

Background: Cerebral dopamine neurotrophic factor (CDNF) is a promising therapeutic agent for treating Parkinson disease.
Results: We determined the solution structure of CDNF and demonstrated its neuroprotective effects against insults caused by α-synuclein oligomers.
Conclusion: We identified structural features of CDNF that might correspond with its physiological activity.
Significance: This work strengthens the therapeutic relevance of using CDNF to treat neurodegenerative diseases.
Keywords: α-synuclein (a-synuclein), neurodegeneration, neurotrophic factor, Parkinson disease, protein dynamic, protein structure
Abstract
Cerebral dopamine neurotrophic factor (CDNF) is a promising therapeutic agent for Parkinson disease. As such, there has been great interest in studying its mode of action, which remains unknown. The three-dimensional crystal structure of the N terminus (residues 9–107) of CDNF has been determined, but there have been no published structural studies on the full-length protein due to proteolysis of its C-terminal domain, which is considered intrinsically disordered. An improved purification protocol enabled us to obtain active full-length CDNF and to determine its three-dimensional structure in solution. CDNF contains two well folded domains (residues 10–100 and 111–157) that are linked by a loop of intermediate flexibility. We identified two surface patches on the N-terminal domain that were characterized by increased conformational dynamics that should allow them to embrace active sites. One of these patches is formed by residues Ser-33, Leu-34, Ala-66, Lys-68, Ile-69, Leu-70, Ser-71, and Glu-72. The other includes a flexibly disordered N-terminal tail (residues 1–9), followed by the N-terminal portion of α-helix 1 (residues Cys-11, Glu-12, Val-13, Lys-15, and Glu-16) and residue Glu-88. The surface of the C-terminal domain contains two conserved active sites, which have previously been identified in mesencephalic astrocyte-derived neurotrophic factor, a CDNF paralog, which corresponds to its intracellular mode of action. We also showed that CDNF was able to protect dopaminergic neurons against injury caused by α-synuclein oligomers. This advises its use against physiological damages caused by α-synuclein oligomers, as observed in Parkinson disease and several other neurodegenerative diseases.
Introduction
Parkinson disease (PD)3 is a degenerative disorder of the central nervous system that presents with characteristic motor symptoms that are a result of the death of dopamine-producing neurons in the substantia nigra (1). A hallmark of this disease is the occurrence of Lewy bodies, which are abnormal aggregates inside of neurons and are primarily composed of α-synuclein (2). The α-synuclein protein is soluble and is abundant within pre-synaptic terminals in the brain. It can also form oligomers, which are believed to be the toxic species that result in the development of PD (3).
Several compounds, including naturally occurring neurotrophic factors (NTFs), have been predicted to control the symptoms of and even lead to recovery in patients afflicted by PD (4).
Cerebral dopamine neurotrophic factor (CDNF) is considered one of the most potent NTFs. It can protect or repair rat dopaminergic neurons that have been exposed to 6-hydroxydopamine (6-OHDA) or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, both of which are synthetic drugs that induce Parkinsonism. Initially described in 2007, CDNF has been used in therapeutic trials conducted on animal models in which PD was induced by 6-OHDA or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (5–7). Moreover, the overexpression of CDNF in rat striatum that had been lesioned using 6-OHDA was found to reduce neuroinflammation and repair parkinsonian behavior (8–10).
In its mature form, human CDNF is a protein comprised of 163 amino acids and is a paralog of mesencephalic astrocyte-derived NTF (MANF) (61% amino acid sequence identity and 82% similarity) (11); there are also homologous genes in invertebrates (12). As with other NTFs, human CDNF can be secreted from transiently transfected neurosecretory cells (13). It has been demonstrated that CDNF is widely expressed by neurons in the brain cortex, cerebellum, hippocampus, midbrain, striatum, and substantia nigra, a distribution that corroborates the important neurotrophic activity of this protein (7).
The crystal structures of the N-terminal domain of CDNF (PDB code 2W50), encompassing residues 1–107, and of full-length MANF (PDB codes 2W51 and 2KVD) have revealed homologous N-terminal domains that are each composed of five α-helices and a 310 helix (14). The structure of the CDNF N-terminal domain is conserved among saposins, a class of proteins that interact with lipids and membranes. At the time of this writing, such activity has not been reported for CDNF or for MANF.
Although the structure of the C-terminal portion of CDNF has never been determined, MANF has been found to have a well folded C-terminal domain. The two domains of MANF, an N-terminal domain (residues 7–91) and a C-terminal domain (residues 112–147), appear to have distinct activities (14–16).
The C-terminal domain of MANF has three-dimensional structural homology to the SAP (SAF-A/B Acinus and PIAS) domain of Ku70 (15), an intracellular inhibitor of Bax (Bcl-2-associated X protein) (17). Accordingly, cytoplasmic injection of either full-length MANF or its C-terminal domain alone leads to an inhibition of Bax-dependent apoptosis in mouse superior cervical ganglion neurons, which suggests that this NTF possesses an intracellular mode of action (15). Indeed, exogenously applied CDNF is able to prevent apoptosis in PC12 cells by modulating Bcl-2/Bax and caspase-3 activation (18). MANF also contains a functional KDEL endoplasmic reticulum retention signal at its C terminus, which binds to KDEL receptors both intracellularly and on the cell membrane (19). The removal of this C-terminal tetrapeptide motif can increase the secretion of MANF, which contributes to the hypothesis that MANF has at least two distinct targets: an intracellular target that is associated with the endoplasmic reticulum and an extracellular target.
It is notable that although MANF possesses two domains that are distinct in their activities, the intact, full-length protein is biologically necessary. For example, only mature MANF (and none of its individual domains) has been shown to rescue larval lethality in Drosophila, as determined by a transgenic rescue approach (20). More striking is the fact that, in the same study, CDNF was not able to rescue the lethal phenotype, which suggests that these paralogs perform distinct actions. The synergic effect of MANF and CDNF intranigral overexpression in a rat model for PD supports this hypothesis (21).
It is still not clear why the crystallized structure of CDNF lacks a 6-kDa C-terminal domain, which was intriguingly cleaved either during or after purification even when the solution of the protein appeared to be pure (14, 22). It has been speculated that CDNF requires proteolytic cleavage of the linker region that connects its two domains to produce a mature protein that can function through these domains separately (14). Additionally, it has been suggested that the C-terminal domain of CDNF, which spans residues 108–161, is intrinsically unfolded (14).
In this paper, we addressed two eminent uncertainties regarding the structure and function of CDNF by addressing the following questions. 1) Does the three-dimensional structure of CDNF differ from that of MANF, specifically with respect to the structure of the C-terminal domain? 2) Can CDNF actively protect or rescue neurons against a physiological effector of neuronal injury that is associated with PD, namely, oligomers of α-synuclein?
To address the first question, we determined the NMR solution structure of recombinant, full-length CDNF (amino acids 1–161) in solution at pH 6. This task was made possible because of an improved purification protocol that prevents the proteolysis of CDNF. The three-dimensional structure of CDNF revealed a monomeric protein with two well folded domains: an N-terminal domain of ∼100 amino acids and a C-terminal domain of ∼51 amino acids. More strikingly, we found two dynamic surface patches on the N-terminal domain that could represent active sites. The structure of the C-terminal domain resembles the structure of MANF, and the intracellular effects of these paralogs, including those associated with organelle distribution and anti-apoptotic effects, are therefore most likely conserved (15). In answering the second question above, we demonstrated that recombinant CDNF protects and rescues both primary cultures of dopaminergic neurons, which were isolated from E14 mouse mesencephalon, and cultures of differentiated neuron 2a (N2a) cells from toxicity induced by α-synuclein oligomers.
Experimental Procedures
Protein Production
CDNF was produced as described by Latgé et al. (22), with some modifications. After CDNF was eluted from HiTrap SP, with a linear gradient of NaCl (0–1 m) at 67 mm per min, fractions containing the protein as checked by monitoring absorption at 280 nm and denaturing polyacrylamide gel electrophoresis were loaded onto a Zn2+ chelating Hitrap column (GE Healthcare) equilibrated with 20 mm MES, pH 6.5, and 100 mm NaCl. The unbound material was loaded onto a Superdex 75 column (16 × 100 mm, GE Healthcare) equilibrated with 20 mm sodium phosphate, pH 6, 100 mm NaCl, 3 mm NaN3, and 1 mm EDTA. A major well defined peak around the expected elution volume according to the calculated molecular mass of CDNF (∼18 kDa) was observed by monitoring absorption at 280 nm. The fractions of this main peak were further analyzed by denaturing polyacrylamide gel electrophoresis, MALDI-TOF, and the protective activity on mesencephalic primary culture, which confirmed the presence of pure active CDNF-(1–161). This sample was concentrated by ultrafiltration using Amicon ultracentrifuge device (Millipore) with cutoff of 10 kDa. The sample for structure determination contained 0.65 mm CDNF in 20 mm sodium phosphate, pH 6, 100 mm NaCl, 3 mm NaN3, 1 mm EDTA, and 10% (v/v) D2O. The oligomers of recombinant α-synuclein were produced as described previously (23).
NMR Experiments
NMR measurements were performed at 25 °C on Bruker Avance III spectrometer operating at 1H frequency of 1000 and 600 MHz using z axis gradient 5-mm triple resonance cryogenic probe and 800 MHz, using z axis gradient 5-mm triple resonance probe. 1H chemical shifts were referenced to internal 3-(trimethyl-silyl)-1-propanesulfonic acid, sodium salt. Using the absolute frequency ratios, the 13C and 15N chemical shifts were referenced indirectly to 3-(trimethyl-silyl)-1-propanesulfonic acid sodium salt. Two-dimensional [1H,15N]-HSQC, three-dimensional HNCO, three-dimensional HNCACB, and three-dimensional CBCA(CO)NH, three-dimensional HBHA(CO)NH, three-dimensional 15N-edited [1H,1H]-TOCSY and three-dimensional 15N-edited [1H,1H]-NOESY spectra (24) were used to obtain sequence-specific assignments for the polypeptide backbone. The sequence-specific assignment of amino acids without detectable HN signal was completed through sequential NOEs identified in the three-dimensional 13C-resolved [1H,1H]-NOESY spectrum optimized for aliphatic moieties. 1H and 13C assignments of all aliphatic side chains were obtained using three-dimensional HBHA(CO)NH, three-dimensional 15N-resolved [1H,1H]-TOCSY, three-dimensional HC(C)H-TOCSY, and three-dimensional HC(C)H-COSY spectra. The spin systems of the aromatic rings were identified using the three-dimensional 13C-resolved [1H,1H]-NOESY spectrum optimized for aromatic moieties. The resonances of the side chain ϵ-NH groups of the arginines, and the side chain amide groups of the asparagines and glutamines were identified in the two-dimensional [15N,1H]-HSQC spectrum, and sequence-specific assignments were then obtained using the three-dimensional 15N-resolved [1H,1H]-NOESY spectrum. The ϵ-CH3 groups of the methionines were initially identified in the two-dimensional [13C,1H]-HSQC spectrum, and sequence specifically assigned using the three-dimensional 13C-resolved [1H,1H]-NOESY spectrum optimized for aliphatic moieties. All backbone resonances were assigned except the 13CO chemical shifts of Asp-10, Cys-11, Glu-12, Val-13, Cys-14, Lys-15, Lys-68, Ile-69, Leu-70, Ser-71, Val-73, Thr-74, Arg-75, Met-81, Cys-87, Glu-99, Leu-100, Glu-131, Cys-132, Glu-137, Ala-149, His-156, Leu-161, the N-terminal amino group, 15N for all prolines, and HN of Cys-11, Glu-12, Cys-14, Lys-15, Glu-16, Ile-69, Leu-70, Ser-71, Glu-72, Thr-74, Arg-75, Lys-101, Cys-132, Arg-133, Ala-134, and Lys-138. Assignments were obtained for all CH moieties, except CHζ of Phe-17, CHα and CH2β of Cys-132, Cα and Cβ of Cys-135. The NHϵ resonances of arginines 20 and 28 and the chemical shifts of amide side chain groups of all asparagine and glutamine residues, except for Asn-143, were assigned. The imidazole ring of histidines, primary amine side chain groups for lysines and arginines, oxygen and sulfur atoms, and the hydrogens bound to these atoms were not assigned. For the NOESY peak picking and NOE assignment we used the software package UNIO version 10 (25). The input consisted of the chemical shift lists obtained from the resonance assignment, a three-dimensional 15N-resolved [1H,1H]-NOESY spectrum, and two three-dimensional 13C-resolved [1H,1H]-NOESY spectra optimized for the aliphatic and aromatic regions. The NOE data were measured at 800 MHz or 1 GHz with a mixing time of 80 ms. The three NOESY spectra were peak picked and assigned automatically with the standard ATNOS/CANDID/CYANA protocol including seven cycles of peak picking, NOE assignment, and three-dimensional structure calculation with simulated annealing in torsion angle space (26). The final target function was 2.6 Å2. The disulfide bridges were identified from the conformers generated using this calculation protocol and then the protocol was run with the disulfide pairs pre-formed.
Steady-state 15N{1H}-NOEs were measured at 1H frequency of 600 MHz using TROSY-based experiments (27, 28). The interscan delay was 5 s, including a saturation period of 3s. The R1 and R2 values were obtained from spectra measured at 1H frequency of 800 MHz, using the following recovery delays (in milliseconds): 0, 50, 100, 200, 300, 500, 750, 1000, and 1500 for R1 and 0, 16, 32, 48, 64, 80, 96, 112, 144, and 176 for R2 (29–31). The spectra with a delay of 500 ms for R1 and 80 ms for R2 were recorded three times and the peak intensities measured in these spectra were used to calculate the standard deviation of the experimental data. The peak intensities obtained from each spectra for each residue was fitted to a single exponential decay equation to extract the R1 and R2 data: I(t) = A × exp(−R1,2 × t), where I(t) is the intensity obtained from peak height measurements at a given recovery delay t (ms), and A is the intensity at t = 0. The NMR spectra were processed with Topspin 3 and analyzed with Cara 1.8.4.2.
Structure Calculation
The structure calculations were performed using the standard procedure recommended by the RECOORD database initiative (32). For the structure calculation we used the software package CNS_SOLVE version 1.3 (33). The input consisted of the NOE upper distance limits and disulfide bonds obtained by the UNIO automated analysis of the NOESY spectra and structure determination, backbone φ and ψ dihedral angle constraints derived from the Cα chemical shifts (34, 35), and an extended structure generated with the primary sequence of CDNF. The 20 conformers with the lowest total CNS energy, of 100 calculated, were energy-minimized in a water shell. The structure quality was assessed by the software package available at the AutoDep protein structure deposition interface (36).
Three-dimensional Structure Analysis
The figures with structures were prepared with Molmol (37). The backbone average local displacements were calculated with Molmol, using the overlap of three residues, centered on each amino acid within the range of 2–160. Exposure for the amino acid side chains were calculated for each monomer in crystallized proteins and for the conformer with the lowest total energy for the NMR solution structures, using standard setup in Molmol.
Small Angle X-ray Scattering Data Collection and Shape Restoration
SAXS data were acquired using the SAXS1 small-angle beam line of the National Synchrotron Light Laboratory, Campinas, Brazil, using a two-dimensional position-sensitive detector, with a mica sample holder and a wavelength of 0.155 nm at 20 °C. 5 frames of 5 s were taken for CDNF (2 mg/ml), however, because of x-ray effects, solely the first frame was considered for further analysis. The modulus of scattering vector q was calculated using the following equation: q = (4π/λ) sinθ, where λ is the wavelength used and 2θ is the scattering angle. Data were subtracted from the buffer contribution (20 mm sodium phosphate, pH 6, 100 mm NaCl, 3 mm NaN3, and 1 mm EDTA). The sample-detector distance was set at 941.76 mm, allowing detection of a q in a range from 0.107 to 4.07 nm−1.
Low resolution shape restoration was performed for CDNF with 10 independent calculation trials using the DAMMIN program (38) and averaged in DAMAVER (39). The precision of the averaged envelope was available from the normalized spatial discrepancy parameter (NSD for CDNF equal to 1.066 ± 0.354). Superposition between the high resolution three-dimensional structure obtained by NMR and the low resolution model obtained by SAXS was done using SUPCOMB (40).
α-Synuclein Oligomer Formation
Recombinant human α-synuclein (α-syn) wild type and its mutant A30P (41) were expressed and purified as previously described (42). Solutions of 140 μm A30P monomer in phosphate-buffered saline (PBS), pH 7.5, were filtered in a sterile 0.22-μm filter and incubated at 37 °C, with agitation at 800 rpm for 48 h, to allow oligomer formation.
Thioflavin T Binding Assays
For thioflavin T (ThT) binding assays, the samples were diluted to 2 μm in PBS at pH 7.5 containing 20 μm ThT and binding was monitored using a spectrofluorimeter (ISS Inc., Champaign, IL) to measure the fluorescence increase (excitation at 450 nm and fluorescence emission at 465–520 nm) (43).
Atomic Force Microscopy (AFM)
Topographic AFM images were obtained in air at room temperature using TappingMode® AFM with Dimension ICON. Images were acquired using oxide sharpened Si3N4 AFM tips (K∼3 N/m) (model OTESPA) at 0.5 Hz scan rates and scan resolution of 512 samples per line. Images were subjected to 2nd order polynomial flattening as needed to reduce the effects of image bowing and tilt. For imaging methods, 5 μl of 140 μm α-syn in PBS was applied to freshly cleaved mica and allowed to remain in contact for 5 min, followed by two 5-min washes with 100 μl of MilliQ water. The samples were then allowed to dry for 10 min.
Dot Blot Assay
Samples of α-syn (140 μm) before and at 48 h or 10 days after incubation (37 °C with agitation at 800 rpm) were spotted onto nitrocellulose membrane. The membrane was blocked using blocking solution (Odyssey) for 1 h. The membranes were incubated with anti-oligomer A11 (1:1000; AB9234, Millipore), anti-human α-syn (1:1000; AB5336P, Millipore), or anti-amyloid fibrils OC (1:1000; AB2286; Millipore) diluted in blocking solution for 2 h, washed 3 times with PBS, 0.1% Tween 20, and then incubated for 1 h with secondary antibody conjugated to IRDye 700 or 800 nm (1:5,000, 926-32210 or 925-68071, Odyssey). The images were visualized using an Odyssey Infrared Imaging System.
Dopaminergic Neurons Culture
Primary cultures of dopaminergic neurons were prepared from mesencephalon of 14-day-old Swiss Mice embryos (E14), as previously described (44). Pregnant Swiss mice were anesthetized and decapitated; the brain structures of the 14-day-old embryos were removed and dissociated cells were plated on coverslips treated with poly-l-ornithine (1.5 mg/ml; Sigma) in serum-free neurobasal medium. The cultures were incubated at 37 °C in a humidified 5% CO2 and 95% air chamber for 7 days. These neuronal cell cultures were assayed by immunocytochemistry with anti-TH (1:100; AB152, Millipore) antibody and DAPI (4′,6-diamidino-2-phenylindole) to identify tyrosine hydroxylase (TH) and nuclei, respectively. Then, the cultures were preincubated with CDNF or its buffer (vehicle) for 1 h, after which oligomers of α-synuclein were added to test the protective activity of CDNF. The cell viability assays were performed after 12 h. The manipulation of mice has been reviewed and approved by the Animal Use Ethics Committee of the Federal University of Rio de Janeiro (CAAE number 03102012.4.1001.5257).
Cell Viability Assays
MTT reduction assay for the monolayer culture was carried out using the previously described methods (45) using 10 μl of MTT solution/well. The plate was incubated at 37 °C in a 5% CO2 atmosphere for 2 h. The amount of formazan crystals formed was quantified using a microplate reader (SpectraMax® Paradigm® Multi-Mode reader, Molecular Devices). The measures were performed at 570 nm and subtracted by the measures at 630 nm.
Lactate dehydrogenase release assay was quantified in N2a cells using the Cytotox-ONE assay (Promega) according to manufacturer's protocol. LIVE/DEAD assay (Molecular Probes) was used according to the manufacturer's instructions. Cells were visualized using an EVOS microscope (Advanced Microscopy Group), and the percentage of live cells was calculated.
Differentiated Neuro 2a (N2a) Culture Cells
Mouse neuro 2a (N2a) cells (ATCC® CCL-131TM) were plated at a concentration of 12,000 cells/well in 24-well plates with coverslips for immunocytochemistry and 6,000 cells/well in 96-well plates for viability assay. Cells were maintained in DMEM high glucose supplemented with 4 mm l-glutamine (Sigma), 100 μg/ml of penicillin and streptomycin, and 10% FBS (Invivocell) at 37 °C in 5% CO2 for 24 h. For differentiation, medium was changed to DMEM + 0.5% FBS with 1 mm Br-cAMP (Sigma) for 3 days at 37 °C in 5% CO2. Differentiation was confirmed by immunocytochemistry by the presence of TH.
Western Blotting
The samples were heated at 95 °C for 5 min in the presence of Laemmli buffer (46). SDS-PAGE was performed under reducing conditions using 15% polyacrylamide gels. Samples were transferred to nitrocellulose membranes and probed with rabbit polyclonal anti-TH antibody (1:100; AB152, Millipore) and anti-β-actin (1:10,000; A2066, Millipore). Blots were then probed with secondary antibody conjugated to IRDye 700 or 800 nm (1:5,000; 926-32210 or 925-68071, Odyssey) and visualized using an Odyssey Infrared Imaging System.
Immunocytochemistry
N2a cells were fixed with 4% paraformaldehyde for 20 min at room temperature, permeabilized in Triton X-100 (1% in PBS) for 4 min at room temperature, and preincubated with blocking buffer OdysseyTM for 1 h. Cells were then incubated with rabbit polyclonal anti-TH (1:100; AB152, Millipore) and mouse anti-MAP2 (1:100; AB2290B, Millipore) in blocking buffer OdysseyTM + 0.1% Triton X-100. After washing with PBS, the cells were incubated with secondary antibodies conjugated with Alexa Fluor 488 (anti-rabbit 1:1000; A-11008, Molecular Probes) and Alexa Fluor 546 (anti-mouse 1:500; A-11003, Molecular Probes) overnight at 4 °C. The cells were then washed with PBS, stained with DAPI, washed three times with PBS, and mounted. The cells were imaged using an Avos (AMG) microscope with ×20 lens. The images were processed using ImageJ software.
Results
Three-dimensional Structure of Full-length CDNF (FL-CDNF) in Solution
The solution structure of recombinant FL-CDNF is presented (Fig. 1). To obtain the full-length protein, a new protocol for recombinant expression (in Escherichia coli) and purification of FL-CDNF was developed by our group, which included an additional step of Zn2+ affinity chromatography to eliminate any metalloprotease activity that might explain why CDNF undergoes a spontaneous cleavage near position 105, freeing the C terminus. In this purification step, CDNF was eluted in the non-retained fraction and was then stored in a buffer containing 1 mm EDTA. With these modifications to the purification protocol, CDNF demonstrated stability at room temperature (∼22 °C) for months without noticeable proteolysis (data not shown). This enabled us to solve the solution structure of the full-length protein and to test its neuroprotective activity on cell lines (described below).
FIGURE 1.
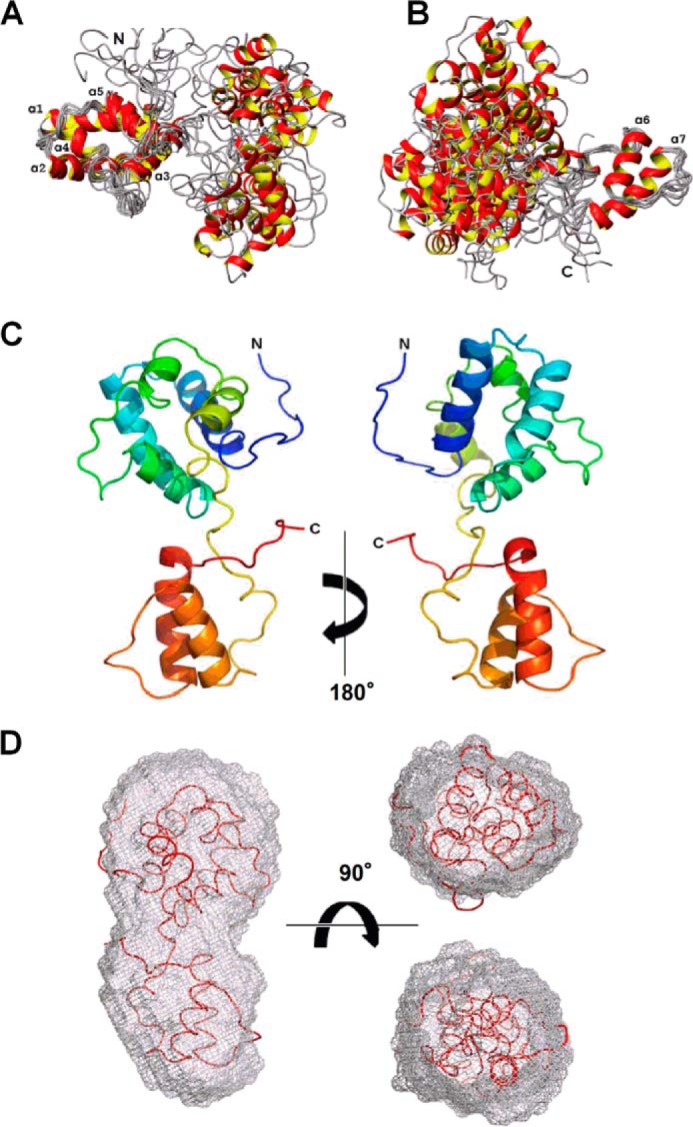
Solution structure of CDNF. Two-domain structure revealed by superposition of a bundle of 10 conformers that represent the NMR solution structure. In A, the superposition was performed on residues 10–100, which lie within the N-terminal domain, and in B, the superposition was performed on residues 110–150 of the C-terminal domain. Seven α-helices are indicated by labels. The representative conformer of the three-dimensional structure of full-length CDNF is shown in C. This is the conformer that possessed the lowest r.m.s. deviation relative to a bundle of 20 models. The color change from blue to red corresponds with a change in sequence from the N terminus to the C terminus. A fitting of the CDNF conformer shown in C to the molecular envelope determined by SAXS is shown in D.
The solution structure of FL-CDNF is represented in Fig. 1, A and B, in which we illustrate the superimposition of 10 conformers on the N-terminal domain and again on the C-terminal domain. It is evident that both domains have considerable freedom of orientation relative to each other. This finding is addressed in greater detail in the following section. In Fig. 1C, we depict the conformer with the lowest r.m.s. deviation of the bundle of 20 structures that were calculated for CDNF; the two domains of CDNF are readily identifiable. Using the SUPCOMB software package, we also constructed a molecular envelope based on SAXS data that were collected for the full-length protein (40). The high resolution solution structure that we obtained for FL-CDNF was able to be fit into this molecular envelope (Fig. 1D), thereby cross-validating the data obtained by these two distinct techniques and proving that CDNF is monomeric when in solution and contains two structured domains.
The three-dimensional structure of CDNF is defined by 1,902 NOEs and 159 dihedral angle constraints, and its high quality is indicated by the very few residual violations that are present, its low total energy, and Ramachandran plot statistics that indicated ∼77% of its residues to be within the most favorable regions and only 1.6% of its residues as having disallowed phi and psi angles (Table 1). Despite having superior structural statistics, the N-terminal domain was considered to have only a fair degree of definition, with an r.m.s. deviation of ∼1.1 Å for the backbone and of 1.6 Å for all heavy atoms. The C-terminal domain was better defined and had r.m.s. deviation values of ∼0.7 and 1.3 Å for backbone atoms and heavy atoms, respectively (Table 1).
TABLE 1.
Input for the structure calculation and characterization of the energy-minimized NMR structure of full-length CDNF
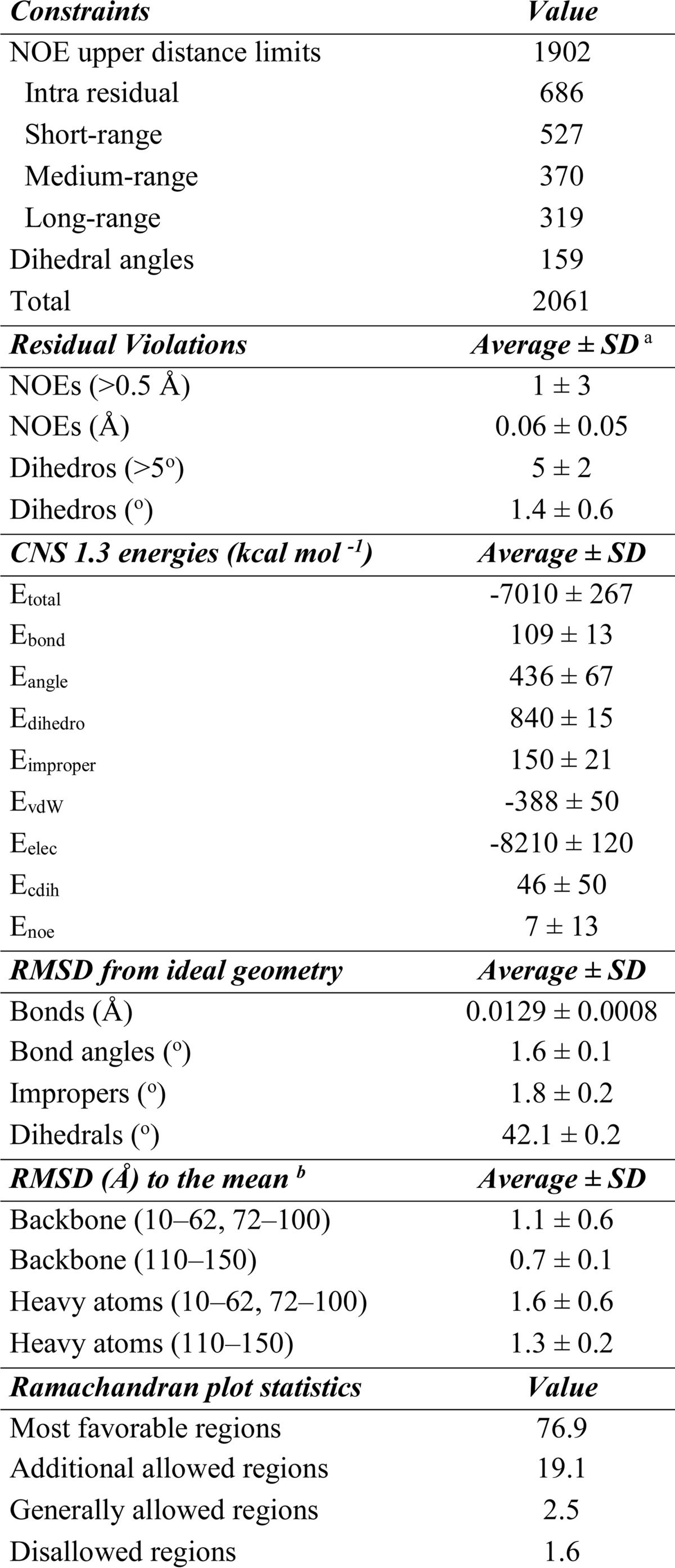
a The average values for the 20 energy-minimized conformers with the lowest residual CYANA target function values and the standard deviations among them are listed.
b The numbers in parentheses indicate the residues for which the r.m.s. deviation was calculated.
Two Dynamic Patches on the Surface of the N-terminal Domain of CDNF
Proteins have many dynamic regions, which are often represented by their solvent-exposed loops and termini. Many studies have shown a strong correlation between conformational entropy and protein activity (reviewed in Ref. 47). Dynamic structural regions primarily serve as interfaces of protein-protein interactions or catalytic sites. To a lesser extent, these regions can act as allosteric sites, or are at least adjacent to binding sites, which facilitate protein-protein interactions or catalysis by decreasing penalties to entropy loss during binding to a substrate or interaction partner.
To investigate the structural dynamics of CDNF, we collected NMR relaxation data (line widths, R1, R2, and {1H}-15N NOE) that enabled main chain dynamics to be identified in the range of picosecond to millisecond, and we compared this data with the average local displacements of the backbone, which describe the local fitting of each of the conformers in the bundle (Fig. 2).
FIGURE 2.

Dynamics of CDNF revealed by 1H line width, R2/R1, and 15N{1H}-NOE values, in addition to backbone average local displacements plotted against residue number. The data were collected at a 1H frequency of 600 MHz at 298 K, except for the 1H line width, which was collected at 1 GHz. Shaded areas identify residues where no Hn signal could be identified due to extensive line broadening. In A, helices are depicted as green bars. In B, blue and red solid lines identify average line width values for the indicated residues. In C and D, blue and red dashed lines identify the average value of R2/R1 and the 15N{1H}-NOE intensity, respectively, whereas dotted lines indicate standard deviations of the indicated residues. The dynamics data are compiled on the surface map shown in E and F, using a color scheme to indicate residues with low flexibility (no significant change of measured values: blue), intermediate flexibility (line width broadening: yellow), intermediate to fast flexibility (variation in R2/R1 but not in {1H}-15N NOE: orange), and fast to very fast flexibility (significant decrease of {1H}-15N NOE: red).
CDNF is composed of two semi-independently oriented domains and because of this it is impossible to obtain a fair fit among all residues of the 20 best structures that were obtained (backbone r.m.s. deviation = 7.16 ± 1.51 Å, see Fig. 1). As shown in Table 1, there was a higher degree of definition at residue positions 10–100 and 110–150 in the N-terminal and C-terminal domains, respectively (numbered according to the mature protein).
Four different lines of experimental evidence support a model of CDNF with two independent domains: 1) there were no NOEs present between the two domains (constraints were deposited in PDB code 4BIT); 2) there were no significant differences between the HN chemical shifts of residues 2–100 in the full-length protein that is presented here compared with those that have been previously obtained using a CDNF1–105 construct (average combined δΔ = 0.005 ± 0.005 ppm, maximum δΔ = 0.040 ppm; chemical shifts deposited in the BMRB with numbers 19164 and 18269) (22, 71); 3) as shown in Fig. 2B, the line widths of 1H that were observed in the two-dimensional [15N,1H]-HSQC were significantly different, averaging 30 and 25 Hz for the most structured regions (with an average local displacement of their backbone atoms below 0.1 Å; Fig. 2A) of the N-terminal (residues 17–24, 34–37, 39–46, 52–58, 74–78, identified by blue lines) and C-terminal domains (residues 113, 118–129, and 139–150, identified by red lines), respectively (p = 0.0001 from unpaired t test with Welch's correction); 4) the R2/R1 averages of the N- and C-terminal domains were significantly different (14.4 ± 5.7 and 10.7 ± 3.2 s−1, respectively, where the error represents standard deviation; p = 0.0013 from unpaired t test with Welch correction, Fig. 2C). The final two findings indicate that CDNF possesses a C-terminal domain with a slightly higher molecular tumbling rate than the N-terminal domain, which is compatible with the fact that the molecular weight of the C-terminal domain is ∼1.8 times less than the N-terminal domain. The significantly different tumbling rates of N- and C-terminal domains that were described for CDNF have been similarly indicated in a three-dimensional solution structure of MANF (16).
It is notable that the secondary structural elements of CDNF (α-helices 1–7, identified by green bars in Fig. 2A) are the most well defined regions of the protein, having the lowest average local displacements. The dynamics data are in close agreement with this evidence, as the residues that comprise these α-helices have an above average R2/R1 (Fig. 2C) and intense positive {1H}-15N NOEs (Fig. 2D). The 310 helix that spans residues 95–102 (green shaded box in Fig. 2A) has a higher degree of flexibility and is therefore underrepresented among the CDNF conformers (present in 8 out 20 conformers). We speculate that this is because the 310 helix is contained within the relatively flexible linker region (residues 99–113) that connects the two domains. The N-terminal regions of α-helices 1 (residues 13–16) and 4 (residues 70–73) are also less well defined, as shown in Fig. 2A.
We found that the majority of the regions that lacked backbone resolution (average local displacements above 0.1 Å) possessed clear indications of flexibility across diverse time scales. For example, residues 11–16, 68–73, 88, 99–101, 132–133, and 138 (identified by gray bars in Fig. 2, B–D) all had extreme broadening of their amide proton line widths, a phenomenon that typically accounts for chemical exchange processes that occur over a millisecond time scale. Furthermore, the poorly defined N-terminal residues 4–9 and C-terminal residues 158–161 (Fig. 2A) were all demonstrated to experience thermal fluctuations in their conformations (pico- to nanosecond time scale), as indicated by R2/R1 and {1H}-15N NOE values that were below the average by a difference that was larger than the standard deviation (Fig. 2, C and D). Values for residues 2 and 3 were not included due to signal overlap, whereas residue Gln-1 does not possess a backbone amide. The majority of the residues comprising the central linker between the two domains in CDNF have {1H}-15N NOE values that are lower than the overall average (0.70), indicating that this region of the polypeptide chain has a considerably dynamic nature (Fig. 2D, residues 103–113).
In addition to the above discussed dynamic regions, significant flexibility was also found with respect to individual residues of the polypeptide chain, as indicated by the conformational fluctuations of Ser-33, Leu-34, Thr-48, Leu-55, and Ala-66 that occurred over an order of pico- to nanoseconds. These fluctuations were identified by significantly decreased {1H}-15N NOEs and by motions occurring on the order of nano- to milliseconds in Leu-41, Cys-45, Leu-46, Gly-60, Asp-112, Leu-125, Tyr-141, Tyr-152, Ala-154, and Thr-155, all of which had significant variations in R2/R1 but not in {1H}-15N NOE.
These dynamics data are compiled on the surface map shown in Fig. 2, E and F, using a color scheme of blue, yellow, orange, and red to, respectively, indicate residues with low flexibility (no significant change of measured values), intermediate flexibility (line width broadening), intermediate to fast flexibility (variation in R2/R1 but not in {1H}-15N NOE), and fast to very fast flexibility (significant decrease of {1H}-15N NOE). Residues that possessed a solvent exposure that was lower than 10% across all conformers were not labeled in the surface plot (residues Cys-14, Leu-41, Cys-45, Thr-48, Leu-55, Val-73, Leu-125, and Tyr-141). It is notable that this surface plot represents a conformation in which linker residues 103–113 are partially shielded, although these residues were found to have a mean exposure to solvent ranging from 7.6 ± 2.6 to 46.9 ± 9.7%.
We found two major dynamic regions on the surface of the N-terminal domain of CDNF. One of them includes residues Ser-33, Leu-34, Gly-60, Ala-66, Lys-68, Ile-69, Leu-70, Ser-71, and Glu-72 (Fig. 2E, upper). On the opposite side of the structure, there is a patch of nearly sequential residues that span from the N terminus to residue Glu-16, with Glu-88 being the only non-sequential residue included (Fig. 2F, lower). We speculate that flexibility in this region is necessary for proteolytic processing of pre-CDNF, during which the 24-amino acid long signal peptide is predicted to be cleaved from the N terminus of the mature protein. Although the dynamic region of the C-terminal domain that comprises residues Ala-154, Thr-155, and Lys-158 through Leu-161 appears to be in close proximity to the dynamic N terminus, the distance and orientation between them is actually quite variable as a result of the above discussed freedom of orientation that exists between these domains. Several other residues that possess significant flexibility were found on the surface of CDNF, including a portion of the linker between the two domains (Glu-99, Leu-100, and Lys-101), residue Asp-112, and a small patch formed by residues Cys-132, Arg-133, and Lys-138. The above average levels of flexibility and solvent exposure in the linker region (residues 99–113) corroborate its susceptibility to proteolysis, which occurs more frequently on the C-terminal side of residue Thr-105 (22). The exact functions of the flexible regions that are located on the surface of CDNF have not yet been determined; however, based on the findings of several previous studies, we speculate that these dynamic patches might delimit binding or catalytic sites that undergo induced fit or conformational selection (47–49).
Key Structural Elements Identified by the Comparison of CDNF Solution Structure and Crystallographic Structure
We compared the three-dimensional structure described here with the three-dimensional structure that was solved by x-ray crystallography (Fig. 3). Parkash and co-workers (14) previously reported an x-ray structure corresponding to the N-terminal domain of human CDNF (residues 1–107; PDB code 2W50), which indicated the presence of five α-helixes (α1-α5) and a 310 helix.
FIGURE 3.

A, comparison of the three-dimensional structure of full-length CDNF that was solved by solution NMR (shown in blue) with the three-dimensional structure of the N-terminal-truncated CDNF that was solved by x-ray crystallography (shown in red). Labels identify the N and C termini as well as the helices. The global r.m.s. deviation between the backbones of these structures was 1.15 Å for the more ordered residues (10–62, 72–100, see Fig. 2), which is in the range of the value found for the NMR conformers (see Table 1). The average local displacements (B) were calculated for each three residues. The helices in the graph are depicted using the same color scheme that was adopted for the three-dimensional structures (A). The difference in average area of solvent exposure for each residue (C) was calculated using Molmol software (green points and line). The error bars represent the S.D. of solvent exposure among the bundle of 20 NMR structures that represent CDNF in solution. The dotted lines indicate the standard deviation calculated for all averages of residual solvent exposure. Residues with distinct levels of exposure between structures solved by NMR in solution and x-ray crystallography are labeled.
CDNF formed a dimer under the crystallization conditions used above, which included 100 mm sodium acetate at pH 4.6, 200 mm ammonium acetate, and 25–30% (w/v) MME-PEG 2000; however, in the conditions that were used in our experiments to determine its solution structure (see ”Experimental Procedures“), CDNF remained monomeric (illustrated in Fig. 1D and by gel filtration chromatography data, not shown). It has also been described that MANF, a CDNF paralog, exists as a monomer in a solution containing 10 mm BisTris, pH 6.8 and either 50 mm NaCl or 10 mm sodium phosphate at pH 6.0 (15, 16). MANF was also found to exist as a monomer in a crystal lattice that was prepared using 100 mm sodium cocadylate at pH 6.5, 200 mm magnesium acetate, and 12–18% (w/v) PEG 8000 (14). We speculate that these discrepancies in oligomeric states result from the different solution conditions used.
As can be observed in Fig. 3, structures solved by solution NMR (colored in blue) and x-ray crystallography (colored in red) contained similar secondary structural elements, and differences between the structures were primarily found in regions with high dynamic behavior, including residues 1–15 and 99–103, as described in the previous section (Fig. 2).
Of special attention is the region spanning residues 65 to 73, which encompasses the N-terminal region of α-helix 4 (Fig. 3, A and B) and has been noted as an important dynamic site in CDNF (Fig. 2). As discussed above, the amide proton resonances of residues 68–75 were absent from the NMR spectrum due to intermediate chemical exchange. Although we were able to assign the side chains of these eight amino acids, which include the hydrophobic residues Ile-69, Leu-70, and Val-73, they all had weak signals (not shown) and very few NOEs when compared with the rest of the sequence (constraints deposited in PDB code 4BIT). This set of experimental data provides the main evidence that, in solution, these hydrophobic side chains are not tightly packed into the hydrophobic core of CDNF and that they undergo conformational exchange on the order of milliseconds. We solved the three-dimensional structure of the N-terminal domain alone (residues 1–105) and found that its characteristics are similar to those of the N-terminal domain of FL-CDNF, primarily with respect to the absence of NMR signals corresponding to the N-terminal region of α-helix 4 (22).
We compared the average solvent exposure of each residue in the NMR and x-ray structures and identified 19 residues with differences in exposure that were larger than the standard deviation (Fig. 3C). In the NMR structure, the N-terminal region of α-helix 4 exhibited overexposures at residues Ala-66 (25.1 ± 9.0%) and, more prominently, Leu-70 (36.4 ± 10.0%) and the burial of residues Thr-67 and Lys-68 compared with the x-ray structure (Fig. 3C).
Because the segment of CDNF that comprises residues 65–73 is a dynamic region when in solution, we believe that this region varies in conformation from being partially folded with solvent-exposed hydrophobic residues (Ala-66 and Leu-70) to α-helical, as identified in the crystal structure. As discussed above, we believe this region is of particular interest, as dynamic regions of proteins are frequently involved in protein activity and frequently delimit active sites.
C Terminus of CDNF Possesses Active Sites That Are Conserved in MANF
As shown in Fig. 1B and Table 1, the structure of the C terminus is well structured and contains two α-helices (residues 118–127 and 138–152 for α-helices 6 and 7, respectively). This fold closely resembles a fold that is found in human MANF (r.m.s. deviation of 3.29 Å; PDB code 2KVD).
Henderson et al. (19) showed that the C terminus of MANF (-RTDL) is essential for its retention in the endoplasmic reticulum and for its ability to bind plasma membranes, which depends on KDEL receptors. CDNF has a highly homologous sequence (-KTEL), which is exposed to solvent and has high conformational flexibility (Fig. 2F). As such, it is reasonable to assume that the C-terminal domains of both MANF and CDNF possess active sites that are necessary for the regulation of their secretion from the endoplasmic reticulum.
Additionally, Hellman et al. (15) demonstrated that the C-terminal domain of MANF is structurally homologous to the SAP domains of diverse proteins, including the anti-apoptotic protein Ku70, which binds and inhibits the proapoptotic protein Bax through the motif 578VPMLKE583 (50). The surfaces of the C-terminal domains of both MANF and CDNF possess a homologous motif that encompasses residues 113VKELKK118 and 118VAELKQ123, respectively (Fig. 4, residues shown in red). Indeed, it has previously been demonstrated that the C-terminal domain of MANF has anti-apoptotic activity and provides an intracellular protection mechanism to superior cervical ganglion neurons (15).
FIGURE 4.
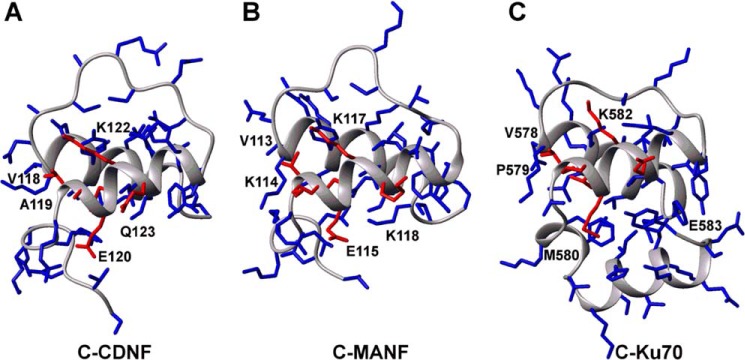
Comparison of three-dimensional structures of CDNF C-terminal domain (A) with its homologs MANF (B) and Ku70 (C). C-terminal domains are shown as backbone drawings; side chains are highlighted as sticks. Amino acids conserved in the active site of Ku70 are labeled and shown in red sticks. The structures were taken from the PDB codes 4BIT, 2KVD, and 1JJR.
Residues Val-118, Leu-121, and Lys-122 (numbering relative to CDNF) are strictly conserved between MANF, CDNF, and Ku70. Residue Leu-121 in CDNF, which corresponds to Leu-581 in Ku70 and Leu-116 in MANF, is the only residue that is predicted to have a prominent structural role in its active site, as it is completely buried in the hydrophobic core of the C-terminal domain. A structural characterization of the interaction between Ku70 and Bax is not available and therefore it is not possible to discern the importance of individual residues to the binding activity of this hexapeptide. Nevertheless, based on the high degree of structural similarity within this motif, we propose that CDNF possesses similar anti-apoptotic activity to what has been proposed for Ku70 and MANF.
CDNF Protects Dopaminergic Neurons against Toxic α-Synuclein Oligomers
In 2007, Lindholm et al. (7) described the neuroprotective activity of CDNF against the toxicity of 6-OHDA. Although this is a widely accepted model of Parkinsonism, no other injury model has been used to investigate the neuroprotective abilities of CDNF. In the present study, we evaluated the protective potential of CDNF against toxicity induced by α-synuclein oligomers on primary neuron cultures isolated from E14 mouse mesencephalon (Fig. 6) and on AMPc-butyryl-treated N2a cells (Fig. 7).
FIGURE 6.
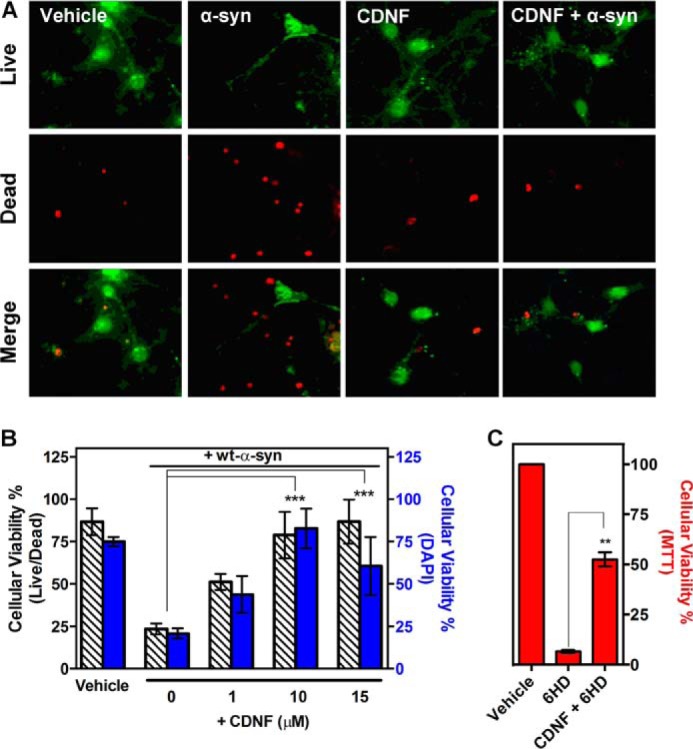
CDNF protects primary cultures of mesencephalic neurons against toxicity induced by α-synuclein oligomers. Primary cultures were maintained for 7 days and then pretreated with different concentrations of CDNF for 1 h before oligomers of α-synuclein are added. After 24 h of treatment, cell viability was assessed using a LIVE/DEAD assay in which live cells are visualized in green and dead cells in red. A, photomicrography of dopaminergic neurons treated with either vehicle (20 mm MES, pH 6.0, and 100 mm NaCl) or with 10 μm α-synuclein oligomers, in the absence or presence of 10 μm CDNF or with 10 μm CDNF alone. B, quantification of dead cells relative to the total number of cells in each well. Bars represent the mean with the S.D. for three independent experiments performed in triplicate. C, CDNF protects primary cultures of mesencephalic neurons against 6-OHDA. Primary cultures were maintained for 7 days and then pretreated with 10 μm CDNF 1 h before addition of 10 μm 6-OHDA to the culture. After 24 h of treatment, cell viability was assessed using an MTT assay. Statistical test used was one-way analysis of variance by Tukey's multiple comparison test, where **, p < 0.01 and ***, p < 0.001.
FIGURE 7.

CDNF protects dopaminergic neurons that were differentiated from N2a cells against toxicity induced by α-synuclein oligomers. Neuroblastoma cultures (N2a) were differentiated as described under “Experimental Procedures.” A, immunocytochemistry for DAPI, TH, and MAP2 of N2a cells in the absence and presence of 1 mm AMPc-butyryl. B, Western blot analysis of the indicated cell extracts using anti-TH and anti-β-actin antibodies. Bands corresponding to tyrosine hydroxylase (62 kDa) are visualized only in primary dopaminergic culture and N2a cells that were differentiated into dopaminergic neurons. β-Actin (42 kDa) was used as a control of protein load. C and D, differentiated N2a cells were maintained for 3 days in culture and then pretreated with 10 μm CDNF for 1 h before the addition of α-synuclein. After 24 h, cell viability was assessed using lactate dehydrogenase and MTT assays. The statistical test used was one-way analysis of variance by Tukey's multiple comparison test, where *, p < 0.05 and ***, p < 0.001.
α-Synuclein is abundant in human brain tissue and has been correlated with the pathogenesis of PD (51). Familial forms of PD have been found to be associated with variants of α-synuclein (52, 53), including an A30P mutant, which is the variant that was used in the present study. In vitro, α-synuclein primarily behaves as an intrinsically disordered protein and only forms amyloidogenic fibers under specific conditions (54). During α-synuclein aggregation, on-pathway oligomers are formed, which have been found to possess enhanced toxicity against cell lines in culture (3, 55).
In Fig. 5A, the aggregation kinetics of wild-type (WT) α-synuclein at pH 7.5 following ThT binding is presented. A subtle increase in ThT fluorescence after 50 h indicates the progressive appearance of amyloid fibrils; prior to this, oligomers are the prominent species, as observed by atomic force microscopy (inset, panel A) and dot blot analysis (Fig. 5B). In the beginning states of aggregation, only monomeric α-synuclein is present (left blot), whereas oligomers predominated the solution at 48 h (right blot), as revealed by A11 antibody staining. Oligomers grown for 48 h were used in further experiments.
FIGURE 5.
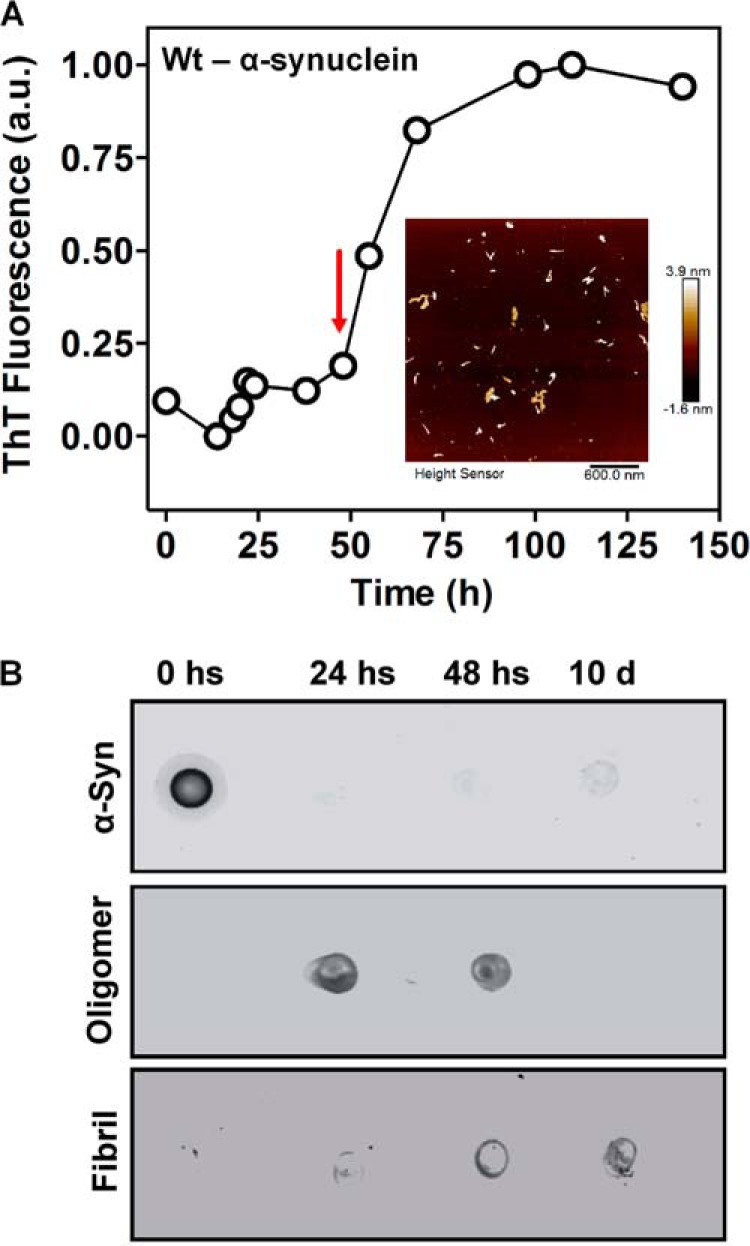
Characterization of α-synuclein aggregation. A, aliquots of α-synuclein monomers were taken following incubation at 37 °C and centrifugation at 800 rpm. They were diluted to 2 μm in PBS, pH 7.5, containing 20 μm ThT, to perform a fluorescence assay to detect amyloid aggregation. For this assay, the wavelengths that were used included the following: excitation = 450 nm, emission = 465–520 nm. The red arrow highlights a sample with pre-fibril oligomers that was used for activity assays in neurons. Inset: atomic force microscopy identifying α-synuclein oligomers at 48 h. α-Synuclein particles were electrostatically adsorbed to mica, as described under “Experimental Procedures.” A representative AFM image was acquired in peak force tapping mode. A topographical image of the oligomers shows oblate structures of different heights (color scale bar). B, a dot blot was used to identify α-synuclein oligomerization using an A11 antibody, a human α-synuclein antibody, or an anti-amyloid fibrils OC antibody.
Following the above, we challenged mesencephalon neuron cultures that were isolated from E14 mice with a 10 μm concentration of WT-α-synuclein oligomers, with or without preincubating the cells in 10 μm CDNF for 1 h (Fig. 6A). As observed in the results from LIVE/DEAD assays, CDNF protects these cells against insults induced by the addition of oligomers. Following these promising results, the concentration of CDNF was varied to produce a dose-response curve, the results of which are presented in panel B. Although the addition of a 10 μm concentration of oligomers resulted in only ∼25% cell viability, the addition of 1 μm CDNF increased cell viability to ∼50%, and the addition of 10 and 15 μm CDNF protected primary dopaminergic neurons from oligomer toxicity completely. Fig. 6C illustrates the results following the treatment of these neurons with 10 μm 6-OHDA, which resulted in death of the entire treated population. However, pre-treatment with 10 μm CDNF for 1 h considerably protected the cells against the toxic effects of 6-OHDA, as previously shown by Lindström et al. (20).
Finally, N2a cells were differentiated into dopaminergic neurons by the addition of AMPc-butyryl (Fig. 7, A and B). Following this treatment, cells became TH and MAP2 positive, which are markers of dopaminergic neuron (56) differentiation and neuritogenesis (57), respectively.
These dopaminergic cells (∼58% of the cells are TH positive) were incubated with oligomers of WT- and A30P-α-synuclein with or without pre-treatment with 10 mm CDNF for 1 h and cell viability was evaluated (Fig. 7, C and D). We found that CDNF almost completely protected the cells against oligomer-induced toxicity.
Discussion
In addition to the present work, four other studies have been performed to analyze the functions of different regions found within the three-dimensional structures of CDNF and its paralog MANF (14–20). In 2011 Hellman et al. (15) indicated that the C-terminal domain of MANF contains an active site similar to that of the anti-apoptotic protein Ku-70; this site in MANF was found to have anti-apoptotic activity when expressed within or injected into mouse superior cervical ganglion neurons. MANF also possesses an active endoplasmic reticulum retention signal at its C terminus, corroborating its anti-apoptotic activity (19).
The N-terminal domains of CDNF and MANF remain mysterious. They are structurally homologous to saposin-like proteins (SAPLIPS), a group of proteins with varied activities that include membrane binding, however, there is very little conservation of amino acids between these structural homologs (14–16, 58). No membrane binding activity has been detected for CDNF, and exogenous MANF does not bind to superior cervical ganglion neurons in vitro (15). Additionally, mutational analysis of basic residues on the surface of MANF, which are conserved on the surface of CDNF and might enable these proteins to interact with negatively charged phospholipids on cell membranes in a similar manner as SAPLIPS, revealed that they are not essential for its activity (20). This finding can be added to the recurrent cases of an ancient fold being used by proteins possessing very different activities.
Our NMR data on CDNF dynamics indicated two patches on the surface of CDNF that possessed dynamic properties equivalent to active sites, which may participate in either protein-protein interactions or catalysis (Fig. 2). The abundance of water-exposed hydrophobic residues (Leu-34, Leu-70, Ile-69, and Ala-66) in one of these sites indicates that it might serve as a protein-protein interaction interface (59–61). The N-terminal region of α-helix 4 (residues Ala-65 to Val-73), which is the central portion of this active site, was found to have intermediate dynamics and therefore might experience conformations ranging from the solvent exposure of residues Ala-66 and Leu-70 to a conformation that resembles or is identical to the crystallographic structure, which has a well defined helix with buried Ala-66 and Leu-70 side chains (Fig. 3).
The C-terminal domain of CDNF, which was absent in the constructs used for the crystallization, has been presumed to be unstructured and to therefore introduce a long, disordered segment into CDNF (14); however, in this study, we show that this domain is well folded and possesses high structural similarity to the MANF C-terminal domain, including to its two active sites (Fig. 4).
We demonstrate that CDNF prevents the toxic effects produced by α-synuclein oligomers from affecting dopaminergic neurons (Figs. 6 and 7). It has already been reported that CDNF protects dopaminergic neurons against lesions induced by 6-OHDA and that this protection is dose-dependent (7). We also observed that CDNF protects against 6-OHDA-induced injury in primary cell cultures prepared from mouse mesencephalon, which supports the relevance of using this model to assess the neurotrophic properties of this protein.
The toxicity of α-synuclein has primarily been related to its propensity to form prefibrillar oligomers. The lethal effect observed in our experimental setup is consistent with the most accepted model for the toxicity of α-synuclein, which is the gain of toxic function by the oligomers. It is worth noting that the neurotoxicity can be caused by exogenous α-synuclein that is taken up through endocytotic mechanism (62). Interestingly, the formation of toxic aggregates seems to be triggered by factors such as the dopamine (63). In fact, we tested α-synuclein against undifferentiated N2a cells and it did not exert any toxic effect (data not shown). The downstream mechanism of toxicity induced by the oligomers is still not well known, and may involve proteasomal dysfunction, down-regulation of mitochondrial complex I activity, Ca2+ influx across the plasma membrane, apoptosis, and necrosis (62). The cellular clearance of α-synuclein is performed by the ubiquitin-proteasome system and chaperone-mediated autophagy (64), and interventions that stimulate chaperone-mediated autophagy can prevent α-synucleinopathy (65–67). Furthermore, aberrant α-synuclein inhibits the chaperone-mediated autophagy, which adds to the multitude of proposed mechanisms for its toxicity (68).
Because CNDF contains putative protein-protein interaction sites, we first hypothesize that the protective effect of CDNF could arise by direct binding to α-synuclein, which could avoid the formation of toxic oligomers or block their uptake by neurons. Using steady-state tryptophan intrinsic fluorescence polarization (69) and far dot blotting (70) we did not find any evidence of interaction among CDNF and neither monomeric nor oligomeric α-synuclein (data not shown). Notwithstanding, other possible mechanisms of action for CDNF might include: 1) binding to a transmembrane receptor, like other neurotrophic factors, activating survival pathways that surpasses the toxic effect of α-synuclein; 2) stimulating the cellular clearance pathways such as chaperone-mediated autophagy; and 3) inhibiting apoptosis by interaction with BAX via its Ku70-like active site (15, 18). Because CDNF has two domains, it can use at least two mechanisms to protect dopaminergic neurons against toxic prefibrillar oligomers of α-synuclein.
CDNF was also found to be active in cultures of differentiated TH- and MAP2-positive N2a cells, which is a straightforward model for studying neuroprotective mechanisms. Moreover, this model may be useful in subsequent studies aimed at identifying the active site that resides within the N terminus of CDNF, as well as for the discovery of its receptor.
Author Contributions
D. F., F. L. P., and M. S. A. conceived and coordinated the study. D. F., D. P. R., F. L. P., K. M. S. C., and M. S. A. wrote the paper. C. L., M. S. A., and T. H. designed, performed, and analyzed the NMR data. C. L., D. P. R., J. A. F., K. M. S. C., and L. J. prepared the protein samples and provided technical assistance. G. A. P. O. designed, performed, and analyzed the SAXS data. D. P. R., J. A. F., and L. F. R. designed, performed, and analyzed experiments with cell cultures. D. R. P. and J. A. F. designed, performed, and analyzed the dot blot, Western blotting, ThT binding, and AFM. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
The 1 GHz NMR spectra were collected at the RALF-NMR large scale facility for high-field NMR in Lyon was supported by proposal European Union FP7 IRSES Grant 247546 (WW-NMR). SAXS data were collected at the Brazilian Synchrotron Light Laboratory (LNLS) under proposal SAXS1-14200. We thank Dr. Luciana Ferreira Romão for the primary cultures of dopaminergic neurons. The Centro Nacional de Ressonância Magnética Nuclear Jiri Jonas (CNRMN) is gratefully acknowledged for providing access to NMR instrumentation.
This work was supported by Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro and Conselho Nacional de Desenvolvimento Científico e Tecnológico Grants Universal 470349/2012-3, APQ1/E-26/111.771/2012 (to M. S. A.) and Universal 442991/2014-2, APQ1/E-26/111.415/2013 (to F. L. P.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 4BIT) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The 1H, 13C, and 15N chemical shifts of CDNF have been deposited in the BioMagResBank (71) under BMRB code 19164.
- PD
- Parkinson disease
- NTF
- neurotrophic factor
- CDNF
- cerebral dopamine neurotrophic factor
- 6-OHDA
- 6-hydroxydopamine
- MANF
- mesencephalic astrocyte-derived NTF
- HSQC
- heteronuclear single quantum coherence
- SAXS
- small angle x-ray scattering
- ThT
- thioflavin T
- AFM
- atomic force microscopy
- TH
- tyrosine hydroxylase
- PDB
- Protein Data Bank
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- r.m.s.
- root mean square.
References
- 1. Fahn S. (2003) Description of Parkinson's disease as a clinical syndrome. Ann. N.Y. Acad. Sci. 991, 1–14 [DOI] [PubMed] [Google Scholar]
- 2. Baba M., Nakajo S., Tu P. H., Tomita T., Nakaya K., Lee V. M., Trojanowski J. Q., Iwatsubo T. (1998) Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884 [PMC free article] [PubMed] [Google Scholar]
- 3. Winner B., Jappelli R., Maji S. K., Desplats P. A., Boyer L., Aigner S., Hetzer C., Loher T., Vilar M., Campioni S., Tzitzilonis C., Soragni A., Jessberger S., Mira H., Consiglio A., Pham E., Masliah E., Gage F. H., Riek R. (2011) In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. U.S.A. 108, 4194–4199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lindsay R. M., Altar C. A., Cedarbaum J. M., Hyman C., Wiegand S. J. (1993) The therapeutic potential of neurotrophic factors in the treatment of Parkinson's disease. Exp. Neurol. 124, 103–118 [DOI] [PubMed] [Google Scholar]
- 5. Airavaara M., Harvey B. K., Voutilainen M. H., Shen H., Chou J., Lindholm P., Lindahl M., Tuominen R. K., Saarma M., Hoffer B., Wang Y. (2012) CDNF protects the nigrostriatal dopamine system and promotes recovery after MPTP treatment in mice. Cell Transplant. 21, 1213–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Voutilainen M. H., Bäck S., Peränen J., Lindholm P., Raasmaja A., Männistö P. T., Saarma M., Tuominen R. K. (2011) Chronic infusion of CDNF prevents 6-OHDA-induced deficits in a rat model of Parkinson's disease. Exp. Neurol. 228, 99–108 [DOI] [PubMed] [Google Scholar]
- 7. Lindholm P., Voutilainen M. H., Laurén J., Peränen J., Leppänen V.-M., Andressoo J.-O., Lindahl M., Janhunen S., Kalkkinen N., Timmusk T., Tuominen R. K., Saarma M. (2007) Novel neurotrophic factor CDNF protects and rescues midbrain dopamine neurons in vivo 2. Nature 448, 73–77 [DOI] [PubMed] [Google Scholar]
- 8. Nadella R., Voutilainen M. H., Saarma M., Gonzalez-Barrios J. A., Leon-Chavez B. A., Jiménez J. M., Jiménez S. H., Escobedo L., Martinez-Fong D. (2014) Transient transfection of human CDNF gene reduces the 6-hydroxydopamine-induced neuroinflammation in the rat substantia nigra. J. Neuroinflammation 11, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bäck S., Peränen J., Galli E., Pulkkila P., Lonka-Nevalaita L., Tamminen T., Voutilainen M. H., Raasmaja A., Saarma M., Männistö P. T., Tuominen R. K. (2013) Gene therapy with AAV2-CDNF provides functional benefits in a rat model of Parkinson's disease. Brain Behav. 3, 75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ren X., Zhang T., Gong X., Hu G., Ding W., Wang X. (2013) AAV2-mediated striatum delivery of human CDNF prevents the deterioration of midbrain dopamine neurons in a 6-hydroxydopamine induced parkinsonian rat model. Exp. Neurol. 248, 148–156 [DOI] [PubMed] [Google Scholar]
- 11. Petrova P., Raibekas A., Pevsner J., Vigo N., Anafi M., Moore M. K., Peaire A. E., Shridhar V., Smith D. I., Kelly J., Durocher Y., Commissiong J. W. (2003) MANF: a new mesencephalic, astrocyte-derived neurotrophic factor with selectivity for dopaminergic neurons. J. Mol. Neurosci. 20, 173–188 [DOI] [PubMed] [Google Scholar]
- 12. Palgi M., Lindström R., Peränen J., Piepponen T. P., Saarma M., Heino T. I. (2009) Evidence that DmMANF is an invertebrate neurotrophic factor supporting dopaminergic neurons. Proc. Natl. Acad. Sci. U.S.A. 106, 2429–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sun Z.-P., Gong L., Huang S.-H., Geng Z., Cheng L., Chen Z.-Y. (2011) Intracellular trafficking and secretion of cerebral dopamine neurotrophic factor in neurosecretory cells. J. Neurochem. 117, 121–132 [DOI] [PubMed] [Google Scholar]
- 14. Parkash V., Lindholm P., Peränen J., Kalkkinen N., Oksanen E., Saarma M., Leppänen V. M., Goldman A. (2009) The structure of the conserved neurotrophic factors MANF and CDNF explains why they are bifunctional. Protein Eng. Des. Sel. 22, 233–241 [DOI] [PubMed] [Google Scholar]
- 15. Hellman M., Arumäe U., Yu L. Y., Lindholm P., Peränen J., Saarma M., Permi P. (2011) Mesencephalic astrocyte-derived neurotrophic factor (MANF) has a unique mechanism to rescue apoptotic neurons. J. Biol. Chem. 286, 2675–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoseki J., Sasakawa H., Yamaguchi Y., Maeda M., Kubota H., Kato K., Nagata K. (2010) Solution structure and dynamics of mouse ARMET. FEBS Lett. 584, 1536–1542 [DOI] [PubMed] [Google Scholar]
- 17. Cohen H. Y., Lavu S., Bitterman K. J., Hekking B., Imahiyerobo T. A., Miller C., Frye R., Ploegh H., Kessler B. M., Sinclair D. A. (2004) Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol. Cell 13, 627–638 [DOI] [PubMed] [Google Scholar]
- 18. Mei J.-M., Niu C.-S. (2014) Effects of CDNF on 6-OHDA-induced apoptosis in PC12 cells via modulation of Bcl-2/Bax and caspase-3 activation. Neurol. Sci. 35, 1275–1280 [DOI] [PubMed] [Google Scholar]
- 19. Henderson M. J., Richie C. T., Airavaara M., Wang Y., Harvey B. K. (2013) Mesencephalic astrocyte-derived neurotrophic factor (MANF) secretion and cell surface binding are modulated by KDEL receptors. J. Biol. Chem. 288, 4209–4225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lindström R., Lindholm P., Kallijärvi J., Yu L.-Y., Piepponen T. P., Arumäe U., Saarma M., Heino T. I. (2013) Characterization of the structural and functional determinants of MANF/CDNF in Drosophila in vivo model. PLoS ONE 8, e73928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cordero-Llana Ó., Houghton B. C., Rinaldi F., Taylor H., Yáñez-Muñoz R. J., Uney J. B., Wong L. F., Caldwell M. A. (2015) Enhanced efficacy of the CDNF/MANF family by combined intranigral overexpression in the 6-OHDA rat model of Parkinson's disease. Mol. Ther. 23, 244–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Latgé C., Cabral K. M., Almeida M. S., Foguel D. (2013) 1H-, 13C- and 15N-NMR assignment of the N-terminal domain of human cerebral dopamine neurotrophic factor (CDNF). Biomol. NMR Assign. 7, 101–103 [DOI] [PubMed] [Google Scholar]
- 23. Braga C. A., Follmer C., Palhano F. L., Khattar E., Freitas M. S., Romão L., Di Giovanni S., Lashuel H. A., Silva J. L., Foguel D. (2011) The anti-Parkinsonian drug selegiline delays the nucleation phase of α-synuclein aggregation leading to the formation of nontoxic species. J. Mol. Biol. 405, 254–273 [DOI] [PubMed] [Google Scholar]
- 24. Bax A. D., Grzesiek S. (1993) Methodological advances in protein NMR. Acc. Chem. Res. 26, 131–138 [Google Scholar]
- 25. Guerry P., Herrmann T. (2012) Comprehensive automation for NMR structure determination of proteins. Methods Mol. Biol. 831, 429–451 [DOI] [PubMed] [Google Scholar]
- 26. Serrano P., Pedrini B., Mohanty B., Geralt M., Herrmann T., Wüthrich K. (2012) The J-UNIO protocol for automated protein structure determination by NMR in solution. J. Biomol. NMR 53, 341–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu G., Xia Y., Nicholson L. K., Sze K. H. (2000) Protein dynamics measurements by TROSY-based NMR experiments. J. Magn. Reson. 143, 423–426 [DOI] [PubMed] [Google Scholar]
- 28. Renner C., Schleicher M., Moroder L., Holak T. A. (2002) Practical aspects of the 2D 15N-[1H]-NOE experiment. J. Biomol. NMR 23, 23–33 [DOI] [PubMed] [Google Scholar]
- 29. Meiboom S., Gill D. (1958) Modified spin-echo method for measuring nuclear relaxation times. Rev. Sci. Instrum. 29, 688–691 [Google Scholar]
- 30. Vold R. L., Waugh J. S., Klein M. P., Phelps D. E. (1968) Measurement of spin relaxation in complex systems. J. Chem. Phys. 48, 3831–3832 [Google Scholar]
- 31. Peng J. W., Wagner G. (1994) Investigation of protein motions via relaxation measurements. Methods Enzymol. 239, 563–596 [DOI] [PubMed] [Google Scholar]
- 32. Nederveen A. J., Doreleijers J. F., Vranken W., Miller Z., Spronk C. A., Nabuurs S. B., Güntert P., Livny M., Markley J. L., Nilges M., Ulrich E. L., Kaptein R., Bonvin A. M. (2005) RECOORD: a recalculated coordinate database of 500+ proteins from the PDB using restraints from the BioMagResBank. Proteins 59, 662–672 [DOI] [PubMed] [Google Scholar]
- 33. Brunger A. T. (2007) Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2, 2728–2733 [DOI] [PubMed] [Google Scholar]
- 34. Spera S., Bax A. (1991) Empirical correlation between protein backbone conformation and C.α and C.β 13C nuclear magnetic resonance chemical shifts. J. Am. Chem. Soc. 113, 5490–5492 [Google Scholar]
- 35. Luginbühl P., Szyperski T., Wüthrich K. (1995) Statistical basis for the use of 13Cα chemical shifts in protein structure determination. J. Magn. Reson. B 109, 229–233 [Google Scholar]
- 36. Lin D., Manning N. O., Jiang J., Abola E. E., Stampf D., Prilusky J., Sussman J. L. (2000) AutoDep: a web-based system for deposition and validation of macromolecular structural information. Acta Crystallogr. D Biol. Crystallogr. 56, 828–841 [DOI] [PubMed] [Google Scholar]
- 37. Koradi R., Billeter M., Wüthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55, 29–32 [DOI] [PubMed] [Google Scholar]
- 38. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Volkov V. V., Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Cryst. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kozin M. B., Svergun D. I. (2001) Automated matching of high- and low-resolution structural models. J. Appl. Cryst. 34, 33–41 [Google Scholar]
- 41. Krüger R., Kuhn W., Müller T., Woitalla D., Graeber M., Kösel S., Przuntek H., Epplen J. T., Schöls L., Riess O. (1998) Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat. Genet. 18, 106–108 [DOI] [PubMed] [Google Scholar]
- 42. Shaltiel-Karyo R., Frenkel-Pinter M., Rockenstein E., Patrick C., Levy-Sakin M., Schiller A., Egoz-Matia N., Masliah E., Segal D., Gazit E. (2013) A blood-brain barrier (BBB) disrupter is also a potent α-synuclein (α-syn) aggregation inhibitor: a novel dual mechanism of mannitol for the treatment of Parkinson disease (PD). J. Biol. Chem. 288, 17579–17588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. LeVine H., 3rd (1993) Thioflavine T interaction with synthetic Alzheimer's disease β-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 2, 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Romão L. F., Sousa Vde O., Neto V. M., Gomes F. C. (2008) Glutamate activates GFAP gene promoter from cultured astrocytes through TGF-β1 pathways. J. Neurochem. 106, 746–756 [DOI] [PubMed] [Google Scholar]
- 45. Mosmann T. (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63 [DOI] [PubMed] [Google Scholar]
- 46. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 47. Jarymowycz V. A., Stone M. J. (2006) Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem. Rev. 106, 1624–1671 [DOI] [PubMed] [Google Scholar]
- 48. Boehr D. D., Wright P. E. (2008) How do proteins interact? Science 320, 1429–1430 [DOI] [PubMed] [Google Scholar]
- 49. Csermely P., Palotai R., Nussinov R. (2010) Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem. Sci. 35, 539–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sawada M., Hayes P., Matsuyama S. (2003) Cytoprotective membrane-permeable peptides designed from the Bax-binding domain of Ku70. Nat. Cell. Biol. 5, 352–357 [DOI] [PubMed] [Google Scholar]
- 51. Iwai A., Masliah E., Yoshimoto M., Ge N., Flanagan L., de Silva H. A., Kittel A., Saitoh T. (1995) The precursor protein of non-Aβ component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron 14, 467–475 [DOI] [PubMed] [Google Scholar]
- 52. Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E. S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson W. G., Lazzarini A. M., Duvoisin R. C., Di Iorio G., Golbe L. I., Nussbaum R. L. (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047 [DOI] [PubMed] [Google Scholar]
- 53. Narhi L., Wood S. J., Steavenson S., Jiang Y., Wu G. M., Anafi D., Kaufman S. A., Martin F., Sitney K., Denis P., Louis J. C., Wypych J., Biere A. L., Citron M. (1999) Both familial Parkinson's disease mutations accelerate α-synuclein aggregation. J. Biol. Chem. 274, 9843–9846 [DOI] [PubMed] [Google Scholar]
- 54. van Rooijen B. D., van Leijenhorst-Groener K. A., Claessens M. M., Subramaniam V. (2009) Tryptophan fluorescence reveals structural features of α-synuclein oligomers. J. Mol. Biol. 394, 826–833 [DOI] [PubMed] [Google Scholar]
- 55. Wan O. W., Chung K. K. (2012) The role of alpha-synuclein oligomerization and aggregation in cellular and animal models of Parkinson's disease. PLoS ONE 7, e38545 10.1371/journal.pone.0038545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Smeyne R. J., Jackson-Lewis V. (2005) The MPTP model of Parkinson's disease. Mol. Brain Res. 134, 57–66 [DOI] [PubMed] [Google Scholar]
- 57. Fraichard A., Chassande O., Bilbaut G., Dehay C., Savatier P., Samarut J. (1995) In vitro differentiation of embryonic stem cells into glial cells and functional neurons. J. Cell Sci. 108, 3181–3188 [DOI] [PubMed] [Google Scholar]
- 58. Bruhn H. (2005) A short guided tour through functional and structural features of saposin-like proteins. Biochem. J. 389, 249–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chakrabarti P., Janin J. (2002) Dissecting protein-protein recognition sites. Proteins 47, 334–343 [DOI] [PubMed] [Google Scholar]
- 60. Bordner A. J., Abagyan R. (2005) Statistical analysis and prediction of protein-protein interfaces. Proteins 60, 353–366 [DOI] [PubMed] [Google Scholar]
- 61. De S., Krishnadev O., Srinivasan N., Rekha N. (2005) Interaction preferences across protein-protein interfaces of obligatory and non-obligatory components are different. BMC Struct. Biol. 5, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lashuel H. A., Overk C. R., Oueslati A., Masliah E. (2013) The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 14, 38–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Conway K. A., Rochet J. C., Bieganski R. M., Lansbury P. T., Jr. (2001) Kinetic stabilization of the α-synuclein protofibril by a dopamine-α-synuclein adduct. Science 294, 1346–1349 [DOI] [PubMed] [Google Scholar]
- 64. Webb J. L., Ravikumar B., Atkins J., Skepper J. N., Rubinsztein D. C. (2003) α-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278, 25009–25013 [DOI] [PubMed] [Google Scholar]
- 65. Spencer B., Potkar R., Trejo M., Rockenstein E., Patrick C., Gindi R., Adame A., Wyss-Coray T., Masliah E. (2009) Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of Parkinson's and Lewy body diseases. J. Neurosci. 29, 13578–13588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Decressac M., Mattsson B., Weikop P., Lundblad M., Jakobsson J., Björklund A. (2013) TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl. Acad. Sci. U.S.A. 110, E1817–E1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Xilouri M., Brekk O. R., Landeck N., Pitychoutis P. M., Papasilekas T., Papadopoulou-Daifoti Z., Kirik D., Stefanis L. (2013) Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain 136, 2130–2146 [DOI] [PubMed] [Google Scholar]
- 68. Xilouri M., Vogiatzi T., Vekrellis K., Park D., Stefanis L. (2009) Abberant α-Synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE 4, e5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jameson D. M., Croney J. C., Moens P. D. (2003) Fluorescence: basic concepts, practical aspects, and some anecdotes. Methods Enzymol. 360, 1–43 [DOI] [PubMed] [Google Scholar]
- 70. Chambraud B., Sardin E., Giustiniani J., Dounane O., Schumacher M., Goedert M., Baulieu E. E. (2010) A role for FKBP52 in Tau protein function. Proc. Natl. Acad. Sci. U.S.A. 107, 2658–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ulrich E. L., Akutsu H., Doreleijers J. F., Harano Y., Ioannidis Y. E., Lin J., Livny M., Mading S., Maziuk D., Miller Z., Nakatani E., Schulte C. F., Tolmie D. E., Kent Wenger R., Yao H., Markley J. L. (2008) BioMagResBank. Nucleic Acids Res. 36, D402–D408 [DOI] [PMC free article] [PubMed] [Google Scholar]