

Abstract
Holm oak is a dominant tree in the western Mediterranean region. Despite being well adapted to dry hot climate, drought is the main cause of mortality post-transplanting in reforestation programs. An active response to drought is critical for tree establishment and survival. Applying a gel-based proteomic approach, dynamic changes in root proteins of drought treated Quercus ilex subsp. Ballota [Desf.] Samp. seedlings were followed. Water stress was applied on 20 day-old holm oak plantlets by water limitation for a period of 10 and 20 days, each followed by 10 days of recovery. Stress was monitored by changes in water status, plant growth, and electrolyte leakage. Contrary to leaves, holm oak roots responded readily to water shortage at physiological level by growth inhibition, changes in water status and membrane stability. Root proteins were extracted using trichloroacetate/acetone/phenol protocol and separated by two-dimensional electrophoresis. Coomassie colloidal stained gel images were analyzed and spot intensity data subjected to multivariate statistical analysis. Selected consistent spots in three biological replicas, presenting significant changes under stress, were subjected to MALDI-TOF mass spectrometry (peptide mass fingerprinting and MS/MS). For protein identification, combined search was performed with MASCOT search engine over NCBInr Viridiplantae and Uniprot databases. Data are available via ProteomeXchange with identifier PXD002484. Identified proteins were classified into functional groups: metabolism, protein biosynthesis and proteolysis, defense against biotic stress, cellular protection against abiotic stress, intracellular transport. Several enzymes of the carbohydrate metabolism decreased in abundance in roots under drought stress while some related to ATP synthesis and secondary metabolism increased. Results point at active metabolic adjustment and mobilization of the defense system in roots to actively counteract drought stress.
Keywords: holm oak, roots, drought, recovery, proteomics
Introduction
Forest trees are of enormous ecological and economic value in global and local scale. They will be among the species most harmfully affected by the predicted climate changes with frequent temperature and precipitation extremes; however for a number of reasons our knowledge of the biochemical mechanisms to counteract inevitable environmental stresses is still very limited, especially concerning trees (Plomion et al., 2006; Abril et al., 2011). Quercus ilex is a dominant tree in western Mediterranean region, one of the main plant species of the so-called savannah-type woodland ecosystems (dehesas) which cover more than 4 million ha in western Mediterranean and northern African countries (David et al., 2007; Corcobado et al., 2013). Holm oak has double importance both from ecological and economic points of view, the latter coming from the fact that its acorns are major constituents in the diet of free range domestic animals; the rich nutrient composition and tannins of acorns give the original and specific taste of local meat products (David et al., 2007). According to climate model simulations, the Mediterranean region is expected to be a hot-spot which will be particularly affected by long term drought and warming episodes (Giorgi and Lionello, 2008).
Holm oak is very well adapted to dry hot climate, due to morphological particularities like a deep and well-structured root system with relatively large surface area and rapid development, which allows efficient capture of water from deepest soil layers, and small evergreen sclerophylous leaves with minimal transpiration and economical water use efficiency (David et al., 2007; Tsakaldimi et al., 2009). More biomass in this species is allocated in roots, forming larger below-ground starch and lipid reserves (Sanz-Pérez et al., 2009). Physiologically, holm oak is considered to be a typical water-spender species which maintains low leaf water potential. Stomatal closure upon water stress does not inhibit carbon assimilation in holm oak contrary to plants with water-saving strategy like pines (Baquedano and Castillo, 2006). It is predicted that the natural habitats of Quercus ilex will not be greatly affected or will even be expanded in the context of future climate changes (David et al., 2007; Bussotti et al., 2014). The overall good adaptation potential and huge economic importance of holm oak make it principal and valuable species for Mediterranean reforestation programs, which fostered research on Quercus ilex variability and stress response, especially at the proteome level (Jorge et al., 2006; Echevarría-Zomeño et al., 2009; Valero Galván et al., 2011, 2013). Main limitation in reforestation practices is the very low field survival rate of planted seedlings. Early seedling establishment is an extremely vulnerable phase of plant development and drought is considered to be the main cause of mortality post-transplanting (Navarro Cerrillo et al., 2005; Tsakaldimi et al., 2009). Besides, prolonged drought weakens tree defense systems against fungal pathogens. The forest decline is attributed to fungal attacks (e.g., Hypoxylon mediterraneum (De. Not.) Mill., Biscogniauxia mediterranea (De Not.) (Kuntze), or Phytophthora cinnamomi Rands.) after severe drought combined with high temperatures (Corcobado et al., 2013; Sghaier-Hammami et al., 2013). An active response to drought is critical for tree establishment and survival, however, our knowledge concerning the biochemical mechanisms of trees to counteract drought stress is still very limited, especially at the protein level (Abril et al., 2011).
Drought is one of the most deleterious abiotic stresses with respect to plant survival (Wang et al., 2003). Plants counteract drought using a combination of survival strategies like drought escape (through adjustment in the developmental program and ending reproductive cycle before severe drought development), drought avoidance (maintaining the internal water balance under drought conditions mainly through morphological and physiological adjustments), and drought tolerance (coping with water limitation mainly at cellular and biochemical level), which are subordinated to the natural climatic variations (Dolferus, 2014). In the genus Quercus for example, the rapid spring growth and allocation of high quantity of carbon reserves into roots could be regarded as elements of drought avoidance strategy—to develop robust root system and gain access to water in deeper soil layers (Sanz-Pérez et al., 2009). Survival under prolonged drought is more linked to tolerance strategies at the cellular level. Among them should be mentioned: synthesis of compatible solutes, enhanced protection against oxidative damage, protection of membranes, and proteins from denaturation through dehydrins and chaperones, degradation of unnecessary proteins and reusing the building blocks, and others (Wang et al., 2003; Dolferus, 2014). Roots are the plant part directly and primarily exposed to soil drought, so roots play a primordial role in water stress sensing and response. Besides the general anchoring and supportive function, root as a tissue provide water and mineral nutrients to all plant parts, and produce hormones for stress signaling (Ghosh and Xu, 2014). Root functions are indeed impeded upon severe water stress. Root tissue is very actively engaged in the adaptation to drought; for example water scarcity could stimulate root growth contrary to the aboveground plant part where growth is usually inhibited (Yamaguchi and Sharp, 2010). Contrasting metabolic changes have been reported in roots compared to shoots under drought stress—shoots have been metabolically inactivated and have had lower concentrations of sugars, amino acids, nucleosides, whereas roots have been metabolically activated, and more primary metabolites have been allocated to/synthesized in roots under water stress (Gargallo-Garriga et al., 2014). Increasing number of studies is accumulating now concerning root drought response at physiological, molecular biology, biochemical and proteomic levels (reviewed in Ghosh and Xu, 2014). As proteins are the main and direct executors of cellular functions, more pronounced impact of proteomic studies is recently observed in stress research on roots (Sengupta et al., 2011; Swigonska and Weidner, 2013; Oh and Komatsu, 2015). Proteomics along with metabolomics could be very useful tools in the so-called next-generation phenotyping methods of screening for stress tolerance (Dolferus, 2014). Proteomic studies on Quercus face the common problem with orphan species with non-sequenced genome which could be solved by homology-driven cross-species identifications based on high similarity to proteins from other plant species (Valero Galván et al., 2011). Preliminary studies of drought response in 1-year old plantlets of Quercus ilex using a proteomics approach have been performed in our group, analysing the changes in the leaf protein profile (Jorge et al., 2006; Echevarría-Zomeño et al., 2009).
The aim of the present study was to follow the dynamic changes in root proteins of drought treated Quercus ilex seedlings at very early developmental stage, applying a gel-based proteomic approach, and to relate proteome changes to stress severity and to recovery from stress, thus, to build a picture on the functional meaning of the observed protein changes.
Material and methods
Plant material and stress treatment
Holm oak (Quercus ilex subsp. Ballota [Desf.] Samp.) acorns were harvested on place in December 2011 from a region near Cerro Muriano (20 km apart from Cordoba). Mature fruits were dropped down directly from the trees, minimizing contact with soil. Acorns were immediately transported to the laboratory in tightly closed plastic bags, washed, surface-sterilized for 10 min in 10% sodium hypochlorite solution, extensively washed again, and inspected for worm damage. Healthy acorns were blotted dry, put in clean plastic bags and stored at 4–8°C. Spontaneously germinated acorns after 3 months of storage in cold were used in this study. Plants were grown in individual containers (330 ml volume) in perlite at growth chamber conditions: 12/12 h photoperiod, fixed light (360 μE.m−2.s−1), 24/20°C day/night temperature and 70% air humidity, at optimal water supply (120 ml tap water per 30 g dry perlite). Water stress was applied on 20 days-old holm oak seedlings with developed 9 ± 3 leaves on randomly selected sets of 12 plants, by carefully placing plants into new containers filled with perlite which was wetted with limited water quantity (40 ml water per 30 g dry perlite). Water limitation treatment was for a period of 10 and 20 days, followed by a 10 days recovery phase from stress by restoring the optimal water supply. Sufficient or limited water quantity was maintained by gravimetric measurements and daily compensation for the loss of water due to evapotranspiration. Sampling was performed at the appropriate time points—roots from drought treated plants for 10 (D10) and 20 (D20) days, and after 10 days recovery from 10 or 20-days water limitation period (R10, R20), with the respective age controls (C0—at the treatment beginning; C10—for D10, C20—for R10 and D20, and C30—for R20). Water status and electrolyte leakage were monitored on fresh plant material. For proteomic analyses, roots were quickly but thoroughly rinsed with distilled water, blotted dry, quick-frozen in liquid nitrogen, ground to fine powder and stored at −80°C.
Stress estimation parameters—plant growth, water status, electrolyte leakage
Stress was monitored by changes in water status, growth, and electrolyte leakage. These parameters were assessed as previously described (Simova-Stoilova et al., 2008). Time course changes were followed in the water content of roots and leaves along with observations for any visible changes in plants. Biomass reduction and recovery growth were registered gravimetrically. Root and shoot fresh weight was taken at sampling from all the sets of plants (n = 12). Water content was measured in 3 individual plants per treatment, calculated according to the formula (FW-DW)/FW where FW is fresh weight, DW—dry weight of the same sample by drying it at 70°C to constant weight for 48 h, and was expressed in percentage. Water deficit in roots and leaves was estimated as (TW-FW)/TW where TW is the turgid weight after floating the tissues for 4 h at room temperature in deionized water, and expressed in percentage. Electrolyte leakage was estimated by measuring the conductivity of the effusate solution from intact tissue, kept for 4 h at room temperature in deionized water, relative to conductivity of the effusate from the same tissue after boiling it for 10 min and cooling down.
Protein extraction and 2-DE separation
Root samples after 10 and 20 days of drought (D10 and D20) with the respective age controls (C10, C20) and 10-days recovery after 10 days of drought (R10) were analyzed by gel-based proteomics. Proteins were extracted from 3 biological replicas (each replica from 3 individual plants) according to the protocol of Wang et al. (2006) using TCA/acetone-phenol-methanol. Protein content in samples was estimated by the method of Bradford (1976) with bovine serum albumin as a standard. Samples were isoelectrofocused in the range of pI 5–8 using a Protean IEF Cell system (Bio-Rad, Hercules, CA, USA), 17 cm IPG strips, at 400 μg protein load per strip, active rehydration for 16 h at 50 V, rapid voltage ramp to 10,000 V, 50,000 Volt-hours in total, 500 V maintaining focused state. The second dimension was run at 12% SDS (PROTEAN® Plus Dodeca Cell, Bio-Rad, Hercules, CA, USA) and gels were double stained with colloidal Coomassie. Broad range molecular weight standards (Bio-Rad, Hercules, CA, USA) run by side in the same gel were used for estimation of MW.
Image analysis and selection of spots of interest
Images of the gels were captured with a GS-800 densitometer (Bio-Rad, Hercules, CA, USA) and analyzed applying PDQuest software (Bio-Rad, Hercules, CA, USA). Ten-fold over background criterion was used to assess presence/absence of a spot. Data on normalized spot volumes were exported and subjected to multivariate statistical analysis including sample clustering, ANOVA and Principal component analysis using a free online-based software (NIA arrays analysis tools, http://lgsun.grc.nia.nih.gov/ANOVA/index.html). Prior to statistical analysis the normalized values were log transformed to reduce dependence between abundance and standard deviation. Data were statistically analyzed by ANOVA using the following settings: error model max (average, actual), 0.01 proportions of highest variance values to be removed before variance averaging, 10° of freedom for the Bayesian error model, 0.05 FDR threshold, and zero permutations. The principal component analysis (PCA) settings were: covariance matrix type, 4 principal components, 1.5–fold change threshold for clusters, and 0.5 correlation threshold for clusters. PCA results were represented as a biplot, with consistent proteins in those experimental situations located in the same area of the graph. Selected variable spots of interest (90 in total), well defined and presenting statistically significant changes (drought and/or recovery compared to the respective age controls), with at least 1.5-fold difference in abundance ratio, were manually cut for subsequent MS analysis.
Mass spectrometry analysis, protein identification, and functional annotation
The MALDI-TOF/TOF Mass Spectrometry and Protein identification analysis was carried out in the UCO-SCAI proteomics facility, a member of Carlos III Networked Proteomics Platform, ProteoRed-ISCIII. The excised spots of interest were digested with porcine trypsin (sequencing grade) and loaded onto a MALDI plate, by using a ProPrep II station (Digilab Genomic Solutions Inc., Cambridgeshire, UK). The gel specimens were destained twice over 30 min at 37°C with 200 mM ammonium bicarbonate/40% acetonitrile. Gel pieces were then subjected to three consecutive dehydratation/rehydratation cycles with pure acetonitrile and 25 mM ammonium bicarbonate in 50% acetonitrile, respectively, and finally dehydrated for 5 min with pure acetonitrile and dried out over 4 h at room temperature. Then, 20 μl trypsin, at a concentration of 12.5 ng/μl in 25 mM ammonium bicarbonate was added to the dry gel pieces and the digestion proceeded at 37°C for 12 h. Peptides were extracted from gel plugs by adding 1 μl of 10% (v/v) trifluoracetic acid (TFA) and incubating for 15 min. Then, extracted peptides were desalted and concentrated by using μC-18 ZipTip columns (Millipore, Billerica, MA, USA) and were directly loaded onto the MALDI plate using α-cyano hydroxycinnamic acid as a matrix. Mass analysis of peptides (MS) of each sample was performed with a MALDI-TOF/TOF 4800 Proteomics Analyzer (Applied Biosystems, Foster City, CA, USA) mass spectrometer in the m/z range 800–4000, with an accelerating voltage of 20 kV. Spectra were internally calibrated with peptides from trypsin autolysis (M+H+ = 842.509, M+H+ = 2211.104). The most abundant peptide ions were then subjected to fragmentation analysis (MS/MS), providing information that can be used to determine the peptide sequence. Proteins were assigned identification by peptide mass fingerprinting and confirmed by MS/MS analysis. Mascot 2.0 search engine (Matrix Science Ltd., London, UK; http://www.matrixscience.com) was used for protein identification running on GPS ExplorerTM software v3.5 (Applied Biosystems, Foster City, CA, USA) over non-redundant NCBI protein and Uniprot databases. The following parameters were allowed: taxonomy restrictions to Viridiplantae, one missed cleavage, 100 ppm mass tolerance in MS and 0.5 Da for MS/MS data, cysteine carbamidomethylation as a fixed modification, and methionine oxidation as a variable modification. The confidence in the peptide mass fingerprinting matches (p < 0.05) was based on the MOWSE score, and confirmed by the accurate overlapping of the matched peptides with the major peaks of the mass spectrum. Proteins with statistically significant (p < 0.05) hits were positively assigned identification after considering Mr and pI values. Annotation of their biological function was consistent with Bevan et al. (1998). To predict the most probable intracellular localization of proteins, results from different software were compared—WolfPSORT (Horton et al., 2007, http://www.genscript.com/psort/wolf_psort.html), TargetP (Emanuelsson et al., 2000, http://www.cbs.dtu.dk/services/TargetP/), PredSL (http://aias.biol.uoa.gr/PredSL/input.html, Petsalaki et al., 2006), MultiLoc (http://abi.inf.uni-tuebingen.de/Services/MultiLoc/, Höglund et al., 2006). To construct a heat map the identified protein intensity, values were log10 transformed (base-10 logarithm of 1 + mean intensity values) and mean-centered to rescale them. Hierarchical clustering of samples and proteins were performed using MeV4.8 (http://www.tm4.org/mev.html) with agglomeration method set to average and the distances were calculated based on Pearson's correlation.
Data submission
Starting from each individual search (Mascot.dat file), the identified spots were translated to PRIDE XML using the PRIDE Converter 2.0 software (Côté et al., 2012). A total of 90 PRIDE XML files together with the corresponding raw MS files, mzXML peak lists and the Mascot search results (.dat file) were submitted to the ProteomeXchange repository (Vizcaíno et al., 2014) following the ProteomeXchange submission guidelines. Data are available with identifier PXD002484.
Results
Holm oak morphological and physiological response to drought
In this study water stress was imposed on 20 days-old holm oak plantlets with well-developed root system consisting of small white tip part and larger brown lignified part, and well developed 9 ± 3 leaves. At this time the tissue linking cotyledon and plantlet was practically desiccated, and even lost somewhere, thus, cotyledons were not considered as influencing the plantlet. Water limitation was maintained for a period of 10 and 20 days, each followed by 10 days recovery. Appropriate age controls were used for comparison of stress treatments and recovery—C20 was for R10 and D20, C30 was for R20. The experimental design is presented schematically in Supplementary Material Figure S1. Leaf number was not significantly increased during the whole experimental period both in control and in treated plants. Stressed plants had longer and thinner roots with diminished white root part, which was more evident after 20 days of treatment and was not restored in recovery from the longer stress. Photos of control, stressed and recovered plants are presented in Supplementary Material Figure S2. Stress development was monitored by changes in plant growth parameters (Figure 1, dark columns ‘part—root FW, white columns’ part—shoot FW), in water status [relative water content Figure 2—separate columns for roots (dark) and leaves (white columns), and water deficit—Table 1 left part], and in electrolyte leakage (Table 1 right part) as an indicator of membrane damage. Biomass was significantly reduced only after 20 days of stress, more in roots (by 37.5%) than in shoots (by 20.9%). Water content in control plants was in the limits: root white part—87–78% (slightly diminishing with plant age), leaves—38–45%. Drought stress induced change in relative water content only in roots (diminution by about 10–15%), not in leaves. In roots, this parameter did not decrease further with stress prolongation and was completely restored in recovery. Water deficit increased only in roots after drought treatment, along with increase in membrane instability. A 2-fold rising in relative electrolyte leakage (EL) was registered in roots under 10 days of drought stress. In leaves EL remained very low, possibly linked to establishment of xeromorphic leaf structure. Based on these data, an active strategy for metabolic adaptation to drought is expected to be found in root tissue at the proteome level.
Figure 1.
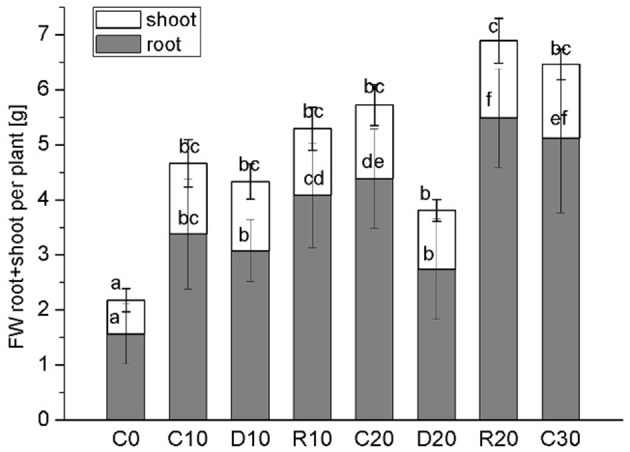
Plant growth parameters. Dark columns‘ part—root FW, white columns’ part—shoot FW. Mean values (n = 12) are given. Vertical bars represent standard deviations. C-control plants, C0, C10, C20, C30—the respective age controls (of days of treatment). D—plants subjected to water limitation treatment for 10 days (D10) or 20 days (D20). R—recovery by resuming optimal water supply for 10 days after 10 days (R10) or 20 days (R20) of water stress. Different letters above columns denote statistically significant differences.
Figure 2.
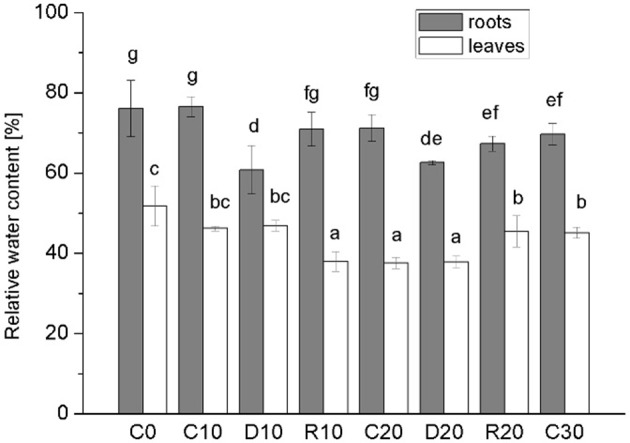
Relative water content. Separate columns for roots (dark) and leaves (white). Mean values (n = 3) are given. Vertical bars—standard deviations. C-control plants, C0, C10, C20, C30—the respective age controls (of days of treatment). D—plants subjected to water limitation treatment for 10 days (D10) or 20 days (D20). R—recovery by resuming optimal water supply for 10 days after 10 days (R10) or 20 days (R20) of water stress. Different letters above columns denote statistically significant differences.
Table 1.
Water deficit (left part), and electrolyte leakage (right part).
| Parameter | Water deficit % | Relative electrolyte leakage % | ||
|---|---|---|---|---|
| Tissue | Roots | Leaves | Root tips | Leaves |
| C0 | 10.1 ± 3.1 | 10.9 ± 1.3 | 16.4 ± 9.5 | 9.9 ± 2.9 |
| C10 | 8.6 ± 4.2 | 10.7 ± 1.1 | 20.9 ± 6.7 | 6.8 ± 1.7 |
| D10 | 29.3 ± 6.9 | 11.5 ± 1.7 | 41.9 ± 4.9 | 7.0 ± 0.9 |
| Remark | 3 times increase | n.s. change | 2 times increase | n.s. change |
Mean ± standard deviations from three independent replicates are shown; n.s.-non-significant changes. C0-control at the beginning of the treatment, D10—water limitation treatment for 10 days, C10—the respective age control.
Quercus ilex root proteome changes after drought and recovery
The protein extraction with modified TCA/acetone/phenol protocol resulted in protein yield of about 370–740 μg protein per g FW of root tissue (Table 2). Protein extraction was also made of samples recovered from D20, but in R20 the quantity of extracted protein dropped substantially. Five variants of samples were analyzed applying gel based proteomics—C10, D10, R10, C20, D20, in biological triplicates. Due to the insufficient protein yield, R20 was not studied. The 2-DE gel images of each of the variants (pI 5–8, and 12% SDS PAGE), 400 μg protein load, Coomassie colloidal staining) are shown in Figure S3, Supplementary. Images were analyzed with PDQuest software. Approximately 359 ± 9 consistent protein spots were clearly resolved on the gels. Concerning variability in abundance on the basis of spot volume ratio (treated to age control variants), relatively more spots were found to be decreased in abundance than increased, and more variability was found in recovery compared to drought treatment (Table 2). Sample clustering and PC analysis data (Figure 3) clearly separated the five sample variants – C10, D10, R10, C20, and D20.
Table 2.
Quercus ilex root proteome changes after drought treatment and recovery.
| Variants | Protein yield (μg.g1FW) | Total number of spots | Consistent spots | Number of Variable spots >2-fold/>1.5-fold change | Spots up under drought >2-fold change/>1.5-fold change | Spots down under drought >2-fold change/>1.5-fold change |
|---|---|---|---|---|---|---|
| C10 | 370 | 384 | 364 | |||
| D10 | 580 | 364 | 353 | 75/156 | 34/75 | 41/81 |
| R10 | 582 | 447 | 361 | 104/184 | 32/66 | 72/118 |
| C20 | 633 | 391 | 369 | |||
| D20 | 740 | 358 | 347 | 80/148 | 30/58 | 50/90 |
Variable spots (>2-fold/>1.5-fold change) are related to the respective age controls. C-control plants, C10, C20—the respective age controls (of days of treatment). D—plants subjected to water limitation treatment for 10 days (D10) or 20 days (D20). R–recovery by resuming optimal water supply for 10 days after 10 days of drought (R10), the age control in this case is C20.
Figure 3.

Sample clustering on the whole dataset of 2-DE spot intensities according to tissue and treatment, principal component analysis biplot (PC1/PC2) and tendencies of changes for the first four PCs. PC1 clearly separates variants depending on age while PC2 –on the treatment. The first four PCs contribute to 83.89% of the variation.
Identification of differently abundant spots and patterns of protein changes
Selected well defined spots, presenting significant changes under stress (at least 1.5-fold change compared to controls) and consistent in the three biological replicas, were subjected to MALDI-TOF PMF and MS/MS. For protein identification, combined search was performed with MASCOT search engine over NCBInr Viridiplantae and Uniprot databases. Detailed tables with identified protein species and their most probable subcellular location are presented in the Supplementary Material (Supplementary Tables S1, S2). There were five cases of low score unreliable identification and three cases of 2–3 different proteins identified in the same spot, which were excluded from these tables. In spite of the fact that Quercus ilex is an orphan species and identification was mainly based on homology, the majority of reliable hits for one given spot were for the same protein in different plant species. A few of the proteins were detected in more than one spot, like actin 2, enolase (EC 4.2.1.11), betaine aldehyde dehydrogenase (EC 1.2.1.8), cysteine synthase (EC 2.5.1.47), pyruvate decarboxylase (EC 4.1.1.1), but with some exceptions they presented similar trends of changes. Differences in protein abundance are clearly distinguished in the heat map built on the basis of normalized spot abundancy ratios of treatment to the respective age control (Figure 4). Different patterns of changes were found when looking at the identification results; however, the majority of spot abundance changes presented the same trend at drought stress irrespective treatment duration—D10 or D20. The following types of dynamic changes were observed—changes in abundance under stress unrelated to how long was the treatment (43 protein spots—24 up and 19 down), increase or decrease in abundance only at D10 (14 protein spots—8 up and 6 down in abundance), decrease only at D20 (2 protein spots), spot abundance changes in different directions comparing D10 and D20 (4 protein spots), prominent abundance changes in recovery, mainly diminution (8 spots), and changes beyond detection level, so-called qualitative changes (11 protein spots). Among the identified proteins with earlier and reversible increase in abundance under drought followed by recovery were found many enzymes related to secondary metabolism such as: caffeoyl CoA 3-O-methyl transferase (EC 2.1.1.104), chalcone synthase (2 spots, EC 2.3.1.74), shikimate dehydrogenase (EC 1.1.1.25), quinone oxidoreductase (EC 1.6.5.5), as well as ubiquitin activating enzyme E1 (EC 6.3.2.19), DEAD box RNA helicase (EC 3.6.4.13), betaine aldehyde dehydrogenase (EC 1.2.1.8). Down-accumulated in D10 were some inducible and hormone responsive proteins, glycyl-tRNA synthetase (EC 6.1.1.14), D-3-phosphoglycerate dehydrogenase (L-serine biosynthesis, (EC 1.1.1.95). There were not up-accumulated proteins typical only for prolonged drought treatment but 2 spots presented decrease in abundancy only at D20—methylmalonate-semialdehyde dehydrogenase (EC 1.2.1.27), and methylene tetrahydrofolate reductase (EC 1.5.1.20), a cytoplasmic enzyme member of one-carbon metabolism. A few proteins had opposite trends of changes comparing D10 and D20—peroxidase (EC 1.11.1.7)—D10 decrease, D20 increase, S-adenosylmethionine synthase (one-carbon metabolism, 2 spots, (EC 2.5.1.6)—D10 increase, D20 decrease, and the mitochondrial NADH-ubiquinone oxidoreductase (EC 1.6.5.3)—D10 increase, D20 decrease. The following proteins presented prominent changes in abundance after recovery, mainly diminution: Pyruvate dehydrogenase E1 (EC 1.2.4.1) subunit beta, component 3 of pyruvate dehydrogenase complex, enolase (EC 4.2.1.11), protein disulfide-isomerase (EC 5.3.4.1), STI like HSP (2 spots). The only protein spot with increase in recovery was proteasome subunit beta (EC 3.4.25.1). The most interesting qualitative differences were in one cysteine synthase isoform (EC 2.5.1.47), which spot was below detection under drought; however, there was another spot identified also as cysteine synthase with increased abundance under drought. Identified proteins were classified into functional groups according to Bevan et al. (1998): primary and secondary metabolism, protein biosynthesis and proteolysis, defense against biotic stress, cellular protection against abiotic stress, intracellular transport. In Table 3 are summarized the main functional groups and subgroups and the tendencies of changes in individual proteins within groups. It is seen that several enzymes of the carbohydrate metabolism were decreased in abundance in roots under drought stress while some related to ATP synthesis and secondary metabolism were increased. Results point at active metabolic adjustment and mobilization of the defense system in roots to actively counteract stress. A summary diagram of molecular mechanisms involved in Quercus ilex root response to water limitation is presented in Figure 5.
Figure 4.

Heat map built on the basis of normalized spot abundancy ratios—treatment to the respective age control. D—plants subjected to water limitation treatment for 10 days (D10) or 20 days (D20). R—recovery by resuming optimal water supply for 10 days after 10 days of drought (R10); C10, C20—control plants (the respective age controls of days of treatment).
Table 3.
Main functional groups and subgroups of identified proteins and the tendencies of changes in individual proteins within groups.
| Protein name/Species of closest homology | SSP | Accession No. | Mr/pI | Mascot scores | Spot volume ratio changes | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Experimental | Theoretical | Protein score/C.I | Total ion score/ C.I. | D10/C10 | R10/C20 | D20/C20 | C20/C10 | |||
| GLYCOLYSIS AND GLYOXYLATE BYPASS | ||||||||||
| Glucose-6-phosphate isomerase | ||||||||||
| Pop.trichocarpa | 1401 | tr|B9GV29|B9GV2 | 45.56/5.8 | 68.1/5.55 | 367/100 | 275/100 | 1.301 | 0.835 | 1.415* | 0.914 |
| Phosphofructokinase | ||||||||||
| Citrus paradisi | 4707 | tr|Q9ZST3|Q9ZST | 65.7/6.71 | 62.23/6.1 | 163/100 | 129/100 | 0.672* | 0.722 | 0.584* | 1.137 |
| Phosphoglycerate mutase | ||||||||||
| Ricinus communis | 1702 | sp|P35493|PMGI_ | 70.51/5.9 | 61.01/5.5 | 249/100 | 205/100 | 0.532* | 0.356* | 0.312* | 1.086 |
| 2701 | tr|B9S1V6|B9S1V | 69.86/6.1 | 306/100 | 255/100 | 0.549* | 0.813 | 0.464* | 0.967 | ||
| Enolase | ||||||||||
| Jatropha curcas | 1504 | tr|E6NU46|E6NU4 | 55.7/5.92 | 52.69/6.3 | 384/100 | 319/100 | 1.799 | 1.089 | 1.719* | 0.911 |
| Spinacia oleracea | 1507 | tr|Q9LEE0|Q9LEE | 55.25/6.0 | 48.37/5.5 | 187/100 | 134/100 | 1.121 | 0.207* | 0.851 | 1.463 |
| Alnus glutinosa | 3503 | sp|Q43321|ENO_A | 56.84/6.4 | 47.79/5.4 | 460/100 | 396/100 | 0.746 | 1.362 | 0.477* | 0.718 |
| Ricinus communis | 5602 | tr|B9R9N6|B9R9N | 58.52/6.8 | 48.15/5.7 | 461/100 | 400/100 | 0.243* | 0.409* | 0.181* | 1.099 |
| Glucose-1-phosphate adenylyl transferase | ||||||||||
| Glycine max | 1407 | tr|I1JJP2|I1JJP | 51.01/6.03 | 56.71/6.3 | 574/100 | 445/100 | 1.565 | 0.393* | 1.413 | 1.235 |
| 6-phospho-fructokinase 3 PFK3 | ||||||||||
| Arabidopsis thaliana | 7605 | sp|Q94AA4|K6PF3 | 61.41/7.4 | 54.09/6.61 | 317/100 | 268/100 | 0.609 | 1.174 | 0.578* | 0.964 |
| Phosphoglucomutase | ||||||||||
| Populus tremula | 3707 | sp|Q9ZSQ4|PGMC_ | 74.14/6.4 | 63.37/5.5 | 240/100 | 145/100 | R+ | |||
| Triosephosphate isomerase | ||||||||||
| Solanum tuberosum | 5106 | tr|Q3HRV9|Q3HRV | 20.82/6.8 | 27.92/5.9 | 378/100 | 321/100 | 0.564 | 0.379* | 0 | 2.005* |
| Malic enzyme | ||||||||||
| Oryza brachyantha | 7701 | tr|J3L3Y9|J3L3Y | 76.28/7.1 | 71.65/8.8 | 123/100 | 116/100 | 0 | 1.564 | 0 | 0.99 |
| Fructose-bisphosphate aldolase, cytoplasmic isozyme 2 | ||||||||||
| Pisum sativum | 7306 | sp|P46257|ALF2_ | 43.39/7.43 | 38.64/6.77 | 104/100 | 83/100 | R+ | |||
| Aconitate hydratase, cytoplasmic | ||||||||||
| Cucurbita maxima | 5806 | sp|P49608|ACOC_ | 98.54/6.9 | 98.57/5.7 | 166/100 | 118/100 | 0.441* | 0.948 | 0.305* | 1.222 |
| TRICARBOXYLIC ACID CYCLE | ||||||||||
| Isocitrate dehydrogenase | ||||||||||
| Ricinus communis | 2303 | tr|B9SRZ2|B9SRZ | 41.8/6.11 | 40.63/7.0 | 327/100 | 258/100 | 1.587* | 0.74 | 1.447 | 0.712 |
| Pyruvate dehydrogenase E1 beta | ||||||||||
| Pisum sativum | 1201 | sp|P52904|ODPB_ | 38.03/5.8 | 38.99/5.9 | 251/100 | 234/100 | 1.085 | 0.681 | 1.522* | 0.811 |
| Dihydrolipoyl lysine-residue acetyltransferase | ||||||||||
| Arabidopsis thaliana | 1502 | sp|Q5M729|OPD23 | 54.32/5.8 | 58.89/7.9 | 350/100 | 324/100 | 0.743 | 0.182* | 0.701 | 1.227 |
| AMINO ACID METABOLISM | ||||||||||
| Glutamine synthetase | ||||||||||
| Camellia sinensis | 3302 | tr|Q762D2|Q762D | 40.11/6.3 | 39.41/5.5 | 206/100 | 188/100 | 1.301 | 0.751 | 1.476* | 0.899 |
| Cysteine synthase | ||||||||||
| Ricinus communis | 2102 | tr|B9RET4|B9RET | 31.66/6.2 | 34.44/ 5.5 | 409/100 | 369/100 | 1.255 | 1.048 | 1.288 | 0.671* |
| Alanine aminotransferase | ||||||||||
| Oryza sativasubsp. japonica | 0606 | tr|Q7X7S9|Q7X7S | 56.56/5.5 | 53.77/5.2 | 227/100 | 199/100 | 0.687* | 0.332* | 0.489* | 0.885 |
| 3-phosphoglycerate dehydrogenase | ||||||||||
| Ricinus communis | 4702 | tr|B9RYA3|B9RYA | 64.92/6.5 | 63.35/7.6 | 210/100 | 162/100 | 0.511* | 1.053 | 0.94 | 1.031 |
| Glutamate dehydrogenase | ||||||||||
| Populus trichocarpa | 7402 | tr|B9IPQ2|B9IPQ | 45.2/7.15 | 44.72/6.84 | 338/100 | 281/100 | 1.729* | 1.555* | 1.308 | 1.587* |
| ONE CARBON METABOLISM | ||||||||||
| S-adenosylmethionine synthase | ||||||||||
| Camellia sinensis | 2402 | sp|Q9LDQ7|METK_ | 48.07/6.1 | 43.23/5.3 | 555/100 | 403/100 | 1.401 | 0.349 | 0.454* | 2.449 |
| Elaeagnus umbellata | 2405 | sp|Q9AT55|METK | 47.84/6.2 | 43.56/5.5 | 731/100 | 563/100 | 1.579* | 0.772 | 0.674 | 1.081 |
| Vit.b12 indep. methionine synthase | ||||||||||
| Pop. trichocarpa | 6801 | tr|B9HQI3|B9HQI | 87.0/6.97 | 85.03/6.2 | 566/100 | 499/100 | 2.555 | 2.484 | 1.998* | 0.889 |
| Formate dehydrogenase | ||||||||||
| Quercus robur | 7302 | tr|Q7XHJ0|Q7XHJ | 43.52/7.1 | 40.85/6.5 | 509/100 | 365/100 | 1.291 | 1.299 | 0.966 | 0.763* |
| 7408 | tr|Q7XHJ0|Q7XHJ | 44.88/7.4 | 489/100 | 368/100 | 1.587 | 2.649* | 1.361 | 0.531* | ||
| Methylene tetrahydro folate reductase | ||||||||||
| Arabidopsis thaliana | 4704 | sp|O80585|MTHR | 65.2/6.62 | 67.39/5.3 | 104/100 | 95/100 | 1.007 | 1.274 | 0.279* | 1.114 |
| AMINE BIOSYNTHESIS | ||||||||||
| Betaine aldehyde dehydrogenase | ||||||||||
| Amaranthus hypochondr. | 1603 | sp|O04895|BADH_ | 63.69/5.8 | 55.4/5.4 | 246/100 | 222/100 | 1.877* | 0.77 | 0.775 | 1.009 |
| Corylus heterophylla | 1605 | tr|E9NRZ9|E9NRZ | 63.72/5.9 | 55.2/5.44 | 152/100 | 127/100 | 0.677 | 0.36 | 0.221* | 1.404 |
| ETHANOL FERMENTATION | ||||||||||
| Pyruvate decarboxylase | ||||||||||
| Prunus armeniaca | 3703 | tr|B0ZS79|B0ZS7 | 69.93/6.3 | 66.2/5.74 | 244/100 | 234/100 | 0.345* | 1.54 | 0.818 | 0.451* |
| 3705 | 70.07/6.4 | 248/100 | 230/100 | 0.209* | 0.348* | 0 | 1.128 | |||
| SECONDARY METABOLISM | ||||||||||
| Chalcone synthase | ||||||||||
| Juglans nigra xJuglans regia Malus domestica | 6403 | tr|Q42864|Q4286 | 46.52/7.1 | 42.9/6.3 | 323/100 | 235/100 | 2.609* | 0.554* | 0.927 | 1.317 |
| 7401 | tr|B8R6A2|B8R6A | 46.7/7.12 | 42.79/5.7 | 494/100 | 396/100 | 2.472* | 0.709 | 0.963 | 1.252 | |
| (+)-Neomenthol dehydrogenase | ||||||||||
| Arabid.thaliana | 0104 | sp|Q9M2E2|SDR1_ | 25.81/5.5 | 33.07/5.4 | 83/99.97 | 68/99.997 | 1.342* | 0.407* | 1.556* | 1.067 |
| Caffeoyl CoA 3-O-methyltransferase | ||||||||||
| Betula platyphylla | 0105 | tr|Q5I2D1|Q5I2D | 23.07/5.5 | 27.95/5.4 | 433/100 | 325/100 | 1.718* | 0.603* | 1.051 | 1.151 |
| Flavanone 3-hydroxylase | ||||||||||
| Ampelopsis grossedentata | 3303 | tr|I6ZTY9|I6ZTY | 40.46/6.4 | 41.2/5.3 | 840/100 | 606/100 | 1.994* | 0.449* | 1.106 | 1.252 |
| Dihydroflavonol 4-reductase | ||||||||||
| P.suffruticosa | 6304 | tr|G4WCQ6|G4WCQ | 41.6/7.03 | 41.4/5.67 | 92/99.996 | 76/100 | 2.738* | 2.012* | 1.594* | 0.731 |
| Shikimate dehydrogenase | ||||||||||
| Populus trichocarpa | 6601 | tr|B9HSF2|B9HSF | 64.05/6.9 | 58.27/5.9 | 96/99.998 | 80/100 | 2.154* | 1.066 | 1.105 | 0.95 |
| UDP-glucosyl transferase | ||||||||||
| Ricinus communis | 4507 | tr|B9S1I8|B9S1I | 54.8/6.61 | 52.97/6.21 | 69/99.224 | 59/99.987 | 0 | 0.194* | 0.382* | 1.814* |
| ATP SYNTHESIS | ||||||||||
| ATP synthase subunit beta | ||||||||||
| Vitis vinifera | 1601 | tr|F6GTT2|F6GTT | 58.71/5.7 | 59.3/5.9 | 941/100 | 741/100 | 1.284 | 0.943 | 1.724* | 0.963 |
| NADH-ubiquinone oxidoreductase | ||||||||||
| Zea mays | 3806 | tr|B6U2J0|B6U2J | 84.5/6.42 | 81.67/6.1 | 235/100 | 212/100 | 1.605* | 0.388* | 0.806 | 0.926 |
| Gamma carbonic anhydrase | ||||||||||
| A. thaliana | 5107 | sp|Q9FWR5|GCA1_ | 23.46/6.91 | 30.12/7.13 | 115/100 | 94/100 | 2.505 | 2.939* | 1.991 | 0.712 |
| PROTEIN SYNTHESIS | ||||||||||
| DEAD box RNA helicase | ||||||||||
| Pisum sativum | 1509 | tr|Q8H1A5|Q8H1A | 50.52/5.5 | 47.14/5.39 | 310/100 | 202/100 | 1.784* | 0.495 | 1.14 | 1.509* |
| Glycyl-tRNA synthetase | ||||||||||
| Arabidopsis lyrata | 3805 | tr|D7KEM8|D7KEM | 78.97/6.5 | 82.49/6.43 | 443/100 | 395/100 | 0.411* | 0.558 | 0.878 | 0.466* |
| PROTEIN FOLDING/PROCESSING | ||||||||||
| Groes chaperonin | ||||||||||
| Ricinus communis | 3007 | tr|B9RR63|B9RR6 | 19.06/6.3 | 26.58/8.89 | 86/99.983 | 53/99.912 | 2.342* | 0.43* | 2.245* | 1.634 |
| Protein disulfide-isomerase | ||||||||||
| Nicotiana tabacum | 1202 | tr|P93358|P9335 | 38.81/5.8 | 40.08/5.99 | 201/100 | 180/100 | 0.884 | 0.409* | 1.2 | 0.834 |
| Chaperonin-60kD | ||||||||||
| Ricinus communis | 1701 | tr|B9S582|B9S58 | 66.69/5.7 | 61.48/6.2 | 393/100 | 332/100 | 0.646 | 0.606* | 0.638 | 0.987 |
| heat shock protein STI-like | ||||||||||
| Glycine max | 4809 | tr|I1LGM2|I1LGM | 77.99/6.6 | 65.86/6.26 | 67/98.652 | 54/99.95 | 0.632* | 0.485* | 0.991 | 0.795 |
| 4810 | tr|I1J9X8|I1J9X | 78.27/6.6 | 66.91/6.07 | 68/99.023 | 50/99.85 | 0.405* | 0.414* | 0.912 | 0.356* | |
| Chaperonin,TCP-1 | ||||||||||
| Malus domestica | 3604 | tr|D9ZJD1|D9ZJD | 63.5/6.33 | 57.4/5.6 | 292/100 | 223/100 | 0.398* | 0.379* | 1.254 | 1.04 |
| Ricinus communis | 4705 | tr|B9RSN1|B9RSN | 70.46/6.6 | 59.56/ 6.08 | 362/100 | 238/100 | 0.917 | 0.77 | 0.692 | 0.854 |
| Ricinus communis | 5701 | tr|B9SUJ3|B9SUJ | 69.96/6.8 | 60.64/5.97 | 157/100 | 135/100 | 0.161* | 0.509 | 0.462 | 1.138 |
| PROTEIN DEGRADATION | ||||||||||
| Proteasome subunit beta type | ||||||||||
| Glycine max | 2004 | tr|C6SVE5|C6SVE | 18.43/6.2 | 25.17/5.3 | 651/100 | 592/100 | 1.363 | 0.671 | 1.456* | 1.013 |
| Ricinus communis | 5004 | tr|B9SJ80|B9SJ8 | 20.5/6.92 | 30.6/7.6 | 58/90.457 | 47/99.745 | 1.58* | 1.167 | 1.216 | 1.133 |
| Picea sitchensis | 7002 | tr|A9NZ27|A9NZ2 | 19.97/7.4 | 29.59/5.58 | 224/100 | 203/100 | 1.281 | 1.723* | 0.903 | 0.951 |
| Xaa-Pro aminopeptidase | ||||||||||
| Solanum lycopersicum | 3803 | tr|Q93X46|Q93X4 | 79.52/6.4 | 73.45/5.98 | 88/99.99 | 81/100 | 1.58 | 1.776* | 1.341 | 0.923 |
| Mitochondrial processing peptidase | ||||||||||
| Cucumis melo | 3607 | tr|Q9AXQ2|Q9AXQ | 62.57/6.5 | 58.9/6.56 | 656/100 | 575/100 | 1.25 | 0.703* | 1.22* | 0.894 |
| Leucine aminopeptidase | ||||||||||
| Medicago truncatula | 2603 | tr|G7JGP9|G7JGP | 58.61/6.1 | 59.86/7.56 | 292/100 | 220/100 | 0.577 | 0.252* | 0.363* | 1.152 |
| 26S protease regulatory subunit 6B | ||||||||||
| M. truncatula | 1402 | tr|G7IV48|G7IV4 | 51.86/5.9 | 46.58/5.4 | 540/100 | 335/100 | 0.786 | 0.552* | 0.673* | 1.322* |
| Ubiquitin-activating enzyme E1 | ||||||||||
| Ric.communis | 1901 | tr|B9SKZ1|B9SKZ | 117.4/5.8 | 123.59/5.0 | 87/99.988 | 67/99.997 | 2.059* | 0.758* | 0 | 2.624* |
| STRESS RESPONSE | ||||||||||
| Aldo/keto reductase | ||||||||||
| Manihot esculenta | 0203 | tr|Q52QX9|Q52QX | 35.02/5.6 | 232/100 | 210/100 | 4.2* | 1.116 | 2.516* | 1.422 | |
| 3205 | tr|Q52QX9|Q52QX | 38.15/6.3 | 38.03/6.38 | 325/100 | 246/100 | 2.025* | 0.785 | 2.41* | 0.738 | |
| 4201 | tr|Q52QX9|Q52QX | 38.37/6.51 | 327/100 | 296/100 | 0.341* | 0.178* | 1.203 | 1.444 | ||
| Alcohol dehydrogenase | ||||||||||
| Pyrus communis | 6302 | tr|G0Z9K2|G0Z9K | 41.76/6.9 | 42.08/6.51 | 611/100 | 545/100 | 0.119* | 1.149 | 0.091 | 0.889 |
| Short-chain dehydrogenase / reductase (SDR) family protein | ||||||||||
| Solanum lycopers. | 0103 | tr|K4AZG5|K4AZG | 25.77/5.4 | 35.46/5.39 | 82/99.95 | 78/100 | 1.066 | 0.323* | 1.185 | 1.675* |
| Aldehyde dehydrogenase | ||||||||||
| Vitis vinifera | 6602 | tr|G9HQG0|G9HQG | 60.16/6.98 | 52.53/6.18 | 77/99.86 | 63/99.993 | 0.464* | 1.572* | 0.668 | 1.039 |
| Peroxidase | ||||||||||
| Linum usitatissimum | 0507 | tr|Q43782|Q4378 | 48.17/5.4 | 38.63/8.07 | 77/99.865 | 68/99.997 | 0.332* | 0.406* | 3.295* | 0.96 |
| Stem-specific protein TSJT1 | ||||||||||
| Ricinus Communis | 0005 | tr|B9RTW0|B9RTW | 20.5/5.54 | 25.5/5.56 | 77/99.87 | 70/99.998 | 0.189* | 0.451* | 0.906 | 1.298 |
| Hypersensitive induced response protein 3 | ||||||||||
| Triticum aestivum | 0108 | tr|B6D9L4|B6D9L | 24.68/5.67 | 31.8/5.68 | 231/100 | 202/100 | 0.395* | 1.238 | 2.499* | 0.949 |
| Al induced protein | ||||||||||
| Arabidopsis thaliana | 2105 | tr|Q9LIL3|Q9LIL | 22.29/5.96 | 27.5/5.84 | 94/99.997 | 85/100 | 0.179* | 0.837 | 0.755 | 0.305* |
| CELL MOTILITY | ||||||||||
| Actin 2 | ||||||||||
| Gossypium hirsutum | 0404 | tr|B8YPL4|B8YPL | 45.11/5.7 | 41.94/5.38 | 787/100 | 597/100 | 1.552 | 1.01 | 1.804* | 1.052 |
| Annona cherimola | 0703 | tr|H9A1W7|H9A1W | 65.5/5.51 | 41.85/5.31 | 788/100 | 562/100 | 0 | 0.411 | 0 | 2.661 |
| PHOTOSYNTHESIS | ||||||||||
| PsbP domain-containing protein | ||||||||||
| M. truncatula | 0113 | tr|G7J6G5|G7J6G | 26.71/5.3 | 27.95/6.92 | 62/95.93 | 55/99.96 | 0.514 | 0.056* | 0.889 | 3.077 |
| Oxygen-evolving enhancer protein | ||||||||||
| Populus euphratica | 0109 | sp|P84989|PSBO_ | 24.86/5.5 | 10.6/5.36 | 423/100 | 361/100 | 0 | 1.935 | 5.33* | 0.605 |
| Rubisco LS | ||||||||||
| Noronhia emarginata | 3706 | tr|Q06QN8|Q06QN | 66.65/6.4 | 53.05/6.22 | 197/100 | 101/100 | 0.354* | 0.727 | 0 | 0.825 |
Spot volume ratio changes of variants: D—plants subjected to water limitation treatment for 10 days (D10) or 20 days (D20). R—recovery for 10 days after 10 days of drought (R10); C10, C20—control plants (the respective age controls of days of treatment). Statistically significant differences between control and treated variants or between age controls (p < 0.05) are indicated by asterix.
Figure 5.
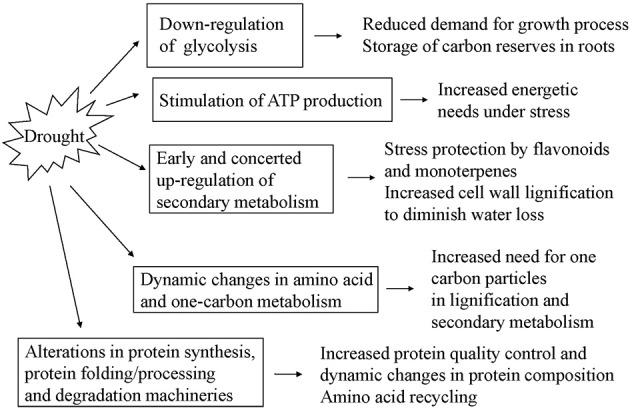
Summary diagram of molecular mechanisms involved in Quercus ilex root response to water limitation.
Discussion
Physiological changes in Quercus ilex roots under water limitation
Holm oak is regarded as relatively drought tolerant plant species, well adapted to Mediterranean type of climate. Its tolerance is due to the large well developed root system, the evergreen sclerophylous leaf anatomy, the very economic use of water resources, as well as some particularities in photosynthesis (David et al., 2007; Tsakaldimi et al., 2009). However, seedling establishment is one of the most stress vulnerable phases in plant development (Tsakaldimi et al., 2009). In our experimental system, much more plant biomass is allocated in roots than in shoots (2.5 to 3-fold more) at early seedling stage. Significant growth inhibition was detected after relatively long period of water limitation (20 days), both for roots and for shoots, which confirms the expected drought resilience of this tree species. Some reports emphasize the active root growth vs. shoot growth inhibition as an adaptive strategy for drought adaptation, which is reflected in changes in root to shoot biomass ratio (Yamaguchi and Sharp, 2010), while in other cases root growth is inhibited in response to progressive water stress (Sengupta et al., 2011), probably linked to stress duration and species peculiarities in stress tolerance. In the case of Quercus ilex seedlings, however, the total root biomass was more negatively affected by prolonged drought than the shoot biomass was, resulting in diminution in root to shoot ratio compared to the respective age controls. In the time course of the experiment, the root to shoot ratio in the age controls was constantly increasing, which supports the more active root growth compared to shoots. Besides root biomass diminution, prolonged drought leaded to changes in the root aspect—longer and thinner roots with less white tip part at D20, which may be linked to increased lignification as a response to water deficit. Increased degree of lignification in the basal part of root elongation zone has been reported in response to drought for other plant species (Yamaguchi et al., 2010). The difference in leaf and root response to water shortage was further supported by the observed changes in water status and membrane stability after drought stress, expressed mainly for the root system. Pronounced change in water status of roots subjected to drought stress, compared to leaves, is also documented for other plant species (Yoshimura et al., 2008; Wendelboe-Nelson and Morris, 2012).
Secondary metabolism is activated in roots under drought treatment
Drought adaptation of plants requires complex rearrangements of the metabolism with interactions between several metabolic pathways. One of the most striking observations in our comparative study was the relatively early increase of several cytoplasmic enzymes engaged in secondary metabolism in roots under water stress, like: shikimate dehydrogenase (EC 1.1.1.25), naringenin-chalcone synthase (EC 2.3.1.74), flavanone 3-hydroxylase (EC 1.14.11.9), dihydroflavonol 4-reductase (EC 1.1.1.219), (+)-neomenthol dehydrogenase (EC 1.1.1.208), and caffeoyl CoA 3-O-methyltransferase (EC 2.1.1.104). Shikimate dehydrogenase, an enzyme of the shikimic acid pathway leading to biosynthesis of aromatic amino acids and simple phenolics, catalyzes the reversible NADP+-dependent reaction of 3-dehydroshikimate to shikimate. Chalcone synthase (or naringenin-chalcone synthase) is a plant enzyme in the initial step and central hub for the pathway of flavonoid biosynthesis, leading to production of flavanoids, isoflavonoid-type phytoalexins, and other metabolites with stress protective functions for plants. Other enzymes of the flavonoid biosynthesis pathway which are found to be up-accumulated in concert with chalcone synthase under drought were: flavanone 3-hydroxylase which catalyzes the stereospecific conversion of flavanones to dihydroflavonols, and dihydroflavonol reductase, which catalyzes the reduction of dihydroflavonols to leucoanthocyanins (Dao et al., 2011). Chalcone synthase is induced under different abiotic and biotic stresses like UV, wounding, herbivory, and microbial pathogens, resulting in the production of compounds with antimicrobial, insecticidial, and antioxidant activity (Selmar and Kleinwächter, 2013). Flavonoids interfere with hormone signaling by inhibiting polar auxin transport (Dao et al., 2011). Increasing evidence suggests that plants exposed to drought accumulate secondary metabolites, and a plausible explanation could be to protect cells from oxidative stress by consuming NADPH+ H+ for the synthesis of highly reduced precursors like aromatic amino acids, monoterpens, alkaloids (Selmar and Kleinwächter, 2013). Increased abundance under drought of the enzyme (+)-neomenthol dehydrogenase, which participates in monoterpenoid biosynthesis, observed in this study, could be linked to possible protective function of monoterpens. Caffeoyl-CoA 3-O-methyltransferase is engaged in the pathway of lignin biosynthesis. The accumulation of this enzyme under drought could be related to increased lignification of the cell wall—a modification in order to avoid water loss. Similar up regulation of Caffeoyl-CoA 3-O-methyltransferase in roots subjected to water stress is reported by several authors (Alam et al., 2010; Yamaguchi et al., 2010; Fulda et al., 2011). In concert with the changes in root growth (longer and thinner roots with less biomass) we observed an increase in abundance of actin 2 (component of microphilaments) as well as early decrease and late increase in peroxidase abundance—a cell wall cross-linking enzyme participating in cell wall lignification, defense against pathogen attack, and activated oxygen consumer. Increased content of peroxidase III in roots of wild watermelon under drought has been reported (Yoshimura et al., 2008).
Carbon metabolism and energy production—glycolysis is down-regulated in roots in water deficit conditions while ATP synthesis is stimulated
In this study we observed concerted decrease in abundance of glycolytic enzymes—glucose-6-phosphate isomerase (EC 5.3.1.9), pyrophosphate-dependent phosphofructokinase (EC 2.7.1.90), 2,3-bisphospho glycerate-independent phosphoglycerate mutase (EC 5.4.2.12), as well as of the enzyme pyruvate dehydrogenase (EC 1.2.4.1)—mitochondrial enzyme which links the glycolysis metabolic pathway to the citric acid cycle. Decrease in the amount of the cytoplasmic aconitate hydratase (EC 4.2.1.3), an enzyme of the glyoxylate bypass in plants for utilization of fatty acids as a carbon source, was detected. As for enzymes of the tricarboxylic acid cycle, an increase in abundance of isocitrate dehydrogenase which catalyzes the rate-limiting step of the cycle, was detected (EC 1.1.1.42). Two protein spots related to ATP production were found to be increased in abundance under drought—ATP synthase (EC 3.6.3.14) subunit beta and NADH-ubiquinone oxidoreductase (EC 1.6.5.3). The general down-regulation of carbohydrate degrading glycolytic enzymes could be linked to reduced root biomass accumulation, and could be regarded as a mechanism to accumulate and store sugars for rapid growth in recovery. Similar decrease of glycolysis-related enzymes in roots under drought stress is reported for soybean (Alam et al., 2010). In the roots of other species—the xerophyte wild watermelon, an up-regulation of glycolysis and tricarboxylic acid cycle was found (Yoshimura et al., 2008). Tricarboxylic acid cycle is embedded into a larger metabolic network, constantly sharing substrates and products with other pathways (Sweetlove et al., 2010). Besides carbohydrates, the TCA cycle may be fuelled by products derived from protein and other macromolecules degradation, in order to produce sufficient ATP to meet the energetic needs under stress. The up-regulation of ATP-synthesis related enzymes could be explained by the need of energy for stress protection and maintaining tissue functional state under water limiting conditions. ATP energy is necessary for many cellular processes, including secondary metabolism and protein quality control.
Dynamic changes are observed in enzymes related to amino acid and one carbon metabolism
We have found increased abundance under drought of some enzymes related to one-carbon and amino acid metabolism—S-adenosyl methionine synthase (EC 2.5.1.6) and formate dehydrogenase (EC 1.2.1.2); glutamine synthetase (EC 6.3.1.2) and cysteine synthase (E.C.2.5.1.47). Decrease in content was detected after prolonged drought for methylene tetrahydrofolate reductase (EC 1.5.1.20), a cytoplasmic enzyme member of one-carbon metabolism. The most interesting qualitative differences were in one cysteine synthase isoform which spot was below detection under drought; however, there was another spot identified also as cysteine synthase with increased abundance under drought. The amino acid cysteine is incorporated into proteins and glutathione (GSH); moreover, it is considered to be the bottleneck for GSH production. The cysteine synthase complex is considered to be the rate-limiting step of cysteine biosynthesis (Chan et al., 2013). Besides, cysteine acts as sulfur donor for methionine (Met) for S-adenosylmethionine and S-methylmethionine synthesis (Ravanel et al., 1998). Glutamine synthetase catalyzes the condensation of glutamate and ammonia to form glutamine, thus playing an essential role in nitrogen metabolism and ammonia assimilation. Ammonia is produced by nitrate reduction or amino acid degradation; on the other hand the amide group of glutamate serves as a readily mobilized nitrogen source for incorporation of amino group in various metabolites. Glutamine synthetase isoforms are reported to be highly responsive to drought (Yoshimura et al., 2008; Alam et al., 2010; Singh and Ghosh, 2013). S-adenosyl methionine synthase catalyzes the conversion, at the expense of ATP, of L-methionine into S-adenosylmethionine—AdoMet or SAM, the major methyl donor for proteins, nucleic acids, carbohydrates, lipids, and small molecules for lignin and many other biosynthesis, precursor for polyamine ant ethylene biosynthesis. Different trends of change in the enzymes related to one-carbon metabolism were reported in drought-stressed roots from different plant species and stress treatment (Yoshimura et al., 2008; Mohammadi et al., 2012; Grebosz et al., 2014). In our study, the accumulation of S-adenosyl methionine synthase and related enzymes in holm oak roots under drought could be linked to enhanced secondary metabolism and lignification which utilize activated one carbon particles.
Protein synthesis, folding/processing and degradation processes are highly responsive to the applied stress
Changes in the protein complement of cells are indispensable for adaptation to stress. Within the large group of proteins related to protein synthesis, folding/processing and degradation, some spots with opposite trends of changes under stress were found, which may reflect the dynamic changes in cell protein profile. A DEAD box RNA helicase was detected among the identified proteins with earlier and reversible increase in abundance under drought. RNA helicases are involved in various aspects of RNA metabolism, including nuclear transcription, pre mRNA splicing, ribosome biogenesis, nucleocytoplasmic transport, translation, RNA decay, and organellar gene expression (Aubourg et al., 1999). Similar increase of DEAD box RNA helicase was reported in osmotically stressed triticosecale roots (Grebosz et al., 2014). On the other hand, glycyl-tRNA synthetase abundancy was found to decrease under drought and in recovery which is not in favor of up-regulation of translation under drought stress and may rather reflect changes in the composition of newly synthesized proteins. Chaperones and proteases are elements of the protein quality control machinery. Maintaining proteins in their functional conformation, preventing aggregation of non-native proteins, refolding of denatured proteins and removal of non-functional and potentially harmful polypeptides are very important for cell survival under drought stress (Vaseva et al., 2012). Some molecular chaperones, which assist protein folding or renaturation, presented complex changes: chaperonin 60 kD, protein disulfide isomerase and chaperone of TCP-1 family decreased in abundance, while GroES chaperonin increased following water stress. TCP-1 family chaperones are related to cpn60/groEL chaperonin family and assist the folding of cytoskeletal proteins in the cytoplasm (Wang et al., 2004). Cpn60 is homologous to E. Coli GroEL found in chloroplasts and mitochondria. It acts in cooperation with GroES chaperonin in assisting proper folding of newly synthesized and membrane-translocated proteins into their native conformation. The observed different abundance changes in GroEL/GroES may reflect changes in composition within the chaperonin family. Under stress the folding capacity of cpn60 is suppressed while its binding affinity toward unfolded proteins is increased, thus protecting proteins from unfavorable conditions by sequestration (Vaseva et al., 2012). Protein disulfide isomerase is an enzyme located in the endoplasmic reticulum with the main function—correct arrangement of disulfide bonds in proteins. Decrease in abundance in this enzyme could reflect inhibition of protein synthesis at the endoplasmic reticulum. The proteasome plays a crucial role in the turnover of regulatory proteins, cellular house-keeping and stress tolerance (Kurepa and Smalle, 2008). Relatively early increase in abundance under stress was found for an ubiquitin activating enzyme E1 which may reflect the importance of stress signaling. The 6B regulatory subunit of 26S proteasome, involved in ATP-dependent protein degradation in the cytosol and nucleus, decreased under drought, while the catalytic proteasome subunit beta type increased. Some evidence suggests that the control of proteasome function may be at the level of subunit composition rather than total increase in proteasome abundance (Kurepa and Smalle, 2008). The proteasome could function in two forms—26S and 20S, the latter containing only the catalytic core subunits and operating without ATP in degradation of oxidized proteins. Thus, 20S may play an important role in tolerance to the secondary oxidative stress developing under drought stress (Vaseva et al., 2012). Up-regulation of the 20S proteasome subunit was found in drought-treated poplar trees (Plomion et al., 2006), alfalfa plants (Aranjuelo et al., 2011), and rapeseed roots (Mohammadi et al., 2012). Opposite changes were observed for some aminopeptidases in roots under applied stress. Leucine aminopeptidase (EC 3.4.11.1) abundancy diminished under drought and in recovery, while Xaa-pro aminopeptidase (EC 3.4.11.9) increased. Aminopeptidases liberate free aminoacids from the N-terminus of the polypeptide chains. Amino acid metabolism was found to be among the top biological processes affected by drought (Kang et al., 2011).
In conclusion, differently abundant identified protein species in holm oak roots subjected to water limitation treatment point at early activation of secondary metabolism, down-regulation of glycolysis and stimulation of ATP synthesis, accumulation of some enzymes related to aminoacid and one-carbon metabolism, and complex changes in protein synthesis, folding/processing and degradation processes, which emphasize the active metabolic adjustment and mobilization of the defense system in roots to actively counteract stress.
Conflict of interest statement
The reviewer Sabine Lüthje declares that, despite having co-hosted the Research Topic INPPO World Congress 2014 with the author Jesús V. Jorrín-Novo, the review process was handled objectively. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Financial support of this study by MC IEF project No 271714 QUIPROD funded by the EU, 7th FP, is greatly acknowledged. Protein identification was carried out at the UCO-SCAI proteomics facility, a member of Carlos III Networked Proteomics Platform, ProteoRed-ISCIII, Cordoba, Spain. MR was supported by a grant from the “Fundación Parque Tecnológico Itaipu-Binacional” and “Centro Multidisciplinario de Investigaciones Tecnológicas—Dirección General de Investigación Científica y Tecnológica-Universidad Nacional de Asunción—Paraguay.”
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.00627
References
- Abril N., Gion J. M., Kerner R., Müller-Starck G., Cerrillo R., Plomion C., et al. (2011). Proteomics research on forest trees, the most recalcitrant and orphan plant species. Phytochemistry 72, 1219–1242. 10.1016/j.phytochem.2011.01.005 [DOI] [PubMed] [Google Scholar]
- Alam I., Sharmin S. A., Kim K.-H., Yang J. K., Choi M. S., Lee B.-H. (2010). Proteome analysis of soybean roots subjected to short-term drought stress. Plant Soil 333, 491–505. 10.1007/s11104-010-0365-7 [DOI] [Google Scholar]
- Aranjuelo I., Molero G., Erice G., Avice J. C., Nogués S. (2011). Plant physiology and proteomics reveals the leaf response to drought in alfalfa (Medicago sativa L.). J. Exp. Bot. 62, 111–123. 10.1093/jxb/erq249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubourg S., Kreis M., Lecharny A. (1999). The DEAD box RNA helicase family in Arabidopsis thaliana. Nucleic Acids Res. 27, 628–636. 10.1093/nar/27.2.628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquedano F. J., Castillo F. J. (2006). Comparative ecophysiological effects of drought on seedlings of the Mediterranean water-saver Pinus halepensis and water-spenders Quercus coccifera and Quercus ilex. Trees 20, 689–700. 10.1007/s00468-006-0084-0 [DOI] [Google Scholar]
- Bevan M., Bancroft I., Bent E., Love K., Goodman H., Dean C., et al. (1998). Analysis of 1.9 Mb of contiguous sequence from chromosome 4 of Arabidopsis thaliana. Nature 391, 485–488. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- Bussotti F., Ferrini F., Pollastrini M., Fini A. (2014). The challenge of Mediterranean sclerophyllous vegetation underclimate change: from acclimation to adaptation. Environ. Exp. Bot. 103, 80–98. 10.1016/j.envexpbot.2013.09.013 [DOI] [Google Scholar]
- Chan K. X., Wirtz M., Phua S. Y., Estavillo G. M., Pogson B. J. (2013). Balancing metabolites in drought: the sulfur assimilation conundrum. Trends Plant Sci. 18, 18–29. 10.1016/j.tplants.2012.07.005 [DOI] [PubMed] [Google Scholar]
- Corcobado T., Cubera E., Moreno G., Solla A. (2013). Quercus ilex forests are influenced by annual variations in water table, soil water deficit and fine root loss caused by Phytophthora cinnamomi. Agricult. Forest Meteorol. 169, 92–99. 10.1016/j.agrformet.2012.09.017 [DOI] [Google Scholar]
- Côté R. G., Griss J., Dianes J. A., Wang R., Wright J. C., van den Toorn H. W. P., et al. (2012). The Proteomics IDEntifications (PRIDE) Converter 2 framework: an improved suite of tools to facilitate data submission to the PRIDE database and the ProteomeXchange consortium. Mol. Cell. Proteomics 11, 1682–1689. 10.1074/mcp.O112.021543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao T. T. H., Linthorst H. J. M., Verpoorte R. (2011). Chalcone synthase and its functions in plant resistance. Phytochem. Rev. 10, 397–412. 10.1007/s11101-011-9211-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David T. S., Henriques M. O., Kurz-Besson C., Nunes J., Valente F., Vaz M., et al. (2007). Water-use strategies in two co-occurring Mediterranean evergreen oaks: surviving the summer drought. Tree Physiol. 27, 793–803. 10.1093/treephys/27.6.793 [DOI] [PubMed] [Google Scholar]
- Dolferus R. (2014). To grow or not to grow: a stressful decision for plants. Plant Sci. 229, 247–261. 10.1016/j.plantsci.2014.10.002 [DOI] [PubMed] [Google Scholar]
- Echevarría-Zomeño S., Ariza D., Jorge I., Lenz C. H., Del Campo A., Jorrín J. V., et al. (2009). Changes in the protein profile of Quercus ilex leaves in response to drought stress and recovery. J. Plant Physiol. 166, 233—245. 10.1016/j.jplph.2008.05.008 [DOI] [PubMed] [Google Scholar]
- Emanuelsson O., Nielsen H., Brunak S., von Heijne G. (2000). Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol., 300, 1005–1016. 10.1006/jmbi.2000.3903 [DOI] [PubMed] [Google Scholar]
- Fulda S., Mikkat S., Stegmann H., Horn R. (2011). Physiology and proteomics of drought stress acclimation in sunflower (Helianthus annuus L.). Plant Biol. 13 632–642. 10.1111/j.1438-8677.2010.00426.x [DOI] [PubMed] [Google Scholar]
- Gargallo-Garriga A., Sardans J., Pérez-Trujillo M., Rivas-Ubach A., Oravec M., Vecerova K., et al. (2014). Opposite metabolic responses of shoots and roots to drought. Sci. Rep. 4:6829. 10.1038/srep06829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D., Xu J. (2014). Abiotic stress responses in plant roots: a proteomics perspective. Front. Plant Sci. 5:6. 10.3389/fpls.2014.00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi F., Lionello P. (2008). Climate change projections for the Mediterranean region. Global Planet. Change 63, 90–104. 10.1016/j.gloplacha.2007.09.005 [DOI] [Google Scholar]
- Grebosz J., Badowiec A., Weidner S. (2014). Changes in the root proteome of Triticosecale grains germinating under osmotic stress. Acta Physiol. Plant. 36, 825–835. 10.1007/s11738-013-1461-0 [DOI] [Google Scholar]
- Höglund A., Dönnes P., Blum T., Adolph H.-W., Kohlbacher O. (2006). MultiLoc: prediction of protein subcellular localization using N-terminal targeting sequences, sequence motifs and amino acid composition. Bioinformatics. 22, 1158–1165. 10.1093/bioinformatics/btl002 [DOI] [PubMed] [Google Scholar]
- Horton P., Park K. J., Obayashi T., Fujita N., Harada H., Adams-Collier C. J., et al. (2007). WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, 585–587. 10.1093/nar/gkm259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorge I., Navarro R. M., Lenz C., Ariza D., Jorrín J. (2006). Variation in the holm oak leaf proteome at different plant developmental stages, between provenances and in response to drought stress. Proteomics 6, S207–S214. 10.1002/pmic.200500364 [DOI] [PubMed] [Google Scholar]
- Kang Y., Han Y., Torres-Jerez I., Wang M., Tang Y., Monteros M., et al. (2011). System responses to long-term drought and re-watering of two contrasting alfalfa varieties. Plant J. 68, 871–889. 10.1111/j.1365-313X.2011.04738.x [DOI] [PubMed] [Google Scholar]
- Kurepa J., Smalle J. (2008). Structure, function and regulation of plant proteasomes. Biochimie 90, 324–335. 10.1016/j.biochi.2007.07.019 [DOI] [PubMed] [Google Scholar]
- Mohammadi P. P., Moieni A., Komatsu S. (2012). Comparative proteome analysis of drought-sensitive and drought-tolerant rapeseed roots and their hybrid F1 line under drought stress. Amino Acids 43, 2137–2152. 10.1007/s00726-012-1299-6 [DOI] [PubMed] [Google Scholar]
- Navarro Cerrillo R. M., Fragueiro B., Ceaceros C., Campo A., Prado R. (2005). Establishment of Quercus ilex L. subsp. Ballota [Desf.] Samp. using different weed control strategies in southern Spain. Ecol. Eng. 25, 332–342. 10.1016/j.ecoleng.2005.06.002 [DOI] [Google Scholar]
- Oh M. W., Komatsu S. (2015). Characterization of proteins in soybean roots under flooding and drought stresses. J. Proteomics 114, 161–181. 10.1016/j.jprot.2014.11.008 [DOI] [PubMed] [Google Scholar]
- Petsalaki E. I., Bagos P. G., Litou Z. I., Hamodrakas S. J. (2006). PredSL: a tool for the N-terminal sequence-based prediction of protein subcellular localization. Geno. Prot. Bioinfo. 4, 48–55. 10.1016/S1672-0229(06)60016-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomion C., Lalanne C., Claverol S., Meddour H., Kohler A., Bogeat-Triboulot M.-B., et al. (2006). Mapping the proteome of poplar and application to the discovery of drought-stress responsive proteins. Proteomics 6, 6509–6527. 10.1002/pmic.200600362 [DOI] [PubMed] [Google Scholar]
- Ravanel S., Gakiere B., Job D., Douce R. (1998). Cystathionine g-synthase from Arabidopsis thaliana: purification and biochemical characterization of the recombinant enzyme overexpressed in Escherichia coli. Biochem. J. 331, 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Pérez V., Castro-Díez P., Joffre R. (2009). Seasonal carbon storage and growth in Mediterranean tree seedlings under different water conditions. Tree Physiol. 29, 1105–1116. 10.1093/treephys/tpp045 [DOI] [PubMed] [Google Scholar]
- Selmar D., Kleinwächter M. (2013). Stress enhances the synthesis of secondary plant products: the impact of stress-related over-reduction on the accumulation of natural products. Plant Cell Physiol. 54, 817–826. 10.1093/pcp/pct054 [DOI] [PubMed] [Google Scholar]
- Sengupta D., Kannan M., Reddy A. R. (2011). A root proteomics-based insight reveals dynamic regulation of root proteins under progressive drought stress and recovery in Vigna radiata (L.) Wilczek. Planta 233, 1111–1127. 10.1007/s00425-011-1365-4 [DOI] [PubMed] [Google Scholar]
- Sghaier-Hammami B., Valero Galvàn J., Romero-Rodríguez M. C., Navarro Cerrillo R. M., Abdelly C., Jorrín Novo J. (2013). Physiological and proteomics analyses of Holm oak (Quercus ilex subsp. ballota [Desf.] Samp.) responses to Phytophthora cinnamomi. Plant Physiol. Biochem. 71, 191–202. 10.1016/j.plaphy.2013.06.030 [DOI] [PubMed] [Google Scholar]
- Simova-Stoilova L., Demirevska K., Petrova T., Tsenov N., Feller U. (2008). Antioxidative protection in wheat varieties under severe recoverable drought at seedling stage. Plant Soil Environ. 54, 529–536. Available online at: http://www.agriculturejournals.cz/publicFiles/02851.pdf [Google Scholar]
- Singh K. K., Ghosh S. (2013). Regulation of glutamine synthetase isoforms in two differentially drought-tolerant rice (Oryza sativa L.) cultivars under water deficit conditions. Plant Cell Rep. 32, 183–193. 10.1007/s00299-012-1353-6 [DOI] [PubMed] [Google Scholar]
- Sweetlove L. J., Beard K. F. M., Nunes-Nesi A., Fernie A. R., Ratcliffe R. G. (2010). Not just a circle: flux modes in the plant TCA cycle. Trends Plant Sci. 15, 462–470. 10.1016/j.tplants.2010.05.006 [DOI] [PubMed] [Google Scholar]
- Swigonska S., Weidner S. (2013). Proteomic analysis of response to long-term continuous stress in roots of germinating soybean seeds. J. Plant Physiol. 170 470–479. 10.1016/j.jplph.2012.11.020 [DOI] [PubMed] [Google Scholar]
- Tsakaldimi M., Tsitsoni T., Ganatsas P., Zagas T. (2009). A comparison of root architecture and shoot morphology between naturally regenerated and container-grown seedlings of Quercus ilex. Plant Soil 324, 103–113. 10.1007/s11104-009-9974-4 [DOI] [Google Scholar]
- Valero Galván J., González-Fernández R., Navarro-Cerrillo R. M., Gil-Pelegrín E., and Jorrín-Novo J. V. (2013). Physiological and proteomic analyses of drought stress response in holm oak provenances. J. Proteome Res. 12, 5110–5123. 10.1021/pr400591n [DOI] [PubMed] [Google Scholar]
- Valero Galván J., Valledor L., Navarro Cerrillo R. M., Gil-Pelegrín E., Jorrín-Novo J. V. (2011). Studies of variability in Holm oak (Quercus ilex subsp. Ballota [Desf.] Samp.) through acorn protein profile analysis. J. Proteomics 74, 1244–1255. 10.1016/j.jprot.2011.05.003 [DOI] [PubMed] [Google Scholar]
- Vaseva I., Sabotič J., Šuštar-Vozlič J., Meglič V., Kidrič M., Demirevska K., et al. (2012). The response of plants to drought stress—the role of dehydrins, chaperones, proteases and protease inhibitors in maintaining cellular protein function, in Droughts: New Research, eds Neves D. F., Sanz J. D. (New York, NY: Nova Science Publishers, Inc.), 1–45. [Google Scholar]
- Vizcaíno J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Ríos D., et al. (2014). ProteomeXchange provides globally co-ordinated proteomics data submission and dissemination. Nat. Biotechnol. 30, 223–226. 10.1038/nbt.2839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Vignani R., Scali M., Cresti M. (2006). A universal and rapid protocol for protein extraction from recalcitrant plant tissues for proteomic analysis. Electrophoresis 27, 2782–2786. 10.1002/elps.200500722 [DOI] [PubMed] [Google Scholar]
- Wang W., Vinocur B., Altman A. (2003). Plant responses to drought, salinity and extreme temperatures:towards genetic engineering for stress tolerance. Planta 218, 1–14. 10.1007/s00425-003-1105-5 [DOI] [PubMed] [Google Scholar]
- Wang W., Vinocur B., Shoseyov O., Altman A. (2004). Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 9, 244–252. 10.1016/j.tplants.2004.03.006 [DOI] [PubMed] [Google Scholar]
- Wendelboe-Nelson C., Morris P. C. (2012). Proteins linked to drought tolerance revealed by DIGE analysis of drought resistant and susceptible barley varieties. Proteomics 12, 3374–3385. 10.1002/pmic.201200154 [DOI] [PubMed] [Google Scholar]
- Yamaguchi M., Sharp R. E. (2010). Complexity and coordination of root growth at low water potentials: recent advances from transcriptomic and proteomic analyses. Plant Cell Environ. 33, 590–603. 10.1111/j.1365-3040.2009.02064.x [DOI] [PubMed] [Google Scholar]
- Yamaguchi M., Valliyodan B., Zhang J., Lenoble M. E., Yu O., Rogers E., et al. (2010). Regulation of growth response to water stress in the soybean primary root. Proteomic analysis reveals region-specific regulation of phenylpropanoid metabolism and control of free iron in the elongation zone. Plant Cell Environ. 33, 223–243. 10.1111/j.1365-3040.2009.02073.x [DOI] [PubMed] [Google Scholar]
- Yoshimura K., Masuda A., Kuwano M., Yokota A., Akashi K. (2008). Programmed proteome response for drou ght avoidance/tolerance in the root of a C3 xerophyte (wild watermelon) under water deficits. Plant Cell Physiol. 49, 226–241. 10.1093/pcp/pcm180 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.