

Abstract
Defects in primary cilium biogenesis underlie the ciliopathies, a growing group of genetic disorders. We describe a whole genome siRNA-based reverse genetics screen for defects in biogenesis and/or maintenance of the primary cilium, obtaining a global resource. We identify 112 candidate ciliogenesis and ciliopathy genes, including 44 components of the ubiquitin-proteasome system, 12 G-protein-coupled receptors, and three pre-mRNA processing factors (PRPF6, PRPF8 and PRPF31) mutated in autosomal dominant retinitis pigmentosa. The PRPFs localise to the connecting cilium, and PRPF8- and PRPF31-mutated cells have ciliary defects. Combining the screen with exome sequencing data identified recessive mutations in PIBF1/CEP90 and C21orf2/LRRC76 as causes of the ciliopathies Joubert and Jeune syndromes. Biochemical approaches place C21orf2 within key ciliopathy-associated protein modules, offering an explanation for the skeletal and retinal involvement observed in individuals with C21orf2-variants. Our global, unbiased approaches provide insights into ciliogenesis complexity and identify roles for unanticipated pathways in human genetic disease.
Keywords: cilia, ciliopathies, reverse genetics, whole-genome siRNA screen, Jeune syndrome, Joubert syndrome
The primary cilium is a microtubule-based organelle present on the apical surface of most vertebrate cells. Primary cilia are regarded as cellular signalling hubs, regulating diverse signalling pathways, with particularly important roles during embryonic development and the patterning of the developing neural tube1. Defects in primary cilia are associated with heterogeneous inherited developmental conditions known as the ciliopathies that often present with cystic kidney disease, and other diverse multi-organ phenotypes that affect the central nervous system, eye and skeleton2. As a group, ciliopathies are comparatively common Mendelian inherited conditions with an overall estimated prevalence of 1 in 20003.
The importance of the primary cilium has only become apparent in the last decade, and therefore processes involved in cilia formation and maintenance remain poorly characterised. To address this in the most comprehensive way, we have carried out a high-throughput cilia characterisation assay to interrogate the involvement of every gene in the formation of the primary cilium. This represents a global, hypothesis-neutral approach to identify genes involved in ciliogenesis. It also provides a complementary approach to traditional linkage analysis, candidate gene, or whole exome sequencing (WES) approaches for gene discovery. Previously, several cell-based medium- or small-scale screens of ciliogenesis have been published, performed on subsets of target genes such as a druggable library (7784 targets)4 and uncharacterised components of the cilia proteome (40 targets)5. Here, we present a whole genome knockdown screen in a ciliated cell line that provides a comprehensive view of critical factors involved in ciliogenesis and offers numerous candidate genes for ciliopathies.
RESULTS
Genome-wide siRNA screen for ciliogenesis
We used small interfering RNA (siRNA) to knockdown and ascertain the function of 19059 Entrez RefSeq (B37.1) mouse genes across the genome. The siRNA screen was carried out in the mouse inner medullary collecting duct (mIMCD3) ciliated cell-line using a Thermo Fisher siGENOME library containing pools of four siRNA duplexes per gene (see On-line Methods and Supplementary Note for further details). We assayed for a loss-of-cilium phenotype 72 hours after transfection by imaging at three focal planes to detect the nucleus (DAPI), cytoplasm (TOTO3) and cilium (acetylated α-tubulin) (Figure 1a). The cilia could be identified as single spots above the nucleus of the cell (Figure 1b,c), allowing the use of simple cilia recognition protocols based on optimised Perkin-Elmer spot detection algorithms (Figure 1b).
Figure 1. Automated high content imaging, validation of siRNA screen controls and calculation of cut-off values.

(a) Schematic of a polarised mIMCD3 cell showing the focal planes used to image nuclei (blue), cytoplasm (pink) and ciliary axonemes (green). (b) mIMCD3 cells imaged using an Operetta high-content imaging system, with representative images from Harmony/Columbus software of cilia recognition (“find spots”). Scale bar = 10 μm. (c) Upper panels: significant effect on ciliogenesis following reverse transfection with positive control siRNA pool against Ift88 compared to non-targeting scrambled siRNA, imaged by immunostaining for acetylated (Ac) α-tubulin (green). Bar graph (green) quantitates the effect on ciliogenesis (% cells with a single cilium) for positive controls (Plk1, Mks1, Rpgrip1l, Ift88) and negative (−ve) controls including MLNR, scrambled (scr.) siRNAs and mock transfection. Lower panels: positive transfection control siRNA pool against Plk1 has a significant effect on cell number. Images show cells stained for acetylated α-tubulin (green), TOTO3 (pink) and DAPI (blue). Bar graph (blue) quantitates the effect on cell number following knock-down with control siRNAs. Scale bar = 20 μm. Significance of pairwise comparisons with all negative controls (#): ns, not significant; * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 (paired two-tailed Student’s t-test), for n=32 independent replicates. (d) Bar graphs showing mean robust z score for % cells with a single cilium (zcilia) and mean robust z scores for cell number (zcell) for positive and negative siRNA controls. (e) Strictly standardised mean difference (SSMD) values for each replicate per batch in the screen. The average SSMD value for all batches was 1.717. Each bar is colour-coded by batch for sub-libraries in the ThermoFisher mouse siGENOME library (m, mouse; GPCR, G protein-coupled receptor; UC, ubiquitin conjugation; IC, ion channel; PP, protein phosphatase; P, protease; PK, protein kinase) or set of 10 plates (DT, druggable targets; G, genome libraries). (f) Robust z score cut-off values for cilia number (zcilia cutoff) calculated for each replicate per batch with error bars indicating median absolute deviation, where zcilia cutoff is the median z score for all positive controls. Dots are colour-coded by batch for sub-libraries as in (e).
The statistical significance of effects on cilia and cell number after siRNA knockdown was tested by calculating robust z scores6 (Figure 1d, Suppl. Table 1). To ensure normalisation of data and exclusion of batch-specific effects, data were analysed within processed batches (Figure 1e,f). Duplicate assays of batches resulted in little variation, with a median Pearson’s correlation coefficient between replicates of 0.71 (Figure 2a), and an average strictly standardised mean difference (SSMD) value for all batches of 1.717 (Figure 1e). Robust z scores for cell number (zcell) and the percentage of cells with a single cilium (zcilia) were calculated for all results, compared to the median and median absolute deviation (MAD) of all positive and negative controls in a processed batch (Figure 2a,b, Suppl. Table 1). This allowed application of meaningful statistical cut-off values (zcilia cutoff and zcell cutoff) for the identification of significant “hits” affecting ciliogenesis based on the median and MAD of positive and negative controls per 10-plate batch7 (Figure 1f).
Figure 2. Analysis and data filtering strategy for the whole genome siRNA screen.

(a) Scatter plot of zcilia values of two screen replicates (rep. 1 and rep. 2). Each dot represents one targeted gene in the Thermo Fisher mouse siGENOME library, colour-coded by batch for either sub-library (m, mouse; GPCR, G protein-coupled receptor; UC, ubiquitin conjugation; IC, ion channel; PP, protein phosphatase; P, protease; PK, protein kinase) or set of 10 plates (DT, druggable targets; G, genome libraries). The median Pearson’s correlation coefficient (R2) between replicates was 0.71, if failed measurements in batch mDT51-60 were excluded (grey region). (b) Scatter plot of mean zcilia values plotted against robust z scores for cell number (zcell) for each targeted gene in the screen with each batch colour-coded as for (a); candidate hit genes are indicated. (c) Schematic to summarise data filtering steps in the primary, secondary and tertiary screens (complete datasets shown in Suppl. Tables 1-3, respectively). The number of genes filtered at each step are shown in bold, with the filter applied highlighted in grey boxes (see main text and On-line Methods for further details). Validated hits are listed in Table 1. (d) Classification enrichment for selected GO terms, or KEGG and BioCarta pathways/processes as determined using DAVID. Left bar graph: n=1829 screen hits with a significant effect on cilia number (zcilia< zcilia cutoff), without filtering for any effect on cell number, that had a human homologue. Right bar graph: 154 filtered screen hits with a human homologue that showed a significant effect on cilia number (zcilia< zcilia cutoff) but no significant effect on cell number (zcell> zcell cutoff), where zcell cutoff is the median z - 2MAD of all negative controls. The significance for terms or pathways was determined by a Benjamini-Hochberg q threshold value <0.05 (red dotted line) to correct for the false discovery rate, with the number of associated human genes shown above each bar.
Filtering strategy, bioinformatics analysis, and validation screens
In total, 2174 siRNA pools significantly decreased the percentage of cells with a single primary cilium (zcilia< zcilia cutoff in a processed batch for both replicates, where zcilia cutoff = median z of all positive controls), with 1956 hits targeting a gene with a human orthologue (Figure 2c, Suppl. Table 2). We filtered out potential non-specific siRNAs comprising those with predicted off-target effects or with microRNA-like effects (see Supplementary Note) leaving a total of 1829 mouse genes with a human orthologue (Suppl. Table 2). The list of 1829 genes was significantly enriched in known ciliary components (the SYSCILIA Gold Standard8; p=6.7×10−4 hypergeometric test, observed 48, expected 31.2). Functional annotation clustering with enrichment analysis using DAVID9,10 and relevant literature information was used to select representative functional candidate hits for further validation in a secondary screen. We selected a total of n=40 hits that were components of significantly enriched pathways (Figure 2d left panel, Suppl. Table 3) including the spliceosome, the proteasome, and the ubiquitin-proteasome system (UPS).
To exclude the effect of proliferation on ciliogenesis, we filtered the 1829 primary screen hits to prioritise those that significantly affected the percentage of cells with a single cilium but did not have a significant effect on cell number (zcell>zcell cutoff) in a processing batch for both replicates, where zcell cutoff=median z-2MAD for all negative controls; see On-line Methods). This filtering gave a total of n=154 hits (Suppl. Table 3) that were significantly enriched in known ciliary components (p=7.7×10−4 hypergeometric test, observed 9, expected 2.5), and were taken forward for validation. DAVID analysis of this list identified enrichment of GPCRs, and components of the non-motile primary cilium and the photoreceptor outer segment (Figure 2d right panel). In total, 194 functional candidates were taken forward for validation in a secondary screen.
Secondary screening in mIMCD3 cells validated a total of n=68/194 mouse genes that had a significant effect on cilia number (zcilia<zcilia cutoff) for n>=1 out of 4 individual siRNAs in duplicate, and n=29/194 for n>=2 out of 4 individual siRNAs in duplicate (Table 1, Suppl. Table 3). Validated hits included genes identified previously in functional genomics of centriole biogenesis in human cells11 including PLK4 and CEP120, and other hits with known ciliary roles including CCDC4112 and OFD111,13. PLK4 is implicated in the regulation of both centriole biogenesis and cilia-dependent processes, with mutations causing microcephaly, growth failure and retinopathy14,15. In addition, mutations in CEP120 are a cause of the skeletal ciliopathy Jeune asphyxiating thoracic dystrophy16, suggesting that our screen has a high predictive value to identify genes involved in ciliary processes. Functional classifications for a selection of these validated genes are shown in Figure 3a. Interestingly, the two hits PRPF8 and PRPF38A have also been implicated in the process of centriolar under-duplication11.
Table 1. Validated hits from secondary and tertiary screens of ciliogenesis.
Secondary screen hits are listed (n=68, validated from a total of 194 functional candidates) that had a significant effect on ciliogenesis in mouse mIMCD3 cells (given by robust z scores if zcilia< zcilia cutoff = −1.34, calculated as the median z of positive controls) for n>=1/4 individual siRNAs duplexes in duplicate. Tertiary screen hits in human hTERT-RPE1 cells for defects in cilia number and/or length (n=37/68, using cut-offs of z<−2 or z>+2) for siRNA pools in duplicate. Human and mouse gene symbols, mouse gene IDs, known or inferred protein function and selected OMIM annotations are also listed.
| Mouse Gene ID |
Mouse Gene Symbol |
Human Gene Symbol |
mIMCD3 siRNA | hTERT-RPE1 siRNAs | Protein function | OMIM examples (number) | |||
|---|---|---|---|---|---|---|---|---|---|
| #1 | #2 | #3 | cilia number |
cilia length |
|||||
| 76719 | 1700081L11Rik | KANSL1 | −2.13 | −5.20 | −5.05 | histone acetyltransferase | Koolen-De Vries syndrome (610443) | ||
| 67884 | 1810043G02Rik | C21orf2 | −1.72 | −1.67 | +4.94 | centriolar protein | |||
| 69612 | 2310037I24Rik | KANSL2 | −2.97 | −2.52 | −2.28 | histone acetyltransferase | |||
| 67217 | 2810055F11Rik | C14orf149 | −1.69 | isomerase activity | |||||
| 71617 | 9130011E15Rik | C10orf76 | −4.20 | membrane protein | |||||
| 30806 | Adamts8 | ADAMTS8 | −1.78 | metalloendopeptidase | |||||
| 268822 | Adck5 | ADCK5 | −2.28 | protein serine/threonine kinase | |||||
| 11768 | Ap1m2 | AP1M2 | −2.15 | −1.62 | +2.05 | post-Golgi vesicle-mediated transport | |||
| 11910 | Atf3 | ATF3 | −2.94 | −2.03 | −1.90 | transcription factor | |||
| 242341 | Atp6v0d2 | ATP6V0D2 | −1.34 | +14.57 | −7.03 | ATP hydrolysis/proton transport | |||
| 14595 | B4galt1 | B4GALT1 | −2.94 | −3.06 | +4.24 | post-translational protein modification | Congenital disorder of glycosylation IId (607091) | ||
| 12419 | Cbx5 | CBX5 | −2.55 | −2.53 | transcription factor | ||||
| 77048 | Ccdc41 | CCDC41 | −4.41 | −2.45 | −11.25 | −3.37 | centriolar protein | ||
| 217232 | Cdc27 | CDC27 | −7.38 | mitotic cell cycle | |||||
| 214498 | Cdc73 | CDC73 | −2.35 | mitotic cell cycle | Hyperparathyroidism, familial primary (145000) | ||||
| 225523 | Cep120 | CEP120 | −1.95 | −1.47 | centrosomal protein | Jeune asphyxiating thoracic dystrophy | |||
| 12922 | Crhr2 | CRHR2 | −4.32 | corticotrophin-releasing factor receptor | |||||
| 227292 | Ctdsp1 | CTDSP1 | −2.46 | CTD phosphatase activity | |||||
| 83603 | Elovl4 | ELOVL4 | −2.16 | +2.14 | detection of visible light | Macular dystrophy, autosomal dominant (600110) | |||
| 14428 | Galr2 | GALR2 | −2.57 | −2.78 | galanin receptor | ||||
| 14563 | Gdf5 | GDF5 | −3.67 | growth factor activity | Brachydactyly, type C (113100) | ||||
| 384009 | Glipr2 | GLIPR2 | −1.37 | Golgi membrane | |||||
| 70771 | Gpr173 | GPR173 | −2.27 | orphan G-protein coupled receptor | |||||
| 239530 | Gpr20 | GPR20 | −2.36 | +6.45 | orphan G-protein coupled receptor | ||||
| 15107 | Hadh | HADH | −2.74 | −2.20 | fatty acid beta-oxidation | 3-hydroxyacyl-CoA dehydrogenase deficiency (231530) | |||
| 15551 | Htr1b | HTR1B | −2.92 | −2.70 | −2.93 | serotonin receptor | |||
| 59026 | Huwe1 | HUWE1 | −6.62 | −2.23 | base-excision repair | Mental retardation, X-linked, Turner type (300706) | |||
| 15959 | Ifit3 | IFIT3 | −2.78 | −1.86 | +2.98 | type I interferon-mediated signaling | |||
| 16535 | Kcnq1 | KCNQ1 | −2.39 | potassium ion export | Long QT syndrome-1 (192500) | ||||
| 16663 | Krt13 | KRT13 | −3.22 | keratin filament | White sponge nevus (193900) | ||||
| 109594 | Lmo1 | LMO1 | −2.70 | −1.99 | −2.58 | metal ion binding | Leukemia, T-cell acute lymphoblastic (613065) | ||
| 27756 | Lsm2 | LSM2 | −3.14 | −2.62 | −2.12 | −7.95 | −2.35 | RNA splicing | |
| 66373 | Lsm5 | LSM5 | −2.32 | −2.05 | −3.36 | RNA splicing | |||
| 17171 | Mas1 | MAS1 | −2.26 | −1.93 | angiotensin type II receptor | ||||
| 51812 | Mcrs1 | MCRS1 | −3.96 | −2.38 | −8.30 | −3.26 | histone acetyltransferase | ||
| 17349 | Mlf1 | MLF1 | −2.23 | +3.33 | transcription, DNA-dependent | Leukemia, acute myeloid (601626) | |||
| 17751 | Mt3 | MT3 | −2.13 | +3.69 | protein import into nucleus | ||||
| 17754 | Mtap1a | MAP1A | −1.88 | −5.55 | microtubule-associated complex | ||||
| 217166 | Nr1d1 | NR1D1 | −2.19 | +2.15 | steroid hormone receptor | ||||
| 192292 | Nrbp1 | NRBP1 | −2.76 | 1.65 | protein kinase | ||||
| 77976 | Nuak1 | NUAK1 | −3.45 | +4.60 | −2.94 | response to DNA damage stimulus | |||
| 237222 | Ofd1 | OFD1 | −3.97 | −2.75 | −2.63 | +2.96 | primary cilium protein | Oral-facial-digital syndrome 1 (311200) | |
| 404315 | Olfr372 | OR2Z1 | −1.57 | +3.97 | olfactory receptor | ||||
| 258961 | Olfr631 | OR51M1 | −3.33 | −1.44 | olfactory receptor | ||||
| 14539 | Opn1mw | OPN1MW | −1.36 | −3.79 | −3.00 | vision perception | Colorblindness, deutan (303800) | ||
| 12057 | Opn1sw | OPN1SW | −2.35 | photoreceptor outer segment | Colorblindness, tritan (190900) | ||||
| 18389 | Oprl1 | OPRL1 | −1.54 | nociceptin receptor | |||||
| 140795 | P2ry14 | P2RY14 | −2.14 | −1.98 | purinergic nucleotide receptor | ||||
| 93714 | Pcdhga6 | PCDHGA6 | −2.98 | calcium ion binding | |||||
| 52023 | Pibf1 | PIBF1 | −3.45 | −1.76 | −6.81 | centrosomal protein | |||
| 20873 | Plk4 | PLK4 | −2.10 | −1.63 | −4.03 | +2.77 | mitotic cell cycle | Microcephaly and chorioretinopathy (616171) | |
| 68988 | Prpf31 | PRPF31 | −10.50 | −6.64 | +2.35 | spliceosome RNA splicing | Retinitis pigmentosa 11 (600138) | ||
| 230596 | Prpf38a | PRPF38A | −8.07 | −8.26 | spliceosome RNA splicing | ||||
| 68879 | Prpf6 | PRPF6 | −9.51 | −6.54 | −5.89 | −3.11 | spliceosome RNA splicing | Retinitis pigmentosa 60 (613983) | |
| 192159 | Prpf8 | PRPF8 | −14.42 | −9.26 | −8.65 | spliceosome RNA splicing | Retinitis pigmentosa 13 (600059) | ||
| 74760 | Rab3il1 | RAB3IL1 | −2.00 | +4.81 | Rab guanyl-nucleotide exchange factor | ||||
| 19387 | Rangap1 | RANGAP1 | −2.71 | −1.53 | Ran GTPase activator | ||||
| 19400 | Rapsn | RAPSN | −2.36 | +4.84 | acetylcholine receptor binding | Myasthenic syndrome, congenital (608931) | |||
| 56438 | Rbx1 | RBX1 | −6.22 | DNA repair | |||||
| 216227 | Slc17a8 | SLC17A8 | −3.28 | −1.41 | +2.35 | L-glutamate transmembrane transporter | Deafness, autosomal dominant 25 (605583) | ||
| 20526 | Slc2a2 | SLC2A2 | −1.60 | D-glucose transmembrane transporter | Fanconi-Bickel syndrome (227810) | ||||
| 83701 | Srrt | SRRT | −3.08 | −2.42 | −1.77 | primary miRNA processing | |||
| 66592 | Stoml2 | STOML2 | −3.62 | mitochondrial inner membrane protein | |||||
| 67902 | Sumf2 | SUMF2 | −2.33 | −1.93 | +3.70 | endoplasmic reticulum lumen protein | |||
| 78829 | Tsc22d4 | TSC22D4 | −2.05 | −1.53 | transcription factor | ||||
| 56085 | Ubqln1 | UBQLN1 | −2.01 | regulator of protein ubiquitination | |||||
| 28035 | Usp39 | USP39 | −6.08 | −4.89 | −3.64 | +8.37 | RNA splicing | ||
| 74716 | Wbp2nl | WBP2NL | −2.12 | −3.16 | meiotic protein | ||||
Figure 3. Validation screens of ciliogenesis genes.

(a) Robust z scores for % cells with a single cilium (zcilia) for selected validated genes from the secondary screen, each assayed in two replicates with four individual siRNAs in mouse mIMCD3 cells. Error bars indicate the range in values for the replicates. Cut-off value of zcilia cutoff = −1.34 is indicated (red dotted line), calculated as the median z of positive controls. Colours indicate selected functional annotations. (b) Bar graphs for quantitative PCR values for knock-down of the indicated target genes, for all assays when negative control (siScr.) cycle threshold (Ct) values<30. Transcript levels expressed as relative quantities, with the statistical significance of the indicted pair-wise comparisons: * p<0.05, ** p<0.01 and **** p<0.0001 (unpaired two-tailed Student’s t-test), for n=3 independent replicates. Error bars indicate s.d. (c) Western immunoblots (IB) showing knockdown of expression for the indicated proteins. Loading control is β-actin (act.), with the ratio of band intensities shown below. (d) IF confocal microscopy of mIMCD3 cells knocked-down with siRNAs for the indicated validated genes from the screen and immunostained for the cognate protein (green) and acetylated (Ac) α-tubulin (red). Magnified insets for selected cells (white arrowheads) are shown in white frames. All knockdowns cause loss of ciliary-associated protein (yellow arrowheads) and cilia. Scale bars = 10μm. (e) Knock-down of Plk4 in three-dimensional spheroid cultures of mIMCD3 cells causes statistically significant ciliogenesis defects (visualised by acetylated (Ac) α-tubulin in white); ** p<0.01 (paired two-tailed Student’s t-test), for n=3 independent experiments. The number of cells analysed were control: 204, 195 and 190; and Plk4 knockdown: 198, 106 and 212, respectively, for each experiment. Error bars indicate s.d. Scale bars = 10μm.
Tertiary screening in hTERT-RPE1 cells using pooled siRNAs enabled the assessment of increase or decrease in both cilia number and/or cilia length. From the hits that were validated by the secondary screen, n=37/68 human genes had defects in cilia number and/or length (using conventional cut-offs of z<−2 or z>+2) including: C21orf2, CCDC41, OFD1, PIBF1 and PLK4, and several PRPFs (PRPF31, PRPF6 and PRPF8; Table 1, Suppl. Table 3). Since the tertiary screen validated representative UPS hits (HUWE1 and USP39; Table 1, Suppl. Table 3), we used hTERT-RPE1 cells and individual siRNAs in a secondary screen to investigate all enriched human UPS components from the primary screen (n=57/1829). This validated a total of n=44/57 genes that had a significant effect on cilia number and/or cilia length (using cut-offs of z<−2 or z>+2) for n>=2 out of 3 individual duplexes (Suppl. Table 4; see Supplementary Note for further details). Interestingly, out of the 24 proteasome subunits represented in this secondary screen, knockdown of the majority (n=17/24) caused a significant increase in cilia number (Suppl. Table 4).
Validation screens identify PRPFs and GPCRs as components of ciliogenesis pathways
Based on our significance analyses, functional categorisation, relevant literature information, and availability of validated reagents, we chose a total of 15 genes encoding GPCRs, PRPFs, and predicted centrosomal proteins for further study. For each gene we measured a reduction in mRNA levels by quantitative PCR (Figure 3b), and, when possible, decrease in protein levels by western immunoblot analysis (Figure 3c). We confirmed the decrease in cilia number was accompanied by a reduction in immunostaining of the knocked-down protein by immuofluoresence (IF) microscopy (Figure 3d, Suppl. Figure 1a). 3D spheroid culture of mIMCD3 cells was also used as a more physiologically relevant model to confirm the effect of Plk4 knockdown on cilia number (Figure 3e).
PRPFs were selected for further analysis since PRPF6, PRPF8 and PRPF31 are all mutated in autosomal dominant retinitis pigmentosa (RP types 60, 13 and 11, respectively). The pathogenic mechanism for these forms of RP remains poorly understood, and none have been characterised as non-syndromic retinal ciliopathies. Although PRPF6, PRPF8 and PRPF31 predominantly localised to the nuclear speckles as expected (Figure 3d, 4a-c), we also detected co-localisation of these proteins to the base of the cilium in diverse human and mouse ciliated cell-lines (Figure 4a) and to the cilium of photoreceptor cells in adult mouse retina (Figure 4b). Immunoelectron microscopy staining showed that PRPF6 and PRPF8 localised to the apical inner segment, basal body complex, apical connecting cilium of photoreceptor cells (Figure 4d) and post synapse of secondary retinal neurons (data not shown).
Figure 4. Ciliary localisation and functional effect on ciliary axonemal formation of pre-mRNA processing factors.

(a) PRPF6, PRPF8 and PRPF31 (green) localise to proximal/basal regions of primary cilia (green arrowheads) and nuclear speckles in the indicated cell-lines. Selected cells (white arrowheads) are magnified (insets). (b) PRPF6, PRPF8 and PRPF31 (green) localise to the ciliary regions of photoreceptor cells (connecting cilium, CC) and nuclei of inner nuclear layer (INL) in longitudinal sections of adult murine retinas. Retinal layers are depicted schematically: photoreceptor outer segment (OS), connecting cilium (CC), and inner segment (IS); secondary neurons (2nd); ganglion cell layer (GC); synaptic region (S) in the outer and inner plexiform layer (OPL and IPL). (c) Immunoelectron microscopy confirms localisation of PRPF6 and PRPF8 in nuclear speckles in the INL (arrowheads). Frames indicate the magnified insets. (d) Immunofluorescence of PRPFs (green) and the ciliary marker centrin-3 (red), and immunoelectron microscopy, reveal localisation of PRPF6 and PRPF8 at the basal body (BB) and adjacent centriole (CE) of the CC (arrowheads) and apical CC (arrow). (e) Primary cilia length and number measurements for dermal fibroblasts from normal healthy controls, age-matched disease-control (ARMD), and four patients with RP type 11 of variable severity carrying heterozygous (+/−) PRPF31 frame-shift mutation c.1115_1125delGAAGCAGGCCA. Significance of pair-wise comparisons with the disease negative control (#): ns, not significant; * p<0.05, *** p<0.001, **** p<0.0001 (paired two-tailed Student’s t-test), for n=4 independent experiments. Error bars indicate s.d. (f) Transmission EM images of sensory cilia from sequential cross sections (numbered arrows in schematics) of C. elegans amphid pore in wild-type (N2) and prp-8(rr40) mutants. Wild-type amphid pores contain 10 ciliary axonemes, each consisting of a distal segment (DS; singlet A microtubules), middle segment (MS; doublet A/B microtubules), transition zone (TZ) and periciliary membrane compartment (PCMC). In prp-8(rr40) mutants, axonemes are missing in DS and DS/MS boundary, and microtubule (MT) number is reduced. Images are representative of four analysed amphid pores for each strain. Scale bars: (a) 20 μm and 5 μm; (b) 50 μm; (c) 800 nm; (d) 1 μm and 0.4 μm; (f) 200 nm (low magnification images), 100 nm (high magnification images).
We obtained adult dermal fibroblasts from three RP11 families carrying the heterozygous PRPF31 frame-shift mutation c.1115_1125del17. Fibroblast lines from individuals either mildly or severely affected with RP had statistically significant decreases in the length and/or number of cilia, compared to an age-matched disease-control individual with age-related macular degeneration (ARMD) and healthy control individuals (Figure 4e). Furthermore, a Caenorhabditis elegans strain (MR247; see On-line Methods) containing a homozygous splice-site mutation (rr40) in the PRPF8 orthologue (prp-8) previously associated with reduced spliceosome function in the germline18, had a partial ciliogenesis defect (Figure 4f). The microtubule singlet (A-tubule)-containing distal segments (DS) of at least 4 (of 10) amphid channel sensory cilia were truncated or missing entirely, and in those distal segments that remained, A-tubule numbers were abnormally low (Figure 4f). A few axonemes in the distal segment/middle segment (MS; microtubule doublets) boundary also had reduced microtubule numbers (Figure 4f).
Many of the human GPCRs identified as hits in our validation screen were neuroactive GPCRs. We therefore investigated the localisation of these proteins in a human ciliated neuroblastoma cell-line (SH-SY5Y) with neuronal-like characteristics that differentiate by extending neurites. We identified the localisation of most of these GPCRs (HTR1B, P2RY14, MAS1, OPRL1, and CRHR2) to the base of the cilium in differentiated SH-SY5Y cells (Figure 5a, Suppl. Figure 1a) and to the ciliary region of photoreceptors of murine retinas (Figure 5b). Higher resolution fluorescence and immunoelectron microscopy of a sub-set revealed localisation at the basal body and adjacent centriole of the connecting cilium, at the apical part of the connecting cilium (outer segment base), and in the axoneme of the outer segment of photoreceptor cells (Figure 5c,d). We confirmed the localisation of several GPCRs (SREB3, MAS1, P2RY14 and CRHR2) to the base of primary cilia in the marginal zone (layer I), sub-plate zone (layer IV) and ventricular zone (layer VI) of embryonic mouse neocortex (Suppl. Figure 1b). In knock-out mouse embryos mutated for the transition zone orphan receptor TMEM67 (meckelin)19, the ciliary localisation of these GPCRs was lost (Suppl. Figure 1b) suggesting that they require the structural or functional integrity of the transition zone to maintain their ciliary localisation. We hypothesised that ligand binding to GPCRs may promote the earliest stages of ciliogenesis. However, treatment of proliferating mIMCD3 cells and hTERT-RPE1 cells with a range of concentrations of antagonists against CRHR2, HTR1B and OPRL1 over 6-72 hours had no effect on ciliogenesis (data not shown). Inhibition of ligand binding was therefore not sufficient to inhibit cilia formation, suggesting that other mechanisms regulate ciliogenesis through GPCRs e.g. endocytic membrane trafficking20.
Figure 5. Ciliary localisation of G protein-coupled receptors.

(a) Localisation of selected indicated GPCRs (green) to proximal or basal regions of primary cilia (polyglutamylated α-tubulin; red) in differentiated SH-SY5Y neuronal cells. Magnified insets for selected cells (white arrowheads) are shown in white frames. Scale bar = 10μm. (b) Immunofluorescence (IF) labelling of MAS1 and OPRL1 (green) in the ciliary regions of photoreceptor cells (CR) (red; yellow arrowheads in magnified insets) in adult mouse retina. Abbreviations: GCL, ganglion cell layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer. Scale bar = 50μm (c) High magnification immunofluorescence for GPCRs (green) and centrin-3 (red) reveals localisation of GPR20, SREB3, and MAS1 at the adjacent centriole and the basal body (arrowheads) of the connecting cilium. Scale bars = 1μm (d) IEM reveals SREB3, MAS1, and HTR1B at the basal body (BB) of the connecting cilium (CC) (arrowheads). Immunoelectron microscopy of MAS1 shows additional labelling throughout the CC and at the ciliary tip (arrow; two different images of photoreceptors are merged at the dashed line). Abbreviations: CE, centriole; IS, inner segment; OS, outer segment. Scale bars = 400nm.
Validated screen hits PIBF1 and C21orf2 predict new ciliopathy disease genes for Joubert syndrome and Jeune syndrome
We next investigated whether our list of validated ciliogenesis effectors could be used to prioritise predicted pathogenic variants identified from WES of ciliopathy patients including Joubert syndrome and Jeune syndrome. Joubert syndrome (JBTS; OMIM #213300) represents a classic ciliopathy characterised by hypotonia, ataxia, cognitive impairment, and a distinctive brain malformation (the so-called “molar tooth sign”), with retinal dystrophy, cystic kidney disease, liver fibrosis and polydactyly occurring in subsets of patients21. Jeune asphyxiating thoracic dystrophy (JATD; Jeune syndrome, OMIM #611263) is a chondrodysplasia within the short-rib polydactyly syndrome ciliopathy spectrum, characterised by shortened limbs and ribs and a narrowed chest. Additional features include polydactyly, kidney cysts and renal failure, retinal degeneration and liver disease22.
A rare, homozygous missense variant (NM_006346.2: c.1910A>C, p.Asp637Ala) in PIBF1 (also known as C13orf24 or CEP90) was identified by WES in an affected JBTS individual of Schmiedeleut Hutterite descent (H1-3) and a similarly affected brother (H1-4; Figure 6a-c, Suppl. Figure 2a-c, Suppl. Table 5), in the absence of pathogenic mutations in known JBTS genes (Suppl. Table 5). The c.1910A>C variant segregated with the phenotype (Suppl. Figure 2b), was absent in the Exome Variant Server (EVS) and Exome Aggregation Consortium (ExAC) datasets, and was identified as homozygous in the affected individuals from a further three Hutterite families (H2, H3 and H4; Suppl. Figure 2b) suggesting a founder effect. Imaging findings for PIBF1-mutated individuals ranged from the classic molar tooth sign to moderate vermis hypoplasia with mildly thick superior cerebellar peduncles and characteristic superior cerebellar dysplasia (Figure 6a-c, Suppl. Figure 3, Suppl. Table 6). All individuals for whom information was available had ataxia and developmental delay, ranging from mild to moderate (Suppl. Table 7). Targeted molecular inversion probe sequencing (MIPS) of known JBTS genes and WES (Suppl. Table 5) identified a further seven families in which the affected individuals carried heterozygous truncating PIBF1 variants (out of 643 additional families sequenced) in the absence of pathogenic mutations in known JBTS genes. Although exogenous expression of human wild-type PIBF1 rescued ciliogenesis in mIMCD3 cells following siRNA knockdown of endogenous Pibf1, expression of PIBF1 containing the p.Asp637Ala Hutterite variant was unable to rescue normal ciliogenesis suggesting that it is a pathogenic missense mutation (Suppl. Figure 4).
Figure 6. Clinical features of ciliopathy patients with mutations in validated hits PIBF1 or C21orf2.

(a-c) Brain MRI findings for PIBF1-related Joubert syndrome in individual H1-4. Molar tooth sign with moderate vermis hypoplasia, elevated and thickened superior cerebellar peduncles (arrowhead), and superior cerebellar dysplasia (arrow) indicated. (a) is a T1-weighted and (b-c) are T2-weighted images. (d-l) Clinical features of individuals with C21orf2 mutations including narrow and deformed thorax in UCL 78.1 (d-f; child), UCL-111.1 (g; child) and CR-F024.1 (i-j; adult). UCL78.1 presented with a slightly dysplastic pelvis (h) in later childhood. Examples of fundoscopy images revealing mild pigmentary depositions and some mottling shown for CR-F024.1 (k) and 111.2 (l); both patients were clinically blind at the time of examination. (m) Upper panel: schematic of the PIBF1 protein (NP_006337.2) showing the approximate positions of coiled coil domains (green boxes) and the p.Asp637Ala mutation. Lower panel: schematic of the C21orf2 protein (NP_004919.1) showing the positions of three leucine-rich repeats (LRR1-3, blue boxes), an LRRCT motif occurring C-terminal to LRRs (green box) and mutations.
We next cross-compared WES data from patients with JATD to validate ciliogenesis candidate genes from the screen. Compound heterozygous missense changes in C21orf2 (NM_004928.2: c.218G>C, p.Arg73Pro and c.671T>C, p.Leu224Pro) were identified in two affected siblings of non-consanguineous Caucasian northern European descent (UCL-111.1 and UCL111.2; Suppl. Table 5, Suppl. Figure 5a) with a clinical diagnosis of JATD. Compound heterozygous mutations (p.Leu161Serfs*9 and predicted intronic splice site mutation c.96+6T>A) were also found in a non-consanguineous Caucasian northern European individual UCL78.1 (Suppl. Table 7, Suppl. Figure 5f). All variants were either very rare or absent from the EVS and ExAC datasets, predicted damaging by Polyphen2 and/or Mutationtaster, and/or were non-conservative changes in highly conserved regions (see On-line Methods for further details). The three affected individuals presented with narrow thorax (Figure 6d-g,i,j, Suppl. Table 7) and pelvic bone malformation (Figure 6h) in skeletal radiological surveys. All developed retinal degeneration due to cone-rod dystrophy in childhood but none had impairment of renal function (Figure 6k,l, Suppl. Table 7). Further screening of C21orf2 identified a homozygous missense change shared with the UCL-111 family (p.Arg73Pro) in individual GC4693.1 and four siblings, all of whom have cone-rod dystrophy, severe scoliosis and hip dysplasia (Suppl. Figure 5b, Suppl. Table 7). Sanger sequencing confirmed that all variants segregate with the disease (Suppl. Figure 5a-f). Exogenous expression of both the p.Arg73Pro and p.Leu224Pro variants in C21orf2 partially rescued ciliogenesis in mIMCD3 cells following siRNA knockdown of endogenous C21orf2, suggesting that they are hypomorphic mutations (Suppl. Figure 4).
Homozygous mutations in C21orf2 were also found in patients that initially presented with isolated cone-rod dystrophy (Figure 6l), consistent with a previous report that C21orf2 is mutated in non-syndromic retinal degeneration23. Re-assessment of these individuals revealed previously unidentified skeletal involvement in CR-F024.1 (c.103delA, p.Ile35Phefs*10; Figure 6I,j, Suppl. Table 7, Suppl. Figure 5c). These findings indicate that mutations in C21orf2 (Figure 6m) cause both syndromic and non-syndromic forms of retinal dystrophy, with a variable spectrum of skeletal involvement.
The validated screen hit C21orf2 forms a ciliary functional module with NEK1 and SPATA7
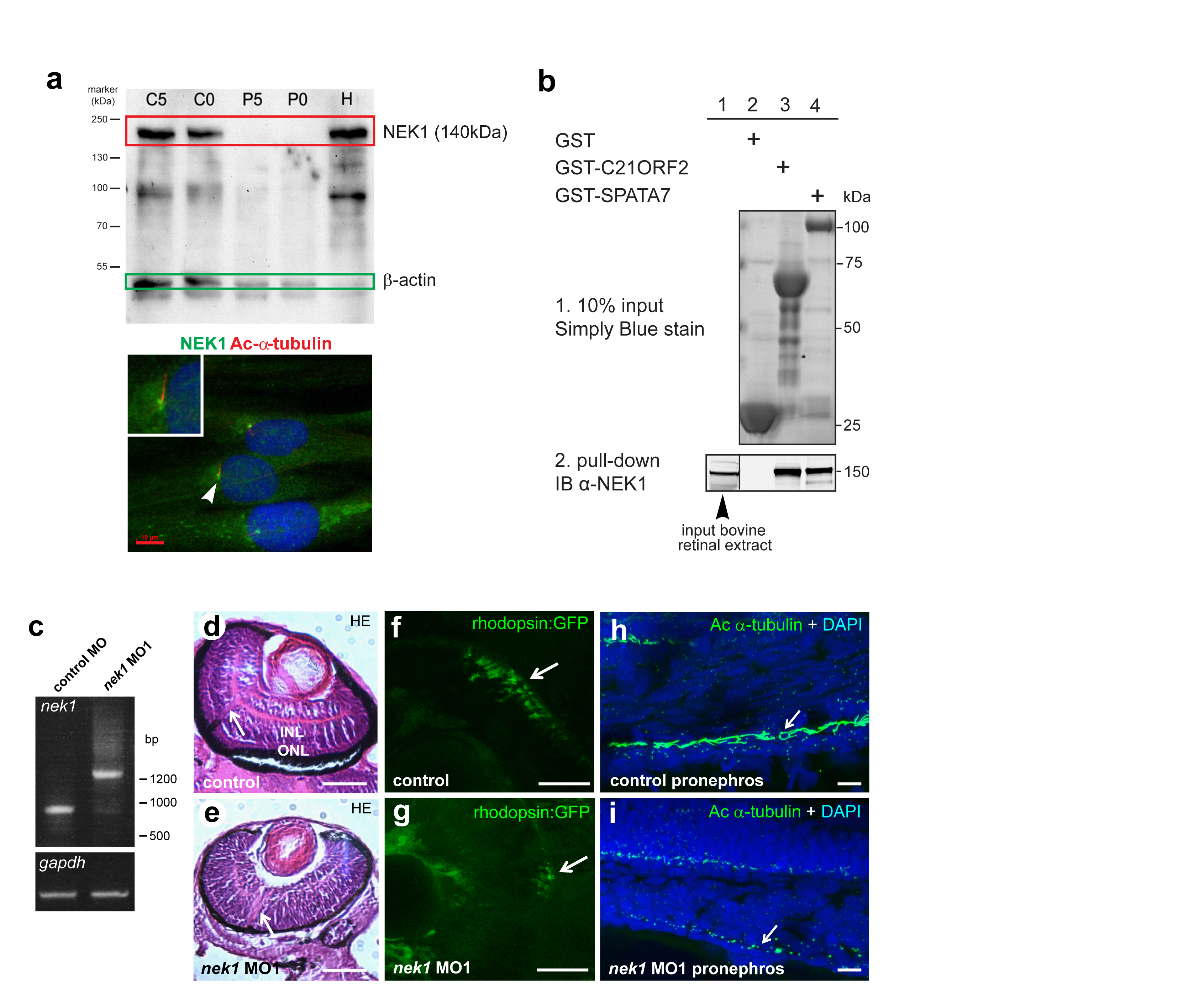
C21orf2 (also known as LRRC76) is an LRR-containing protein conserved from C. elegans to human (Figure 6m). Although a small-scale siRNA screen suggested that knockdown impairs ciliogenesis and Hedgehog (Hh) signalling in mammalian cell cultures5, the exact role of this protein in ciliogenesis is unclear. C21orf2 localises to the basal body in mIMCD3 and hTERT-RPE1 cells and to the base of the connecting cilium in mouse photoreceptors (Figure 3d, Suppl. Figure 6a). Immunostaining confirms expression in tissues consistent with the phenotype of JATD, including developing ribs and hip joint in mouse embryos (Suppl. Figure 6b). Tandem affinity purification followed by mass spectrometry (MS) analysis identified a total of 88 unique proteins co-purifying with Strep-tag II/FLAG (SF-TAP) tagged C21orf2 in n≥2/3 experiments (Suppl. Table 8). Affinity proteomics (Figure 7a) and co-immunoprecipitation assays (Figure 7b,c) revealed that C21orf2, NEK1, and a retinal ciliopathy protein, SPATA724, are part of the same protein complex. Furthermore, co-immunoprecipitations with anti-NEK1 (Suppl. Figure 7a) followed by MS analysis of unique peptides (for three concurrent repeats of the experiment) showed that NEK1 pulled down endogenous C21orf2 from bovine retinal lysates (Suppl. Table 9). Immunofluorescence studies showed that C21orf2 co-localises with NEK1 and SPATA7 at the basal body in hTERT-RPE1 cells (Suppl. Figure 6c), and GST pull-down of SPATA7 from bovine retinal lysates efficiently recovered endogenous C21orf2 and NEK1 (Suppl. Figure 7b, Suppl. Table 9). Collectively, these data indicate the existence of a ciliary functional module that includes NEK1-C21orf2-SPATA7, which occurs endogenously in ciliated cells (Suppl. Table 9). Importantly, both the p.Arg73Pro and p.Leu224Pro mutations in C21orf2 abolished the interaction with NEK1 (Figure 7c).
Figure 7. Validated hit C21orf2 forms a ciliary functional module with NEK1 and SPATA7.

(a) Interactions between C21orf2, NEK1 and SPATA7 were identified by tandem affinity purification. The values indicate sequence coverage of identified prey peptides (horizontal) co-purified with specific bait proteins (vertical); see Suppl. Table 8 for details. (b) Reciprocal coimmunoprecipitation of HA-C21orf2 and both NTAP-NEK1, and NTAP-SPATA7 (5% input: panels 1 and 2; immunoprecipitation (IP) assays in panels 3 to 6), but not the negative control HA-LRRK2 (LRR; panels 4 and 6: lanes 2 and 4) following immunoblot (IB) detection of either the HA or FLAG (Strep-tag II-FLAG, NTAP) epitopes. (c) FLAG-NEK1 coimmunoprecipitates wild-type (WT) C21orf2 (panel 4, lane 1; indicated by arrow), but not p.Arg73Pro or p.Leu224Pro mutant C21orf2 (panel 4, lanes 2 & 3) or the negative control HA-LRRK2 (LRR; lanes 4 to 6). Positive control co-IP (NPHP4-RPGRIP1L) is in lane 7, 10% input is shown in panels 1 and 2 and IP assays in panels 3 to 6. IgG heavy chain (HC) is indicated in panel 4. (d) Antisense morpholino oligonucleotide (MO1) injection against zebrafish nek1 resulted in ciliopathy phenotypes (ventrally curved body axis, hydrocephalus, smaller eyes and otolith defects at 3-4 dpf; upper and middle panels, otoliths indicated by arrowheads; scale bars = 500 or 50 μm, respectively). Lower panels: cartilage visualisation using Alcian Blue staining 4.5 dpf revealed impaired craniofacial cartilage development in nek1 morphants compared to control MO-injected embryos. These defects resembled, but were less severe, than those for zebrafish smo mutant embryos. Scale bar = 200 μm. (e) Co-injection of 1.5ng nek1 MO1 with 150pg human wild-type (WT) C21orf2 RNA (three independent experiments, total n=76 embryos) caused significant but incomplete phenotypic rescue, whereas RNA expressing the C21orf2 missense mutation p.Arg73Pro (C21orf2-p.Arg73Pro; three experiments, total n=72 embryos) rescued less effectively compared to negative control (#). Co-injection of 150pg C21orf2-p.Leu224Pro (three experiments, total n=91 embryos) had a marginal effect on rescue. Significance of pair-wise comparisons with the negative nek1 MO1-only control (#), or other comparisons indicated by braces, are: n.s., not significant; * p<0.05, ** p<0.01, **** p<0.0001 (unpaired two-tailed Student’s t-test).
The association of C21orf2 with NEK1 could provide an explanation for the variable JATD skeletal phenotype observed in C21orf2-mutated individuals (Figure 6d-j), since mutations in NEK1 cause short-rib polydactyly syndrome type II (SRPS type II; Majewski syndrome, OMIM #263520) in humans25, a ciliary chondrodysplasia related to JATD. Furthermore, the association of C21orf2 with SPATA7 could explain the prominent retinal phenotype (which tends to be only an occasional feature of typical JATD) in individuals with C21orf2 mutations, since mutations in SPATA7 cause the retinal dystrophies Leber congenital amaurosis type 3 (LCA3) and juvenile RP (OMIM #604232). Knockdown of nek1 in zebrafish using antisense morpholino oligonucleotides (MO; verified in Suppl. Figure 7c) produced classical ciliopathy phenotypes, with a ventrally curved body axis, hydrocephalus, otolith abnormalities and abnormal craniofacial cartilage development (Figure 7d). Knockdown of nek1 also resulted in retinal defects including loss of photoreceptors (Suppl. Figure 7fd-g) and cilia length defects in the pronephros (Suppl. Figure 7h,i). Co-injection of human wild-type (WT) C21orf2 RNA with the nek1 MO partially rescued these phenotypes (Figure 7e), consistent with the functional interaction between C21orf2 and NEK1. RNA expressing C21orf2 with the missense mutation p.Arg73Pro rescued the nek1 morphant phenotype less effectively than wild-type, whereas C21orf2-p.Leu224Pro had little effect (Figure 7e), confirming the predicted hypomorphic effect of both variants observed in vitro (Suppl. Figure 4).
DISCUSSION
Here we present a whole genome siRNA-based functional genomics screen to identify genes whose activity is required for ciliogenesis and/or cilia maintenance. By interrogating every gene for a role in these processes, we obtained a global resource for investigation and interventions into the processes that are critical for the ciliary system. Our visual assay was primarily designed to test the presence or absence of cilia in the primary screen, and we provide all numerical features for this cellular phenotype as a functional resource to investigate aspects of cilia biology (Suppl. Table 1). In addition, we also imaged other cellular phenotypes during data acquisition (cilia intensity and nuclear morphology) and include these other numerical features (Suppl. Table 1) to enable the scientific community to interrogate and further annotate our screen data-set.
To minimise the false positive rate, we extensively characterised a widely used cellular model system (ciliated mIMCD3 cells) by arrayCGH, and our siRNA library was filtered for off-target effects, partial on-target effects and microRNA-like effects (Figure 2c, Suppl. Table 1). Using stringent filters and robust statistical cut-offs, we identified 194 genes which significantly affected ciliogenesis and were of particular functional interest, and took these on for validation by further knock down experiments in mIMCD3 and hTERT-RPE1 cells.
In total, we present a high confidence list of 68 validated genes that play significant roles in ciliogenesis in mIMCD3 cells, of which 37 were further found to affect ciliogenesis in hTERT-RPE1 cells (Table 1, Suppl. Table 3). A parallel secondary screen validated a further 44 genes encoding components of the UPS (Suppl. Table 4). Validated hits include CEP120, KCNQ1, several GPCRs (CRHR2, HTR1B and OPRL1) and PRPFs (PRPF6, PRPF8, PRPF31 and PRPF38A) (Figure 3a-b, Table 1, Suppl. Table 3). The specificity of the screen for targeting mediators of ciliogenesis is demonstrated by the number of genes that have been independently implicated in this process. These include CCDC4112, OFD113, CEP12016 and PIBF126. C21orf2 has been suggested to have a role in ciliogenesis from a previous small-scale siRNA screen5. In a complementary functional genomics study, PLK4, CEP120, PRPF8 and PRPF38A have all been implicated in the process of centriolar under-duplication11. Furthermore, PLK4, OFD1, CDC27, PRPF6 and PRPF8 were identified in the Mitocheck screen as important effectors of cell division27. Shared hits across multiple independent functional genomics studies indicate that our screen is a useful resource for the identification of proteins and functional modules that are essential for cilia biology and related cell biological processes.
This study also demonstrated the power of an unbiased screen to discover cellular or molecular processes that play a role in cilium biology. To illustrate this point, our screen unveiled neuroactive GPCRs as one of the functionally enriched groups of proteins affecting ciliogenesis (Figure 3a, Table 1, Suppl. Figure 1). GPCRs may play sensory or signalling roles in the ciliary vesicle or procilium at the earliest stages of ciliogenesis both in the developing brain and in other tissues28. In support of this hypothesis, adenylyl cyclase III co-localises with GPCRs at proximal ciliary regions in the developing neocortex, and over-expression of specific ciliary GPCRs, including the 5-HT6 serotonin receptor, in cortical neurons causes cilia elongation29.
Although the role of the PRPFs in the spliceosome is well-known and well-studied, we identified a group of seven splicing factors including PRPF6, PRPF8, PRPF31 and PRPF38a (Table 1) that are required for ciliogenesis. We demonstrate that PRPFs mutated in retinitis pigmentosa localise specifically to the base of the primary cilium in several different cell-types (Figure 3d, Figure 4a) and to the basal body complex and connecting cilium in photoreceptors (Figure 4b,d). We speculate that some of the splicing factors fulfil an additional ciliary function independent of their nuclear role in splicing. In support of this notion, two previous studies of the centrosomal proteome have suggested that splicing factors, including two of the splicing factors identified in our screen (PRPF6 and PRPF8), may be true centrosomal proteins30,31. However, it seems less speculative to hypothesise that these proteins may be required for the correct splicing of an unidentified subset of genes that are involved in cilia formation. In support of the possible involvement of pre-mRNA processing factors in ciliogenesis, previous screens have identified that several splicing factor hits are important in related cellular processes of microtubule formation and regulation, namely centriolar biogenesis (PRPF8, PRPF38A)11 and cell division (PRPF6, PRPF8)27.
Several hits in our screen have a known function in the ubiquitin proteasome system (UPS), reflecting an importance of specific proteostasis mechanisms in mediating or regulating ciliogenesis which could be mediated through, for example, ubiquitination or SUMOylation of ciliary transcription factors or chaperoning of ciliary proteins to the base of the cilium. Remarkably, 5/7 of our splicing factor hits (LSM2, PRPF6, PRPF8, PRPF31 and USP39) are implicated in the ubiquitin-dependent regulation of the spliceosome32. It is interesting to note that the interaction of PRPF8 with ubiquitinated PRPF3 is regulated by the deubiquitinating enzyme USP4, and that loss of USP4 prevents the correct splicing of mRNAs including those for α-tubulin32. In support of this mechanism, we observe that a viable C. elegans strain possessing a splice site mutation in PRPF8/prp-8 has structural and numerical defects of axonemal microtubules in the primary cilia of amphid sensory neurons (Figure 4f). Thus, UPS and/or PRPF proteins could act as multifunctional “nexus molecules” that are involved in multiple aspects of proteostasis of ciliary proteins or their trafficking (for example, the ubiquitin-mediated internalisation and endosomal sorting of GPCRs). Alternatively, or additionally, these specific PRPFs could also ensure the correct splicing of transcripts encoding proteins important for ciliogenesis, including structural components of the cilium such as α-tubulin32,33.
We also show the specificity and clinical utility of the screen as a tool for disease-gene discovery. When combined with large variant datasets, for example WES of ciliopathy patients, the functional data from our siRNA screen allowed the filtering and prioritisation of variants to identify pathogenic mutations. Although previous studies have indicated both PIBF1 and C21orf2 as functional candidate genes for ciliopathies5,23, we formally demonstrate the utility and validity of a systems biology approach by the identification of mutations in these genes in JBTS and JATD patients, respectively (Figure 6, Suppl. Table 7). Furthermore, our affinity proteomics data provides an explanation of the mechanism for the variable phenotype in this form of JATD. We propose that the retinal phenotype observed in C21orf2-mutated patients may result from a dysfunctional SPATA7-C21orf2-containing module in the photoreceptors of these patients. Conversely, the skeletal phenotype observed in both C21orf2- and NEK1-mutated human patients probably has a common origin in the disruption of the C21orf2-NEK1 interaction. NEK1 is a serine-threonine kinase and two serines (positions 136 and 177) in C21orf2 have been previously found to be phosphorylated34. In light of this, our affinity proteomics findings, and the incomplete rescue of NEK1 knockdown effects in zebrafish by C21orf2 overexpression, C21orf2 might represent a substrate of NEK1.
In conclusion, our findings highlight the utility of combining different systems biology approaches, namely high-content functional genomic screening, WES, and affinity proteomics. Our approach has identified ciliary roles for well-studied proteins, identified disease-causing genes, allowed the refinement of patient phenotypes, and highlighted potential disease pathways that could provide deeper insights into cilium biology. This work demonstrates the utility of a systems biology approach in providing a compendium of candidate genes and offering insights into pathogenic mechanisms for an increasingly important group of Mendelian conditions, the ciliopathies.
ON-LINE METHODS AND MATERIALS
Human subjects
All samples used in this study were obtained with informed consent according to the protocols approved by the ethical committees of the Institute of Child Health/Great Ormond Street Hospital (REC ref. no. 08/H0713/82), South Yorkshire Research Ethics Committee (11/H1310/1), King Faisal Hospital, Saudi Arabia (RAC# 2070 023), and the University of Washington, Seattle WA (Protocol #28853). Informed consent was obtained for the publication of all patient photographs.
Cell culture
Mouse inner medullary collecting duct (mIMCD3), human telomerase reverse transcriptase-transformed retinal pigment epithelium (hTERT-RPE1) and human neuroblastoma (SH-SY5Y) cells were derived from American Type Culture Collection (ATCC). The genomic status of the cell-lines were assessed by array CGH and karyotyping, and SH-SY5Y cells were authenticated by STR profiling (May 2013). All cell-lines were tested every three months for mycoplasma. Cell-lines were maintained in DMEM/Ham’s F12 medium supplemented with 10% foetal calf serum (FCS), under standard conditions (37°C, 5% CO2). Cells were passaged at a split ratio of 1:10 twice a week, with low passages (<25) for both mIMCD3 and hTERT-RPE1 cells. mIMCD3 cells were obtained from ATCC at passage 13 and were used for screening purposes between passage 17 and 25. SH-SY5Y cells were differentiated over 6 days by retinoic acid (RA) treatment followed by brain derived neurotrophic factor (BDNF) treatment. Cells were plated in standard culture medium for 24 hours, in standard medium containing 10μM RA for 3 days and in serum-free medium containing 50ng/ml BDNF cells for 3 days. Human dermal fibroblasts were derived from skin biopsies and cultured in fibroblast growth medium (Genlantis). hTERT-RPE1 cells were serum starved in normal media with 0.2% FCS for 48 hours to induce ciliogenesis.
Mice
C57Bl/6J wild-type mice and the B6;129P2-Tmem67tm1Dgen/H line were kept on a 12 h light-dark schedule with unlimited access to food and water. For all experiments a minimum of n=3 wild-type animals were used, and, where required, compared to n=3 age-matched Tmem67−/− knock-out mutants from the same litter. For retinal immunofluoresence microscopy studies, adult mice (n=2 male, n=2 female) were used at post-natal day 30 (P30) as described previously35. Tmem67 mouse embryos were used at embryonic ages E14.5 or E18.5, but unselected for gender since this has no influence on mutant phenotype, localization of transition zone proteins or dysregulation of ciliary signalling19. No statistical method was used to predetermine sample size, the experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment. All procedures were in accordance with local laws on animal protection, the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and under the guidance issued by the Medical Research Council in Responsibility in the Use of Animals for Medical Research (July 1993) in accordance with UK Home Office regulations under the Project Licence no. PPL40/3349.
Plasmids
Full-length Gateway Entry clones for human C21orf2 (IOH23207), SPATA7 (IOH62585), PRPF6 (IOH3880), PRPF31 (IOH41469) and PRPF38A (IOH46080) were kindly provided by Nico Katsanis (Duke University, Durham, USA). Human PIBF1 (kindly provided by Kym Boycott) and NEK1 (NM_012224.2, kindly provided by Andreas Gießl) were cloned into the Gateway system (LifeTechnologies). Full-length NPHP4, amino acids 611-1286 of RPGRIP1 and the LRR domain of LRRK2 constructs were generated as previously described31 and cloned into the Gateway system. Entry clones were sequence-verified and further cloned into HA-, GST-, FLAG-, mRFP, eCFP and strep-II tag/FLAG-tandem affinity purification (SF-TAP)-tagged destination vectors using the Gateway system. Missense mutations for PIBF1 and C21orf2 were introduced with the Q5 Site-Directed Mutagenesis Kit (New England Biolabs), using conditions recommended by the manufacturer.
Generation of hNEK1 specific antibody
A 444 bp hNEK1 (NM_012224) cDNA (hNEK1 H3T1; 1759-2202 bp, encoding amino acids 587-734)36 was cloned into the pGEX-4T3 expression vector (GE Healthcare Life Sciences) using BamHI and XhoI restriction sites. Expression and purification of the GST fusion protein was performed according to the manufacturer’s instructions (GE Healthcare Life Sciences). After cleavage of the fusion protein with thrombin on the column, hNEK1H3 was eluted with 1 × PBS followed by peptide immunization of rabbits by a commercial supplier (Pineda Antikörper-Service, Berlin, Germany). Polyclonal antisera against recombinantly-expressed hNEK1H3T1 were affinity-purified on high trap N-hydroxysuccinimide columns (HiTrap NHS-Activated HP; product code:17-0716-01; GE Healthcare Life Sciences). Validation experiments for antisera are shown in Suppl. Figure 7a.
Antibodies and staining reagents for immunofluorescence
The following antibodies were used with the indicated titres for immunofluorescent staining: rabbit anti-PRP6 (Santa Cruz sc-48786) 1:100, mouse anti-PRPF8 (Santa Cruz sc-55534) 1:100, rabbit anti-PRPF8 (Santa Cruz sc-030207) 1:100, goat anti-PRPF31 (Abnova PAB7154) 1:100, PIBF1/CEP90 (Novus NBP2-19823) 1:100, rabbit anti-PLK4 (Cell Signalling Technologies 3258) 1:100, rabbit anti-GPR20 polyclonal (Thermo Scientific, PA3-068) 1:100, rabbit anti-SREB3 (Novus Biologicals NBP1-71064) 1:100, rabbit anti-MAS1 (Novus Biologicals NBP1-78444) 1:100, rabbit anti-HTR1B (Novus Biologicals NBP1-01013) 1:100, rabbit anti-P2RY14 (Novus NBP1-70970) 1:100, rabbit anti-CRHR2 (Novus NBP1-00767) 1:100, rabbit anti-OPRL1 (Abcam ab66219) 1:50, rabbit anti-C21orf2 (GeneTex [N1C2], GTX119046) 1:100, mouse anti-acetylated alpha tubulin (Sigma Aldrich clone 6-11 B1 T6793) 1:1000, rat anti-ZO1 (1:400, Santa Cruz), rabbit anti-β-catenin (1:400, BD Bioscience), rabbit anti-gamma tubulin (AbCam ab11317) 1:1000, rabbit anti-ARL13B (Proteintech 17711-1-AP) 1:1000, mouse anti-GT335 monoclonal (Enzo Life Sciences ALX-804-885-C100) 1:1000 and mouse anti-FLAG (Sigma F3165) 1:1000. The mouse monoclonal antibody against centrin-3 has been described previously35. Secondary antibodies were Alexa-Fluor-conjugated goat anti-mouse IgG and goat anti-rabbit IgG (Life Technologies Ltd.)
siRNA rescue experiments in mIMCD3
Rescue experiments followed our standard siRNA transfection and image analysis protocols for pooled siRNA mouse siGENOME siRNAs (Thermo Scientific) in mIMCD3 cells for either Pibf1 or C21orf2. Reverse transfections in 96-well assay plates (PerkinElmer View Plates) included 100ng of wild-type or missense mutated FLAG-tagged human PIBF1 or C21orf2 constructs. Our standard high-content image analysis protocol was adapted to include immunofluorescent staining with anti-FLAG to enable quantitation of transfection efficiency.
Immunofluorescence and confocal microscopy of hTERT-RPE1 and mIMCD3 cells
hTERT-RPE1 cells were cultured as described above. To induce ciliogenesis, cells were seeded on coverslips and serum starved (culture medium with 0.2% FCS) for 24 hrs prior to transfection. Subsequently, Lipofectamine2000 (LifeTechnologies) was used to transfect cells with the appropriate vector according to the manufacturer’s protocol. 24 hrs post-transfection, ciliated cells were fixed in 2% PFA for 20 minutes, treated with 1% Triton X-100 in PBS for 5 minutes, and blocked in 2% bovine serum albumin (BSA) in PBS for 30 minutes. Fixed cells were stained for 1 hour with the corresponding primary antibodies: rabbit anti-FLAG (Sigma-Aldrich) 1:1000 and mouse anti-acetylated alpha tubulin (Sigma-Aldrich) 1:1000. Coverslips were then washed in PBS and stained for 45 minutes with AlexaFluor-405 or AlexaFluor-647-conjugated secondary antibodies (1:500, LifeTechnologies). Coverslips were washed again with PBS and briefly with milliQ water before mounting in Vectashield with or without DAPI (Vector Laboratories). mIMCD3 cells were seeded at 2.5×105 cells/well on sterile glass coverslips in six-well plates and fixed in ice-cold methanol. Cells were blocked in 1% non-fat milk for 5 min each. Immunofluorescence staining was performed as described previously37.
Confocal images were obtained using a Zeiss Axio Imager ZI fluorescence microscope (Zeiss), equipped with a 63x objective oil lens, or a Nikon A1R confocal microscope with x100 oil objective lens controlled by NIS-Elements AR 4.20.01 (Nikon) software. Optical sections were generated through structured processed using Axiovision 4.3 (Zeiss) or NIS-Elements AR 4.20.01 (Nikon) software. Images were analysed using Adobe Photoshop CS and FIJI software. Images were assembled with Adobe Illustrator CS.
mIMCD3 spheroid assay
1×104 mIMCD3 cells per well were seeded in a 48 well-plate and transfected using Lipofectamine RNAiMax (LifeTechnologies) according to the manufacturer’s protocol. Cells were trypsinized 24 h post-transfection and resuspended cells were then mixed 1:1 with growth factor-depleted Matrigel (BD Bioscience) in Labtech chamber slides. After polymerization for 20 min at 37°C, warm medium was dripped over the matrix until just covered. The mIMCD3 cells formed spheroids with cleared lumens 3 days later. Medium was removed by pipetting and the gels were washed three times for 10 min with warm PBS supplemented with Ca2+ and Mg2+. The gels were then fixed in fresh 4% PFA for 30 min at room temperature. After washing three times in PBS, the cells were permeabilized for 15 min in gelatin dissolved in warm PBS (350 mg/50 ml) with 0.5% Triton X-100. Primary antibody (mouse anti-acetylated tubulin, Sigma, 1:20,000), rat anti-ZO1 (1:400, Santa Cruz) and rabbit anti-β-catenin (1:400, BD Bioscience) was diluted in permeabilization buffer and incubated overnight at 4°C. After washing the spheroids three times for 30 min in permeabilization buffer, goat anti-mouse-Cy5, donkey anti-rabbit-TRITC, and goat anti-rat-FITC secondary antibodies (1:500, LifeTechnologies) were each diluted in permeabilization buffer and incubated with the spheroids for 4 hr at room temperature. Spheroids were washed three times in permeabilization buffer for 10 min per wash and then incubated for 1 hr with DAPI, before being washed an additional three times in PBS, and finally mounted in Fluoromount-G (Cell Lab, Beckman Coulter). Images and z-stacks were taken with a Zeiss LSM700 confocal microscope. Cilia were counted manually from maximum projections of z-stacks, with axonemes >1μm in length scored as positive, as described previously37.
Confocal immunofluorescence microscopy and high resolution immunofluorescence analysis of mouse photoreceptor cells
Images were obtained using a Nikon Eclipse A1 scanning confocal microscope, controlled and processed using Nikon Elements software. For high magnification and high resolution immunofluorescence analysis eyes of C57Bl/6J mice were cryofixed, sectioned, and immunostained as previously described38,39 for two independent experiments. Cryosections were double stained for PRPF6, PRPF8 (A5463), GPR20, MAS1, SREB3, HTR1B (1:200) and centrin-3 (1:100) as a molecular marker for the connecting cilium, the basal body, and the adjacent centriole of photoreceptor cells40. Subsequently, cryosections were incubated with the according secondary antibodies conjugated to Alexa 488 or Alexa 555 (LifeTechnologies), counterstained with DAPI (Sigma-Aldrich), and mounted in Mowiol 4-88 (Hoechst). Specimens were analysed and deconvoluted in a Leica LEITZ DM6000B microscope (Leica). Images were processed with Adobe Photoshop CS using different tools including contrast and colour correction as well as pixel extrapolation.
Immunoelectron microscopy of mouse photoreceptor cells
For immunoelectron microscopy of mouse photoreceptor cells, the pre-embedding labelling protocol was used, as described previously41,42. Ultrathin sections were analysed in a transmission electron microscope (TEM) (Tecnai 12 BioTwin, FEI). Images were obtained with a charge-coupled device camera (SIS MegaView3, Olympus) and processed with Adobe Photoshop CS.
Transmission electron microscopy on C. elegans amphid channel cilia
TEM was performed on the amphid channel cilia of strain MR247: prp-8(rr40);cdc-25.1(rr31) unc-13(e450); rrIs01. This strain was obtained in a forward genetic screen for suppressors of the cell cycle phosphatase cdc-25.1(rr31), which is a dominant maternal effect gain-of-function missense allele (G47D) sensitizing the intestinal lineage to an extra cell division43. The rr40 suppressor is a homozygous donor splice site mutation (hypomorphic) in intron 8 of prp-8. MR247 also contains an undefined (likely missense) mutation (e450) in the neurotransmitter vesicle release regulator unc-13 (used to map the prp-8(rr40) mutation), and an integrated intestinal GFP marker (rrIs01)44. Although the prp-8 splice-site mutation is the likely cause of the ciliogenesis defect in this strain, we cannot formally exclude the possibility that a dominant missense mutation in cdc-25.1 or an undefined (likely missense) mutation in unc-13 could be causative for these defects. Young adult worms were processed and imaged essentially as described previously45, except that MR247 worms were fixed overnight at 4°C in 2.5% gluteraldehyde and 1% paraformaldehyde in Sørensen’s buffer.
Confirmation of siRNA knockdown by qRT-PCR
QRT-PCR was conducted as previously described46. The following primers were used: Plk4: 5′-CGTTGTCACTCAGTAGAAATGCT, 5′-GGTGTCTGGTCTGCAAATGG; Prpf6: 5′-AAGCTGACTCCTGTTCCTGA, 5′-TCATTCCACCTGGGTATGGAG; Prpf8: 5′-ACTGGAAGCCATTCAGCTAGAG, 5′-AATGTGAACTGCCAGCGTTG; Prpf31: 5′-TATCAGCAAGCAAGCCAACG, 5′-CTATCTCCACAGTCAGGTTGTTG; Prpf38a: 5′-AAGAGCCAGAACCGAAATGG, 5′-GACTCTCTCACTGTGCAGGA; Pibf1: 5′-CCCAGAAAAACATGATGATCGAC, 5′-CTAATCGCAAAGTTAGCAGCTG; C21orf2: 5′-ACCTGCTGAGCTACACAGAG, 5′-TTCTTGTGACTCCTCATCGTGT; β-actin: 5′-GCTTCTTTGCAGCTCCTTCGT, 5′-AGCGCAGCGATATCGTCAT.
Western blotting
Whole cell extract and western blots were conducted as previously described46. Antibodies were used at the following concentrations: rabbit anti-PRPF6 (Santa Cruz sc-48786) 1:500, mouse anti-PRPF8 (Santa Cruz sc-55534) 1:200, rabbit anti-PRPF8 (Santa Cruz sc-030207) 1:200, goat anti-PRPF31 (Abnova PAB7154) 1:500 , rabbit anti-PLK4 (Cell Signalling Technologies 3258) 1:500, rabbit anti-C21orf2 1:500 and rabbit anti-NEK1 (H3T1; validated in Suppl. Figure 8a) 1:2000 incubated overnight at 4°C; and mouse anti-FLAG (Sigma Aldrich clone M2 F9291) 1:2000, mouse anti-HA (Sigma Aldrich H9658) 1:1000 and mouse anti-β actin (Abcam ab6276) 1:5000 incubated for 1 hour at room temperature. Membranes were incubated with HRP-conjugated secondary antibodies (Dako Cytomation P0447 and P0448) for 1 hour at room temperature and immunopositive bands visualized with Supersignal West Femto chemiluminescent substrate (Pierce) on a BioRad ChemiDoc MP system. Full unprocessed western blotting scans are shown in Suppl. Figure 9, with the regions used in main display items indicated by red frames.
Zebrafish experiments
Zebrafish husbandry was undertaken to UK Home Office Rules under standard conditions. Antisense morpholino oligonucleotide (MO; Gene Tools Inc.) for nek1 (ENSDART00000035239 zv9) were designed against the splice donor sites of exon 6 and exon 8 (exon 6 sequence MO1: AGCATTAAAACACATCTGACCTATT, exon 8 sequence MO2: GATAAGAAAGACAAGCGCACCTTCC). MO injection into embryos was performed at the one- to two-cell stage and embryos were incubated at 28.5°C. Effective doses were defined as the MO concentration that resulted in the least dead fish and the most fish with the phenotypes of interest. nek1 knockdown resulted in a marked embryonic lethality of about 25% even at low doses due to developmental defects during gastrulation. MO specificity was confirmed by RT-PCR (Suppl. Figure 7c). RNA was extracted at 48 hpf using TRIzol (LifeTechnologies). First-strand complementary DNA was synthesized using random nonamers (Sigma-Aldrich) and Omniscript transcriptase (Qiagen). Standard PCR was carried out using primers in exons 4 and 10 for the nek1 exon 6 splice MO (expected wild-type product size approx. 750 bp, intron 6/7 length 575 bp), with 60 bp. RT-PCR result suggesting the inclusion of the downstream intron (Suppl. Figure 7c). Amplification of gapdh was used as a quantification control. Primer sequences are available on request. For rescue experiments, 150 pg of human C21orf2 RNA were co-injected with nek1 MO and embryos assessed for phenotypic features 3 dpf by light microscopy. RNA was generated using the mMessage Machine kit (Ambion) according to the manufacturer’s protocol.
For whole-mount immunofluorescence analysis, embryos were dechorionated and fixed in 100% methanol overnight at −20°C. After rehydration, embryos were washed several times in PBST (0.5% Triton X-100/PBS) then blocked in 5% BSA for 1 hour and incubated with anti-acetylated tubulin antibody (Sigma) overnight at 4°C followed by incubation with anti-mouse IgG 488 secondary antibody (Sigma) and staining with DAPI. Embryos were flat mounted on glass slides in Citifluor (Citifluor Ltd) and immunofluorescence analysis was performed using a Zeiss confocal microscope. Image analysis was performed using ImageJ software. Images represent z-stack maximum intensity projections. Alcian Blue staining for cartilage visualization was performed after over-night incubation of dechorionated embryos in 100% methanol at −20°C using a 1% Alcian Blue 70% acidic ethanol solution. Histology of the eye was performed using paraffin embedded embryos and a standard H&E staining protocol. Transgenic zebrafish lines included rhodopsin:GFP (in house heterozygous out-cross of cu3Tg to TULP long fin wild-type) and smo mutant (in-house heterozygote × heterozygote cross of smob641/b641).
Tandem affinity-purification and mass spectrometry
HEK293T (human embryonic kidney) cells were transfected with constructs encoding either C21orf2, or NEK1, or SPATA7 fused to a strep-II tag/FLAG tag for tandem affinity purification (SF-TAP-tag) using polyethylenimine (PEI, PolySciences) as a transfection reagent. 48 hours after transfection the cells were lysed in lysis buffer containing 30mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5% Nonidet-P40 (NP40), freshly supplemented with protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany), phosphatase inhibitor cocktail 2 (Sigma-Aldrich) and PhosphataseArrest III (GBiosciences) for 20 minutes at 4°C. The streptavidin- and FLAG-based tandem affinity purification steps were performed as previously described47. 5% of the final elute was resolved by SDS-PAGE and analysed by silver staining according to standard protocols while the rest was subjected to protein precipitation with chloroform and methanol. Protein precipitates were subsequently subjected to mass spectrometry analysis and peptide identification as previously described47.
Exogenous co-immunoprecipitation
Plasmids expressing N-terminal SF-TAP-tagged SPATA7 and N-terminal SF-TAP-tagged NEK1 were co-transfected with plasmids expressing HA-tagged wild-type C21orf2 or mutant constructs C21orf2-p.Arg73Pro or C21orf2-p.Leu224Pro in HEK293T cells. A plasmid expressing functionally unrelated HA-tagged LRR (leucine-rich repeat) domain of LRRK2 was used as a negative control. FLAG-NPHP4 and HA-RPGRIP1 (aa611-1286) were included as positive control as described previously47. 24 hrs post-transfection, cells were lysed on ice in lysis buffer (50 mM Tris-HCl [pH7.5], 150 mM NaCl, 1% NP-40) supplemented with complete protease inhibitor cocktail (Roche Diagnostics). Lysates were incubated with either α-FLAG M2-agarose from mouse (Sigma-Aldrich) or anti-HA affinity matrix (Roche) for 2-3 hrs at 4°C. After incubation, beads with bound protein complexes were washed in ice-cold lysis buffer. Subsequently, 4x NuPAGE sample buffer was added to the beads and heated for 10 minutes at 70°C. Beads were precipitated by centrifugation, and supernatant was run on NuPAGE Novex 4%-12% Bis-Tris SDS-PAGE gels. After blotting overnight at 4°C, blots were stained with either rabbit anti-FLAG (Sigma, 1:1000), mouse anti-FLAG (Sigma, 1:1000), rabbit anti-HA (Sigma, 1:1000) or mouse anti-HA (Sigma, 1:1000). Goat anti-rabbit IRDye800 (Li-COR, 1:20,000) or goat anti-mouse IRDye800 (Li-COR, 1:20,000) were used as secondary antibodies. Fluorescence was analysed on a Li-COR Odyssey 2.1 infrared Scanner. Full unprocessed western blotting scans are shown in Suppl. Figure 8 with the regions used in main display items indicated by red frames.
Endogenous co-immunoprecipitation from retinal extracts
Protein A/G PLUS-agarose beads (Santa Cruz Biotechnology) were washed three times in extraction buffer (10mM HEPES [pH7.9], 10mM NaCl, 3mM MgCl2, freshly added 1mM DTT, 1mM Na3VO4) supplemented with complete protease inhibitor cocktail (Roche) and then coupled to polyclonal anti-NEK1 (H3T1) or polyclonal anti-FLAG (Sigma, F7425) overnight at 4°C. Coupled beads were washed three times in extraction buffer and incubated with fresh bovine retinal lysates overnight at 4°C. Retinas were homogenized by sonication on ice two times for 30 seconds in extraction buffer (10mM HEPES [pH7.9], 10mM NaCl, 3mM MgCl2, freshly added 1mM DTT, 1mM Na3VO4) supplemented with complete protease inhibitor cocktail (Roche). Beads were washed five times with extraction buffer, transferred to microspin columns (GE Healthcare) and washed three times in washing buffer (1xTBS, 0.12% NP-40, supplemented with complete protease inhibitors (Roche)). Then, neutralization buffer (1M Tris/HCl, [pH8.0]) was added. Eluates were retrieved by adding 3x elution buffer (200 mM glycine [pH2.5]) for 20 min. at 4°C and subjected to protein precipitation with chloroform and methanol. Protein precipitates were subsequently subjected to mass spectrometry analysis and peptide identification as previously described48.
GST-pulldown from retinal extracts
In order to produce GST fusion proteins, BL21-DE3 bacteria were transformed with either pDEST15-SPATA7, pDEST15-C21orf2 or pDEST15 alone. Protein expression was induced at 28°C for 3 hours with 0.5 mM IPTG (Sigma-Aldrich) and harvested by centrifugation. Subsequently, cells were lysed with STE buffer (10 mM Tris-HCl, pH 8.0; 1 mM EDTA; and 150 mM NaCl) supplemented with 10 mg/ml lysozyme, 0.5% Sarkosyl, 1% Triton X-100, and Complete protease inhibitor cocktail (Roche). Lysates were then incubated for 2 hours at 4°C with glutathione-sepharose 4B beads (Amersham Biosciences). After incubation, beads were washed with STE and TBSTD (TBS with 1% Triton X-100 and 2 mM DTT). The amount of GST fusion proteins bound to the beads was verified on a NuPAGE Novex 4%–12% Bis-Tris SDS-PAGE gel by staining with SimplyBlue SafeStain (LifeTechnologies). Beads with bound GST-fusion proteins were then incubated over-night with bovine retinal extracts at 4°C. After overnight incubation, samples were washed three times with extraction buffer. Protein were eluted from beads in 4X NuPAGE sample buffer and heated for 10 minutes at 70°C. Beads were precipitated afterward by centrifugation. The supernatants were run on NuPAGE Novex 4%-12% Bis-Tris SDS-PAGE gels, followed by immunoblotting overnight at 4°C and staining with rabbit polyclonal α-NEK1 as primary antibody (H3T1; 1:1000) and goat anti-rabbit IRDye800 (LiCOR, 1:20,000) as secondary antibody. Fluorescence was analysed on a Li-COR Odyssey 2.1 infrared Scanner. Full unprocessed western blotting scans are shown in Suppl. Figure 8 with the regions used in main display items indicated by red frames.
For mass spectrometry analysis of the GST pull down, beads were transferred to microspin columns (GE Healthcare), and washed 2 times with filtered TBS(1X). Eluates were retrieved by adding 300 μl of 100 mM reduced Glutathione (GSH) (Sigma-Aldrich) in 50mM Tris-HCl [pH=8.0] and incubating overnight at 4°C on an Intelli mixer. Eluates were then subjected to protein precipitation with chloroform and methanol. Protein precipitates were subsequently subjected to mass spectrometry analysis and peptide identification as previously described48.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
Our deepest thanks go to all of the families and individuals with Joubert syndrome, Jeune syndrome and retinitis pigmentosa. We thank Paul Barker, Martin Jackson, Kieran Roberts and Amanda Jones from PerkinElmer Inc. for technical support. The research received funding from the European Community’s Seventh Framework Programme FP7/2009 under grant agreement no: 241955 SYSCILIA towards C.A.J., R.R., M.U., T.G., P.L.B, O.E.B, U.W., R.H.G., M.A.H. and H.O. This work was supported by a Sir Jules Thorn Award for Biomedical Research (JTA/09, to C.F.I. and C.A.J.) and a Medical Research Council grant (MR/K011154/1, to C.A.J.) M.S. was funded by an Action Medical Research Clinical Training Fellowship (RTF-1411), a Radboud University Excellence fellowship, a Radboud UMC Hypatia Tenure Track fellowship and acknowledges funding from the Deutsche Forschungsgemeinschaft (DFG; grant SFB 1140 (KIDGEM)). R.R. is funded by he Netherlands Organization for Scientific Research (NWO Vici-865.12.005), and by the Foundation Fighting Blindness (C-CMM-0811-0546-RAD02). P.L.B. is a Wellcome Trust Senior Research Fellow, and P.L.B., M.S., R.H.G. and R.R. acknowledge funding from the Dutch Kidney Foundation (CP11.18, “Kouncil”). C.T.T. is funded by DFG grant TH 896/3-3 and IZKF (Interdisciplinary Centre for Clinical Research of the Universität of Erlangen-Nürnberg) grant F4. U.W. is funded by the FAUN foundation, Nuernberg. Z.A.A. and W.H. are supported by grants from the Rosetree’s Trust (A210 and A465). H.M.M. is supported by the Great Ormond Street Hospital Children’s Charity and received grants from the Milena Carvajal Pro-Kartagener Foundation and Action Medical Research (GN2101). H.R. is funded by the Canadian Institutes of Health Research (CIHR) Training Program in Genetics, Child Development and Health, Alberta Children’s Hospital Research Institute (ACHRI) and Alberta Children’s Hospital Foundation. A.R.W. is funded by RP Fighting Blindness, Moorfields Eye Hospital Special Trustees, the National Institute for Health Research (NIHR; Moorfields Eye Hospital and Institute of Ophthalmology, London, U.K.) and the Foundation Fighting Blindness (U.S.) D.D. is funded by NINDS grant R01NS064077, the University of Washington Intellectual and Developmental Disabilities Research Center Genetics Core P30HD002274, and private donations from families of individuals with Joubert syndrome. J.S. is funded by NCI grant R21CA160080. Access to the B6;129P2-Tmem67tm1Dgen/H line was funded by the Wellcome Trust Knock-out Mouse Resource scheme (C.A.J. and C.F.I.; grant ME041596). The UK10K project is funded by the Wellcome Trust under grant agreement WT091310. A full list of UK10k investigators can be accessed at http://www.uk10k.org. The University of Washington Center for Mendelian Genomics is supported by NHGRI grant U54HG006493.
AUTHOR CONTRIBUTIONS
K.S., G.W., S.N., W.H., M.A. and J.H. performed the primary and secondary siRNA screens. K.S., G.W., D.A.M., T-M.T.N. and S.J.F.L. performed and analysed the tertiary and other validation screens. L.W. wrote scripts for UPS validation screen analysis. C.T., S.M.B., J.B., E.E.M., D.C.T. and C.F.I supervised and managed the BioScreening Technology Group and edited the manuscript. S.N. did qRT-PCR analyses, and K.S., G.W. and W.H. did western blot analyses in validation screens. G.W. and Z.A.A. performed and interpreted IF and IHC staining of mouse tissue sections. D.A.P. wrote Perl scripts for automated analysis of siRNA screen data and re-analysed UK10k WES data. K.S. and G.W. performed confocal IF microscopy of primary fibroblasts and cell-lines. G.T. and T.G. developed software and analysed array CGH data, and G.T. analysed siRNA sequences. G.G.S. and R.H.G. performed 3D spheroid cultures of mIMCD3 cells. J.K. and O.B. performed and interpreted all experiments with C. elegans. N.S., U.W., and K.A.W. analysed mouse photoreceptor cells by high magnification IF and immunoelectron microscopy. K.K., A.G. and C.T.T. developed and characterised the NEK1 antibody. H.R., C.L.B., P.G., R.T.P., R.L. and F.P.B ascertained and recruited subjects, performed linkage analysis, WES and Sanger sequencing, and analysed data for Hutterite participants. J.S.P. and A.M.I. diagnosed and enrolled participants, collated clinical documentation and supervised research on Hutterite participants. C.O. analysed and provided data for Schmiedeleut controls. R.H. analysed and provided data for Dariusleut and Leherleut controls. L.H. and K.M.B. provided reagents for work on PIBF1. I.G.P. analysed the Joubert syndrome exomes, designed the MIPS capture and performed the MIPS sequencing and analysis. J.S. conceived of the MIPS method and E.A.B. contributed to MIPS method development. D.D. diagnosed and enrolled participants, collated clinical documentation, supervised the sequencing and edited the manuscript. A.M, B.N.C, A.E.C., C.E.W., K.M.B., P.L.B., F.S.A., M.M., C.T., C.F.I, H.O., C.W., F.S., S.A.H. and P.F. diagnosed and ascertained participants and collated clinical information. A.R.W. diagnosed and ascertained participants, collated clinical information and analysed SNP genotyping data. P.I.S. collated clinical information, prepared samples for WES and analysed WES data. M.A. and S.A. performed RT-PCR experiments. T-M.T.N., D.A.M. and J.v.R. performed and analysed C21orf2 PPI experiments and IF microscopy. N.H., K.B. and M.U. performed, analysed and supervised TAP MS experiments. M.S. performed WES analysis, zebrafish experiments, performed and supervised Sanger sequencing analysis and analysed the data. M.S. and H.M.M. conceived the human genetics and zebrafish experiments and wrote the manuscript. R.R. and C.A.J. conceived the study, directed and supervised the research, analysed and collated data, and wrote the manuscript.
Web Resources
Online Mendelian Inheritance in Man (OMIM) (http://www.ncbi.nlm.nih.gov/Omim)
University of California, Santa Cruz (UCSC) Genome Browser (http://www.genome.ucsc.edu)
Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (http://evs.gs.washington.edu/EVS/) date accessed Dec. 2014
Exome Aggregation Consortium (ExAC), Cambridge, MA, USA (http://exac.broadinstitute.org) date accessed Dec. 2014
The Consensus CDS (CCDS) project (http://www.ncbi.nlm.nih.gov/projects/CCDS/)
Copy Number Analyzer for Affymetrix GeneChip (http://www.genome.umin.jp/)
GraphPad, Quick Calcs (http://www.graphpad.com/quickcalcs/chisquared2.cfm)
Polyphen2 (http://genetics.bwh.harvard.edu/pph2/)
Mutationtaster (http://www.mutationtaster.org/)
Sorting intolerant from tolerant (SIFT) (http://sift.jcvi.org/)
Genomic evolutionary rate profiling (GERP) (http://mendel.stanford.edu/SidowLab/downloads/gerp)
PhyloP (http://genome.ucsc.edu)
Alamut (http://www.interactive-biosoftware.com/software/alamut/features
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare that that they do not have competing financial interests.
Accession numbers
PIBF1: NM_006346.2 and NP_006337.2; C21orf2: NM_004928.2 and NP_004919.1.
Primary accessions
Whole exome sequencing data is available from the UK10k Consortium (UK10kCIL5236549 and UK10kCIL5236550) and dbGaP (phs000205.v5.p2)
Please refer to Supplementary Note for methodological details of the primary, secondary and tertiary screens.
Please refer to Supplementary Note for methodological details of patient ascertainment and sample preparation, and genetic analyses including SNP genotyping arrays, linkage analysis, whole exome sequencing and Sanger sequencing.
REFERENCES
- 1.Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–44. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams M, Smith UM, Logan CV, Johnson CA. Recent advances in the molecular pathology, cell biology and genetics of ciliopathies. Journal of Medical Genetics. 2008;45:257–267. doi: 10.1136/jmg.2007.054999. [DOI] [PubMed] [Google Scholar]
- 3.Quinlan RJ, Tobin JL, Beales PL. Modeling ciliopathies: Primary cilia in development and disease. Curr Top Dev Biol. 2008;84:249–310. doi: 10.1016/S0070-2153(08)00605-4. [DOI] [PubMed] [Google Scholar]
- 4.Kim J, et al. Functional genomic screen for modulators of ciliogenesis and cilium length. Nature. 2010;464:1048–51. doi: 10.1038/nature08895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lai CK, et al. Functional characterization of putative cilia genes by high-content analysis. Mol Biol Cell. 2011;22:1104–19. doi: 10.1091/mbc.E10-07-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birmingham A, et al. Statistical methods for analysis of high-throughput RNA interference screens. Nat Methods. 2009;6:569–75. doi: 10.1038/nmeth.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang XD. A pair of new statistical parameters for quality control in RNA interference high-throughput screening assays. Genomics. 2007;89:552–61. doi: 10.1016/j.ygeno.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 8.van Dam TJ, et al. The SYSCILIA gold standard (SCGSv1) of known ciliary components and its applications within a systems biology consortium. Cilia. 2013;2:7. doi: 10.1186/2046-2530-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 11.Balestra FR, Strnad P, Fluckiger I, Gonczy P. Discovering regulators of centriole biogenesis through siRNA-based functional genomics in human cells. Dev Cell. 2013;25:555–71. doi: 10.1016/j.devcel.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 12.Joo K, et al. CCDC41 is required for ciliary vesicle docking to the mother centriole. Proc Natl Acad Sci U S A. 2013;110:5987–92. doi: 10.1073/pnas.1220927110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrante MI, et al. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat Genet. 2006;38:112–7. doi: 10.1038/ng1684. [DOI] [PubMed] [Google Scholar]
- 14.Martin CA, et al. Mutations in PLK4, encoding a master regulator of centriole biogenesis, cause microcephaly, growth failure and retinopathy. Nat Genet. 2014;46:1283–92. doi: 10.1038/ng.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaheen R, Al Tala S, Almoisheer A, Alkuraya FS. Mutation in PLK4, encoding a master regulator of centriole formation, defines a novel locus for primordial dwarfism. J Med Genet. 2014;51:814–6. doi: 10.1136/jmedgenet-2014-102790. [DOI] [PubMed] [Google Scholar]
- 16.Shaheen R, et al. A founder CEP120 mutation in Jeune asphyxiating thoracic dystrophy expands the role of centriolar proteins in skeletal ciliopathies. Hum Mol Genet. 2015;24:1410–9. doi: 10.1093/hmg/ddu555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vithana EN, et al. A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11) Mol Cell. 2001;8:375–81. doi: 10.1016/s1097-2765(01)00305-7. [DOI] [PubMed] [Google Scholar]
- 18.Hebeisen M, Drysdale J, Roy R. Suppressors of the cdc-25.1(gf)-associated intestinal hyperplasia reveal important maternal roles for prp-8 and a subset of splicing factors in C. elegans. RNA. 2008;14:2618–33. doi: 10.1261/rna.1168408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdelhamed ZA, Wheway G, Szymanska K, Natarajan S, Toomes C, Inglehearn C, Johnson CA. Variable expressivity of ciliopathy neurological phenotypes that encompass Meckel-Gruber syndrome and Joubert syndrome is caused by complex de-regulated ciliogenesis, Shh and Wnt signalling defects. Hum Mol Genet. 2013;22:1358–72. doi: 10.1093/hmg/dds546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mukhopadhyay S, et al. The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell. 2013;152:210–23. doi: 10.1016/j.cell.2012.12.026. [DOI] [PubMed] [Google Scholar]
- 21.Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013;12:894–905. doi: 10.1016/S1474-4422(13)70136-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huber C, Cormier-Daire V. Ciliary disorder of the skeleton. Am J Med Genet C Semin Med Genet. 2012;160C:165–74. doi: 10.1002/ajmg.c.31336. [DOI] [PubMed] [Google Scholar]
- 23.Abu-Safieh L, et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013;23:236–47. doi: 10.1101/gr.144105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eblimit A, et al. Spata7 is a retinal ciliopathy gene critical for correct RPGRIP1 localization and protein trafficking in the retina. Hum Mol Genet. 2014:pii: ddu573. doi: 10.1093/hmg/ddu573. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thiel C, et al. NEK1 mutations cause short-rib polydactyly syndrome type Majewski. Am J Hum Genet. 2011;88:106–14. doi: 10.1016/j.ajhg.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim K, Lee K, Rhee K. CEP90 is required for the assembly and centrosomal accumulation of centriolar satellites, which is essential for primary cilia formation. PLoS One. 2012;7:e48196. doi: 10.1371/journal.pone.0048196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neumann B, et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature. 2010;464:721–7. doi: 10.1038/nature08869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arellano JI, Guadiana SM, Breunig JJ, Rakic P, Sarkisian MR. Development and distribution of neuronal cilia in mouse neocortex. J Comp Neurol. 2012;520:848–73. doi: 10.1002/cne.22793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guadiana SM, et al. Arborization of dendrites by developing neocortical neurons is dependent on primary cilia and type 3 adenylyl cyclase. J Neurosci. 2013;33:2626–38. doi: 10.1523/JNEUROSCI.2906-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andersen JS, et al. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426:570–4. doi: 10.1038/nature02166. [DOI] [PubMed] [Google Scholar]
- 31.Jakobsen L, et al. Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. EMBO J. 2011;30:1520–35. doi: 10.1038/emboj.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song EJ, et al. The Prp19 complex and the Usp4Sart3 deubiquitinating enzyme control reversible ubiquitination at the spliceosome. Genes Dev. 2010;24:1434–47. doi: 10.1101/gad.1925010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pelisch F, Risso G, Srebrow A. RNA metabolism and ubiquitin/ubiquitin-like modifications collide. Brief Funct Genomics. 2013;12:66–71. doi: 10.1093/bfgp/els053. [DOI] [PubMed] [Google Scholar]
- 34.Oppermann FS, et al. Large-scale proteomics analysis of the human kinome. Mol Cell Proteomics. 2009;8:1751–64. doi: 10.1074/mcp.M800588-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giessl A, Pulvermüller A, Trojan P, Park JH, Choe HW, Ernst OP, Hofmann KP, Wolfrum U. Differential expression and interaction with the visual G-protein transducin of centrin isoforms in mammalian photoreceptor cells. J Biol Chem. 2004;279:51472–81. doi: 10.1074/jbc.M406770200. [DOI] [PubMed] [Google Scholar]
- 36.Thiel C, et al. NEK1 mutations cause short-rib polydactyly syndrome type Majewski. Am J Hum Genet. 2011;88:106–14. doi: 10.1016/j.ajhg.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dawe HR, et al. Nesprin-2 interacts with meckelin and mediates ciliogenesis via remodelling of the actin cytoskeleton. J Cell Sci. 2009;122:2716–2726. doi: 10.1242/jcs.043794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arts HH, et al. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet. 2007;39:882–8. doi: 10.1038/ng2069. [DOI] [PubMed] [Google Scholar]
- 39.Overlack N, et al. Direct interaction of the Usher syndrome 1G protein SANS and myomegalin in the retina. Biochim Biophys Acta. 2011;1813:1883–92. doi: 10.1016/j.bbamcr.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 40.Trojan P, et al. Centrins in retinal photoreceptor cells: regulators in the connecting cilium. Prog Retin Eye Res. 2008;27:237–59. doi: 10.1016/j.preteyeres.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 41.Maerker T, et al. A novel Usher protein network at the periciliary reloading point between molecular transport machineries in vertebrate photoreceptor cells. Hum Mol Genet. 2008;17:71–86. doi: 10.1093/hmg/ddm285. [DOI] [PubMed] [Google Scholar]
- 42.Sedmak T, Sehn E, Wolfrum U. Immunoelectron microscopy of vesicle transport to the primary cilium of photoreceptor cells. Methods Cell Biol. 2009;94:259–72. doi: 10.1016/S0091-679X(08)94013-9. [DOI] [PubMed] [Google Scholar]
- 43.Hebeisen M, Drysdale J, Roy R. Suppressors of the cdc-25.1(gf)-associated intestinal hyperplasia reveal important maternal roles for prp-8 and a subset of splicing factors in C. elegans. RNA. 2008;14:2618–33. doi: 10.1261/rna.1168408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kostic I, Roy R. Organ-specific cell division abnormalities caused by mutation in a general cell cycle regulator in C. elegans. Development. 2002;129:2155–65. doi: 10.1242/dev.129.9.2155. [DOI] [PubMed] [Google Scholar]
- 45.Cevik S, et al. Joubert syndrome Arl13b functions at ciliary membranes and stabilizes protein transport in Caenorhabditis elegans. J Cell Biol. 2010;188:953–69. doi: 10.1083/jcb.200908133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang L, et al. TMEM237 is mutated in individuals with a Joubert Syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am J Hum Genet. 2011;89:713–30. doi: 10.1016/j.ajhg.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roepman R, et al. Interaction of nephrocystin-4 and RPGRIP1 is disrupted by nephronophthisis or Leber congenital amaurosis-associated mutations. Proc Natl Acad Sci U S A. 2008;102:18520–5. doi: 10.1073/pnas.0505774102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coene KL, et al. The ciliopathy-associated protein homologs RPGRIP1 and RPGRIP1L are linked to cilium integrity through interaction with Nek4 serine/threonine kinase. Hum Mol Genet. 2011;20:3592–605. doi: 10.1093/hmg/ddr280. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.