs, SQSTM1 proteins and ubiquitinated proteins were double-labeled. Both proteins were rarely detected in the Co-BSA-treated group (data not shown). Compared with the nontreated group, the ubiquitinated proteins significantly accumulated in the cytoplasm after exposure to AGE-BSA, particularly at 12 and 24 h. Interestingly, these proteins predominantly co-localized with the SQSTM1-positive puncta (Fig. 9A). indicating the disruption of the autophagy-lysosome pathway because autophagic-lysosomal degradation is required for removing ubiquitinated proteins (24). Furthermore, the Western blot assay revealed the accumulation of SQSTM1 and ubiquitinated proteins after treatment with AGE-BSA (Fig. 9B).
FIGURE 9.
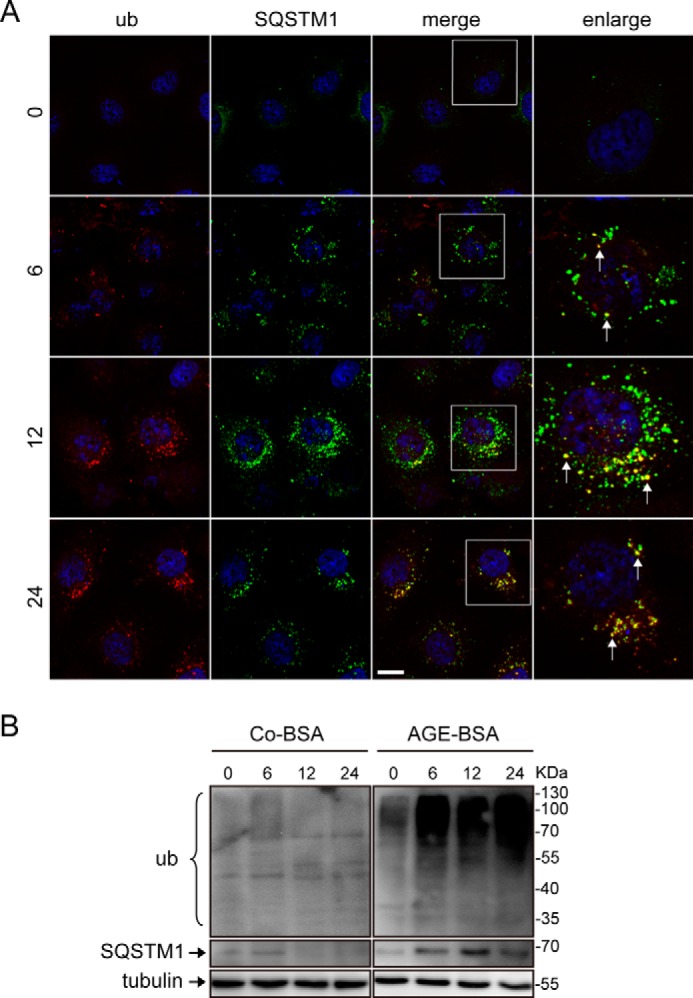
Effect of AGE-BSA on ubiquitinated protein and autophagy substrate degradation in HK-2 cells. A, immunofluorescent staining of ubiquitinated (ub) proteins and SQSTM1-positive aggregates after exposure to AGE-BSA for 0, 6, 12, and 24 h. The accumulated ubiquitinated proteins were primarily co-localized with SQSTM1-positive aggregates. Scale bar, 20 μm. B, Western blot analysis of ubiquitinated proteins and SQSTM1-positive proteins after exposure to Co-BSA and AGE-BSA for different times.
Discussion
Diabetes-induced decrease in the lysosomal enzyme activity (25, 26) and reduction in the renal lysosome-mediated degradation of albumin (27) have been implicated in several studies. We found that AGEs-RAGE interaction, but not endocytosis of AGEs, induced a significant decline of lysosomal activity and degradative potential for DQ-ovalbumin. In addition to the cathepsin leakage and irregular LAMP1 expression, the decreased LTR fluorescence also suggested the occurrence of LMP (20), which is likely a primary cause of lysosomal dysfunction. LMP is susceptible to oxidative stress, suggesting that overproduced ROS plays a key role in governing LMP (28). Furthermore, the cytoplasmic active cathepsins released from lysosomes might play an important role in triggering TEC apoptosis as reported previously (29).
Nearly all intrinsic renal cells have a basal level of autophagy (14, 30, 31). However, the manner in which the autophagy process changes in these cells in DN remains unclear. In TECs from diabetic animals, inhibited autophagy was implicated by the investigation of Zhan et al. (32), whereas autophagy activation was suggested by the study of Zhao et al. (33). In podocytes, Fang et al. (34) reported that hyperglycemia induced impaired autophagy, whereas Ma et al. (35) demonstrated that high glucose promoted autophagy. This discrepancy is most likely because these studies mainly assessed the upstream but not the downstream of autophagic pathway. In our study, we found that autophagic vacuoles accumulate in TECs during the progression of DN. However, the increase in autophagic vacuoles may be due to an increased formation and/or decreased clearance of autophagic vacuoles (36). A separate assessment of autophagosome/autolysosome accumulation and an accurate examination of LC3-II turnover are the principal methods for monitoring autophagic flux (16, 19). The inhibition of the lysosomal degradation of autophagosomes and the blockage of the lysosomal turnover of LC3-II were shown in our study of TECs exposed to AGE-BSA, suggesting a decrease in autophagic activity. Therefore, the lysosomal degradation pathway may be more important for assessing the autophagy activity in DN patients.
Diabetes-induced changes in renal lysosomal processing were reported to link several initial events of DN, including reduced albumin reabsorption in lysosomes and an increase in lipid peroxidation in the tubules (25, 37). Further study showed that lysosomal impairment contributed to a decrease in kidney protein degradation and, hence, cellular hypertrophy in diabetes (26). However, it is not clear whether autophagy inactivation induced by lysosomal impairment is involved in the accumulation of abnormal proteins. In our study, the ubiquitinated proteins was assessed in TECs, because the ubiquitination of mutant huntingtin or ubiquitin-related degradation is one of the most important ways to detoxify an abnormal cellular protein. As expected, we found that ubiquitinated proteins were accumulated after exposure of HK-2 cells to AGE-BSA. It is well known that ubiquitinated proteins target SQSTM1/p62 for autophagic degradation, and disruption of the autophagy-lysosome pathway results in ubiquitinated protein aggregates (6, 38). In our study, an obvious co-localization of ubiquitinated proteins and SQSTM1 was observed, indicating that the ubiquitinated protein bodies were delivered to the autophagy machinery but not degraded by the impaired lysosomes (39). Therefore, the impaired autophagic-lysosomal degradation of ubiquitinated and aggregated proteins may cause abnormal protein accumulation, which probably recapitulates many features of DN including diabetic renal hypertrophy.
Taken together, the results show that the TEC autophagy-lysosome pathway is disrupted by AGEs during the development of DN, which results in the accumulation of abnormal proteins (Fig. 10). The autophagy-lysosome pathway should be emphasized to elucidate the mechanisms underlying DN.
FIGURE 10.

Schematic representation of the disruption of the autophagy-lysosome pathway in TECs during the development of DN. Autophagosomes in which the cargo is enclosed accumulate in TECs after the interaction of AGEs with RAGE/oxidative stress pathway. Halting LMP and normalizing lysosomal function to degrade the accumulated autophagic vacuoles may be a promising approach for the treatment of DN.
Author Contributions
W. J. L. and H.-F. L conceived and coordinated the study and wrote the paper. T. T. S. and R. H. C. designed, performed, and analyzed the experiments shown in Figs. 3 and 5–7. H.-L. W. and Y. J. W. designed, performed, and analyzed the experiments shown in Figs. 1, 2 and 4. J. K. D., Q. H. C., Q. P., and C.-M. H. F. designed, performed, and analyzed the experiments shown in Figs. 8–10. J.-L. T. and D. L. provided technical assistance and contributed to the preparation of the figures. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by National Natural Science Foundation of China Grant 81270798, Guangdong Natural Science Foundation Grants 2014A030313540 and S2012040006276, Zhanjiang Science and Technology Planning Project 2013B01056, and Grants B2012080 and BK201201 from the Project Sponsored by the Scientific Research Foundation for the Returned Overseas Doctors in Guangdong Medical College and the Affiliated Hospital of Guangdong Medical College. The authors declare that they have no conflicts of interest with the contents of this article.
- DN
- diabetic nephropathy
- TEC
- tubular epithelial cell
- AGE
- advanced glycation end product
- RAGE
- receptor for AGE
- LMP
- lysosomal membrane permeabilization
- CB
- cathepsin B
- CD
- cathepsin D
- CL
- cathepsin L
- LAMP
- lysosomal-associated membrane protein
- LC3
- microtubule-associated protein 1 light chain 3
- LTR
- Lyso-Tracker Red
- tfLC3
- mRFP-GFP tandem fluorescent-tagged LC3
- ROS
- reactive oxygen species
- Co-BSA
- control BSA
- NAC
- N-acetyl cysteine.
References
- 1. Kume S., Kitada M., Kanasaki K., Maegawa H., Koya D. (2013) Anti-aging molecule, Sirt1: a novel therapeutic target for diabetic nephropathy. Arch. Pharm. Res. 36, 230–236 [DOI] [PubMed] [Google Scholar]
- 2. Daroux M., Prévost G., Maillard-Lefebvre H., Gaxatte C., D'Agati V. D., Schmidt A. M., Boulanger E. (2010) Advanced glycation end-products: implications for diabetic and non-diabetic nephropathies. Diabetes Metab. 36, 1–10 [DOI] [PubMed] [Google Scholar]
- 3. Furukawa M., Gohda T., Tanimoto M., Tomino Y. (2013) Pathogenesis and novel treatment from the mouse model of type 2 diabetic nephropathy. ScientificWorldJournal 2013, 928197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mizushima N. (2007) Autophagy: process and function. Genes Dev. 21, 2861–2873 [DOI] [PubMed] [Google Scholar]
- 5. Malicdan M. C., Noguchi S., Nishino I. (2007) Autophagy in a mouse model of distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Autophagy 3, 396–398 [DOI] [PubMed] [Google Scholar]
- 6. Yu C., Huang X., Xu Y., Li H., Su J., Zhong J., Kang J., Liu Y., Sun L. (2013) Lysosome dysfunction enhances oxidative stress-induced apoptosis through ubiquitinated protein accumulation in HeLa cells. Anat. Rec. (Hoboken) 296, 31–39 [DOI] [PubMed] [Google Scholar]
- 7. Pan T., Kondo S., Le W., Jankovic J. (2008) The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain 131, 1969–1978 [DOI] [PubMed] [Google Scholar]
- 8. Phillips A. O., Steadman R. (2002) Diabetic nephropathy: the central role of renal proximal tubular cells in tubulointerstitial injury. Histol. Histopathol. 17, 247–252 [DOI] [PubMed] [Google Scholar]
- 9. Pallet N., Bouvier N., Legendre C., Gilleron J., Codogno P., Beaune P., Thervet E., Anglicheau D. (2008) Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy 4, 783–791 [DOI] [PubMed] [Google Scholar]
- 10. Zeng Y., Yang X., Wang J., Fan J., Kong Q., Yu X. (2012) Aristolochic acid I induced autophagy extenuates cell apoptosis via ERK 1/2 pathway in renal tubular epithelial cells. PLoS One 7, e30312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takahashi A., Kimura T., Takabatake Y., Namba T., Kaimori J., Kitamura H., Matsui I., Niimura F., Matsusaka T., Fujita N., Yoshimori T., Isaka Y., Rakugi H. (2012) Autophagy guards against cisplatin-induced acute kidney injury. Am. J. Pathol 180, 517–525 [DOI] [PubMed] [Google Scholar]
- 12. Howell G. M., Gomez H., Collage R. D., Loughran P., Zhang X., Escobar D. A., Billiar T. R., Zuckerbraun B. S., Rosengart M. R. (2013) Augmenting autophagy to treat acute kidney injury during endotoxemia in mice. PLoS One 8, e69520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu W. J., Luo M. N., Tan J., Chen W., Huang L. Z., Yang C., Pan Q., Li B., Liu H. F. (2014) Autophagy activation reduces renal tubular injury induced by urinary proteins. Autophagy 10, 243–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tanaka Y., Kume S., Kitada M., Kanasaki K., Uzu T., Maegawa H., Koya D. (2012) Autophagy as a therapeutic target in diabetic nephropathy. Exp. Diabetes Res. 2012, 628978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu W. J., Xie S. H., Liu Y. N., Kim W., Jin H. Y., Park S. K., Shao Y. M., Park T. S. (2012) Dipeptidyl peptidase IV inhibitor attenuates kidney injury in streptozotocin-induced diabetic rats. J. Pharmacol. Exp. Ther. 340, 248–255 [DOI] [PubMed] [Google Scholar]
- 16. Kimura S., Noda T., Yoshimori T. (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460 [DOI] [PubMed] [Google Scholar]
- 17. Shi L., Yu X., Yang H., Wu X. (2013) Advanced glycation end products induce human corneal epithelial cells apoptosis through generation of reactive oxygen species and activation of JNK and p38 MAPK pathways. PLoS One 8, e66781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Klionsky D. J., Abdalla F. C., Abeliovich H., Abraham R. T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J. A., Ahn H. J., Ait-Mohamed O., Ait-Si-Ali S., Akematsu T., Akira S., et al. (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rubinsztein D. C., Cuervo A. M., Ravikumar B., Sarkar S., Korolchuk V., Kaushik S., Klionsky D. J. (2009) In search of an “autophagomometer.” Autophagy 5, 585–589 [DOI] [PubMed] [Google Scholar]
- 20. Boya P., Kroemer G. (2008) Lysosomal membrane permeabilization in cell death. Oncogene 27, 6434–6451 [DOI] [PubMed] [Google Scholar]
- 21. Sakurai S., Yonekura H., Yamamoto Y., Watanabe T., Tanaka N., Li H., Rahman A. K., Myint K. M., Kim C. H., Yamamoto H. (2003) The AGE-RAGE system and diabetic nephropathy. J. Am. Soc. Nephrol. 14, S259–S263 [DOI] [PubMed] [Google Scholar]
- 22. Lim C. H., Bijvelds M. J., Nigg A., Schoonderwoerd K., Houtsmuller A. B., de Jonge H. R., Tilly B. C. (2007) Cholesterol depletion and genistein as tools to promote F508delCFTR retention at the plasma membrane. Cell Physiol. Biochem. 20, 473–482 [DOI] [PubMed] [Google Scholar]
- 23. Sevillano N., Girón M. D., Salido M., Vargas A. M., Vilches J., Salto R. (2009) Internalization of the receptor for advanced glycation end products (RAGE) is required to mediate intracellular responses. J. Biochem. 145, 21–30 [DOI] [PubMed] [Google Scholar]
- 24. Rubinsztein D. C. (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443, 780–786 [DOI] [PubMed] [Google Scholar]
- 25. Osicka T. M., Kiriazis Z., Pratt L. M., Jerums G., Comper W. D. (2001) Ramipril and aminoguanidine restore renal lysosomal processing in streptozotocin diabetic rats. Diabetologia 44, 230–236 [DOI] [PubMed] [Google Scholar]
- 26. Shechter P., Boner G., Rabkin R. (1994) Tubular cell protein degradation in early diabetic renal hypertrophy. J. Am. Soc. Nephrol. 4, 1582–1587 [DOI] [PubMed] [Google Scholar]
- 27. Osicka T. M., Houlihan C. A., Chan J. G., Jerums G., Comper W. D. (2000) Albuminuria in patients with type 1 diabetes is directly linked to changes in the lysosome-mediated degradation of albumin during renal passage. Diabetes 49, 1579–1584 [DOI] [PubMed] [Google Scholar]
- 28. Johansson A. C., Appelqvist H., Nilsson C., Kågedal K., Roberg K., Ollinger K. (2010) Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis 15, 527–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Repnik U., Stoka V., Turk V., Turk B. (2012) Lysosomes and lysosomal cathepsins in cell death. Biochim. Biophys. Acta 1824, 22–33 [DOI] [PubMed] [Google Scholar]
- 30. Lee A. S., Kim D. H., Lee J. E., Jung Y. J., Kang K. P., Lee S., Park S. K., Kwak J. Y., Lee S. Y., Lim S. T., Sung M. J., Yoon S. R., Kim W. (2011) Erythropoietin induces lymph node lymphangiogenesis and lymph node tumor metastasis. Cancer Res. 71, 4506–4517 [DOI] [PubMed] [Google Scholar]
- 31. Pangare M., Makino A. (2012) Mitochondrial function in vascular endothelial cell in diabetes. J. Smooth Muscle Res. 48, 1–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhan M., Usman I. M., Sun L., Kanwar Y. S. (2015) Disruption of renal tubular mitochondrial quality control by myo-inositol oxygenase in diabetic kidney disease. J. Am. Soc. Nephrol. 26, 1304–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao X., Liu G., Shen H., Gao B., Li X., Fu J., Zhou J., Ji Q. (2015) Liraglutide inhibits autophagy and apoptosis induced by high glucose through GLP-1R in renal tubular epithelial cells. Int. J. Mol. Med. 35, 684–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fang L., Zhou Y., Cao H., Wen P., Jiang L., He W., Dai C., Yang J. (2013) Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PLoS One 8, e60546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ma T., Zhu J., Chen X., Zha D., Singhal P. C., Ding G. (2013) High glucose induces autophagy in podocytes. Exp. Cell Res. 319, 779–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mizushima N., Yoshimori T., Levine B. (2010) Methods in mammalian autophagy research. Cell 140, 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tojo A., Onozato M. L., Ha H., Kurihara H., Sakai T., Goto A., Fujita T., Endou H. (2001) Reduced albumin reabsorption in the proximal tubule of early-stage diabetic rats. Histochem. Cell Biol. 116, 269–276 [DOI] [PubMed] [Google Scholar]
- 38. Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I., Ezaki J., Mizushima N., Ohsumi Y., Uchiyama Y., Kominami E., Tanaka K., Chiba T. (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., Johansen T. (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]