

Abstract
Phorbol-12-myristate-13-acetate (PMA), a protein kinase C (PKC) activator, can modulate 1α, 25 dihydroxyvitamin D3 (1,25(OH)2D3)-induced expression of the 24-hydroxylase (CYP24A1) gene but this has not been studied in differentiated enterocytes, a primary 1,25(OH)2D3 target cell. We found that in differentiated Caco-2 cells, an established model of the mature absorptive epithelial cell, PMA significantly enhanced 1,25(OH)2D3-induced human CYP24A1 (hCYP24A1) mRNA accumulation and hCYP24A1 promoter-luciferase reporter gene activation by 150%. Reporter gene studies further identified the region between −298 to +74 bp in the hCYP24A1 promoter as critical for the PMA enhancing effect and chromatin immunoprecipitation (ChIP) analysis showed that PMA enhanced 1,25(OH)2D3-induced binding of vitamin D receptor to this region. PMA can activate PKC, ERK1/2 and p38 MAP kinases and inhibition of these signaling pathways reduced both 1,25(OH)2D3-induced hCYP24A1 gene transcription and the enhancing effect of PMA. The PMA enhancing effect on 1,25(OH)2D3 action was evident in a minimal promoter with three osteocalcin VDREs and was reduced after mutation of a putative vitamin D stimulatory site in the hCYP24A1 promoter. In contrast, mutation of a Ets binding site in the hCYP24A1 promoter had no impact on 1,25(OH)2D3 action or the PMA enhancing effect. These data suggest that in the differentiated enterocyte PMA-induced activation of several signaling pathways contribute to 1,25(OH)2D3-induced hCYP24A1 gene expression through multiple regulatory motifs within the proximal hCYP24A1 promoter.
Keywords: Vitamin D, 25-hydroxyvitamin D3 24-hydroxylase, Transcription, Kinases
Introduction
1,25-dihydroxyvitamin D3 (1,25(OH)2D3), the biologically active form of vitamin D, plays important roles in several aspects of intestinal function, including stimulation of calcium absorption in small intestine (Bronner et al., 1986), regulation of intestinal immune responses (Cantorna et al., 2000;Froicu et al., 2003) and as a chemopreventive agent against colorectal cancer (Fedirko et al., 2009;Fichera et al., 2007). The action of 1,25(OH)2D3 is mediated through transcriptional activation of target genes via binding of ligand-activated vitamin D receptor (VDR)-retinoic acid X receptor (RXR) heterodimers to vitamin D response elements (VDRE) (Pike et al., 2007).
The molecular action of 1,25(OH)2D3 depends on a balance between synthesis and degradation of the hormone. 1,25(OH)2D3 is synthesized from the precursor 25(OH)D3 by the cytochrome P450 enzyme 25-hydroxyvitamin D3 1α-hydroxylase (CYP27B1) while its degradation is regulated by the mitochondrial cytochrome P450 enzyme, 25-hydroxyvitamin D3 24-hydroxylase (CYP24A1). The most powerful inducer of CYP24A1 gene expression is 1,25(OH)2D3 (Omdahl et al., 2002). This effective regulatory system forms a natural negative feedback loop for controlling cellular 1,25(OH)2D3 levels and actions (Ly et al., 1999;Miller et al., 1995;St-Arnaud et al., 2000).
Others have shown that the phorbol-12-myristate-13-acetate (PMA) enhances the effect of 1,25(OH)2D3 on CYP24A1 mRNA level in rat renal cells (Chen et al., 1993) and intestinal epithelial cells (Armbrecht et al., 1993). PMA has traditionally been utilized as a tool to activate the protein kinase C (PKC) signaling pathway. PKC inhibitors such as H-7, stauroporin, and bisindolylmaleimide I (BIS) block the enhancing effect of PMA on 1,25(OH)2D3 action in rat intestinal epithelial cells (Armbrecht et al., 2001;Koyama et al., 1994). However, in addition to its effects on PKC, PMA can also activate other kinases. Thus other signaling pathways in addition to PKC might contribute to the effect of PMA on vitamin D-mediated CYP24A1 regulation.
The human CYP24A1 (hCYP24A1) promoter has two functional vitamin D response elements (VDRE) located on the anti-sense strand at positions −293/−272 (distal VDRE, VDREd) and −172/−143 (proximal VDRE, VDREp) (Chen and DeLuca, 1995). Studies have identified other regions in the promoter that can modulate the transactivation of the CYP24A1 gene by 1,25(OH)2D3 including putative AP-1 sites between the two VDREs (Chen and DeLuca, 1995), C/EBP binding sites in the distal promoter (Dhawan et al., 2005), an Ets-1 binding site (EBS) downstream of the proximal VDRE in the rat CYP24A1 promoter (Dwivedi et al., 2000;Cui et al., 2009) and a vitamin D stimulatory element (VSE site) on the rat CYP24A1 promoter (Nutchey et al., 2005). However, the existence and roles of these sites in the hCYP24A1 promoter have not been examined.
In the present study, we found that in addition to PKC, PMA can activate the mitogen activated protein kinases (MAPK) ERK1/2 and p38 kinase in differentiated Caco-2 cells. Our data show that all three signaling pathways contribute to 1,25(OH)2D3-induced and PMA-enhanced effects on hCYP24A1 gene expression. We also identify a sequence in the hCYP24A1 promoter that is similar to the VSE site in rat CYP24A1 promoter and show that it partially accounts for the synergistic effects of PMA on 1,25(OH)2D3-induced hCYP24A1 gene expression.
Material and Methods
Reagents
1,25(OH)2D3 (BioMol International, Plymouth Meeting, PA) was dissolved in ethanol. Phorbol-12-myristate-13-acetate (PMA, PKC activator), Go6976 (classic PKC inhibitor), U0126 (specific inhibitor for MEK1/2), SB202190 (p38 kinase inhibitor) were obtained from EMD Biosciences, Inc. (San Diego, CA) and dissolved in dimethyl sulfoxide (DMSO). Antibodies against total and phosphorylated ERK1/2 as well as total and phosphorylated p38 were obtained from Cell Signaling Technology, Inc. (Beverly, MA).
Cell culture and Treatments
The parental line of the human colonic adenocarcinoma cell line Caco-2 was obtained from American Type Culture Collection (HTB-37, Rockville, MD) and was cultured as described elsewhere (Fleet et al., 2002). Experiments on differentiated Caco-2 cells were carried out at 15 days in culture (11 d post confluent). Differentiation was confirmed by observing high levels of expression of sucrose-isomaltase and calbindin D9k mRNA, two markers of enterocyte differentiation (Wang et al., 2004). Treatment solutions were prepared with medium containing 0% FBS for protein expression studies and 5% FBS for mRNA and reporter gene studies.
In studies for mRNA analysis or Western blot analysis, cells were first pretreated with either 100 nM PMA or vehicle (0.002% DMSO) for 5 min and the cells then were treated for an additional 2 hours with either 10 nM 1,25(OH)2D3 or vehicle (0.01% ethanol) in the absence of PMA. For kinase inhibitor studies, cells were first pretreated with either vehicle (0.1% ethanol) or various kinase inhibitors, including 2 or 10 μM Go6976, 10 μM U0126, or 8 μM SB202190 for 30 minutes. Afterwards cells were treated with PMA (5 min), followed by 1,25(OH)2D3 (2 h treatment) in the presence or absence of inhibitors but with PMA removed.
RNA Isolation and Analysis
RNA was isolated using TriReagent following the manufacturer’s instructions (Molecular Research Center Inc., Cincinnati, OH). Total RNA (2 μg) was reverse transcribed to make cDNA and analyzed by real-time PCR using primers and conditions previously described by our group (Klopot et al., 2006;Wang et al., 2004)
Chromatin Immunoprecipitation
Differentiated Caco-2 cells were pretreated with either vehicle or 100 nM PMA for 5 minutes and then incubated with 10 nM 1,25(OH)2D3 or vehicle for 2 hours in the absence of PMA. Chromatin immunoprecipitation was performed as we have described previously (Cui et al., 2009). This study was repeated twice.
Western Blot Analysis
Differentiated Caco-2 cells were changed to serum free media overnight, pretreated with U0126 (10, 20 or 100 μM, 30 min) or Go6976 (2 or 10μM, 30 min) and then treated with 100 nM PMA or vehicle in the presence of the inhibitors for 5 minutes. For ERK1/2 analysis, samples were harvested immediately after the 5 minute PMA treatment. For p38 kinase analysis, medium was replaced with serum-free medium containing 0.002% DMSO and harvested 1 hour after PMA treatment. Cells were prepared and cell lysates (45 μg) were analyzed for phosphorylated ERK1/2 and p38 kinase as using methods we have previously described (Cui et al., 2009). After detection of phosphoproteins, the membrane was stripped and re-probed for total ERK1/2 or p38 kinase. Antigen-antibody complexes were detected using the ECL Plus Western blotting detection reagents (Pierce, Rockford, IL).
Plasmids
The −5500 to −22 bp hCYP24A1 promoter luciferase construct (−5500 to −22 bp hCYP24A1-luciferase) was a gift from Dr. Sylvia Christakos (Barletta et al., 2004). The hCYP24A1 promoter region from −298 to +74 was amplified from human genomic DNA via PCR and this fragment was then cloned upstream from the firefly luciferase reporter gene into the pGL3 basic vector to create the −298 to +74 bp hCYP24A1-luciferase construct. A reporter gene construct containing three copies of the VDRE from the mouse osteopontin gene (3X VDRE-luciferase) was provided by Dr. Sunil Nagpal (Eli Lilly and Company, Indianapolis, IN). A promoterless pRL-null Renilla luciferase vector was purchased from Promega (Madison, WI).
Mutations were made in the −298 to +74 bp hCYP24A1-luciferase reporter gene in EBS site between −138 and −130 bp (Dwivedi et al., 2000) or in the putative VSE site −192 to −184 bp of the hCYP24A1 promoter using the Quikchange XL site-directed mutagenesis kit (Stratagene) following the manufacturer’s instructions to create the reporter gene constructs hCYP24A1 mEBS-luciferase and hCYP24A1 mVSE-luciferase. Primers for mutating the EBS site were 5′CTCACCTCGCTGACTCCAAAATCCTTCCACCCCCCCCTCCA3′ and 5′GGAGGGGGGGGTGGAAGGATTTTGGAGTCAGCGAGGTGAG3′; Primers for mutating the VSE were: 5′CGCCCAGCGAACATAGCCCAAGCTTCCCCAGGCCCGGACGCCC3′ and 5′GGGCGTCCGGGCCTGGGGAAGCTTGGGCTATGTTCGCTGGGCG3′ (letters underlined are the mutated sequence). The mutations were verified by sequencing at the Purdue Genomics Core Facility.
Reporter gene assay
Cells seeded in 24-well plates were transfected using Liopofectamine Plus (Invitrogen Corp., Carlsbad, California) following the protocol provided by the manufacturer. Serum-free OPTImem medium (Invitrogen) was used for transfection studies. Differentiated Caco-2 cells were transfected using a 1:10:10 ratio of DNA:Plus:Lipofectamine. In vector comparison studies, cells were transfected with 300 ng (per well) of following vectors, −5500 to −22 bp hCYP24A1 -luciferase, −298 to +74 bp hCYP24A1 -luciferase (wild-type, mEBS, mVSE versions) or 3X VDRE-luciferase for 15 hours. 5 ng (per well) pRL-null renilla luciferase plasmid was co-transfected in all experiments for normalization of transfection efficiency. A vector for expression of human VDR, PCR3.1-hVDR (Shao et al., 2001), was co-transfected at 5 ng per well to facilitate the hCYP24A1 promoter response. 15 h after transfection cells were pre-incubated with 100 nM PMA or vehicle (0.002% DMSO) for 10 min followed by treatment with ethanol vehicle (0.01%) or 10 nM 1,25(OH)2 D3 for an additional 4 hours in the absence of PMA. After treatment, cells were collected in 1X passive lysis buffer (Promega Corp., Madison, WI) and luciferase activity in the samples was determined using the Dual-Luciferase Reporter Assay System (Promega Corp., Madison, WI) with a luminometer (TD-20/20, Turner Design).
Statistical Analysis
Data are expressed as the mean ± the standard error of the mean (SEM). The data were tested for fitting a normal distribution. If the data were not normally distributed a log transformation was performed and normalization of the distribution confirmed. Statistical analysis of the data was performed by ANOVA followed by Fisher’s protected LSD (p<0.05). P-values less than 0.05 were considered statistically significant.
Results
PMA enhances 1,25(OH)2D3-induced hCYP24A1 gene expression in differentiated Caco-2 cells
After 2 h 1,25(OH)2D3 treatment significantly increased hCYP24A1 mRNA levels (6.5-fold, Figure 1A). Treatment with PMA alone has no effect on hCYP24A1 mRNA expression. However, pretreatment with PMA, followed by 1,25(OH)2D3 treatment resulted in a 17-fold increase in hCYP24A1 mRNA level that was 2.6-fold higher (P<0.05) than the effect of 1,25(OH)2D3 alone. The effect of PMA on vitamin D-induced CYP24A1 mRNA induction was associated with an increase in VDR and RXR binding to the CYP24A1 proximal promoter (Figure 1B). PMA is a well-established regulator of protein kinase C (PKC) isoforms so we examined whether the enhancement of 1,25(OH)2D3-mediated hCYP24A1 mRNA level by PMA was dependent upon PKC. Treatment with PMA and the PKC inhibitor Go6976 had no impact on the low, basal expression of hCYP24A1 mRNA in differentiated Caco-2 cells (data not shown). However, the induction of hCYP24A1 mRNA by 1,25(OH)2D3 was reduced 73% by Go6976 (10 μM) and the enhancement of this induction by PMA was reduced by 60% (Figure 1B).
Figure 1.
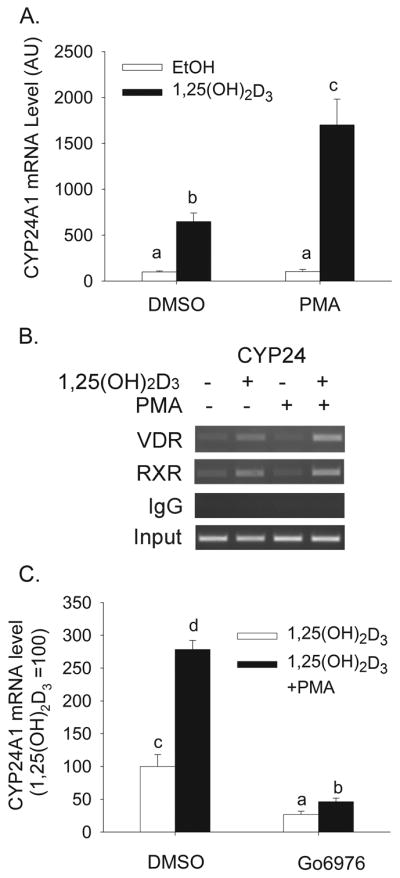
(A) Effect of PMA on 1,25(OH)2D3-induced CYP24A1 mRNA expression in differentiated Caco-2 cells. Cells were pretreated for 5 min with 100 nM PMA followed by treatment for 2 h with 10 nM 1,25(OH)2D3 or vehicle in the absence of PMA. Data are expressed as mean±sem (n=4) with the vehicle treated group as a reference value (100). (B) Effect of PMA on 1,25(OH)2D3-induced association of VDR and RXR on the proximal human CYP24A1 gene promoter in differentiated Caco-2 cells. ChIP assays were performed with non-immune rabbit immunoglobulin (IgG), or antibodies against VDR or RXR. ChIP enriched DNA was amplified with primers recognizing the proximal CYP24 promoter (−252 to −52 bp). (C) Effect of PKC inhibition on PMA enhanced CYP24A1 induction by 1,25(OH)2D3. Cells were pretreated with 10 μM Go6976 for 30 min and 100 nM PMA was added for the last 5 min. Afterwards inhibitor and PMA were removed and cells were treated with 10 nM 1, 25(OH)2D3 or vehicle in the absence of PMA. Data are expressed relative to the 1,25(OH)2D3-induced expression of CYP24A1 (calculated as 1,25(OH)2D3/vehicle = 100). Bars with different letter superscripts are significantly different from one another (p< 0.05, Fisher’s protected LSD). Data are representative of three independent experiments.
PMA independently activates ERK1/2 and p38 MAPK in differentiated Caco-2 cells
Others have shown that in addition to classic and novel isoforms of PKC, PMA can also activate ERK1/2 and p38 kinase (Almog and Naor, 2008;Nomura et al., 2007). We confirmed that short-term treatment with 100 nM PMA can activate both ERK1/2 (Figure 2A, B) and p38 kinase (Figure 2 C,D). While ERK1/2 activation occurred within 5 min, the activation of p38 kinase was seen 1 hour after the PMA treatment. This suggests that p38 is a downstream signal of another PMA-regulated signal transduction pathway(s).
Figure 2.
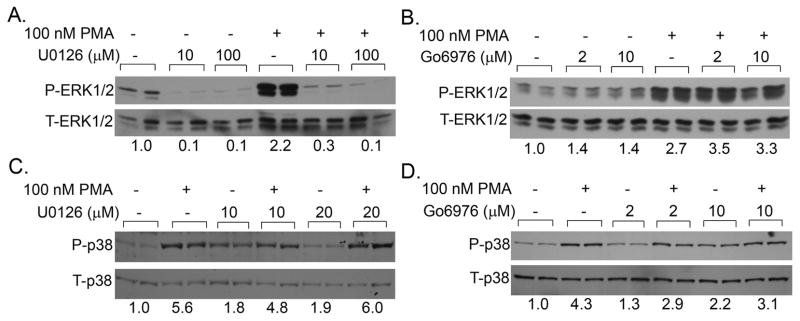
Effect of PMA on MAPK activation in differentiated Caco-2 cells. (A,B) Effect of MEK (U0126) or PKC inhibition (Go6976) on PMA-induced ERK1/2 phosphorylation. Cells were treated with inhibitors or vehicle for 30 min and PMA or vehicle was added for the last 5 min. Proteins from whole cell extracts were isolated and analyzed for total and phosphorylated ERK1/2. (C,D) Effect of MEK (U0126) or PKC inhibition (Go6976) on PMA-induced p38 kinase phosphorylation. Cells were pretreated with inhibitors or vehicle for 30 min and PMA or vehicle was added for the last 5 min. Inhibitor treatments were continued for an additional 1h in the absence of PMA. Proteins from whole cell extracts were isolated and analyzed for total and phosphorylated p38 kinase. Data are representative of three independent experiments. Number on the bottom is the ratio between phosphorylated and total protein levels.
Go6976 did not inhibit PMA-induced ERK1/2 (Figure 2 B) or but partially reduced p38 kinase activation by PMA (Figure 2 D) demonstrating full (ERK1/2) or partial (p38 kinase) independence of these effects from PKC activation. Pretreatment with the MEK inhibitor U0126 (10 μM, 20 μM or 100 μM) completely abolished PMA-induced ERK1/2 activation and partially reduced p38 kinase activation by PMA (Figure 2 A.C). This suggests that PMA-activated ERK1/2 is MEK-dependent, whereas the activation of p38 kinase has both MEK-dependent and MEK-independent components.
Inhibition of p38 kinase and ERK1/2 reduces 1,25(OH)2D3-induced and PMA-enhanced hCYP24A1 mRNA gene expression in differentiated Caco-2 cells
We next investigated whether PMA-induced activation of p38 kinase or ERK1/2 are involved in the enhancing effect of PMA on 1,25(OH)2D3-induced hCYP24A1 gene expression. Treatment with PMA and various inhibitors had no impact on the low, basal expression of hCYP24A1 mRNA in differentiated Caco-2 cells (data not shown). However, the induction of hCYP24A1 mRNA by 1,25(OH)2D3 (2h, 10 nM) was significantly reduced 65% by the MEK inhibitor U0126 (Figure 3 A) and 67% by p38 kinase inhibitor SB202190 (Figure 3 B). Consistent with data in Figure 1, PMA enhanced 1,25(OH)2D3-mediated hCYP24A1 mRNA accumulation by >3-fold. Both the MEK and p38 kinase inhibitors reduced the synergistic effect of PMA on 1,25(OH)2D3-induced hCYP24A1 mRNA accumulation by approximately 40%. Although U0126 did not reduce the activation of a −298 to +74 bp hCYP24A1 promoter-luciferase reporter gene construct by 1,25(OH)2D3 alone (Figure 3C), this inhibitor, as well as the p38 kinase inhibitor SB202190, reduced the enhancing effect of PMA on 1,25(OH)2D3-induced hCYP24A1 promoter activity. These data demonstrate that the impact of PMA mediated through the MAPK kinase signaling pathway on the hCYP24A1 mRNA is occurring at the transcriptional level.
Figure 3.

Inhibitors of ERK1/2, classic PKC and p38 kinase independently reduce 1,25(OH)2D3-induced and PMA-enhanced CYP24A1 gene expression in differentiated Caco-2 cells. (A, B) Cells were preincubated with inhibitors (A: 10 μM U0126, B: 8 μM SB202190) for 30 min and PMA (100 nM) or vehicle was added for the last 5 min. Afterwards inhibitor treatment was continued with 10 nM 1,25(OH)2D3 or ethanol vehicle for an additional 2 h in the absence of PMA. (C) Cells were transfected with a −298 to +74 bp hCYP24A1 promoter-luciferase reporter gene. 15 h after transfection, cells were pre-treated with or without inhibitors (10 μM U0126,8 μM SB202190) for 30 min and PMA (100 nM) or vehicle was added for the last 10 min. Afterwards inhibitor treatment was continued with 10 nM 1,25(OH)2D3 or ethanol vehicle for an additional 4 h in the absence of PMA. Data are expressed as mean±sem (n=4) relative to the 1,25(OH)2D3-induced expression of CYP24A1 (calculated as 1,25(OH)2D3/vehicle = 100). Bars with different letter superscripts are significantly different from one another (P< 0.05, Fisher’s protected LSD). Data in each panel are representative of three independent experiments.
The −298 to +74 bp hCYP24A1 promoter contains the elements necessary for enhancement of 1,25(OH)2D3 transactivation by PMA
To determine which regions of the hCYP24A1 promoter are critical for the synergy between PMA and 1,25(OH)2D3 in regulating hCYP24A1 mRNA expression, reporter gene studies were done using two hCYP24A1 promoter constructs: −5500 to −22 bp hCYP24A1-luciferase and −298 to +74 bp hCYP24A1-luciferase. Both constructs contain the two previously identified functional VDREs, at −172/−143 bp and at −293/−273 bp (Chen and DeLuca, 1995) and −5500 to −22 bp hCYP24A1-luciferase also contains putative VDREs reported to occur at −4000 bp (Sinkkonen et al., 2005).
Regardless of the promoter length, 1,25(OH)2D3 induced a similar 28-fold increase in reporter gene activity. This indicates that elements within the −298 bp hCYP24A1 promoter are sufficient for 1,25(OH)2D3 to stimulate the hCYP24A1 gene transcription. Similarly, treatment with PMA enhanced 1,25(OH)2D3-induced hCYP24A1 promoter activity by 2-fold on both constructs (Figure 4). This demonstrates that PMA also utilizes DNA sequences that are within the −298 bp promoter to modify 1,25(OH)2D3 effects on hCYP24A1 gene transcription. This is consistent with previous research showing the proximal −316 bp of the hCYP24A1 promoter is sufficient for the synergistic effect of PMA on 1,25(OH)2D3-induced expression of the hCYP24A1 gene in COS-7 cells (Barletta et al., 2004).
Figure 4.

The proximal CYP24A1 promoter contains the elements necessary for enhancement of 1,25(OH)2D3 transactivation by PMA. Differentiated Caco-2 cells were transfected with one of two different hCYP24A1 promoter-luciferase plasmids (−298 to +74 bp or −5500 to −22 bp). 15 h after transfection, cells were pre-treated with 100 nM PMA for 10 min followed by 10 nM 1,25(OH)2D3 for 4 h in the absence of PMA. Data are expressed as mean±sem (n=6) relative to the 1,25(OH)2D3-induced expression of the −298 to +74 bp hCYP24A1 promoter (calculated as 1,25(OH)2D3/vehicle = 100). Bars with different letter superscripts are significantly different from one another (P< 0.05, Fisher’s protected LSD).
Specific promoter elements are required for the enhancing effect of PMA on 1,25(OH)2D3-mediated hCYP24A1 gene transcription
We first investigated whether PMA modulates vitamin D action through the VDRE required for 1,25(OH)2D3-mediated hCYP24A1 gene transcription. To do this we compared the impact of PMA on 1,25(OH)2D3-induced activation of the −298 to +74 bp hCYP24A1 promoter-luciferase and a minimal 3X VDRE-promoter-luciferase constructs. As expected, the induction of the minimal promoter by 1,25(OH)2D3 was lower than that seen for the natural promoter (Figure 5). However, the impact of PMA on 1,25(OH)2D3-induced transcription occurred on both the minimal 3X VDRE and natural hCYP24 promoter, demonstrating that at least part of the PMA effect is mediated through events directly on the VDRE. In addition, the effect of PMA on the minimal promoter was not as strong as that observed on the natural promoter construct (43% lower, Figure 5). This suggests that while some of the PMA effect is mediated through the VDRE, other sequences in the promoter are also required.
Figure 5.
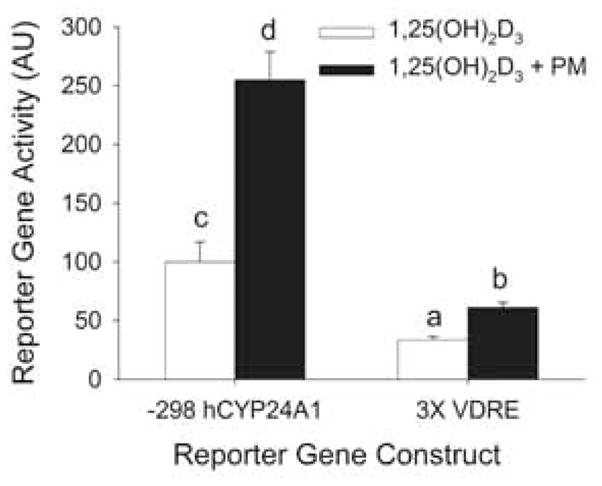
The enhancement of PMA is partially mediated through VDREs in the CYP24A1 promoter. Differentiated Caco-2 cells were transfected with a −298 to +74 bp- human CYP24A1 promoter-luciferase or 3X osteopontin VDRE thymidine kinase-luciferase (3X VDRE) reporter gene construct. 15 h after transfection cells were pre-treated with 100 nM PMA for 10 min followed by vehicle or 10 nM 1, 25(OH)2D3 for 4 h in the absence of PMA. Data are expressed as mean±sem (n=4) relative to the 1,25(OH)2D3-induced expression of the −298 hCYP24A1 promoter (calculated as 1,25(OH)2D3/vehicle = 100). Bars with different letter superscripts are significantly different from one another (P<0.05, Fisher’s protected LSD). Data are representative of three independent experiments.
We subsequently tested the role of two previously described CYP24A1 promoter elements, an Ets Binding Site (EBS) (Dwivedi et al., 2000) and a Vitamin D Stimulatory Element (VSE, (Nutchey et al., 2005)).
By aligning the rat and hCYP24A1 promoters, we identified a conserved EBS sited located on the hCYP24A1 promoter at −138/−130 bp (Figure 6 A). Consistent with our previous report using the rat CYP24A1 promoter (Cui et al., 2009), mutation of the EBS on hCYP24A1 promoter did not alter 1,25(OH)2D3-induced reporter gene activity in differentiated Caco-2 cells. In addition, EBS mutation had no effect on PMA enhanced hCYP24A1 reporter gene activation (Figure 6 B).
Figure 6.

A VSE site but not the EBS site is involved in 1,25(OH)2D3-induced and PMA-enhanced human CYP24A1 promoter activity in differentiated Caco-2 cells. (A) Schematics showing the location of the EBS (Ets protein binding site) and VSE (vitamin D stimulation element) in the rat and human CYP24A1 promoters. Differentiated Caco-2 cells were transfected with wild type (WT), EBS mutated (B, mEBS) or VSE mutated (C, mVSE) −298 to +74 bp human CYP24A1 promoter-luciferase reporter gene constructs. 15 h after transfection, cells were pre-treated with 100 nM PMA for 10 mins followed by 10 nM 1, 25(OH)2D3 for 4 h in the absence of PMA. Data are expressed as mean±sem (n=6) relative to the 1,25(OH)2D3-induced expression of the wild-type −298 to 74 bp hCYP24A1 promoter (calculated as 1,25(OH)2D3/vehicle = 100). In (A) * = Significantly different from the 1,25(OH)2D3-induced group within each reporter gene construct (p< 0.05, Fisher’s protected LSD). In (B) Bars with different letter superscripts are significantly different from one another (P<0.05, Fisher’s protected LSD). Data are representative of three independent experiments.
A VSE site located at −171/−163 bp of rat CYP24A1 promoter (5′TGTCGGTCA3′) has been shown to be vital for the synergy between PMA and 1,25(OH)2D3 (Nutchey et al., 2005). Aligning promoter sequences from the −298 to +74 bp region of rat and hCYP24A1 promoters revealed 65% identity between two sequences and revealed a putative VSE site in hCYP24A1 promoter at positions −192 to −184 bp (5′-CCCCGGTCA-3′) (Figure 6 A). When this putative VSE site was mutated in the hCYP24A1 promoter construct, 1,25(OH)2D3-induced expression was reduced by 69% and the PMA enhancing effect was reduced by 56% (Figure 6 C).
Discussion
In this report, we have demonstrated that a short 5 minute pre-treatment with PMA enhances the transcriptional regulation of the hCYP24A1 gene induced by 1,25(OH)2D3 within differentiated cultures of the human intestinal cell line, Caco-2 (Figure 1). This is consistent with earlier observations by the Armbrecht group documenting that 1,25(OH)2D3- regulated expression of CYP24A1 mRNA was enhanced by phorbol ester treatment (last 30 min to 2 h in a 7–24 h 1,25(OH)2D3 treatment period) in primary rat renal cells (Chen et al., 1993), in the rat renal cell line NRK-52E (Armbrecht et al., 1997), and in the intestinal crypt cell-like (i.e. undifferentiated) lines IEC-6 and IEC-18 derived from the rat ileum (Armbrecht et al., 2001;Armbrecht et al., 1993). Phorbol esters also enhance 1,25(OH)2D3-regulated induction of a rat CYP24A1-promoter reporter gene in HEK 293T human embryonic kidney cells (2 h pretreatment followed by 22 h with 1,25(OH)2D3 (Nutchey et al., 2005)) as well as 1,25(OH)2D3-regulated induction of a hCYP24A1-promoter reporter gene in COS-7 and LLCRK-1 pig kidney cells (24 h co-treatment with phorbol ester and 1,25(OH)2D3 (Barletta et al., 2004)). Our studies are novel in that we are the first to report phorbol ester-enhanced, 1,25(OH)2D3-induced accumulation of hCYP24A1 mRNA and induction of a hCYP24A1 promoter-reporter gene in differentiated human intestinal cells (a primary vitamin D target cell) and because we used short treatment periods (5 min PMA prior to 2–4 h with 1,25(OH)2D3) to minimize the potential for secondary effects due to prolonged treatment periods. In addition, our ChIP analysis demonstrates that the enhancing effect of PMA is due at least in part to enhanced binding of VDR and RXR to the human CYP24A1 promoter (Figure 1B).
The phorbol ester PMA is a natural analog of the potent PKC activator diacylglycerol (DAG) (Newton, 2001); it activates both classical and non-classical isoforms of PKC. Because of this, PMA-elicited cellular responses are often attributed to activation of PKC signaling pathways. Consistent with this, we (Figure 1B) and others (Nutchey et al., 2005) have shown that inhibitors of classical PKC isoforms with Go6976, Calphostin C, and GF109203X reduce, but do not eliminate either 1,25(OH)2D3-mediated accumulation of hCYP24A1 mRNA or the enhancement of this induction by PMA. Using dominant negative expression vectors for the PKC isoforms α, β1, δ, and ε, Nutchey et al. (Nutchey et al., 2005) showed that the effect of 1,25(OH)2D3 on rat CYP24A1 gene transcription was dependent upon PKC β1 while all the isoforms tested contribute to the enhancing effect of PMA. However, their ability to suppress PMAs enhancing effect using PKC inhibitors was incomplete. This suggests that PMA-induces events in addition to activating PKC that modulate vitamin D-induced CYP24A1 gene expression.
Our data show that PMA treatment activated the MAPK family members ERK1/2 and p38 kinase. Others have previously shown that ERK1/2 is activated by PMA in HEK293T cells (Nutchey et al., 2005); this is likely due to activation of RasGRP, an exchange factor for Ras/Rap1 that contains the DAG-binding C1 domains first identified in PKC (Kazanietz, 2002;Duncia et al., 1998). The induction of p38 kinase by PMA was delayed and could be partially blocked by inhibitors of MEK1/2 and conventional PKC, indicating additional kinases may contribute to p38 kinase activation after PMA treatment. One such kinase could be PKCδ, a novel PKC activated by PMA that can phosphorylate p38 kinase in HT-1080 cells (Hwang YP, 2010).
When we examined the role of the two MAP kinase pathways on 1,25(OH)2D3-mediated hCYP24A1 gene transcription, we found that inhibitors of p38 kinase a/b and MEK1/2 each partially suppress the enhancement of vitamin D action by PMA. While chemical inhibitors of kinases can lack specificity, our findings are consistent with other approaches. Nutchey et al. (Nutchey et al., 2005) previously showed that constructs expressing dominant negative versions of ERK1 or MKK4 could reduce the effects of PMA on 1,25(OH)2D3-mediated induction of a rat CYP24A1 promoter reporter gene construct. They viewed the data from the dominant negative MKK4 construct as supportive of a role for JNK in the PMA enhancing effect. However, since MKK4 can also directly phosphorylate p38 kinase (Dhanasekaran and Premkumar, 1998), these data are also consistent with our observation that inhibition of p38 kinase can significantly reduce the PMA enhancing effect of 1,25(OH)2D3 on the hCYP24A1 promoter.
The effects of PMA are presumably mediated through the phosphorylation of transcription factors or their co-activators which then work through specific elements in the DNA to regulate hCYP24A1 gene transcription. We conducted additional studies to determine the DNA binding regions that were affected by PMA treatment. Others have identified a C/EBP binding site at −964 bp in the rat CYP24A1 promoter (Dhawan et al., 2005) and a putative AP-1 binding site at −1166 bp in the hCYP24A1 promoter (Chen and DeLuca, 1995). However, neither of these distal binding sites is necessary for the enhancing effect of PMA. Similar to findings previously reported by others (Barletta et al., 2004;Nutchey et al., 2005), we found that 1,25(OH)2D3-mediated expression of a −298 bp and a −5500 bp hCYP24A1 promoter construct were each enhanced 2-fold by PMA pretreatment. Within the −298 bp hCYP24A1 promoter there are several potential regulatory regions where PMA could influence transcription.
First, the hCYP24A1 promoter contains two vitamin D responsive elements (VDRE) (at −172/−143 bp and −293/−272 bp) (Chen and DeLuca, 1995). Our data show that PMA enhanced 1,25(OH)2D3-mediated expression on both a minimal 3X osteopontin VDRE-luciferase reporter gene construct as well as the −298 bp hCYP24A1 promoter construct. Although the effect of PMA was lower on the minimal 3X VDRE reporter gene, this indicates there are effects of PMA mediated in part through the VDR interactions with its traditional binding site. Similarly, Barletta et al. (Barletta et al., 2004) found that PMA had the same enhancing effect on vitamin D action on both a full-length hCYP24A1 promoter (−5500 bp) and a minimal promoter that contained the proximal VDR from the hCYP24A1 promoter. This suggests that either VDR binding to the VDRE or assembly of the transcriptional complex on the CYP24A1 promoter is sensitive to phosphorylation events dependent upon PMA. However, Barletta et al. (Barletta et al., 2004) reported that PMA did not alter the VDRE binding ability of nuclear extracts from vitamin D-treated LLCPK-1 cells. This suggests that the impact of PMA-mediated phosphorylation may be through co-activators. We have previously shown that the co-activator MED1 is phosphorylated by ERK1/2 and is necessary for 1,25(OH)2D3 mediated CYP24A1 gene expression in differentiated Caco-2 cells (Cui et al., 2009). Since ERK1/2 is one of the signaling pathways activated by PMA treatment in differentiated Caco-2 cells (Figure 2), we propose that the synergy between PMA and 1,25(OH)2D3 through VDRE site is mediated through the recruitment of ERK1/2 phosphorylated MED-1 to the CYP24A1 promoter or its function at the promoter. Consistent with this hypothesis, Misra et al. (Misra et al., 2002) previously showed that MED-1 (also called PBP or DRIP205) is subject to MAPK-dependent phosphorylation and that this enhanced the transcriptional potential of PPARγ in HeLa cells. However, MED-1 phosphorylation did not increase the interaction between MED-1 and PPARγ or with its heterdimeric partner RXR (Misra et al., 2002) nor does PMA treatment alter the interaction between MED-1 and VDR in HEK 293T cells (Barletta et al., 2004). This suggests MED-1 phosphorylation contributes to 1,25(OH)2D3-mediated expression of hCYP24A1 by enhancing interactions of the VDR-RXR-MED-1 complex with the basal transcription unit containing RNA polymerase II. This hypothesis remains to be tested.
Another potential site that could be influenced by MAPK signaling is the Ets binding site (EBS) at −128/−116 previously characterized in the rat CYP24A1 promoter (Cui et al., 2009;Dwivedi et al., 2000). Ets binding to the EBS is dependent upon ERK5-mediated phosphorylation of Ets-1 at threonine 38 (Dwivedi et al., 2002) and mutation of the core EBS motif reduces 1,25(OH)2D3-dependent regulation of the rat CYP24A1 promoter by 50% in COS-1 cells (Dwivedi et al., 2000) and by 40% in proliferating Caco-2 cells (Cui et al., 2009). In proliferating cultures of HEK-293T cells, mutation of the EBS site in the rat CYP24A1 promoter reduced the impact of PMA on 1,25(OH)2D3–mediated transcription by 50% (Nutchey et al., 2005). The hCYP24A1 promoter has an EBS site at −138/−130 (5′-TCCATCCTCC-3′) that is identical to the rat EBS. However, mutation of human EBS had no effect on either 1,25(OH)2D3-induced nor PMA-enhanced hCYP24A1 promoter activity in the non-proliferating, differentiated cultures of Caco-2 cells we used for these experiments. We previously reported that the EBS has no role in 1,25(OH)2D3-mediated induction of the rat CYP24A1 promoter in differentiated Caco-2 cells (Cui et al., 2009). Our new data extend this observation to the regulation of the human CYP24A1 promoter. In addition, because the EBS is not involved in the PMA enhanced regulation of the human CYP24A1 promoter in differentiated Caco-2 cells, our data suggest that ERK5 is not activated by PMA in differentiated Caco-2 cells.
A final sequence we tested was the VSE site previously characterized as important for the enhancing effects of PMA on vitamin D-regulated expression of the rat CYP24A1 promoter in COS-1 cells (Nutchey et al., 2005). Nutchey et al. (Nutchey et al., 2005) showed that mutation of the VSE site in rat CYP24A1 promoter reduced 1,25(OH)2D3 induction of the gene by 50% and led to an almost complete loss of the PMA/1,25(OH)2D3 synergy. Kumar et al. (Kumar et al., 2010) previously identified the same putative VSE in the human CYP24A1 as our alignment revealed. However, Kumar et al. found that the human VSE was not necessary for vitamin D induced expression of a human CYP24A1 reporter gene in the absence of PMA in rat osteoblast-like ROS17/2.8 cells and in human embryonic kidney HEK293T cells. In contrast, we found that mutating the human VSE site reduced 1,25(OH)2D3-induced activation of the hCYP24A1 promoter by 70% and strongly suppressed (but did not eliminate) the enhancing effect of PMA on 1,25(OH)2D3-induced hCYP24A1 gene expression. Nutchey and coworkers (Nutchey et al., 2005) used EMSA to show that the VSE site in the rat CYP24A1 promoter can specifically bind to a nuclear protein complex from HEK-293T cells; this binding was present in nuclear extracts from untreated cells and it was not altered by treatment with PMA, 1,25(OH)2D3 or the two compounds combined. Although PMA can function through the AP-1 complex in other cellular contexts and the VSE site shares sequence identity with the binding site for the AP1 family member c-Jun, supershift assays using antibodies against the AP-1 family members c-Jun, Jun B, Jun D, c-Fos, Fos B, Fra-1 and Fra-2 and to the cAMP-responsive-element-binding proteins ATF2 and CREB-1 revealed these do not bind the VSE in the rat promoter (Nutchey et al., 2005). Thus, although our data support the importance of the VSE as a modulator of vitamin D action on the hCYP24A1 gene, the transcription factor mediating this effect is still unknown. We examined a VSE positional weight matrix built from the rat and human sequences for similarity to other known transcription factor binding sites using JASPAR (Wasserman and Sandelin, 2004). The GGTCA sequence in the VSE site (CCCCGGTCA) was identical to the proximal half-site of both the PPAR gamma and Estrogen Receptor binding sites, but monomeric binding of these nuclear receptors to DNA is not likely. The VSE site also showed similarity to the binding site for ROR alpha (AT/ANNA/GGGTCA). This transcription factor can be phosphorylated on serine 35 by PKC alpha in HCT116 cells (Lee et al., 2010) so it is a candidate that deserves further testing for its ability to enhance vitamin D-induced CYP24A1 gene regulation in response to PMA treatment.
In summary, our data both confirm and extend observations regarding the enhancement of 1,25(OH)2D3-mediated CYP24A1 induction by PMA. First, we show that modulation of vitamin D-induced CYP24A1 gene regulation by PMA extends to a primary vitamin D target cell – the differentiated intestinal absorptive epithelial cell. Next, we show that three signaling pathways, classic PKC, ERK1/2, and p38 kinase each contribute to the 1,25(OH)2D3/PMA synergism on the hCYP24A1 gene. We also identified a VSE site in the hCYP24A1 gene promoter that is functionally similar to the one previously identified in the rat CYP24A1 gene promoter and we demonstrate that events occur on the VDRE and the VSE within the −298 bp hCYP24A1 promoter are necessary for the enhancing effect of PMA on vitamin D-mediated hCYP24A1 gene transcription. In contrast, the EBS that binds Ets-1 does not participate in the PMA effect in non-proliferating, differentiated Caco-2 cells.
Acknowledgments
Contract grant sponsor: National Institutes of Health;
Contract grant number: DK054111 to JCF
Reference List
- Almog T, Naor Z. Mitogen activated protein kinases (MAPKs) as regulators of spermatogenesis and spermatozoa functions. Molecular and Cellular Endocrinology. 2008;282:39–44. doi: 10.1016/j.mce.2007.11.011. [DOI] [PubMed] [Google Scholar]
- Armbrecht HJ, Boltz MA, Hodam TL, Kumar VB. Differential responsiveness of intestinal epithelial cells to 1,25- dihydroxyvitamin D3--role of protein kinase C. J Endocrinol. 2001;169:145–151. doi: 10.1677/joe.0.1690145. [DOI] [PubMed] [Google Scholar]
- Armbrecht HJ, Chen ML, Hodam TL, Boltz MA. Induction of 24-hydroxylase cytochrome P450 by 1,25-dihydroxyvitamin D and phorbol exters in normal rat kidney (NRK-52E) cells. J Endocrinol. 1997;153:199–205. doi: 10.1677/joe.0.1530199. [DOI] [PubMed] [Google Scholar]
- Armbrecht HJ, Hodam TL, Boltz MA, Chen ML. Phorbol ester markedly increases the sensitivity of intestinal epithelial cells to 1,25-dihydroxyvitamin D3. FEBS Lett. 1993;327:13–16. doi: 10.1016/0014-5793(93)81028-x. [DOI] [PubMed] [Google Scholar]
- Barletta F, Dhawan P, Christakos S. Integration of hormone signaling in the regulation of human 25(OH)D3 24-hydroxylase transcription. Am J Physiol Endocrinol Metab. 2004;286:E598–E608. doi: 10.1152/ajpendo.00214.2003. [DOI] [PubMed] [Google Scholar]
- Bronner F, Pansu D, Stein WD. An analysis of intestinal calcium transport across the rat intestine. Am J Physiol. 1986;250:G561–G569. doi: 10.1152/ajpgi.1986.250.5.G561. [DOI] [PubMed] [Google Scholar]
- Cantorna MT, Munsick C, Bemiss C, Mahon BD. 1,25-dihydroxycholecalciferol prevents and ameliorates symptoms of experimental murine inflammatory bowel disease. Journal of Nutrition. 2000;130:2648–2652. doi: 10.1093/jn/130.11.2648. [DOI] [PubMed] [Google Scholar]
- Chen KS, DeLuca HF. Cloning of the human 1 alpha, 25-dihydroxyvitamin D-3 24-hydroxylase gene promoter and identification of two vitamin D-responsive elements. Biochim Biophys Acta. 1995;1263:1–9. doi: 10.1016/0167-4781(95)00060-t. [DOI] [PubMed] [Google Scholar]
- Chen ML, Boltz MA, Armbrecht HJ. Effects of 1,25-dihydroxyvitamin D3 and phorbol ester on 25- hydroxyvitamin D3 24-hydroxylase cytochrome P450 messenger ribonucleic acid levels in primary cultures of rat renal cells. Endocrinology. 1993;132:1782–1788. doi: 10.1210/endo.132.4.7681765. [DOI] [PubMed] [Google Scholar]
- Cui M, Zhao Y, Hance KW, Shao A, Wood RJ, Fleet JC. Effects of MAPK signaling on 1,25-dihydroxyvitamin D-mediated CYP24 gene expression in the enterocyte-like cell line, Caco-2. J Cell Physiol. 2009;219:132–142. doi: 10.1002/jcp.21657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanasekaran N, Premkumar RE. Signaling by dual specificity kinases. Oncogene. 1998;17:1447–1455. doi: 10.1038/sj.onc.1202251. [DOI] [PubMed] [Google Scholar]
- Dhawan P, Peng X, Sutton AL, MacDonald PN, Croniger CM, Trautwein C, Centrella M, McCarthy TL, Christakos S. Functional cooperation between CCAAT/enhancer-binding proteins and the vitamin D receptor in regulation of 25-hydroxyvitamin D3 24-hydroxylase. Mol Cell Biol. 2005;25:472–487. doi: 10.1128/MCB.25.1.472-487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncia JV, Santella JB, Higley CA, Pitts WJ, Wityak J, Frietze WE, Rankin FW, Sun JH, Earl RA, Tabaka AC, Teleha CA, Blom KF, Favata MF, Manos EJ, Daulerio AJ, Stradley DA, Horiuchi K, Copeland RA, Scherle PA, Trzaskos JM, Magolda RL, Trainor GL, Wexler RR, Hobbs FW, Olson RE. MEK inhibitors: The chemistry and biological activity of U0126, its analogs, and cyclization products. Bioorganic & Medicinal Chemistry Letters. 1998;8:2839–2844. doi: 10.1016/s0960-894x(98)00522-8. [DOI] [PubMed] [Google Scholar]
- Dwivedi PP, Hii CST, Ferrante A, Tan J, Der CJ, Omdahl JL, Morris HA, May BK. Role of MAP kinases in the 1,25-dihydroxyvitamin D-3-induced transactivation of the rat cytochrome P450C24 (CYP24) promoter - Specific functions for ERK1/ERK2 and ERK5. Journal of Biological Chemistry. 2002;277:29643–29653. doi: 10.1074/jbc.M204561200. [DOI] [PubMed] [Google Scholar]
- Dwivedi PP, Omdahl JL, Kola I, Hume DK, May BK. Regulation of rat cytochrome P450C24 (CYP24) gene expression - Evidence for functional cooperation of Ras-activated Ets transcription factors with the vitamin D receptor in 1,25-dihydroxyvitamin D-3-mediated induction. Journal of Biological Chemistry. 2000;275:47–55. doi: 10.1074/jbc.275.1.47. [DOI] [PubMed] [Google Scholar]
- Fedirko V, Bostick RM, Flanders WD, Long Q, Sidelnikov E, Shaukat A, Daniel CR, Rutherford RE, Woodard JJ. Effects of Vitamin D and Calcium on Proliferation and Differentiation In Normal Colon Mucosa: a Randomized Clinical Trial. Cancer Epidemiology Biomarkers & Prevention. 2009;18:2933–2941. doi: 10.1158/1055-9965.EPI-09-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fichera A, Little N, Dougherty U, Mustafi R, Cerda S, Li YC, Delgado J, Arora A, Campbell LK, Joseph L, Hart J, Noffsinger A, Bissonnette M. A vitamin D analogue inhibits colonic carcinogenesis in the AOM/DSS model. J Surg Res. 2007;142:239–245. doi: 10.1016/j.jss.2007.02.038. [DOI] [PubMed] [Google Scholar]
- Fleet JC, Eksir F, Hance KW, Wood RJ. Vitamin D-inducible calcium transport and gene expression in three Caco-2 cell lines. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2002;283:G618–G625. doi: 10.1152/ajpgi.00269.2001. [DOI] [PubMed] [Google Scholar]
- Froicu M, Weaver V, Wynn TA, McDowell MA, Welsh JE, Cantorna MT. A crucial role for the vitamin D receptor in experimental inflammatory bowel diseases. Molecular Endocrinology. 2003;17:2386–2392. doi: 10.1210/me.2003-0281. [DOI] [PubMed] [Google Scholar]
- Hwang YPYH. Suppression of phorbol-12-myristate-13-acetate-induced tumor cell invasion by bergamottin via the inhibition of protein kinase Cdelta/p38 mitogen-activated protein kinase and JNK/nuclear factor-kappaB-dependent matrix metalloproteinase-9 expression. Molecular nutrition & food research. 2010;54:977–990. doi: 10.1002/mnfr.200900283. [DOI] [PubMed] [Google Scholar]
- Kazanietz MG. Novel “nonkinase” phorbol ester receptors: The C1 domain connection. Molecular Pharmacology. 2002;61:759–767. doi: 10.1124/mol.61.4.759. [DOI] [PubMed] [Google Scholar]
- Klopot A, Hance KW, Peleg S, Barsony J, Fleet JC. Nucleo-cytoplasmic cycling of the vitamin D receptor in the enterocyte-like cell line, Caco-2. J Cell Biochem. 2006 doi: 10.1002/jcb.21087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama H, Inaba M, Nishizawa Y, Ohno S, Morii H. Protein-Kinase-C Is Involved in 24-Hydroxylase Gene-Expression Induced by 1,25(Oh)(2)D-3 in Rat Intestinal Epithelial-Cells. Journal of Cellular Biochemistry. 1994;55:230–240. doi: 10.1002/jcb.240550210. [DOI] [PubMed] [Google Scholar]
- Kumar R, Iachini DN, Neilsen PM, Kaplan J, Michalakas J, Anderson PH, May BK, Morris HA, Callen DF. Systematic characterisation of the rat and human CYP24A1 promoter. Mol Cell Endocrinol. 2010;325:46–53. doi: 10.1016/j.mce.2010.04.023. [DOI] [PubMed] [Google Scholar]
- Lee JM, Kim IS, Kim H, Lee JS, Kim K, Yim HY, Jeong J, Kim JH, Kim JY, Lee H, Seo SB, Kim H, Rosenfeld MG, Kim KI, Baek SH. RORalpha attenuates Wnt/beta-catenin signaling by PKCalpha-dependent phosphorylation in colon cancer. Mol Cell. 2010;37:183–195. doi: 10.1016/j.molcel.2009.12.022. [DOI] [PubMed] [Google Scholar]
- Ly LH, Zhao ZY, Holloway L, Feldman D. Liarazole acts synergistically with 1a,25-dihydroxyvitamin D3 to inhibit growth of DU145 human prostate cancer cells by blocking 24-hydroxylase activity. Endocrinology. 1999;140:2071–2076. doi: 10.1210/endo.140.5.6698. [DOI] [PubMed] [Google Scholar]
- Miller GJ, Stapleton GE, Hedlund TE, Moffat KA. Vitamin D receptor expression, 24-hydroxylase activity, and inhibition of growth by 1 alpha, 25-dihydroxyvitamin D3 in seven human prostatic carcinoma cell lines. Clin Cancer Res. 1995;1:997–1003. [PubMed] [Google Scholar]
- Misra P, Owuor ED, Li W, Yu S, Qi C, Meyer K, Zhu YJ, Rao MS, Kong AN, Reddy JK. Phosphorylation of transcriptional coactivator peroxisome proliferator-activated receptor (PPAR)-binding protein (PBP). Stimulation of transcriptional regulation by mitogen-activated protein kinase. J Biol Chem. 2002;277:48745–48754. doi: 10.1074/jbc.M208829200. [DOI] [PubMed] [Google Scholar]
- Newton AC. Protein kinase C: Structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chemical Reviews. 2001;101:2353–2364. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- Nomura N, Nomura M, Sugiyama K, Hamada JI. Phorbol 12-myristate 13-acetate (PMA)-induced migration of glioblastoma cells is mediated via p38MAPK/Hsp27 pathway. Biochemical Pharmacology. 2007;74:690–701. doi: 10.1016/j.bcp.2007.06.018. [DOI] [PubMed] [Google Scholar]
- Nutchey BK, Kaplan JS, Dwivedi PP, Omdahl JL, Ferrante A, May BK, Hii CST. Molecular action of 1,25-dihydroxyvitamin D-3 and phorbol ester on the activation of the rat cytochrome P450C24 (CYP24) promoter: role if MAP kinase activities and identification of an important transcription factor binding site. Biochemical Journal. 2005;389:753–762. doi: 10.1042/BJ20041947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omdahl JL, Morris HA, May BK. Hydroxylase enzymes of the vitamin D pathway: expression, function, and regulation. Annu Rev Nutr. 2002;22:139–166. doi: 10.1146/annurev.nutr.22.120501.150216. [DOI] [PubMed] [Google Scholar]
- Pike JW, Zella LA, Meyer MB, Fretz JA, Kim S. Molecular actions of 1,25-dihydroxyvitamin D3 on genes involved in calcium homeostasis. J Bone Miner Res. 2007;22(Suppl 2):V16–V19. doi: 10.1359/jbmr.07s207. [DOI] [PubMed] [Google Scholar]
- Shao A, Wood RJ, Fleet JC. Increased vitamin D receptor level enhances 1,25-dihydroxyvitamin D3- mediated gene expression and calcium transport in Caco-2 cells. J Bone Miner Res. 2001;16:615–624. doi: 10.1359/jbmr.2001.16.4.615. [DOI] [PubMed] [Google Scholar]
- Sinkkonen L, Malinen M, Saavalainen K, Vaisanen S, Carlberg C. Regulation of the human cyclin C gene via multiple vitamin D-3-responsive regions in its promoter. Nucleic Acids Research. 2005;33:2440–2451. doi: 10.1093/nar/gki502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Arnaud R, Arabian A, Travers R, Barletta F, Raval-Pandya M, Chapin K, Depovere J, Mathieu C, Christakos S, Demay MB, Glorieux FH. Deficient mineralization of intramembranous bone in vitamin D-24-hydroxylase-ablated mice is due to elevated 1,25-dihydroxyvitamin D and not to the absence of 24,25-dihydroxyvitamin D. Endocrinology. 2000;141:2658–2666. doi: 10.1210/endo.141.7.7579. [DOI] [PubMed] [Google Scholar]
- Wang L, Klopot A, Freund JN, Dowling LN, Krasinski SD, Fleet JC. Control of Differentiation-Induced Calbindin-D9k Gene Expression in Caco-2 Cells by Cdx-2 adn HNF-1α. Am J Physiol. 2004;287:G943–G953. doi: 10.1152/ajpgi.00121.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman WW, Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet. 2004;5:276–287. doi: 10.1038/nrg1315. [DOI] [PubMed] [Google Scholar]