

Background: The role of autophagy in colonic homeostasis is not clear.
Results: Colonic epithelial cell-specific and autophagy-deficient mice showed exacerbation of colitis and abnormal gut microflora with less abundant antimicrobial peptide production and secretion of mucus.
Conclusion: Autophagy protects against colitis by the maintenance of normal gut microflora and secretion of mucus.
Significance: These findings may ultimately lead to an efficient intervention for inflammatory bowel diseases.
Keywords: antimicrobial peptide (AMP), autophagy, bacteria, colitis, intestinal epithelium, mucin
Abstract
Genome-wide association studies of inflammatory bowel diseases identified susceptible loci containing an autophagy-related gene. However, the role of autophagy in the colon, a major affected area in inflammatory bowel diseases, is not clear. Here, we show that colonic epithelial cell-specific autophagy-related gene 7 (Atg7) conditional knock-out (cKO) mice showed exacerbation of experimental colitis with more abundant bacterial invasion into the colonic epithelium. Quantitative PCR analysis revealed that cKO mice had abnormal microflora with an increase of some genera. Consistently, expression of antimicrobial or antiparasitic peptides such as angiogenin-4, Relmβ, intelectin-1, and intelectin-2 as well as that of their inducer cytokines was significantly reduced in the cKO mice. Furthermore, secretion of colonic mucins that function as a mucosal barrier against bacterial invasion was also significantly diminished in cKO mice. Taken together, our results indicate that autophagy in colonic epithelial cells protects against colitis by the maintenance of normal gut microflora and secretion of mucus.
Introduction
Inflammatory bowel diseases (IBDs),2 including Crohn disease (CD) and ulcerative colitis (UC), are chronic remittent or progressive inflammatory disorders in the gastrointestinal tract associated with an increased risk of colon cancer (1). UC affects part of the colon or the entire colon in an uninterrupted pattern (1). In contrast, CD generally affects any region of the intestine, including the ileum and colon, often discontinuously (2, 3). In CD, the ileum alone was affected in ∼45% of cases (ileitis), the ileum and colon (ileocolitis) in ∼19% of cases, and the colon alone (colitis) in ∼32% of cases (4). Therefore, colonic symptoms are present in the majority of IBD patients. Although the etiology of IBDs is not well understood, extensive previous studies have suggested the involvement of both environmental and genetic factors that lead to dysfunction of the epithelial barrier, mucosal immune system, and responses to gut microflora (5).
Genome-wide association studies of CD identified susceptible loci containing the autophagy-related gene ATG16L1 (6, 7). Autophagy is a conserved process for the bulk degradation of cytoplasmic components. Triggering of autophagy results in the formation of double membrane-bound vesicles termed autophagosomes. The ATG5-ATG12/ATG16L1 complex and LC3-II are essential for autophagosome formation (8, 9). ATG7 is an autophagy-related E1-like enzyme that is essential for two ubiquitination-like reactions (ATG12 conjugation and LC3 lipidation). Therefore, ATG7 is also essential for autophagy (10–12). Recent studies have revealed that autophagy is important for the clearance of intracellular microbes, including adherent-invasive Escherichia coli (13), Salmonella enterica serovar Typhimurium (7, 14), and Mycobacterium tuberculosis (15, 16). It was reported that ATG16L1-deficient macrophages exhibited elevated endotoxin-induced IL-1β production (17), indicating that autophagy is also important for the control of endotoxin-induced responses. In agreement with this notion, others have reported that autophagy in the small intestinal epithelium reduced LPS-induced proinflammatory responses by inhibiting NF-κB activation (18).
A number of studies have demonstrated the function of autophagy-related genes in the gastrointestinal tract. In hypomorphic Atg16L1 mice that were generated with a Gene-trap mediated method, Paneth cells exhibited notable abnormalities in the granule exocytosis pathway (19). Macrophages harboring T300A variants of ATG16L1 showed defective clearance of the ileal pathogen Yersinia enterocolitica and elevated cytokine production (20). Intestinal epithelium-specific Atg16L1-deficient mice exhibited constitutive endoplasmic reticulum stress in Paneth cells and are highly susceptible to experimental colitis (21). Intestinal epithelium-specific Atg7 deficiency in Villin-Cre transgenic mice (22–24) showed enhanced susceptibility against Citrobacter rodentium infection (used as murine models of EHEC and EPEC infection) (25). In these studies, Atg16L1-deleted fetal liver chimeric mice (17), Atg16L1-hypomorphic mice (19), Atg16L1flox/flox Villin-Cre mice (21), and Atg7flox/flox Villin-Cre mice (25) were used. However, it should be noted that Atg16L1 is more abundantly expressed in the colon than in the small intestine (6). Additionally, colonic Cre recombinase expression in Villin-Cre transgenic mice was much lower than expression in the small intestine (22–24). Therefore, it is unlikely that previous studies using these mutant mice could have clarified the role of autophagy in the colon, which is a major affected area in IBDs.
In this study, we took advantage of the specific Cre recombinase expression in colonic epithelial cells in a GlcNAc6ST-2-Cre transgenic mouse model (26) to delete Atg7 in a colonic epithelial cell-specific manner. By using these mutant mice, we analyzed the function of autophagy in the maintenance of gut commensal microflora and protection against UC-like colitis.
Experimental Procedures
Generation of cKO Mice
cKO mice were generated by crossing GlcNAc6ST-2-Cre transgenic (26) and Atg7flox/flox mice (27). The GlcNAc6ST-2-Cre+/+/Atg7flox/flox and GlcNAc6ST-2-Cre+/−/Atg7flox/flox mice were used as conditional knock-out mice. The Atg7flox/flox mice were used as WT controls throughout the study unless otherwise indicated. To detect Cre recombinase expression, GlcNAc6ST-2-Cre+/+ mice were crossed with R26R reporter mice (28). The experimental protocol was approved by the Animal Research Committee of Hoshi University and University of Shizuoka.
X-gal Staining
X-gal staining was performed as described previously (26). Briefly, frozen sections (7 μm) were fixed in PBS containing 1.5% glutaraldehyde and incubated with X-gal solution and Nuclear Fast Red solution (Sigma).
Quantitative RT-PCR for mRNA Expression
Tissue RNA was extracted with TRIzol reagent (Life Technologies, Inc.). The cDNA was synthesized with the PrimeScript RT-PCR kit with gDNA Eraser (TaKaRa) and subjected to quantitative RT-PCR using SYBR Premix Ex TaqII (Tli RNase H Plus; TaKaRa). The expression of each mRNA was normalized to the expression of β-actin with the ΔΔCt method according to the manufacturer's instructions (TaKaRa Thermal Cycler Dice TP870). The primer sequences are given in Table 1.
TABLE 1.
Primers for quantitative RT-PCR
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Actb | CATCCGTAAAGACCTCTATGCCAAC | ATGGAGCCACCGATCCACA |
| Chst4 | TGGGCCCTGAGGAAGCCTAA | GCCTCCTGGACTCCTCCCTCT |
| Vil1 | TCAAAGGCTCTCTCAACATCA | AGCAGTCACCATCGAAGAAG |
| Atg7 | CAGCAGTGATGACCGCATGA | CAAATGCCAGGCTGACAGGA |
| Ang4 | CTCTGGCTCAGAATGAAAGGTACGA | GAAATCTTTAAAGGCTCGGTACCC |
| Retnlb | ATGGTTGTCACTGGATGTGCTT | AGCACTGGCAGTGGCAAGTA |
| Itln1 | TGACAATGGCCCAGCATTACC | ACGGGGTTACCTTCTGGGA |
| Itln2 | GCGCTTGGGCCATAATCTGT | CGGCCAGAGGGAGAGTAATAA |
| Ifng | AGCGGCTGACTGAACTCAGATTGTAG | GTCACAGTTTTCAGCTGTATAGGG |
| Il4 | ACAGGAGAAGGGACGCCAT | GAAGCCCTACAGACGAGCTCA |
| Il5 | AGCACAGTGGTGAAAGAGACCTT | TCCAATGCATAGCTGGTGATTT |
| Il13 | AGACCAGACTCCCCTGTGCA | TGGGTCCTGTAGATGGCATTG |
| Tslp | AGGCTACCCTGAAACTGAGA | GGAGATTGCATGAAGGAATAC |
| Il25 | CAGCAAAGAGCAAGAACC | CCCTGTCCAACTCATAGC |
| Il33 | CATGCCAACGACAAGGACTA | GCTCTCATCTTTCTCCTCCA |
| Muc2 | GGCATCCACTCTAACATCTCCG | CATAGATGGGCCTGTCCTCAGG |
| Il1b | TCGCTCAGGGTCACAAGAAA | CATCAGAGGCAAGGAGGAAAAC |
| Il6 | CCACTTCACAAGTCGGAGGCTTA | GCAAGTGCATCATCGTTGTTCATAC |
| Il10 | ACCTGGTAGAAGTGATGCCCCAGGCA | CTATGCAGTTGATGAAGATGTCAAA |
| Il17A | ACGCGCAAACATGAGTCCAG | CTCAGCAGCAGCCAACAGCATC |
| Tnfa | AGGCTGCCCCGACTACGT | GACTTTCTCCTGGTATGAGATAGCAAA |
Western Blotting
Mouse colons were homogenized in homogenization buffer (0.25 m sucrose, 10 mm HEPES, pH 7.4, protease inhibitors (1:200 dilution; Sigma)). The lysates were centrifuged at 700 × g for 5 min at 4 °C. The supernatants were collected, and their protein concentrations were determined using a BCA protein assay kit (Thermo Scientific). The obtained lysates were stored at −80 °C until use. Western blotting was performed according to standard procedures using rabbit anti-β-actin polyclonal antibody (bs-0061R, Bioss, 0.6 μg/ml), rabbit anti-mouse ATG7 polyclonal antibody (A2856, Sigma, 0.25 μg/ml), rabbit anti-mouse p62 polyclonal antibody (PM045, MBL, diluted 1:1,000), and rabbit anti-cow ubiquitin polyclonal antibody (Nr.Z0458, DakoCytomation, 0.3 μg/ml). The bands were detected with 0.5 μg/ml horseradish peroxidase-conjugated anti-rabbit IgG (H+L) polyclonal antibody (65-6120, Zymed Laboratories Inc., diluted 1:20,000) and West Pico SuperSignal Chemiluminescent Substrate (Thermo Scientific). Western blot band intensities were quantified using the ImageJ program (National Institutes of Health).
Antibiotic Treatment
For antibiotic treatment, mice were given drinking water containing either a combination of 0.5 g/liter vancomycin (Wako), 1 g/liter ampicillin (Wako), 1 g/liter neomycin (Nacalai Tesque), and 1 g/liter metronidazole (Wako) (4Abx) or a combination of 0.2 g/liter ciprofloxacin (Wako) and 1 g/liter metronidazole (Wako) (2Abx) for 4 or 8 weeks.
Cohousing Experiment
For cohousing experiments, age- and gender-matched WT and cKO mice were cohoused in new cages at 1:1 ratios for 4 weeks before dextran sulfate sodium (DSS) administration. In some experiments, C57BL/6 WT mice (7-week-old, female) obtained from Japan SLC, Inc., were given 4Abx for 8 weeks and then cohoused with gender-matched WT and cKO mice for 4 weeks before DSS administration.
DSS-induced Colitis
In acute colitis model, mice were given 3% (w/v) DSS (molecular mass = 36,000–50,000 Da; MP Biomedicals) in their drinking water for 7 days, which was then replaced with normal water. Each experimental set of mice was administered the same lot of DSS to minimize the effect of lot difference. In some experiments, mice were given 2Abx for 4 weeks in their drinking water. DSS exposure was begun 2 days later. The disease activity index (DAI) was determined according to the criteria described previously (29) with some modifications as follows: diarrhea score (0 point = normal stool, 1 point = soft stool, 2 points = very soft stool, and 3 points = watery stool); bleeding score (0 point = normal color stool, 1 point = brown color stool, 2 points = reddish color stool, and 3 points = bloody stool). The DAI was calculated as the sum of the diarrhea score and the bleeding score, resulting in the total DAI score ranging from 0 (unaffected) to 6 (severe colitis). In the chronic colitis model, mice were injected intraperitoneally with 10 mg/kg azoxymethane (Sigma) in saline at day −7, followed by the three cycles of 3% DSS in drinking water for 7 days and regular water for 14 days. On day 70, the colonic length was measured.
Histological Assessment of Colitis
Excised colons were opened, rinsed with 50 ml of PBS, fixed overnight in 10% formalin or Methanol-Carnoy fixative, and then embedded in paraffin. Paraffin sections (4 μm) were stained with hematoxylin (Sigma) and eosin (Wako). As described previously (29), histological scores were determined by the following parameters: epithelial damage score (0 point = none, 1 point = minimal loss of goblet cells, 2 points = extensive loss of goblet cells, 3 points = minimal loss of crypts and extensive loss of goblet cells, and 4 points = extensive loss of crypts); infiltration score (0 point = none, 1 point = infiltrate around crypt bases, 2 points = infiltrate in muscularis mucosa, 3 points = extensive infiltrate in muscularis mucosa with edema, and 4 points = infiltration of submucosa). The histological score was calculated as the sum of the epithelial damage score and infiltration score, resulting in the total score ranging from 0 (unaffected) to 8 (severe colitis). All of the histopathological assessments were performed blind by two experienced scientists who had specialized in histopathology for more than 6 years. In several experiments, the thickness of the mucosa and the muscularis propria was measured using a BZ-9000 fluorescence microscope (KEYENCE).
Fluorescence in Situ Hybridization (FISH)
FISH was performed according to the method described previously with some modification (30). Paraffin sections (4 μm) were dewaxed and washed in 95% ethanol. The tissue sections were incubated with 5 μg/ml Texas Red-conjugated EUB338 (5′-GCTGCCTCCCGTAGGAGT-3′, Sigma) in hybridization buffer (20 mm Tris-HCl, 0.9 m NaCl, 0.1% SDS, and 10% formamide, pH 7.4) at 50 °C overnight. The sections were rinsed in washing buffer (20 mm Tris-HCl, 0.9 m NaCl, pH 7.4) at 50 °C for 10 min and stained with 1 μg/ml DAPI. After staining, the sections were mounted with Fluoromount (Diagnostic BioSystems). All images were obtained and analyzed with a BZ-9000 fluorescence microscope.
Microarray Analysis
Colonic total RNA was extracted with RNAqueous-4PCR (Life Technologies, Inc.). For microarray analysis, labeled cRNA was synthesized from the extracted total RNA and hybridized to Illumina BeadArray using the Mouse-6 V1.2 chip. Data were analyzed with GeneSpring (Agilent Technologies). For biological process ontology analysis, probe lists were analyzed in a Gene Viewer (GP Biosciences, Japan, and genes were selected based on p values smaller than 0.01.
Bacterial 16S rRNA Analysis
Fecal DNA extractions and quantitative PCR were performed with a previously validated protocol (31). Briefly, fecal samples (0.1 g) were suspended in 0.9 ml of PBS. Next, 0.2 ml of the fecal suspensions were added to 0.3 ml of extraction buffer (167 mm Tris-HCl, 67 mm EDTA, 10% SDS, pH 9.0) and 0.5 ml of TE buffer-saturated phenol. The samples were vortexed vigorously with 300 mg of glass beads (diameter, 0.1 mm) for 30 s using a FastPrep (MP Biomedicals) at a power level of 5.0. After centrifugation at 15,000 × g for 5 min, 0.4 ml of the supernatant was collected. Subsequently, phenol/chloroform extractions were performed, and 0.25 ml of the supernatant was subjected to isopropyl alcohol precipitation. Finally, the fecal DNA was suspended in 1 ml of 10 mm Tris-HCl buffer, pH 8.0. For bacterial 16S rRNA pyrosequensing, the V4 region of 16S ribosomal RNA was amplified by PCR using primers 5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCTNGTGCCAGCMGCCGCGGTAA-3′ and 5′-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNGGACTACHVGGGTWTCTAAT-3′ (where N indicates any nucleotide; M, A, or C; H, A, or C or T; V, A, or C or G; W, A, or T). Purified PCR products of V4 were sequenced by MiSeq sequencer (Illumina). The pyrosequencing reads were compared with the sequences in the GenBankTM database by BLAST search. Taxonomical analysis was performed on the BLAST results using MEGAN5 program (31). For quantitative PCR of bacterial 16S rRNA genes, the fecal DNA was subjected to real time quantitative PCR using the SYBR Premix Ex Taq (TaKaRa). The primers were reported previously (32). The quantity of each bacterial DNA was calculated with the standard DNA preparations of each bacterial species. The primer sequences are in Table 2.
TABLE 2.
Primers for quantitative PCR of bacterial 16S rRNA genes
| Gene | Forward primera | Reverse primer |
|---|---|---|
| Clostridium coccoides group | AAATGACGGTACCTGACTAA | CTTTGAGTTTCATTCTTGCGAA |
| C. leptum subgroup | GCACAAGCAGTGGAGT | CTTCCTCCGTTTTGTCAA |
| Bifidobacterium | CTCCTGGAAACGGGTGG | GGTGTTCTTCCCGATATCTACA |
| Atopobium cluster | GGGTTGAGAGACCGACC | CGGRGCTTCTTCTGCAGG |
| B. fragilis group | AYAGCCTTTCGAAAGRAAGAT | CCAGTATCAACTGCAATTTTA |
| Prevotella | CACRGTAAACGATGGATGCC | GGTCGGGTTGCAGACC |
| E. cylindroides group | GTGAYGGTAKCTTACCAGA | CTTGCGTGCATACTCCC |
| Lactobacillus group | AGCAGTAGGGAATCTTCCA | CACCGCTACACATGGAG |
| Total bacteria | GTGCCAGCMGCCGCGGTAA | GACTACCAGGGTATCTAAT |
a Nucleotide base codes used are as follows: Y, C, or T; R, A, or G; K, G, or T; M, A, or C.
Histology and Immunostaining
Distal colonic tissues were fixed in methanol/Carnoy's fixative. Paraffin-embedded sections (2 μm thick) were dewaxed and hydrated. For Alcian blue-PAS staining, sections were fixed with 0.5% glutaraldehyde in PBS for 15 min. The fixed sections were stained with 1% Alcian blue 8GX (Sigma) in 0.1 n HCl, pH 1.0, for 30 min and then with periodic acid (Wako) and Schiff's reagent (Wako). For immunohistochemical analyses, antigens were retrieved by L.A.B. Solution (Polyscience, Inc.). Sections were then incubated with anti-mouse MUC2 polyclonal antibody (H-300; Santa Cruz Biotechnology), followed by Alexa Fluor 594 F(ab′)2 fragment of goat anti-rabbit IgG (H+L) (Molecular Probes) and Hoechst 33342 solution (DOJINDO). All images were obtained and analyzed with a BZ-9000 fluorescence microscope.
Enzyme-linked Immunosorbent Assay (ELISA) for Fecal MUC2
Freshly isolated feces were homogenized in PBS, which contained 1% NaN3, 20 mm dithiothreitol, and a protease inhibitor mixture (P8340; Sigma, 1:200 dilution), using a vortex mixer for 30 min at 4 °C. The fecal suspensions were centrifuged at 15,000 × g for 10 min at 4 °C. The supernatants were collected, and their protein concentrations were determined using a BCA protein assay kit (Thermo Scientific). A 96-well ELISA plate (Corning) was coated with 10 ng/well of feces extract. After blocking with PBS containing 3% BSA, 0.5 μg/ml rabbit anti-mouse MUC2 polyclonal antibody (H-300; Santa Cruz Biotechnology) or IgG from rabbit serum (Sigma) was added to the wells and incubated overnight. After washing, horseradish peroxidase-conjugated anti-rabbit IgG (H+L) polyclonal antibody (65-6120, Zymed Laboratories Inc., diluted 1:4,000) was added to the wells and incubated for 2 h. After washing, 1-Step Ultra TMB-ELISA Substrate Solution (Thermo Scientific) was added to the wells, and the absorbance at 450 nm was measured using a 96-well spectrometer (SUNRISE Rainbow RC-R, TECAN). Values obtained from control experiments using rabbit IgG were subtracted from those obtained from test samples using the anti-MUC2 antibody described above.
Statistical Analysis
Data are presented as means ± S.D. Student's t test or one-way ANOVA, followed by the Tukey-Kramer, Steel-Dwass, or Dunnett post hoc test, was used to determine the statistical significance of the differences between the experimental groups.
Results
Generation of Colonic Epithelial Cell-specific Atg7 Conditional Knock-out (cKO) Mice
To generate cKO mice, we first examined the expression of Chst4 (encoding GlcNAc6ST-2) and Villin1, whose regulatory elements were used to drive Cre expression in GlcNAc6ST-2-Cre transgenic mice (26) and in Villin-Cre transgenic mice, respectively. The latter mice have been widely used for the generation of intestinal tissue-specific knock-out mice (22–24). As shown in Fig. 1A, the expression of Chst4 was much more abundant in the colon, whereas Villin1 expression was more abundant in the small intestine. These results indicated that GlcNAc6ST-2-Cre transgenic mice may be more useful than Villin-Cre transgenic mice for determining the role of autophagy specifically in the colon. To examine the expression of Cre recombinase, GlcNAc6ST-2-Cre transgenic mice were crossed with R26R reporter mice in which the lacZ gene is transcribed following Cre-mediated recombination (28). X-gal staining of intestinal tissues showed that Cre recombinase was strongly expressed in colonic epithelia but less abundantly expressed in the ileum of the GlcNAc6ST-2-Cre+/R26R mice (Fig. 1B).
FIGURE 1.

Generation of Atg7 cKO mice. A, expression of Chst4 and Villin1 (Vil1) mRNA in lower gastrointestinal tracts. The relative quantity of each mRNA to that of β-actin was determined by quantitative RT-PCR. Error bars represent the standard deviation (S.D.) of samples within a group. n = 3 per group. B, frozen sections of lower gastrointestinal tracts from GlcNAc6ST-2-Cre+/R26R reporter mice were stained with X-gal solution and Nuclear Fast Red as described previously (26). Bar, 50 μm. C, quantitative RT-PCR analysis of Atg7 mRNA in the colon. Error bars represent the S.D. of samples within a group. An unpaired Student's t test was used to compare WT versus cKO mice. **, p < 0.01. n = 7–9 per group. D, Western blot analysis of whole colonic lysates from WT and cKO mice. The arrowhead in each panel indicates the position of each protein. #, high molecular weight form of p62. E, quantification of the ATG7 protein in the whole colon. Western blot bands (D) were quantified. The error bars represent the S.D. of the samples within a group. An unpaired Student's t test was used to compare WT versus cKO mice. **, p < 0.01. n = 5 per group.
We next crossed GlcNAc6ST-2-Cre transgenic mice with Atg7flox/flox mice (27) to generate cKO mice. Quantitative RT-PCR analysis indicated that expression of Atg7 mRNA in the colon was diminished by 84% compared with wild-type (WT) mice (Fig. 1C). Consistent with this result, the Western blot analysis indicated that the expression of the ATG7 protein in the colon was significantly diminished in the cKO mice (Fig. 1, D and E). Furthermore, p62 protein, a selective substrate of autophagy, and the high molecular weight forms of p62 (33) significantly accumulated in cKO mice. Ubiquitinated proteins that are known to be degraded by autophagy (34) also accumulated in cKO mice. These results indicate that functional autophagy was significantly diminished in the colons of cKO mice. We did not observe any significant differences in the colonic histology between the WT and Atg7 cKO mice according to age (4, 8, and 16 weeks and 1 year) during the steady state.3
Atg7 cKO Mice Are More Susceptible to DSS-induced Colitis
To assess the role of autophagy in colonic inflammation, cKO mice were orally administered 3% DSS in their drinking water to induce UC-like colitis. As an initial experiment, we tested various doses of DSS. A 5% dose of DSS induced severe colitis in both the WT and cKO mice; however, the degree of colitis was too severe to assess differences in the extent of inflammation, and several animals died during treatment. Additionally, we tested 2% DSS, but the inflammatory response was too mild.3 Therefore, we selected 3% DSS for the experiments in this study. In the cKO mice, administration of DSS for 5 days resulted in the exacerbation of colitis as determined by the DAI (Fig. 2A), colonic shortening (Fig. 2B) and histological scores (Fig. 2C), which were based on the loss of colonic crypts and infiltration of leukocytes (Fig. 2D), and increased the thickness of the muscularis propria (Fig. 2E). The medial and distal colons were the most severely affected in the cKO mice, which is consistent with the finding that the distal part of the colon is usually affected in human UC (35). In addition, exacerbation of colitis, including weight loss, was observed 7 days after administration of DSS in the cKO mice (Fig. 2, F–I). Furthermore, chronic colitis induced by the three cycles of 3% DSS in drinking water for 7 days and regular water for 14 days after azoxymethane intraperitoneal injection was also significantly exacerbated in the cKO mice, as assessed by the weight loss, survival rate, and colon length (Fig. 3, B–D).
FIGURE 2.

Exacerbation of acute DSS-induced colitis in cKO mice. Mice were given 3% DSS for 5 days (A–E) or for 7 days (F–I). A and G, DAI was defined by the presence of rectal bleeding and the stool consistency. B and H, colon length. C and I, histological score. D, hematoxylin and eosin staining. E, mucosa and muscularis propria thickness. F, changes in body weight. Data are presented as mean ± S.D. from each group (A–E, n = 3–4; F–I, n = 7–9). An unpaired Student's t test was used to compare WT versus cKO mice. *, p < 0.05; **, p < 0.01 versus WT control.
FIGURE 3.
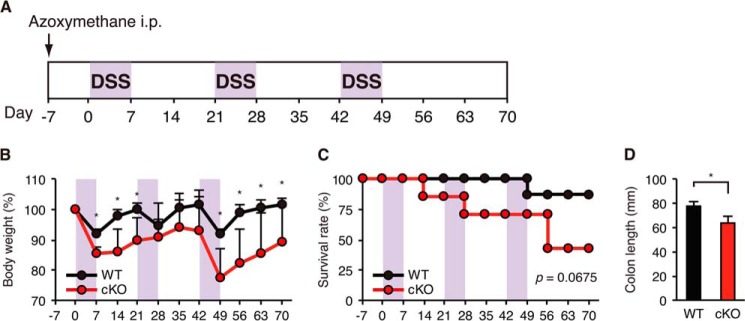
Exacerbation of chronic DSS-induced colitis in cKO mice. A, schematic overview of the azoxymethane/DSS chronic colitis model. After an initial azoxymethane injection (10 mg/kg), 3% DSS was given in drinking water (violet areas) followed by regular water. B, body weight. C, survival rate. D, colonic length at day 70. Each value represents the mean ± S.E. (WT, n = 8; cKO, n = 5). An unpaired Student's t test was used to compare WT versus cKO mice. *, p < 0.05 versus WT control.
Gut microflora is regarded as one of the major factors that determine the sensitivity of colitis (1, 2). Therefore, we next examined the effects of oral administration of antibiotics on DSS-induced colitis in cKO mice (Fig. 4). Oral administration of a combination of two antibiotics (ciprofloxacin and metronidazole) ameliorated weight loss, DAI, and colonic shortening in cKO mice (Fig. 4, A–C). Histological scores determined by epithelial morphology and leukocyte infiltration were also significantly reduced by antibiotic treatment (Fig. 4, D and E). The antibiotic treatment also ameliorated the colitis of cKO mice in the recovery protocol (3% DSS for 7 days, followed by 7 days of water) (Fig. 4, F–H).
FIGURE 4.

Antibiotic treatment ameliorated colitis in Atg7 cKO mice. cKO mice were treated with (Abx-cKO) or without (cKO) a combination of metronidazole and ciprofloxacin for 4 weeks. WT mice were left untreated. 3% DSS exposure was then begun 2 days later. A–E, mice were given 3% DSS for 7 days, and tissues were collected immediately after the treatment (WT, n = 6; cKO, n = 4; Abx-cKO, n = 5). F–H, mice were given 3% DSS for 7 days, which was then replaced with normal water until day 14 (WT, n = 9; cKO, n = 15; Abx-cKO, n = 5). A and F, body weight. B and G, DAI. C, colon length. D, hematoxylin and eosin staining. E and H, histological score. Data are presented as mean ± S.D. from each group. *, p < 0.05; **, p < 0.01 versus the control. One-way ANOVA, followed by the Tukey-Kramer (A and C) or Steel-Dwass (B, E, and H) post hoc test, was used to determine the statistical significance.
Atg7 cKO Mice Have Nontransmissible Abnormal Microflora
Because the combination antibiotic treatment ameliorated colitis in cKO mice, we hypothesized that cKO mice might have abnormal microflora compared with WT mice. To examine gut microflora in WT and cKO mice, bacterial 16S rRNA pyrosequensing was performed. As shown in Fig. 5, A and B, Clostridia was significantly increased in the cKO mice. Further analyses indicated that Prevotellaceae was also significantly increased in the cKO mice (Fig. 5C). To further examine the changes of bacterial composition, we next performed quantitative PCR using group- or genus-specific primers of the bacterial 16S rRNA genes (Table 2). As shown in Fig. 5, D and E, total bacteria and the Clostridum leptum subgroup, the Atopobium cluster, the Bacteroides fragilis group, the Prevotella group, and the Eubacterium cylindroides group were significantly increased in the feces of the cKO mice. These bacterial genera or groups, excluding Prevotella, were significantly diminished following treatment with a combination of metronidazole and ciprofloxacin.
FIGURE 5.

Comparison of major microbial populations in fecal samples. A–C, pyrosequencing analysis was used to examine microbial diversity in feces from WT and cKO mice. Sequences were obtained from pooled stool samples of WT (n = 3; 145,188 reads) and cKO mice (n = 3; 111,639 reads). A, comparisons of the diversity at the phylum or class level. Each chart represents the taxonomic composition. B, comparison of the diversity at the family level in Clostridiales. C, comparison of the diversity at the family level in Bacteroidales. D, total bacteria in fecal samples were quantified by quantitative RT-PCR using universal bacterial 16S rRNA genes (WT, n = 4; cKO, n = 6). E, microbial composition of the feces of WT and cKO mice treated with (Abx-cKO) or without (WT and cKO) a combination of metronidazole and ciprofloxacin for 4 weeks was analyzed by quantitative PCR using group- or genus-specific primers of the bacterial 16S rRNA genes. Error bars represent the S.D. of samples within the group (WT, n = 4; cKO, n = 4; Abx-cKO, n = 8). An unpaired Student's t test and a paired Student's t test were used to compare WT versus cKO mice and cKO versus Abx-cKO mice, respectively. **, p < 0.01; *, p < 0.05. n = 4 per group.
It was reported that abnormal gut microflora was transmissible and correlated with colitis sensitivity in mice lacking specific components of intracellular pattern recognition molecules, such as NLRP6 (36) and NOD2 (37). We thus carried out cohousing experiments. WT mice were cohoused with age- and gender-matched cKO mice for 4 weeks prior to the induction of colitis by DSS. As a result, cKO mice showed more severe weight loss compared with WT mice, similar to what was observed in mice without cohousing (Fig. 6, A and B). Similar differences in bacterial composition between cKO and WT mice were detected after 4 weeks of cohousing (Fig. 6C) compared with each genotype of mice without cohousing (Fig. 5). Next, C57BL/6 female WT mice treated with four antibiotics (vancomycin, ampicillin, neomycin, and metronidazole) were cohoused with gender-matched cKO or WT mice. Four weeks after cohousing, mice were administered 3% DSS for 7 days, and colitis symptoms were assessed. Comparable levels of weight loss and an increase in DAI were observed in C57BL/6 WT mice cohoused with WT or cKO mice (Fig. 6, D and E). No significant differences in bacterial composition were found in WT- and cKO-cohoused WT mice (Fig. 6F), suggesting that the abnormal microflora found in cKO mice was not transmissible by cohousing.
FIGURE 6.

Effects of cohousing on colitis sensitivity and gut microflora. A–C, cKO and WT mice were cohoused (CHd) with the opposite genotypes of mice for 4 weeks, after which DSS colitis was induced. A, body weight. B, DAI. C, comparison of major bacterial populations measured at the end of cohousing by quantitative PCR analysis. An unpaired Student's t test was used to compare WT versus cKO mice. *, p < 0.05; **, p < 0.01. n = 9 per group. D–F, C57BL/6 WT mice were treated with four antibiotics for 8 weeks and then cohoused with cKO or WT mice for 4 weeks, after which DSS colitis was induced. D, body weight. E, DAI. F, comparison of major bacterial population. Error bars represent the S.D. of samples within the group. n = 3–6 per group.
Increased Bacterial Burden in the Colon of Atg7 cKO Mice
Because Atg7 cKO mice had abnormal microflora and colitis sensitivity compared with WT mice, we next assessed bacterial invasion into the colonic epithelium before (Fig. 7, A–C) and after (Fig. 7, D–F) DSS administration by fluorescent in situ hybridization using a ubiquitous eubacterial probe. During the steady state before DSS treatment, significantly more bacteria colonized the crypts of the proximal colon in cKO mice compared with WT mice, whereas only a small number of bacteria invaded the colonic epithelium in either group of mice (Fig. 7, A–C). On day 3 after DSS administration, a massive increase in bacterial colonization of the crypts and invasion into the colonic epithelia were observed in both WT and cKO mice (Fig. 7, D–F). In particular, a significantly higher level of bacterial invasion was detected in the distal colon in cKO mice compared with WT mice. In addition, bacterial colonization in the colonic crypts and bacterial invasion of tissue were significantly diminished in the antibiotic-treated animals (Fig. 7, A–F). Because the colitis symptoms of cKO mice were comparable to those of WT mice on day 3,3 these results indicate that bacterial invasion occurred a few days prior to the onset of differential symptoms of colitis. Therefore, Atg7 cKO mice had more severe bacterial burden in the steady and colitis states compared with WT mice, which is likely the cause of the exacerbated colitis in cKO mice that could be ameliorated by treatment with antibiotics as described above.
FIGURE 7.

Bacterial burden in WT and Atg7 cKO mice. The bacteria in the colonic tissue of WT and cKO mice treated with (Abx-cKO) or without (WT and cKO) a combination of metronidazole and ciprofloxacin for 4 weeks were detected by FISH using general bacterial 16S probes (red, Texas Red-Eub338) during the steady state (A–C, n = 4) or on day 3 of 3% DSS administration (D–F, n = 5). A and D, fluorescence microscopic images. B and E, percentage of bacterially colonized crypts. C and F, number of colonic tissue-invading bacteria per mm2 field. Arrows, bacterially colonized crypts; arrowheads, invading bacteria. Insets, higher magnification of the areas indicated by arrows or arrowheads. Error bars represent the S.D. of samples within a group (n = 3–4 per group). One-way ANOVA, followed by the Tukey-Kramer post hoc test, was used to determine the statistical significance. **, p < 0.01; *, p < 0.05.
Expression of Antimicrobial and Antiparasitic Peptides Was Decreased in the Colon of Atg7 cKO Mice
To explore the reason why Atg7 cKO mice had a higher bacterial burden than WT mice, we next compared gene expression profiles in colonic tissues from WT and cKO mice using microarray analysis. Nine genes were selectively down-regulated by more than 5-fold in cKO mice among differentially expressed genes with a cutoff value of 1,000 (Table 3). Notably, two genes (Ang4 and Retnlb) found in this analysis encode the antibacterial peptide angiogenin-4 (38) and the antiparasitic peptide Relmβ (39), respectively. These genes are induced by intestinal microflora or parasitic infection (38, 40–42). Next, we performed gene ontology analysis of differentially expressed genes induced by bacterial infection (GO:0042742) or nematode infection (GO:0009624) with a cutoff value of 100. As a result, we found that Itln1 encoding intelectin-1, which is known to be involved in bacterial clearance (43), and Itln2 encoding its homologue intelectin-2 (44) fulfilled these criteria.
TABLE 3.
Genes differentially expressed by more than 5-fold in the colon of WT and cKO mice
| Accession | Gene name | Gene symbol | WTa | cKOa | Fold changeb |
|---|---|---|---|---|---|
| NM_023881.2 | Mus musculus resistin-like β | Retnlb | 6565.2 | 934.9 | 0.142402364 |
| XM_132608.2 | Similar to monoclonal antibody κ light chain | LOC232060 | 5594.8 | 338.7 | 0.060538357 |
| XM_001476407.1 | Igkv15-103 immunoglobulin κ chain variable 15-103 | LOC100046552 | 4867 | 86.7 | 0.017813848 |
| NM_177544.2 | M. musculus angiogenin, ribonuclease A family, member 4 | Ang4 | 4750.8 | 506.5 | 0.106613623 |
| NM_010731.3 | M. musculus zinc finger and BTB domain containing 7a | Zbtb7a | 3846.3 | 705.7 | 0.183475028 |
| NM_007520.2 | M. musculus BTB and CNC homology 1 | Bach1 | 2033.8 | 298.6 | 0.146818763 |
| XM_619042.2 | Similar to Ig heavy chain V region 1B43 precursor | LOC630337 | 1548.4 | 204.8 | 0.132265564 |
| IGKV4–90_AJ231224_Ig_ κ _variable_4–90_22 | 1125.1 | −1.1 | 0.00088881 | ||
| IGHV1S30_X02462_Ig_heavy_variable_1S30_12 | 1095.8 | 32.4 | 0.029567439 | ||
| NR_003522.1 | M. musculus abhydrolase domain containing 1 | Abhd1 | 156.2 | 1122.7 | 7.187580026 |
| IGKV4–80_AJ231213_Ig_ κ _variable_4–80_91 | 43.8 | 1050.4 | 23.98173516 |
a Array signal intensity is given.
b Fold change is defined as the ratio of the signal of cKO to that of WT.
To validate differential expression of the four antimicrobial or antiparasitic peptides described above, we next performed quantitative RT-PCR analysis (Fig. 8A). As a result, the four genes were confirmed to be significantly down-regulated in cKO mice compared with WT mice, suggesting that the decline of these antimicrobial and antiparasitic peptide genes might be involved in the higher bacterial burden observed in Atg7 cKO mice.
FIGURE 8.

Quantitative RT-PCR analyses of antimicrobial peptides, antiparasitic peptides, and cytokines in WT and Atg7 cKO mice. Quantitative RT-PCR analyses of antimicrobial peptides, antiparasitic peptides, and cytokines were performed using total RNA samples from the medial colons of WT and cKO mice treated with (Abx-cKO) or without (WT and cKO) a combination of metronidazole and ciprofloxacin for 4 weeks. A, expression of antimicrobial peptides and antiparasitic peptides. B, expression of Th1/Th2 cytokines. C, expression of epithelium-derived cytokines. Error bars represent the S.D. of samples within a group. An unpaired Student's t test and a paired Student's t test were used to compare WT versus cKO mice and cKO versus Abx-cKO mice, respectively. **, p < 0.01; *, p < 0.05. n = 3–4 per group.
Induction of Antimicrobial and Antiparasitic Peptides by Gut Microflora and Th2 Cytokines
It has been reported that Ang4 and Retnlb are induced by gut microflora (38, 40, 41). We speculated that Itln1 and Itln2 might also be bacterially induced. To examine this possibility, we carried out quantitative RT-PCR analysis of the four genes in the medial colon where they are most abundantly expressed3 in WT mice treated with or without combined antibiotics (Fig. 9). A combination of metronidazole and ciprofloxacin (2Abx) and a combination of vancomycin, metronidazole, ampicillin, and neomycin (4Abx) significantly reduced the expression of the four genes in WT mice (Fig. 9A). Major bacterial species were confirmed to be significantly reduced in the stools of the antibiotic-treated WT mice (Fig. 9B). These results indicated that the four antimicrobial and antiparasitic peptide genes are induced by gut microflora in WT mice.
FIGURE 9.

Antibacterial and antiparasitic peptide expression and relative bacterial quantity after antibiotic treatment. A, expression of four antibacterial or antiparasitic peptide genes in the medial colon from antibiotic-treated C57BL/6 WT mice. B, bacterial quantities in feces from C57BL/6 WT mice treated or untreated (Control) with combination antibiotics. 2Abx, a combination of metronidazole and ciprofloxacin. 4Abx, a combination of vancomycin, metronidazole, ampicillin, and neomycin. The quantity of each bacterial genomic DNA was normalized with bacterial genomic DNA from control mice. Data are presented as mean ± S.D. from each group (Control, n = 5; 2Abx, n = 4; 4Abx, n = 4). One-way ANOVA, followed by the Dunnett's post hoc test, was used to determine the statistical significance. *, p < 0.05; **, p < 0.01 versus the control.
We next analyzed Th2 cytokine expression in the medial colon, because Ang4, Retnlb, Itln1, and Itln2 were reportedly induced by Th2 cytokines (40, 42, 45–47). In cKO mice, Il4 and Il13 expression was decreased compared with WT mice, whereas Il5 and Ifng expression was not affected (Fig. 8B). In addition, we examined the basal expression of the following cytokines relative to that of β-actin that was set as 1.0 (Table 4): Tnfa encoding TNF-α; IL1b encoding IL-1β; IL6 encoding IL-6; IL10 encoding IL-10; IL12b encoding IL-12b; and IL17a encoding IL-17a. The difference of the expression levels of these cytokines in the WT versus cKO mice were statistically insignificant.
TABLE 4.
Expression of various cytokines in the colon of WT and cKO mice
| WTa | cKO | |
|---|---|---|
| Tnfa | 9.16 × 10−5 ± 1.97 × 10−5 | 1.28 × 10−4 ± 5.51 × 10−5 |
| Il1b | 4.28 × 10−4 ± 2.45 × 10−4 | 3.28 × 10−4 ± 7.96 × 10−5 |
| Il6 | 2.83 × 10−6 ± 1.85 × 10−6 | 2.31 × 10−6 ± 1.64 × 10−6 |
| Il10 | 1.84 × 10−5 ± 6.44 × 10−6 | 1.79 × 10−5 ± 4.66 × 10−6 |
| Il12b | 1.98 × 10−5 ± 5.71 × 10−6 | 2.64 × 10−5 ± 1.49 × 10−5 |
| Il17a | 6.79 × 10−6 ± 7.24 × 10−6 | 9.41 × 10−6 ± 1.55 × 10−5 |
a The expression of cytokines was shown as a relative amount compared with that of β-actin, which was set as 1.0.
Because epithelium-derived cytokines, such as IL-25, are critical factors for Th2 responses (48–50), their expression was further examined. As shown in Fig. 8C, the expression of Il25, which is known to be induced by gut microflora (51), was decreased in the medial colon of cKO mice compared with WT mice. This decrease might be due to less efficient recognition of gut microflora in cKO mice, resulting in the decrease of Th2 cytokines and antimicrobial peptides. In the antibiotic-treated cKO mice, Il25, Ang4, Itln1, and Itln2 were still less abundantly expressed compared with WT mice, whereas the expression of Il4 and Il13 encoding Th2 cytokines and that of Retnlb encoding an antimicrobial peptide, Relm-β, was restored (Fig. 8, A–C). These findings suggest that induction of Retnlb by Th2 cytokines is independent of the IL-25 signaling pathway, whereas that of Ang4, Itln1, and Itln2 is dependent on this signaling pathway.
Secretion of Colonic Mucin from Goblet Cells Was Diminished in Atg7 cKO Mice
To further explore the reason why Atg7 cKO mice had more bacterial burden and exacerbated colitis than WT mice, we next compared secretion of colonic mucin from goblet cells (Fig. 10). Consistent with a recent report showing that the NLRP6 inflammasome and possibly autophagy regulate secretion of colonic mucins (52), Atg7 cKO mice showed a much less efficient release of mucin stored in the intracellular granules of goblet cells into the intestinal lumen as determined by hematoxylin and eosin and Alcian blue-PAS staining (Fig. 10, A and B). This result was confirmed using immunofluorescent staining with an antibody against a major colonic mucin (MUC2) (Fig. 10C). The percentage of mucus-secreting crypts was significantly decreased in cKO mice (Fig. 10D). Consistently, the thickness of the solid mucus layer (30) was also significantly decreased in cKO mice (Fig. 10E). Furthermore, ELISA showed that the amount of fecal MUC2 mucin was diminished (Fig. 10F) in cKO mice, although mRNA levels were not affected (Fig. 10G). Because colonic mucin functions as a physical barrier against invasion of microbiota (30), less efficient secretion of colonic mucin in cKO mice is likely involved in their severe bacterial burden and exacerbated colitis.
FIGURE 10.

Secretion of colonic mucins in WT and Atg7 cKO mice. Hematoxylin and eosin (A), Alcian blue-PAS (B), and anti-MUC2 (C) staining of the mouse distal colon. D, percentage of mucus-secreting crypts. E, quantification of inner mucus layer thickness. F, detection of fecal MUC2 by ELISA. G, quantitative RT-PCR. The relative quantity of Muc2 mRNA compared with the quantity of β-actin was determined by the ΔΔCt method. Error bars represent the S.D. of samples within a group. Asterisk in A–C, mucus-secreting crypt. Arrows in A–C, mucus-secreting goblet cells. Dashed line in C, mucosal surface. An unpaired Student's t test was used to compare WT versus cKO mice. **, p < 0.01; *, p < 0.05. n = 3 per group.
Discussion
In this study, we generated Atg7 cKO mice that specifically lacked ATG7 in colonic epithelial cells in the gastrointestinal tract and examined the function of autophagy in protection against colitis and maintenance of normal gut microflora. The reduction of Atg7 mRNA expression and accumulation of autophagy substrate p62 and ubiquitinated proteins in the colon in cKO mice were confirmed by quantitative RT-PCR and Western blot analyses (Fig. 1). We found that the Atg7 cKO mice were more susceptible to DSS-induced colitis, an experimental model of UC (Figs. 2–4). In contrast, others reported that intestinal epithelial cell-specific Atg7 deletion did not affect the sensitivity of DSS colitis (53) or the pathogenesis of intestinal tumors (54) using Atg7flox/floxVillin-Cre mice. Therefore, we think that our Atg7 cKO mice are the first ones that could be utilized to assess the function of Atg7 in colitis.
A combination of ciprofloxacin and metronidazole that is clinically used in active CD (55) ameliorated colitis in our cKO mice (Fig. 4). Treatment of WT mice with the two antibiotics decreased all bacterial genera and groups examined with the exception of the Lactobacillus group, which is generally thought to be a part of the healthy microflora (Fig. 9) (56). In contrast, the C. leptum subgroup, Atopobium cluster, B. fragilis group, Prevotella and E. cylindroides group were increased in the feces of cKO mice (Fig. 5). One intriguing possibility is that these bacteria may be involved in the exacerbated colitis observed in the Atg7 cKO mice. Consistent with this hypothesis, the Prevotella and B. fragilis groups that belong to the Bacteroidetes phylum are increased in patients with CD and UC (57). To our knowledge, no connection between IBDs and the increase of the C. leptum subgroup, E. cylindroides group, or Atopobium cluster has been reported. However, the results in Fig. 5 using antibiotics suggest that the levels of these bacterial groups and the B. fragilis group correlated well with the severity of experimental colitis (Fig. 4). Although colitis severity is linked to dysbiotic microflora (36, 37, 58–60), the relationship between gut microflora and deficiency of autophagy-related genes has not been clarified. We believe that the animal model described herein may be useful for the determination of bacterial subgroups involved in the onset of colitis.
It is noteworthy that Atg7 cKO mice suffered from severe bacterial burden during colitis and steady states (Fig. 7). Previous studies showed that abnormal microflora and colitis sensitivity of some gene-targeted mice were transmissible when housed in the same cage (36, 37, 58–60). However, our results indicated that cohousing WT mice with cKO mice did not affect colitis sensitivity and gut microflora (Fig. 6). Therefore, the abnormal microbiota that caused severe colitis in the Atg7 cKO mice may have different properties than the microbiota that were found in previous studies. Seedorf et al. (61) reported that bacteria from diverse habitats can colonize in the germ-free mouse gut after cohousing. However, in that report, several bacterial groups, such as the soil-derived bacteria, dominated even in the presence of other bacterial communities, whereas some other bacteria could not be easily transmitted from one animal to another animal with an established bacterial community. This finding is consistent with our observation that colonic microflora cannot be easily transmitted by cohousing. One possible explanation of our results is that highly anaerobic bacteria that could not be transmitted through feces may have caused severe colitis in the Atg7 cKO mice. Strictly anaerobic bacterial culture and gnotobiotic experiments may clarify this issue.
We found that Ang4, Retnlb, Itln1, and Itln2 were decreased in cKO mice (Fig. 8). These genes are important for protection against microorganisms. Angiogenin-4 (encoded by Ang4) has bactericidal activity against several Gram-positive enteric microbes, such as Enterococcus faecalis (38), although E. faecalis was not detectable in our mice.3 Intelectin-1 (encoded by Itln1) is a host defense lectin that assists phagocytic clearance of microorganisms (43) and is encoded in one of the IBD susceptibility loci (62). This is consistent with our finding that Itln1 was decreased in the colon of Atg7 cKO mice. Intelectin-2 (encoded by Itln2) is presumed to have a similar function to intelectin-1, because it has 91% amino acid sequence identity with intelectin-1 (44) and is induced in the same manner as intelectin-1 (45). Relmβ (encoded by Retnlb) is an important molecule that affects macrophages during parasite-associated intestinal inflammation (63). Relmβ-deficient mice were reportedly protected from DSS-induced colitis (64), whereas our cKO mice expressing lower amounts of Relmβ than WT mice showed more severe colitis. This apparently contradictory result might be explained by our finding that Ang4, Itln1, and Itln2 were decreased in cKO mice. The lower expression of Ang4, Itln1, and Itln2 may have resulted in the exacerbated colitis in the cKO mice and may have hindered the protective effect of the lower expression of Retnlb.
It is reported that Ang4 and Retnlb are induced by microflora (38, 40, 41). Consistently, our results showed that the expression of Ang4 and Retnlb was significantly diminished by broad spectrum antibiotic treatment in WT mice (Fig. 9). In addition, the expression of Itln1 and Itln2 was also diminished by the same treatment. However, the results in Fig. 8 indicate that the antibiotic treatment restored the expression of the Th2 cytokines IL-4 and IL-13 and that of Retnlb in cKO mice. In contrast, the expression of Ang4, Itln1, and Itln2 was minimally restored after the cKO mice were treated with the two antibiotics. We hypothesize that the expression of Retnlb is more dependent on Th2 cytokines than the expression of Ang4, Itln1, and Itln2, which may require signals from certain bacterial genera or groups that were significantly diminished by the antibiotic treatment. The levels of several bacterial species were decreased following the two antibiotic treatments in WT mice (Fig. 9); however, these levels were higher in the cKO mice than in the WT mice. One possibility is that recognition of these bacteria in cKO mice may not be efficient enough to induce antibacterial or antiparasitic peptides because of the lack of functional autophagy. Consistent with this idea, the expression of IL-25, whose expression is dependent on microflora (51), was significantly decreased in cKO mice (Fig. 8C). Another possibility is that these bacteria may not be involved in the induction of the antibacterial or antiparasitic peptide genes and that minor bacterial species that we have not evaluated may have affected their expression, consistent with the keystone hypothesis, which states that keystone species with a low abundance in the bacterial community are considered to have a community-wide impact on health and disease (65). Indeed, Bacteroides thetaiotaomicron, a minor population in the B. fragilis group, is known to induce the expression of Ang4 in Paneth cells in the small intestine (38).
A number of previous studies using autophagy-related gene-deficient mice showed that autophagy is related to the secretion of proteins in various cell systems. Studies using hypomorphic Atg16L1 mice and epithelium-specific Atg5 knock-out mice showed that ileal Paneth cells exhibit abnormal secretory granules (19). Likewise, Atg7-deficient pancreatic β-cells impaired insulin secretion (66). More recently, Nlrp6 knock-out mice and Atg5 heterozygous mice impaired secretion of mucus from goblet cells (52). Consistent with these results, we showed that secretion of colonic mucins from goblet cells was significantly diminished in cKO mice (Fig. 10). Because colonic mucin MUC2 and its glycosylation play a protective role against colitis (67–70), impaired secretion coupled with reduced production of antimicrobial or antiparasitic peptides could be involved in the severe colitis symptoms in our Atg7 cKO mice, as illustrated in Fig. 11.
FIGURE 11.
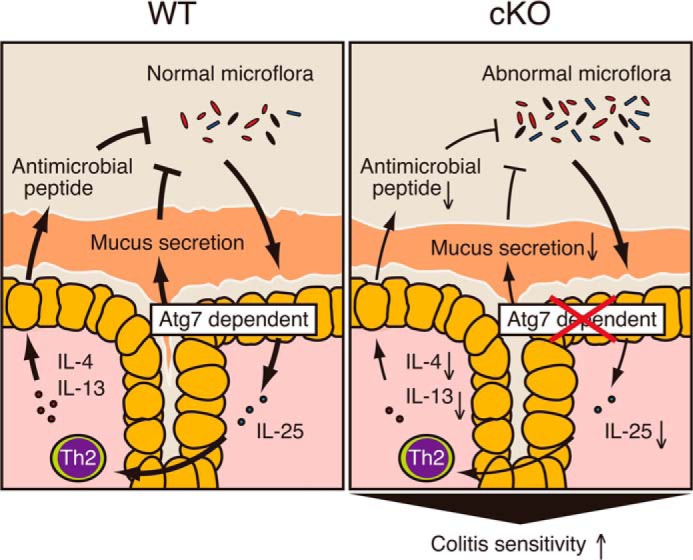
Model depicting the proposed role of ATG7 in protection against colitis. In WT mice, gut commensal bacteria are recognized by autophagy and induce the expression of IL-25. IL-25 then act on Th2 or other cell subsets to induce Th2 cytokines, such as IL-4 and IL-13, which are involved in the induction of four antibacterial or antiparasitic peptides in the colonic epithelial cells. These peptides together with colonic mucins secreted from goblet cells act as a barrier against bacterial invasion and colitis. In cKO mice, this ATG7-dependent physiological cycle might not work properly, causing the expansion of abnormal gut microflora, their invasion into the colonic epithelium, and severe colitis.
In summary, our results indicate that epithelial autophagy is essential for protection against colitis and the maintenance of normal gut microflora, antimicrobial peptide production, and mucin secretion, which may ultimately lead to an efficient intervention for IBDs. Colonic epithelial cell-specific autophagy cKO mice generated in this study may serve as a useful tool for future studies to achieve this goal.
Author Contributions
K. T. and H. K. designed, performed, and analyzed the experiments and wrote the paper. M. N. and A. T. performed the experiments. Y. I. and M. K. provided technical assistance.
Acknowledgments
We thank Drs. Masanobu Nanno, Satoshi Matsumoto, and Mayuko Yamamoto (Yakult Central Institute for Microbiological Research) for supporting quantitative PCR of bacterial 16S rRNA genes and Dr. Makoto Taniguchi (Oral Microbiome Center, Japan) for bacterial 16S rRNA pyrosequensing.
This work was supported in part by grants from the Precursory Research for Embryonic Science and Technology, the Japan Science and Technology Agency (to H. K.), Scientific Research (B) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, Grant 24390018 (to H. K.), Grant-in-aid for JSPS Fellows 26-7438 (to K. T.), and in part by the Institute for Fermentation, Osaka (to H. K.), and the Honjo International Scholarship Foundation (to K. T.). The authors declare that they have no conflicts of interest with the contents of this article.
K. Tsuboi and H. Kawashima, unpublished observations.
- IBD
- inflammatory bowel diseases
- CD
- Crohn disease
- UC
- ulcerative colitis
- DSS
- dextran sulfate sodium
- DAI
- disease activity index
- FISH
- fluorescence in situ hybridization
- cKO
- conditional knockout
- ANOVA
- analysis of variance
- PAS
- periodic acid-Schiff
- DAI
- disease activity index.
References
- 1. Kaser A., Zeissig S., Blumberg R. S. (2010) Inflammatory bowel disease. Annu. Rev. Immunol. 28, 573–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abraham C., Cho J. H. (2009) Inflammatory bowel disease. N. Engl. J. Med. 361, 2066–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mowat C., Cole A., Windsor A., Ahmad T., Arnott I., Driscoll R., Mitton S., Orchard T., Rutter M., Younge L., Lees C., Ho G. T., Satsangi J., Bloom S., IBD Section of the British Society of Gastroenterology. (2011) Guidelines for the management of inflammatory bowel disease in adults. Gut 60, 571–607 [DOI] [PubMed] [Google Scholar]
- 4. Baumgart D. C., Sandborn W. J. (2012) Crohn disease. Lancet 380, 1590–1605 [DOI] [PubMed] [Google Scholar]
- 5. Nguyen H. T., Dalmasso G., Müller S., Carrière J., Seibold F., Darfeuille-Michaud A. (2014) Crohn disease-associated adherent invasive Escherichia coli modulate levels of microRNAs in intestinal epithelial cells to reduce autophagy. Gastroenterology 146, 508–519 [DOI] [PubMed] [Google Scholar]
- 6. Hampe J., Franke A., Rosenstiel P., Till A., Teuber M., Huse K., Albrecht M., Mayr G., De La Vega F. M., Briggs J., Günther S., Prescott N. J., Onnie C. M., Häsler R., Sipos B., et al. (2007) A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 39, 207–211 [DOI] [PubMed] [Google Scholar]
- 7. Rioux J. D., Xavier R. J., Taylor K. D., Silverberg M. S., Goyette P., Huett A., Green T., Kuballa P., Barmada M. M., Datta L. W., Shugart Y. Y., Griffiths A. M., Targan S. R., Ippoliti A. F., Bernard E. J., et al. (2007) Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 39, 596–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mizushima N., Noda T., Yoshimori T., Tanaka Y., Ishii T., George M. D., Klionsky D. J., Ohsumi M., Ohsumi Y. (1998) A protein conjugation system essential for autophagy. Nature 395, 395–398 [DOI] [PubMed] [Google Scholar]
- 9. Kuma A., Mizushima N., Ishihara N., Ohsumi Y. (2002) Formation of the approximately 350-kDa Apg12-Apg5·Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J. Biol. Chem. 277, 18619–18625 [DOI] [PubMed] [Google Scholar]
- 10. Tanida I., Mizushima N., Kiyooka M., Ohsumi M., Ueno T., Ohsumi Y., Kominami E. (1999) Apg7p/Cvt2p: A novel protein-activating enzyme essential for autophagy. Mol. Biol. Cell 10, 1367–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tanida I., Yamasaki M., Komatsu M., Ueno T. (2012) The FAP motif within human ATG7, an autophagy-related E1-like enzyme, is essential for the E2-substrate reaction of LC3 lipidation. Autophagy 8, 88–97 [DOI] [PubMed] [Google Scholar]
- 12. Tanida I., Tanida-Miyake E., Ueno T., Kominami E. (2001) The human homolog of Saccharomyces cerevisiae Apg7p is a protein-activating enzyme for multiple substrates, including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J. Biol. Chem. 276, 1701–1706 [DOI] [PubMed] [Google Scholar]
- 13. Lapaquette P., Glasser A. L., Huett A., Xavier R. J., Darfeuille-Michaud A. (2010) Crohn disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell. Microbiol. 12, 99–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuballa P., Huett A., Rioux J. D., Daly M. J., Xavier R. J. (2008) Impaired autophagy of an intracellular pathogen induced by a Crohn disease associated ATG16L1 variant. PLoS One 3, e3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gutierrez M. G., Master S. S., Singh S. B., Taylor G. A., Colombo M. I., Deretic V. (2004) Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119, 753–766 [DOI] [PubMed] [Google Scholar]
- 16. Deretic V., Singh S., Master S., Harris J., Roberts E., Kyei G., Davis A., de Haro S., Naylor J., Lee H. H., Vergne I. (2006) Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell. Microbiol. 8, 719–727 [DOI] [PubMed] [Google Scholar]
- 17. Saitoh T., Fujita N., Jang M. H., Uematsu S., Yang B. G., Satoh T., Omori H., Noda T., Yamamoto N., Komatsu M., Tanaka K., Kawai T., Tsujimura T., Takeuchi O., Yoshimori T., Akira S. (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456, 264–268 [DOI] [PubMed] [Google Scholar]
- 18. Fujishima Y., Nishiumi S., Masuda A., Inoue J., Nguyen N. M., Irino Y., Komatsu M., Tanaka K., Kutsumi H., Azuma T., Yoshida M. (2011) Autophagy in the intestinal epithelium reduces endotoxin-induced inflammatory responses by inhibiting NF-κB activation. Arch. Biochem. Biophys. 506, 223–235 [DOI] [PubMed] [Google Scholar]
- 19. Cadwell K., Liu J. Y., Brown S. L., Miyoshi H., Loh J., Lennerz J. K., Kishi C., Kc W., Carrero J. A., Hunt S., Stone C. D., Brunt E. M., Xavier R. J., Sleckman B. P., Li E., et al. (2008) A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Murthy A., Li Y., Peng I., Reichelt M., Katakam A. K., Noubade R., Roose-Girma M., DeVoss J., Diehl L., Graham R. R., van Lookeren Campagne M. (2014) A Crohn disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 506, 456–462 [DOI] [PubMed] [Google Scholar]
- 21. Adolph T. E., Tomczak M. F., Niederreiter L., Ko H. J., Böck J., Martinez-Naves E., Glickman J. N., Tschurtschenthaler M., Hartwig J., Hosomi S., Flak M. B., Cusick J. L., Kohno K., Iwawaki T., Billmann-Born S., et al. (2013) Paneth cells as a site of origin for intestinal inflammation. Nature 503, 272–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. el Marjou F., Janssen K. P., Chang B. H., Li M., Hindie V., Chan L., Louvard D., Chambon P., Metzger D., Robine S. (2004) Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39, 186–193 [DOI] [PubMed] [Google Scholar]
- 23. Madison B. B., Dunbar L., Qiao X. T., Braunstein K., Braunstein E., Gumucio D. L. (2002) Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J. Biol. Chem. 277, 33275–33283 [DOI] [PubMed] [Google Scholar]
- 24. Ishikawa T. O., Herschman H. R. (2011) Conditional bicistronic Cre reporter line expressing both firefly luciferase and β-galactosidase. Mol. Imaging Biol. 13, 284–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Inoue J., Nishiumi S., Fujishima Y., Masuda A., Shiomi H., Yamamoto K., Nishida M., Azuma T., Yoshida M. (2012) Autophagy in the intestinal epithelium regulates Citrobacter rodentium infection. Arch. Biochem. Biophys. 521, 95–101 [DOI] [PubMed] [Google Scholar]
- 26. Kawashima H., Hirakawa J., Tobisawa Y., Fukuda M., Saga Y. (2009) Conditional gene targeting in mouse high endothelial venules. J. Immunol. 182, 5461–5468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I., Ezaki J., Mizushima N., Ohsumi Y., Uchiyama Y., Kominami E., Tanaka K., Chiba T. (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Soriano P. (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70–71 [DOI] [PubMed] [Google Scholar]
- 29. Alex P., Zachos N. C., Nguyen T., Gonzales L., Chen T. E., Conklin L. S., Centola M., Li X. (2009) Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm. Bowel Dis. 15, 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Johansson M. E., Phillipson M., Petersson J., Velcich A., Holm L., Hansson G. C. (2008) The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. U.S.A. 105, 15064–15069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huson D. H., Mitra S., Ruscheweyh H. J., Weber N., Schuster S. C. (2011) Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21, 1552–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kanno T., Matsuki T., Oka M., Utsunomiya H., Inada K., Magari H., Inoue I., Maekita T., Ueda K., Enomoto S., Iguchi M., Yanaoka K., Tamai H., Akimoto S., Nomoto K., et al. (2009) Gastric acid reduction leads to an alteration in lower intestinal microflora. Biochem. Biophys. Res. Commun. 381, 666–670 [DOI] [PubMed] [Google Scholar]
- 33. Gamerdinger M., Hajieva P., Kaya A. M., Wolfrum U., Hartl F. U., Behl C. (2009) Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 28, 889–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Komatsu M., Waguri S., Koike M., Sou Y. S., Ueno T., Hara T., Mizushima N., Iwata J., Ezaki J., Murata S., Hamazaki J., Nishito Y., Iemura S., Natsume T., Yanagawa T., et al. (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163 [DOI] [PubMed] [Google Scholar]
- 35. Koutroubakis I. E. (2010) Recent advances in the management of distal ulcerative colitis. World J. Gastrointest. Pharmacol. Ther. 1, 43–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Elinav E., Strowig T., Kau A. L., Henao-Mejia J., Thaiss C. A., Booth C. J., Peaper D. R., Bertin J., Eisenbarth S. C., Gordon J. I., Flavell R. A. (2011) NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Couturier-Maillard A., Secher T., Rehman A., Normand S., De Arcangelis A., Haesler R., Huot L., Grandjean T., Bressenot A., Delanoye-Crespin A., Gaillot O., Schreiber S., Lemoine Y., Ryffel B., Hot D., et al. (2013) NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J. Clin. Invest. 123, 700–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hooper L. V., Stappenbeck T. S., Hong C. V., Gordon J. I. (2003) Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nat. Immunol. 4, 269–273 [DOI] [PubMed] [Google Scholar]
- 39. Herbert D. R., Yang J. Q., Hogan S. P., Groschwitz K., Khodoun M., Munitz A., Orekov T., Perkins C., Wang Q., Brombacher F., Urban J. F., Jr., Rothenberg M. E., Finkelman F. D. (2009) Intestinal epithelial cell secretion of RELM-β protects against gastrointestinal worm infection. J. Exp. Med. 206, 2947–2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. He W., Wang M. L., Jiang H. Q., Steppan C. M., Shin M. E., Thurnheer M. C., Cebra J. J., Lazar M. A., Wu G. D. (2003) Bacterial colonization leads to the colonic secretion of RELMβ/FIZZ2, a novel goblet cell-specific protein. Gastroenterology 125, 1388–1397 [DOI] [PubMed] [Google Scholar]
- 41. Reikvam D. H., Erofeev A., Sandvik A., Grcic V., Jahnsen F. L., Gaustad P., McCoy K. D., Macpherson A. J., Meza-Zepeda L. A., Johansen F. E. (2011) Depletion of murine intestinal microbiota: effects on gut mucosa and epithelial gene expression. PLoS One 6, e17996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Forman R. A., deSchoolmeester M. L., Hurst R. J., Wright S. H., Pemberton A. D., Else K. J. (2012) The goblet cell is the cellular source of the anti-microbial angiogenin 4 in the large intestine post Trichuris muris infection. PLoS One 7, e42248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tsuji S., Yamashita M., Hoffman D. R., Nishiyama A., Shinohara T., Ohtsu T., Shibata Y. (2009) Capture of heat-killed Mycobacterium bovis bacillus Calmette-Guerin by intelectin-1 deposited on cell surfaces. Glycobiology 19, 518–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pemberton A. D., Knight P. A., Gamble J., Colledge W. H., Lee J. K., Pierce M., Miller H. R. (2004) Innate BALB/c enteric epithelial responses to Trichinella spiralis: inducible expression of a novel goblet cell lectin, intelectin-2, and its natural deletion in C57BL/10 mice. J. Immunol. 173, 1894–1901 [DOI] [PubMed] [Google Scholar]
- 45. Voehringer D., Stanley S. A., Cox J. S., Completo G. C., Lowary T. L., Locksley R. M. (2007) Nippostrongylus brasiliensis: identification of intelectin-1 and -2 as Stat6-dependent genes expressed in lung and intestine during infection. Exp. Parasitol. 116, 458–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Steenwinckel V., Louahed J., Lemaire M. M., Sommereyns C., Warnier G., McKenzie A., Brombacher F., Van Snick J., Renauld J. C. (2009) IL-9 promotes IL-13-dependent Paneth cell hyperplasia and up-regulation of innate immunity mediators in intestinal mucosa. J. Immunol. 182, 4737–4743 [DOI] [PubMed] [Google Scholar]
- 47. French A. T., Bethune J. A., Knight P. A., McNeilly T. N., Wattegedera S., Rhind S., Miller H. R., Pemberton A. D. (2007) The expression of intelectin in sheep goblet cells and upregulation by interleukin-4. Vet. Immunol. Immunopathol. 120, 41–46 [DOI] [PubMed] [Google Scholar]
- 48. Fort M. M., Cheung J., Yen D., Li J., Zurawski S. M., Lo S., Menon S., Clifford T., Hunte B., Lesley R., Muchamuel T., Hurst S. D., Zurawski G., Leach M. W., Gorman D. M., Rennick D. M. (2001) IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity 15, 985–995 [DOI] [PubMed] [Google Scholar]
- 49. Saenz S. A., Taylor B. C., Artis D. (2008) Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol. Rev. 226, 172–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ziegler S. F., Artis D. (2010) Sensing the outside world: TSLP regulates barrier immunity. Nat. Immunol. 11, 289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zaph C., Du Y., Saenz S. A., Nair M. G., Perrigoue J. G., Taylor B. C., Troy A. E., Kobuley D. E., Kastelein R. A., Cua D. J., Yu Y., Artis D. (2008) Commensal-dependent expression of IL-25 regulates the IL-23-IL-17 axis in the intestine. J. Exp. Med. 205, 2191–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wlodarska M., Thaiss C. A., Nowarski R., Henao-Mejia J., Zhang J. P., Brown E. M., Frankel G., Levy M., Katz M. N., Philbrick W. M., Elinav E., Finlay B. B., Flavell R. A. (2014) NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 156, 1045–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wittkopf N., Günther C., Martini E., Waldner M., Amann K. U., Neurath M. F., Becker C. (2012) Lack of intestinal epithelial atg7 affects Paneth cell granule formation but does not compromise immune homeostasis in the gut. Clin. Dev. Immunol. 2012, 278059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nishiumi S., Fujishima Y., Inoue J., Masuda A., Azuma T., Yoshida M. (2012) Autophagy in the intestinal epithelium is not involved in the pathogenesis of intestinal tumors. Biochem. Biophys. Res. Commun. 421, 768–772 [DOI] [PubMed] [Google Scholar]
- 55. Greenbloom S. L., Steinhart A. H., Greenberg G. R. (1998) Combination ciprofloxacin and metronidazole for active Crohn disease. Can. J. Gastroenterol. 12, 53–56 [DOI] [PubMed] [Google Scholar]
- 56. Sanders M. E. (2011) Impact of probiotics on colonizing microbiota of the gut. J. Clin. Gastroenterol. 45, S115–S119 [DOI] [PubMed] [Google Scholar]
- 57. Swidsinski A., Weber J., Loening-Baucke V., Hale L. P., Lochs H. (2005) Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J. Clin. Microbiol. 43, 3380–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zenewicz L. A., Yin X., Wang G., Elinav E., Hao L., Zhao L., Flavell R. A. (2013) IL-22 deficiency alters colonic microbiota to be transmissible and colitogenic. J. Immunol. 190, 5306–5312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fuhrer A., Sprenger N., Kurakevich E., Borsig L., Chassard C., Hennet T. (2010) Milk sialyllactose influences colitis in mice through selective intestinal bacterial colonization. J. Exp. Med. 207, 2843–2854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Garrett W. S., Lord G. M., Punit S., Lugo-Villarino G., Mazmanian S. K., Ito S., Glickman J. N., Glimcher L. H. (2007) Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131, 33–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Seedorf H., Griffin N. W., Ridaura V. K., Reyes A., Cheng J., Rey F. E., Smith M. I., Simon G. M., Scheffrahn R. H., Woebken D., Spormann A. M., Van Treuren W., Ursell L. K., Pirrung M., Robbins-Pianka A., et al. (2014) Bacteria from diverse habitats colonize and compete in the mouse gut. Cell 159, 253–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Barrett J. C., Hansoul S., Nicolae D. L., Cho J. H., Duerr R. H., Rioux J. D., Brant S. R., Silverberg M. S., Taylor K. D., Barmada M. M., Bitton A., Dassopoulos T., Datta L. W., Green T., Griffiths A. M., et al. (2008) Genome-wide association defines more than 30 distinct susceptibility loci for Crohn disease. Nat. Genet. 40, 955–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nair M. G., Guild K. J., Du Y., Zaph C., Yancopoulos G. D., Valenzuela D. M., Murphy A., Stevens S., Karow M., Artis D. (2008) Goblet cell-derived resistin-like molecule β augments CD4+ T cell production of IFN-γ and infection-induced intestinal inflammation. J. Immunol. 181, 4709–4715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. McVay L. D., Keilbaugh S. A., Wong T. M., Kierstein S., Shin M. E., Lehrke M., Lefterova M. I., Shifflett D. E., Barnes S. L., Cominelli F., Cohn S. M., Hecht G., Lazar M. A., Haczku A., Wu G. D. (2006) Absence of bacterially induced RELMβ reduces injury in the dextran sodium sulfate model of colitis. J. Clin. Invest. 116, 2914–2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hajishengallis G., Darveau R. P., Curtis M. A. (2012) The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 10, 717–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jung H. S., Chung K. W., Won Kim J., Kim J., Komatsu M., Tanaka K., Nguyen Y. H., Kang T. M., Yoon K. H., Kim J. W., Jeong Y. T., Han M. S., Lee M. K., Kim K. W., Shin J., Lee M. S. (2008) Loss of autophagy diminishes pancreatic β cell mass and function with resultant hyperglycemia. Cell Metab. 8, 318–324 [DOI] [PubMed] [Google Scholar]
- 67. Van der Sluis M., De Koning B. A., De Bruijn A. C., Velcich A., Meijerink J. P., Van Goudoever J. B., Büller H. A., Dekker J., Van Seuningen I., Renes I. B., Einerhand A. W. (2006) Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131, 117–129 [DOI] [PubMed] [Google Scholar]
- 68. Tobisawa Y., Imai Y., Fukuda M., Kawashima H. (2010) Sulfation of colonic mucins by N-acetylglucosamine 6-O-sulfotransferase-2 and its protective function in experimental colitis in mice. J. Biol. Chem. 285, 6750–6760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fu J., Wei B., Wen T., Johansson M. E., Liu X., Bradford E., Thomsson K. A., McGee S., Mansour L., Tong M., McDaniel J. M., Sferra T. J., Turner J. R., Chen H., Hansson G. C., et al. (2011) Loss of intestinal core 1-derived O-glycans causes spontaneous colitis in mice. J. Clin. Invest. 121, 1657–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. An G., Wei B., Xia B., McDaniel J. M., Ju T., Cummings R. D., Braun J., Xia L. (2007) Increased susceptibility to colitis and colorectal tumors in mice lacking core 3-derived O-glycans. J. Exp. Med. 204, 1417–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]