

Abstract
The cardiac action potential (AP) controls the rise and fall of intracellular free Ca2+ (Cai), and thus the amplitude and kinetics of force generation. Besides excitation-contraction coupling, the reverse process where Cai influences the AP through Cai-dependent ionic currents has been implicated as the mechanism underlying QT alternans and cardiac arrhythmias in heart failure, ischemia/reperfusion, cardiac myopathy, myocardial infarction, congenital and drug-induced long QT syndrome, and ventricular fibrillation. The development of dual optical mapping at high spatial and temporal resolution provides a powerful tool to investigate the role of Cai anomalies in eliciting cardiac arrhythmias. This unit describes experimental protocols to map APs and Cai transients from perfused hearts by labeling the heart with two fluorescent dyes, one to measure transmembrane potential (Vm), the other Cai transients. High spatial and temporal resolution is achieved by selecting Vm and Cai probes with the same excitation but different emission wavelengths, to avoid cross-talk and mechanical components.
Keywords: action potential (AP), intracellular free Ca2+ (Cai), photodiode array (PDA), complementary metal oxide silicon (CMOS) camera, Voltage Sensitive Dye (VSD), Cai indicator, Rhod-2AM, Pittsburgh I (PGH I), RH237
INTRODUCTION
Dual optical mapping of Vm and Cai from multiple sites on the heart is a particularly powerful technique to measure the time course of membrane potential changes and cytosolic Ca2+ transients under physiological and pathological conditions. The method has been used to track the sequences of electrical activation and force generation as well as cardiac repolarization and relaxation as a function of rate and sympathetic activity and during electrically induced ventricular fibrillation (VF) and defibrillation. Dual optical mapping has been playing an important role in elucidating arrhythmia mechanisms in models of heart failure (London et al., 2003), long QT syndrome (Choi et al., 2002), and ischemia/reperfusion (Choi and Salama, 2000; Lakireddy et al., 2005; Choi et al., 2006a,b; Lakireddy et al., 2006), and in genetically altered animal models of human cardiac diseases (Katra et al., 2004; Katra and Laurita, 2005; Lan et al., 2007; London et al., 2007; Roell et al., 2007; Hayashi et al., 2008; Laurita and Rosenbaum, 2008; Saba et al., 2008).
The fluorescent probes to measure Vm and Cai and the optical instrument for dual mapping must be carefully selected and designed to achieve the necessary spatial and temporal resolution: (1) Pairs of fluorescent probes for measurements of Vm and Cai should be carefully selected for their specificity to Vm and Cai, their lack of internalization in subcellular organelles, their homogeneity when staining myocytes, and their excitation and emission spectra so that there is minimal risk of cross-talk between them; and (2) the optical instrument should not have any mechanically moving component to change band-pass optical filters, in order to avoid any temporal delays between Vm and Cai readout. An instrument with two separate optical paths, with appropriate filters in each, with separate image sensors to acquire images in synchrony, would in principle provide the highest temporal resolution and efficiency in light collection and thus yield the best signal-to-noise (S/N) ratio. Also, (3) the digitization accuracy and the sampling rate of image acquisition from the heart should be sufficiently high to detect rapidly spreading waves of electrical propagation during pacing, sinus rhythm, and fibrillation of the heart.
Several reviews have previously appeared regarding the advantages and disadvantages of optical instruments and cameras used for optical mapping of electrical activity in heart and nerve networks (Cohen, 1995; Djurisic et al., 2003; Zecevic et al., 2003; Efimov et al., 2004; Entcheva and Bien, 2006; Salama and Choi, 2007). Therefore, this chapter will focus on key features of dual mapping with special attention on detailed protocols used to optimize Vm and Cai signals.
PROTOCOLS
Heart Preparations
A typical study requires a Langendorff apparatus to perfuse the isolated heart in the retrograde direction. The animal is euthanized by a method approved by IACUC (Institutional Animal Care and Use Committee), the chest opened, the heart removed, and the aorta quickly cannulated on a Langendorff apparatus to perfuse the coronary vessels. The isolation of the heart and the cannulation of the aorta should take place in ~1 min. If the dissection cannot be done within a minute or if the Langendorff apparatus is not located adjacent to where the surgical procedure is done, then the isolated heart must be immediately immersed a small beaker with ice-cold perfusate, which in turn is kept in an ice bath. The isolated heart can then be transported, and should be cannulated preferably within 10 min. A stainless steel tube, with a diameter slightly larger than the aorta, serves as the cannula. The aorta is tied to the tip of the cannula, which has ridges or grooves on the tip to help catch the silk thread used to insure a tight seal between the cannula and the aorta.
With a Langendorff apparatus, hearts can be perfused at either constant coronary pressure or constant flow rate. Constant coronary pressure is typically achieved by gravity perfusion, where the perfusate is placed in a flask held at a specific height above the aortic cannula. In such a setup, pressure is constant while perfusion flow rate varies as coronary resistance to flow changes during interventions that alter vascular tone. Alternatively, constant coronary flow rate can be maintained with a peristaltic pump that maintains a constant coronary flow, but with pressure varying in response to the changes in coronary resistance. Note that flow oscillations caused by peristaltic pumps can be diminished by (1) selecting pumps with a larger number of rollers (six to eight) and/or (2) having a compliance chamber in the perfusate delivery route in the form of an air column at a constant pressure. In addition, the pump can be stopped for a few seconds during brief intervals of image acquisition.
Optical Apparatus
The standard dual optical mapping system consists of an epi-illumination system with a dichroic mirror at a 45° angle in front of a condenser lens and the excitation band-pass filter, to reflect the excitation light towards the objective macro lens, which focuses the light on the surface of the heart. The same lens collects the fluorescent light from the heart, which passes through the first dichroic mirror. Then, a second 45°-angle dichroic mirror splits the fluorescent light into two components based on the wavelength—short (for Cai measurements) and long (for Vm measurements). The separated fluorescent lights go through respective emission filters and are refocused on two-image sensors.
Figure 12.17.1 is a picture of an optical apparatus based on two Hamamatsu photodiode arrays. Figure 12.17.2 shows the schematics of an optical instrument with two 16 × 16 photodiode arrays (Hamamatsu Photonics K.K., Model C4675-103) as the image sensors. Important components of the optical instrument are further discussed below.
Figure 12.17.1.

A dual optical mapping instrument based on two Hamamatsu photodiode arrays is designed with sealed optics to reduce noise from thermal convection currents in front of the arrays. The photodiode arrays (PDA 1 and PDA 2) and the first-stage amplifiers are housed in the beige boxes where PDA 1 serves to map Cai signals and PDA 2 maps Vm signals. The first dichroic box is located immediately behind the camera lens and splits the excitation beam coming from the lamp housing. The second dichroic box is located behind the first dichroic box and plots the fluorescence from the heart with the low-wavelength image focused on PDA 1 and the long-wavelength image focused on PDA 2.
Figure 12.17.2.

Schematics of optical apparatus with direct illumination. An alternative design for dual optical mapping was used for large fields of view where the light beam from tungsten-halogen lamps can be directly focused on the heart and fluorescence images from the heart are split with a dichroic element and refocused on the surface of two Hamamatsu PDAs. The dichroic element can be rotated and replaced with a mirror to focus an image of the heart on a graticule placed on a parafocal plane and with the exact dimensions of the surface of the PDA. The image of the heart that is focused on the graticule will be automatically focused on the surfaces of the two PDAs.
Excitation light
The light source is typically either a tungsten-halogen (100- to 300-W) or a mercury (Hg) or xenon (Xe) arc (75- to 200-W) lamp where the light is directly coupled to the instrument via a condensing lens or a light guide. Tungsten halogen lamps provide a broad spectrum spanning the entire visible spectrum, with maximum intensity at deep-red to near-infrared (near-IR or NIR), 700 to 800 nm, while arc lamps do better at blue to green wavelengths. Mercury arc lamps have narrow high-intensity spectral peaks at 435 nm (violet), 546 nm (green), and 578 nm (yellow) from mercury vapor, while xenon arc lamps have relatively smooth and broad spectrum over all visible wavelengths, with its primary peak at yellow to orange (~600 nm) and secondary peak at blue (~480 nm) (http://www.olympusmicro.com/primer/techniques/fluorescence/fluorosources.html). In either case, it is essential to produce an excitation beam of uniform intensity and low noise characteristics. For all light sources, the power supply (dedicated power supply for arc lamps and bench-top-regulated DC power supply for tungsten halogen lamps) must be carefully chosen to minimize AC ripple as well as other low- to medium-frequency noise (<10 kHz), to reduce the noise in the excitation beam (0.1% root mean square or preferably less), because this noise will be injected into the recordings. Light guides have the advantage of placing the light source away from the instrument, but that comes with the trade-off of being inefficient, since a significant portion (typically more than half) of the light is lost at the input end of the guide, and it is also challenging to refocus the output beam on the surface of the heart with uniform intensity. Other options worthy of consideration for (dual) optical mapping are “super luminescent” LEDs (light-emitting diodes) and solid-state lasers. Current super luminescent LEDs are rated at up to 5 W per emitter chip and are available with specific colors (although with somewhat broad spectra, their typical half-widths being anywhere from 20 nm for blue and red LEDs to as much as 40 nm for green LEDs; http://www.lumileds.com/pdfs/DS34.pdf) and stable light outputs. Both offer advantages of low heat generation, stable light intensity during continuous illumination or high-frequency light pulses (turn-on/turn-off times on the order of microseconds), and compact physical dimensions. The relatively low light intensity of LEDs can be overcome by combining several LEDs in a single package. The small physical dimensions of LEDs make it possible to illuminate the heart directly with multiple LEDs (on the order of tens or even hundreds for larger hearts) without resorting to epi-illumination.
Figure 12.17.2 shows an example of an optical apparatus where the excitation beam illuminates the heart at a large angle of incidence and where the first dichroic box is removed. High-intensity diode lasers (0.5 to 1 W of optical power) emit narrow monochromatic beams which can be expanded to match the field of view on the heart and still retain excellent intensity per unit area (>50 mW/mm2). High monochromaticity of diode lasers enables efficient illumination where the excitation wavelength can be selected to be at or near the dye's absorption maximum. Diode lasers, however, are not available around the middle range of visible spectrum, where absorption maxima of the majority of dyes used for optical mapping of the heart lie, but are only available at ultraviolet (UV) to deep blue (440 nm or shorter) or red to near-IR (630 nm or longer). Solid-state lasers, especially diode-pumped solid state (DPSS) lasers, can fill in this range, namely from blue to orange, but they are more expensive and much less efficient than bare diode lasers since they use near-IR diode lasers to pump solid gain media (http://repairfaq.ece.drexel.edu/sam/laserdio.htm). Regardless of the light source used, its noise characteristics should be tested periodically to maximize the S/N ratio of fluorescence recordings.
Selection of lenses and filters
The focal lengths and the aperture sizes of the lenses used for optical mapping depend on the field of view being mapped (for example, mouse hearts, ~4 × 4 mm2; guinea pig hearts, ~1 × 1 cm2; and rabbit hearts, ~2 × 2 cm2) and imaging sensors’ areas. Table 12.17.1 lists the lens combinations most suitable for 16 × 16 element photodiode arrays with a total sensor area of 17.5 × 17.5 mm2. The lens combinations were chosen to maximize the effective numerical aperture (NA) of the system, light collection, and S/N ratio of optical recordings. For a field of view of 4 × 4 mm2, the magnification of the system is 4 ×and for a field of view of 2 × 2 cm2, the magnification of the system is –2×; standard 35-mm, SLR (Single Lens Reflex) camera lenses are best suited for light collection (Fig. 12.17.1) because, at these magnifications, they provide a greater NA and field of view than microscope objectives. Some manufacturers coat their lenses, which may result in significant attenuated light throughput of the excitation beam from the light source to the heart. Such lens coatings would have less impact on light throughput from the heart to the camera and could be used in non-epi-illumination optics, as shown in Figure 12.17.2. All the “secondary” lenses are double-convex or plano-convex, glass-type singlets with high transmission from 420 nm to NIR (near infrared; up to 1 μm), as quartz lenses are not necessary since most common probes have excitation and emission wavelengths in the visible range.
Table 12.17.1.
Lens Selections for Optical Apparatus shown in Figure 12.17.1
| Field of view | Camera lens | Dichroic to array distance | Distance between dichroic boxes | Focal lens of secondary lenses |
|---|---|---|---|---|
| 4 × 4 mm2 | Nikon 50 mm f/1.2a | 75 mm | 65 mm | No lens |
| 5 × 5 mm2 | Nikon 85 mm f/1.4 | 75 mm | 85 mm | No lens |
| 1 × 1 cm2 | Nikon 85 mm f/1.4 | 75 mm | 30 mm | 100 mm |
| 2 × 2 cm2 | Nikon 85 mm f/1.4 | 75 mm | 30 mm | 75 mm |
An alternative is to use an inverted 25-mm Navitar lens.
The band-pass filters (2″ × 2″) are rarely kept in stock by most manufacturers and should be ordered as custom-made components. The transmittance should be at least 80% at peak wavelength, with a 99.99% (OD 5) block of infrared light. The second dichroic, used to split the fluorescence light into short (Cai) and long (Vm) wavelengths, should be precisely placed at a 45° angle between the two cameras and should be thin, 0.5 to 1.0 mm, to minimize secondary reflections from the back surface of the dichroic mirror. The alignment of the dichroic mirror relative to the two cameras is critical; it is necessary to place the two arrays in perfect register such that one photodiode in the Cai array and another at the same location in the Vm array detect fluorescent light from exactly the same area in the field of view. The filters, both long-pass and band-pass, should be of interference type rather than absorptive (i.e., Schott glass filter) to ensure effective transmission as well as to minimize the cross-talk between Cai and Vm signals.
Image sensors
Three types of image sensors have been used for dual mapping in the heart; each type of camera has unique advantages and disadvantages and their properties are summarized in Table 12.17.2.
Table 12.17.2.
Comparison Between Different Image Sensors
| PDA Hamamatsu C4675-103a | PDA RedShirt Imaging Cardio PDA-III | CCD Dalsa CA-D1-0128Ta | CCD Redshirt Imaging Cardio CCD-SMQ | CMOS SciMedia Ultima-L | CMOS RedShirt Imaging Cardio-CMOS | |
|---|---|---|---|---|---|---|
| Quantum efficiency |
80% at 550 nm 85% at 700 nm |
— | ~10%b | 80% at 400-600 nm 75% at 650 nm |
63% at 550 nm 45% at 700 nm |
~60%c |
| Sensor size | 17.5 × 17.5 mm2 | 225 mm2d | 2.0 × 2.0 mm2 | 1.9 × 1.9 mm2 | 10.0 × 10.0 mm2 |
16.4 × 16.4 mm2 |
| No. of pixels | 16 × 16 | 464 (hexagon) | 128 × 128 | 80 × 80 | 100 × 100 | 128 × 128 |
| Pixel size | 0.95 × 0.95 mm2 | 440 μm2 | 16 × 16 μm2 | 24 × 24 μm2 | 100 × 100 μm2 | 128 × 128 μm2 |
| Dynamic range |
104 ~ 106e | 105-107 | 103 | 104-105 | 105 | 105 |
| Digitization | Off-camera (14-bit)f,g |
Off-camera (14-bit)f,h |
12-bit | 14-biti | 14-bit | 14-bit |
| Well depth (e–) |
N/A (108-1010)j | >109k | 300,000 | 215,000 | 1,500,000k,l | 1,000,000k,m |
| AC coupling | Yesn | Yeso | No | No | No | No |
| Max. frame rate |
8000 | 1600 | 490 | 2000p | 10,000 | 2500q |
Obsolete: no longer available from the manufacturers.
Not reported by the manufacturer. Estimation from Laude et al. (2002).
Not reported by the manufacturer. Estimation from private communications between the manufacturer and the authors.
Sensing area is a 13-side packed hexagon, which is relayed to individual photodiodes via optical fibers.
AC coupling in the amplification stage increases effective dynamic range.
Digitization circuit comes after the secondary amplification stage, both of which are custom-made independent of the PDA units.
Digitization board: Microstar Labs DAP-3400/a (Choi and Salama, 2000).
Digitization board: Microstar Labs DAP-4200/a.
Manufacturer offers 16-bit version with slower frame rate.
Not applicable, since the sensor directly converts photon energy into electrical current, instead of into charges. The numbers in parentheses are estimated value based on its sensor responsivity (350 to 500 mA/W) and its transimpedance factor, 10 V/μA, to compare against the other sensor types.
“Effective” electron well size reported by the manufacturer. Like the PDA, the CMOS sensor by itself does not have the concept of electron well due to its operation principle.
An alternative version, Ultima-H, is available with specified electron well size of 10 million, but camera sensitivity is reduced since the digitization remains at 14-bit. With a bright object or at a slower frame rate (i.e., longer sampling time), however, this decreased sensitivity can increase SNR significantly.
Also has an option to select 12 or 100 million electron well sizes in addition to the 1 million via software control, but like Ultima-L vs. Ultima-H, overall camera sensitivity will be reduced, making it suitable for imaging bright object or at slower frame rate.
Implemented in the off-chip custom-made secondary amplification stage, before digitization.
Included in the system transimpedance amplification stage.
At full frame (14-bit version). 5,000 fps can be reached with 3 × 3 binning, effectively resulting in 26 × 26 pixels and 72 μm pixel size, and 10,000 fps by limiting the imaging area to 80 × 12 pixels.
With full frame acquisition. Options of 5,000/10,000 fps are also available by limiting the imaging area to 128 × 64/128 × 32 pixels.
Photodiode arrays (PDAs)
Photodiode arrays (PDAs) have been extensively used for dual mapping of Vm and
Cai to take full advantage of the high quantum efficiency and large pixel size that are typical of silicon photodiodes, particularly in the visible range (Hamamatsu Photonics K.K., Model C4675-103) (Choi and Salama, 2000). At 550 nm, its quantum efficiency (QE) is 80%, with a responsivity of 355 mA/W of optical power; at 700 nm, the QE is 85% at 480 mA/W. Each diode on the array is connected to a channel of the I-V (current-to-voltage) converter array, and the array is read out as 256 parallel voltage-output channels. Model C4675-103 has an I-V conversion ratio (or trans-impedance gain) of 107 V/A and a frequency response of DC to 10 kHz. Two other models are available with greater amplifier gains but with lower frequency responses; C4675-102 has a gain of 108 V/A at DC to 1 kHz and C4673-302 has a gain of 5 × 107 V/A at DC to 3 kHz response. Model C4675-103 was selected for its highest frequency response, so that signals with rise times of 1 msec or less can be faithfully quantified. The flexibility of the system was enhanced by passing the voltage output of each channel through custom-made second-stage amplifiers with variable gain and additional filters with variable cut-on and cut-off frequencies. The 512 channels of photodiode outputs (256 channels/array) after the second-stage amplification, plus 16 instrumentation channels, were digitized in parallel using four multiplexed flash A/D (analog-to-digital) converters at 4000 samples per second per channel—128 channels per converter, with 1 million samples per converter (MicroStar Laboratories Inc., http://www.mstarlabs.com)—then were stored in computer storage. An important advantage of PDA over CCD or CMOS image sensors is its excellent dynamic range and its greater digitization accuracy. These advantages are due to its considerably larger sensor area (0.9 × 0.9 mm2 per diode), and QE of the diode compared to the elements on CCD or CMOS cameras (see Table 12.17.2). In addition, the second-stage amplifiers have the option of having either DC- or AC-coupled outputs. In AC-coupled mode, the background fluorescence is zeroed so that only the fractional fluorescence change is amplified and digitized, resulting in even higher dynamic range and precision in the digitization. The major disadvantage of PDA, namely the low spatial resolution (a total of 256, or 16 × 16 pixels), can also be considered an advantage, since fewer channels make it possible to record Vm uninterruptedly for over 30 min, compared to CCD or CMOS cameras which can only record images for bursts of a few seconds (with the SciMedia Ultima-One CMOS, one can record for 40 sec at 1000 frames/sec).
Charged-coupled devices (CCDs)
More recently, two CCD cameras (Dalsa Inc., model CA-D1-0128T; http://www.dalsa.com/) have been used in place of the PDAs for dual optical mapping of Vm and Cai (Chou et al., 2008). The major advantages of CCD sensors are their higher spatial resolution (128 × 128 pixels) and on-board digitization of the signals, which greatly simplifies the image acquisition setup. However, there are also severe disadvantages like lower dynamic range from the lack of AC-coupling capability, low quantum efficiency, slower frame rate (only up to 500 frames per second, or 2 msec per frame), and small sensor area (2 × 2 mm2; 16 × 16 μm2 per pixel). The small size of CCD sensors requires a considerable amount of demagnification of the field of view from the heart's surface to the CCD's surface, which implies that the fluorescent light is collected with significantly lower effective numerical aperture (NA), and thus results in poor light throughput. Also, while there are specialized low-light CCD sensors with high QE (>90% with back-illumination on thinned-substrate CCDs), most fast-frame-rate CCDs have much lower QE, usually ~10% to 20%. Table 12.17.2 compares the properties of the various sensors, and it should be easy to see that the technology is shifting to CMOS cameras, as CMOS image sensors have advantages over CCD sensors that make them particularly suitable to high-speed, low-light image acquisitions. Dalsa has a detailed comparison of the two technologies with their pros and cons on their Web site (http://www.dalsa.com/).
Complementary metal oxide silicon (CMOS) cameras
The CMOS sensor is a representative type of the so-called “active pixel sensor (APS),” where each pixel consists of a photodetector (typically a photodiode) coupled with a dedicated active amplifier—essentially an integrated-circuit version of PDA with all its accompanying amplification circuitries embedded on a single sensor chip, or, simply put, a hybrid between a CCD and a PDA (Fossum, 1993a,b; Mendis et al., 1993, 1994; Zimmermann, 2000). The name CMOS comes from its manufacturing process, which is the same as many semiconductor integrated circuit (IC) devices. The CMOS sensor was developed as an alternative to the CCD image sensor, which is a type of passive sensor in the sense that each pixel element converts photon energy into electrical charges (integration phase), which are subsequently turned into electrical current (read-out phase) as the final output. The CMOS sensor, on the other hand, is “active” in that it has a dedicated transimpedance amplifier (i.e., with a current-to-voltage conversion stage) for each photodetector that turns electrical current converted by the detector into output voltage. The CMOS sensor solves some of the fundamental problems of CCD sensors, namely the speed and the scalability issue, at the expense of increased design complexity, i.e., even though it is more complicated to design CMOS sensors initially, it is relatively easier to make this type of sensor go faster, increase the number of pixels, and make individual pixels larger. As such, CMOS sensors are used where high spatial and/or temporal resolution is needed. Obviously, optical mapping of the heart is one of the ideal applications where these qualities of the CMOS sensors can clearly be appreciated—currently available CMOS cameras have the advantages of larger sensor area, higher QE (>60%), and greater dynamic range (usually 14 bits), combined with faster scan rates (up to 10,000 frames per second) compared to fast-frame-rate CCD cameras (Table 12.17.2). These advantages over CCD cameras have been recently utilized to map activation patterns during ventricular fibrillation (VF) and to identify wave break locations. (Choi et al., 2007).
Figure 12.17.3 shows an optical mapping system using two CMOS cameras (SciMedia Inc., Model Ultima-L; http://www.scimedia.com/).
Figure 12.17.4 shows an image of a guinea pig heart along with simultaneous measurements of Vm and Cai from three sites on the heart, recorded with the CMOS cameras at a frame rate of 1000 per second.
Figure 12.17.4.
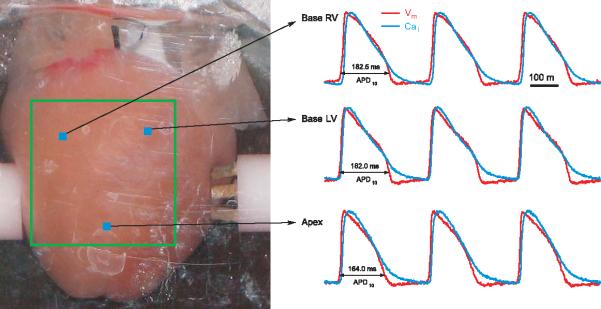
Simultaneous recordings of Vm and Cai from CMOS cameras. With the apparatus shown in Figure 12.17.3, maps of Vm and Cai can be recorded at high spatial and temporal resolution. The left panel shows an image of the heart taken by one of the CMOS cameras with white light illumination. The heart is placed in a chamber designed to abate motion artifacts, and a silhouette of the CMOS sensing surface (square outline) is drawn on the heart to identify the region of the heart that is being mapped by the two CMOS cameras. Each CMOS camera recorded AP or Cai from 100 × 100 pixels at a frame rate of 2 kHz. Examples of simultaneous recordings of APs and Cai transients are shown from three locations on the heart (right traces).
Figure 12.17.5 compares action potentials recorded with the CMOS from SciMedia (Ultima-L) and that made by RedShirt (CardioCMOS; http://www.redshirtimaging.com).
Figure 12.17.5.

APs recorded with CMOS cameras from SciMedia and RedShirt. A guinea pig heart was labeled with PGH1-PEG750 (200 μl of 1 mM dye) and APs were mapped with the Ultima-L camera then the CardioCMOS with areas of 600 and 770 μm, respectively. Frame rate: 1000 frames/sec.
SELECTION OF Vm AND Cai PROBES
Simultaneous mapping of Vm and Cai requires that the heart be uniformly labeled with two fluorescence probes: one to measure voltage and the other to measure intracellular Ca2+. The probes must be carefully selected to avoid cross-talk between these two measurements, which means that the dyes must have either the same excitation (λex) but different emission spectra (λem1 and λem2), or different excitation wavelengths (λex1 and λex2) and the same emission wavelength (λem). Another less desirable option is to choose two dyes where both the excitation and emission spectra differ. Each approach has different requirements:
In the case where the two dyes have similar emissions but different excitations, the optical instrument must rapidly switch the excitation wavelength to sequentially expose the heart to a beam that will excite one dye, then the other, while recording fluorescence images of Vm, and then Cai, intermittently, but in synchrony with the excitation of the Vm and Cai dye. In this configuration, a single camera is needed to measure fluorescence images serially for Vm and Cai in turn, every time there is a change in the excitation wavelength. Rapid wavelength changes have been accomplished with one light source and a high-speed rotating filter wheel which sequentially passes light at multiple wavelengths (up to eight λex). Another approach is based on two light sources; each excitation beam is passed through interference (band-pass) filters that select for λex1 and λex2, followed by a piezoelectric mirror that sequentially blocks one beam then the other, thereby alternating the excitation beam between λex1 and λex2 for dual-wavelength excitation of the preparation. Another approach under development in our laboratory is to rapidly turn “on” and “off” (10 msec on and 10 msec off) super-luminescent LEDs of different color. Square-voltage pulses can be applied to two LEDs that emit at different wavelengths to generate sequential, out-of-phase light pulses at λex1 and λex2. The LEDs can be housed in a box with a 45° dichroic mirror to generate a high-speed light source that alternates between the two excitation wavelengths. A current limitation is the low light intensity that can be obtained from a single LED, but this may be overcome in the near future as more intense LEDs become available commercially. Alternatively, LEDs could be substituted by more powerful solid-state lasers, a practical approach for small fields of view. The main advantage of using dyes with the same emission but different excitation wavelengths is that only one camera is needed to record both images. However, there is a severe disadvantage in response characteristics of the overall system, due to the limitation of how fast the excitation wavelength can be altered without injecting noise into the overall recordings. Even if the light source can switch wavelengths once every 1 msec (such as LEDs or lasers), the camera cannot be reset to read a different wavelength without some dead time, which would slow down the overall response time. Moreover, the synchronization of the camera reading sequence with the alternation of light pulses can further reduce the actual camera integration time to less than the duration of the alternating light pulses, resulting in a decrease of signal quality.
In the case where the two dyes have the same excitation wavelength but different fluorescence emission, the optical instrument has a single, continuous excitation light source at λex to excite both dyes, and fluorescence from the heart is transmitted to a 45° dichroic mirror to split and refocus the emitted light to two cameras. One camera receives the short wavelength λem1, typically the emission of the Cai indicator; the other receives the long wavelength λem2, typically the emission of the voltage-sensitive dye. The advantage of this configuration is substantial because the system has no moving parts—both the light source and the image sensors function continuously with no intermittent alternations, which improves response characteristics of the system and reduces noise injected by mechanical components. Thus, combinations of dyes that have almost identical excitation spectra but different emission spectra (different Stoke's shift) are the most effective, but it remains challenging to make sure that the two emission spectra are well separated, to avoid cross-over between the Vm and Cai signals.
The last and least desirable option is to select two dyes with different excitation and emission wavelength that do not overlap. Here, the optical instrument must include a light source with the capabilities of illuminating the heart serially with λex1 then λex2 as described above, and it must also use either a beam splitter and two cameras as described above, or a single camera with a filter wheel, to sequentially read λem1 and λem2. Thus, instruments designed for dual mapping with probes where both the excitation and emission wavelengths are different will necessarily record Vm and Cai signals with slower response and S/N ratio.
Four combinations of voltage-sensitive dyes and Ca2+ indicator have been thus far been applied to Langendorff perfused hearts, respectively:
RH 237 and Rhod-2AM,
Di-4-ANEPPS and Indo-1AM
Pittsburgh 1 (PGH I) and Rhod-2AM.
RH 237 and the genetically encoded Ca2+ sensor, GCaMP2.
All these dyes are commercially available from Molecular Probes, except for PGH I, which can be obtained from the authors. In cultured rat neonate cardiac myocytes, the voltage-sensitive dye RH 237 was also used in combination with low-affinity (Fluo-4FF and Rhod-2FF) and high-affinity (Rhod-2 and Fluo-4) Ca2+ indicator dyes, and, as expected, Cai measurements from myocytes were highly dependent on the Ca2+ affinity of the dye (Fast, 2005).
RH 237 and Rhod-2AM
The combination of Rhod-2AM and RH237 (Choi and Salama, 2000) was chosen because both dyes have a narrow excitation spectrum that peaks at 540 nm but have different peak emission wavelengths, respectively at 585 and 650 nm.
Figure 12.17.6 (from Choi and Salama, 2000, Fig. 1B) shows the excitation and emission spectra of the two dyes when measured from a stained guinea pig heart. The combination of RH 237 and Rhod-2 is particularly effective at measuring Cai at 585 nm, with no interference from RH 237 fluorescence. However, the fluorescence of nearly ideal Rhod-2 offers a number of advantages: (1) it exhibits >100-fold increase in fluorescence upon binding to Ca2+, with negligible fluorescence in the absence of Ca2+; (2) its peak fluorescence is at 585 nm, where there is negligible tissue autofluorescence; (3) it is stable and not prone to loss of dye by exocytosis; (4) it is resistant to photobleaching; and (5) it is a single-wavelength dye, and hence suitable for simultaneous recordings with a voltage-sensitive dye (VSD) with high temporal resolution.
Figure 12.17.6.
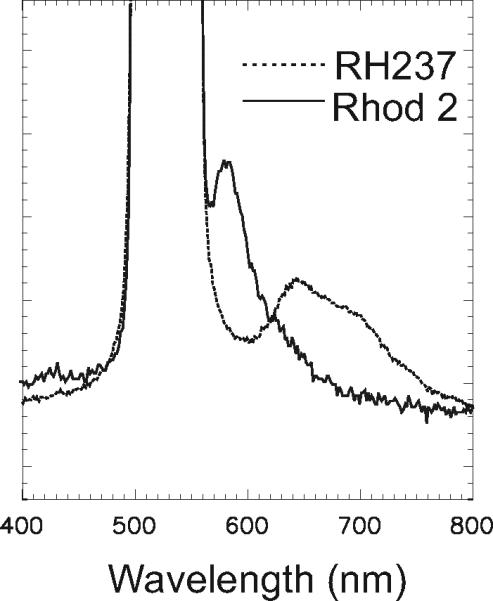
Emission spectra of Rhod-2 and RH 237 in a guinea pig heart. A guinea pig heart was labeled with RH237 or Rhod-2 as previously described (Choi and Salama, 2000), and the fluorescence emission spectra of the dyes were recorded with a high-speed spectrograph. The considerable overlap between the two emission spectra required the careful selection of filters to avoid cross-talk between the two signals.
Rhod-2 also presents certain disadvantages. (1) It is not a ratiometric dye and its fluorescence at 585 nm is a measure of dye bound to Ca2+ or [Rhod-2-Ca2+], and [Rhod-2] fluoresces weakly at the same wavelength. The lack of a reference fluorescence measurement was overcome by using the dye's absorption at 540 nm as a reference, because it is a measure of total Rhod-2 concentration in that both the [Rhod-2-free] and [Rhod-2-Cai] complexes have the same absorption. (2) The peak fluorescence emission at 585 nm is partially quenched by an absorption band of oxy-myoglobin. (3) The fluorescence spectrum of Rhod-2 has a long tail in the red range, which can interfere with VSD measurements. This can be overcome by selecting a VSD with a large Stoke's shift and/or by blocking the Rhod-2 fluorescence well past 640 nm, to avoid injecting the Cai signal into the VSD signals.
Di4-ANEPPS and Indo-1AM
In Langendorff-perfused guinea pig hearts, the combination of the voltage sensitive dye, di-4-ANEPPS, and the Ca2+ indicator, Indo-1 AM, was used to measure Vm and Cai, respectively (Laurita and Singal, 2001). Vm was measured with excitation and emission wavelengths of 515 ± 5 and >695 nm and Cai transients were measured at 365 ± 25 and 485 ± 5 nm, while maps were recorded with a Hamamatsu 16 × 16 element photodiode array. The use of two fluorescent dyes with different excitation and emission wavelengths and a single photodiode array required that maps of Vm and Cai be measured sequentially and not simultaneously (Laurita and Singal, 2001). A set of filters was first used to measure fluorescence maps of Vm, then the filters had to be changed to map Cai. The superposition of Vm and Cai traces was accomplished by aligning the measurements with respect to a reference impulse. The temporal relationship between Vm and Cai could be deduced as long as the preparation was stable from a physiological point of view. Based on the in vitro measurements of dye excitation and emission spectra and the choice of optical filters, the percent error on Vm signals due to spectral overlap of the Cai signal was 1.7 ± 1.0% (n = 3), whereas the percent error on Cai signals was negligible during Cai transients (0%, n = 3). This combination of dyes is not suitable for “simultaneous” recordings of Vm and Cai transients because there is no practical method to rapidly (<1 msec) change the excitation and emission wavelengths without injecting significant noise into the recordings. During dynamic conditions, the sequential measurements of Vm and Cai cannot be used to investigate the role of Cai as a trigger of early afterdepolarizations (Choi et al., 2002) and Torsade de Pointes or reperfusion arrhythmias (Choi et al., 2006a). An additional limitation is that the excitation for Indo-1 is in the far-UV range and has a considerably reduced tissue penetration compared to the excitation for di-4-ANEPPS at 515 nm. As a result, signals for Vm and Cai are recorded from different voxels of tissue, making it difficult to interpret the data.
PGH I and Rhod-2AM
A class of long-wavelength voltage-sensitive dyes was developed to record cardiac action potentials (APs) from deeper layers in the heart by increasing the tissue penetration of the excitation beam by prolonging the dyes’ excitation and emission wavelengths (Salama et al., 2005). The latter was accomplished by causing a red shift in the emission spectrum of styryl dyes by incorporating a thienyl group in the polymethine bridge to lengthen it while retaining the rigidity of the chromophore. PGH dyes exhibited two absorption, two excitation, and two voltage-sensitive emission peaks, with large Stokes shifts (>100 nm). Hearts (rabbit, guinea pig, and frog) were effectively stained by injecting a bolus (10 to 50 μl) of stock solution of dye (2 to 5 mM) dissolved in dimethylsulfoxide (DMSO) plus 16% low-molecular-weight Pluronic L64. PGH I exhibited an increase in fractional fluorescence, ΔF/F = 17.5% per AP at 720 nm with 550 nm excitation and ΔF/F = –6% per AP at 830 nm with 670 nm excitation (Salama et al., 2005). The large Stokes shift makes PGH I a particularly valuable dye for simultaneous mapping of APs and Cai using Rhod-2AM, because it becomes easier to avoid cross-talk through an effective separation of the emissions of the two dyes.
Figure 12.17.7 illustrates examples of APs recorded with PGH I and Cai transients recorded with Rhod-2 recorded from hearts of different species.
Figure 12.17.7.

Simultaneous Vm and Cai recordings from the heart of different species. Simultaneous recordings of Vm and Cai were obtained with PGH1 and Rhod-2 from guinea pig (A), rabbit (B), and mouse (C) hearts. The heart was excited at 540 nm, the emission for Rhod-2 was measured at 585 ± 30 nm, and the PGH I emission was measured above 700 nm. In rabbit hearts, Vm and Cai are shown at slow (a) and fast (b) sweep speeds, after the addition of an IKr blocker (E4031) (c), and PGH1 was measured at long excitation wavelengths (λex = 690 nm and >750 nm).
RH 237 and the Genetically Encoded Ca2+ Sensor, GCaMP2
Molecularly engineered mice (ccGC2 mice) were recently generated to conditionally express a genetically encoded Ca2+-sensitive GFP (green fluorescent protein), GCaMP2, in the heart under the control of the α-myosin heavy chain (α-MHC) promoter (Tallini et al., 2006). GCaMP2 consists of circularly permutated enhanced GFP (eGFP), with the molecule interrupted at residue 145; a 13-residue peptide of myosin light chain kinase (M13) and a calmodulin (CaM) were placed at the new N and C termini, respectively (Tallini et al., 2006). GCaMP2 was found to have the brightness of eGFP with peak excitation and emission wavelengths at 488 and 508 nm, respectively.
The conditional expression of the Ca2+ sensor offers numerous advantages. In studies of cardiac development, the encoded probe makes it possible to measure Cai transients at as early as 8.5 days, and to track cardiac activation patterns without disturbing fragile embryos to inject an organic Ca2+ sensitive dye (Tallini et al., 2006). Cardiac embryonic myocytes from ccGC2 mice can be isolated and implanted in infarcted hearts to track the survival, genotype, phenotype, and coupling of implanted myocytes with the host hearts through the GCaMP2 signals (Roell et al., 2007). GCaMP2 can be selectively expressed in a subclass of cardiac cells; for example it can be placed under the control of endogenous Connexin 40 transcription regulatory elements, resulting in the selective expression of GCaMP2 in the conduction system of the heart (Tallini et al., 2007).
A disadvantage of GCaMP2 comes from the difficulty of generating molecularly engineered animals other than the mouse. Another limitation is the slower response of GCaMP2 compared to organic Ca2+ indicator dyes. At 37°C, the average time constants for the probe to associate (τon) and dissociate (τoff) from free Ca2+ were found to be 14.29 ± 0.30 and 75.19 ± 0.68 msec, respectively (Tallini et al., 2006). The slower response times compared to Rhod-2 (τon and τoff <1 msec; unpub. observ.) has a marked influence on the recovery of Cai transients measured with GCaMP2.
Figure 12.17.8 (from Tallini et al., 2006, Fig. 3d) compares the kinetics of a simultaneously recorded AP and Cai transient measured with RH 237 and GCaMP2, respectively.
Figure 12.17.8.
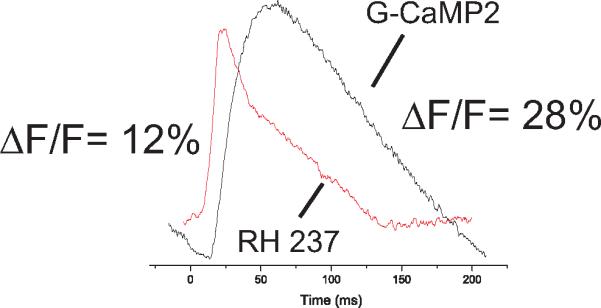
Mouse Vm and Cai recordings from RH 237 and GCaMP2. A heart from a mouse that was genetically encoded with GCaMP2 was isolated, perfused in a Langendorff apparatus, and stained with RH 237. APs were recorded from RH 237 (λex = 540 nm and λem > 630 nm) and Cai from GCaMP2 (λex = 480 ± 15 nm and λem =520 ± 150 nm).
Figure 12.17.9 compares the time course of Cai transients measured with GCaMP2 and Rhod-2 as a function of heart rate (or cycle length) and shows that at short cycle lengths, GCaMP2 fluorescence does not fully recover on a beat-to-beat basis due to its particularly slow rate of dissociation from Ca2+. The slower response is typically not a serious concern, since most mammalian hearts have considerably slower rates where the slower GCaMP2 kinetics will not significantly distort Cai measurements.
Figure 12.17.9.

Kinetics of GCaMP2 versus Rhod-2. The kinetics of GCaMP2 Ca2+c transients (left traces) were slower than those recorded with Rhod-2 (right traces). The effect was negligible at long cycle lengths (top traces) and became increasingly more severe at short cycle lengths (middle and bottom traces) where the GCaMP2 signal fails to recover back to baseline.
PROPERTIES OF RHOD-2AM IN PERFUSED HEARTS
Rhod-2 offers one of the best options among the available organic dyes to measure Cai from Langendorff-perfused hearts. Rhod-2AM is the membrane-permeable acetoxymethyl ester of the dye, which can diffuse into cells based on concentration gradient between the outside and inside of the cell (Minta et al., 1989). Once inside heart cells, the dye is hydrolyzed by native esterases and trapped in the cells. In the absence of Ca2+, the Ca2+-free form of Rhod-2 fluoresces very weakly, and when Ca2+ is bound to the BAPTA-like Ca2+ chelation pocket, its fluorescence increases by over 100-fold. The fluorescence properties of Rhod-2 are related to the tetramethyl-rhodamine fluorophore with a peak excitation, λex, at 540 nm and peak emission, λem, at 585 nm (Tsien, 1980).
Figure 12.17.10 shows the chemical structure of Rhod-2.
Figure 12.17.10.

Chemical structure of Rhod-2.
In intact hearts, the most effective approach to loading the heart is to add a bolus of concentrated dye solution in the perfusate, which results in a uniform delivery of dye throughout the heart via the coronary vessels. A single pass of dye through the coronaries yields the best results within 5 min as the dye courses through the vasculature. A second dye injection or more dye injections rarely improve the signal–to-noise ratio of Rhod-2 Cai signals. It is more important to add the correct concentration in one pass. Too high a concentration results in significant buffering of cytosolic free Ca2+, which can be detected as a decrease in the force of contractions or a reduced left ventricular pressure. Too low a concentration produces Cai transients with poor S/N ratio, as well as short-lived experiments, since subsequent additions of dye produce little improvement in signal quality.
Table 12.17.3 lists the Tyrode's solutions used to perfuse the hearts of various species and the recommended Rhod-2 concentration that yields the optimum S/N ratio without reducing left ventricular pressure (see below).
Table 12.17.3.
Tyrode's Solutions and Recommended Rhod-2 Injections for Different Species
| Component | Mouse | Rabbit | Guinea pig |
|---|---|---|---|
| NaCl | 112 mM | 130 mM | 130 mM |
| KCl | 5 mM | 4 mM | 4.75 mM |
| NaHCO3 | 25 mM | 24 mM | 25 mM |
| KH2PO4 | 1 mM | — | — |
| NaH2PO4 | — | 1.2 mM | — |
| MgSO4 | 1.2 mM | — | 1.2 mM |
| MgCl2 | — | 1.0 mM | — |
| CaCl2a | 1.8 mM | 0.75 mM | 1.25 mM |
| Glucose | 50 mM/(10 mM)b | 50 mM/(10 mM)b | 20 mM |
| Mannitolc | —/(45 mM)b | —/(45 mM)b | 45 mM |
| Rhod-2d | 20-40 μl | 0.4-0.6 ml | 0.1-0.2 ml |
To prevent calcium precipitation, gas the solution with CO2 prior to dissolving CaCl2.
Use the concentration in parentheses when preparing osmotic buffer.
Used as osmolarity buffer to keep the solution isotonic: can be substituted with dextran. This concentration can be changed to manipulate osmolarity by substituting glucose (Choi et al., 2006a,b).
Dye concentration at 1 mM: i.e., 1 mg/ml for rhod-2. Injection should be via bolus injection into the bubble trap, trickling directly into the cannula.
Measurements of Cai with organic dyes require numerous controls to validate the experimental conditions and the interpretation of Cai measurements. There are many pitfalls which can distort Cai measurements:
The dye distribution in various cells in the heart, including nonworking myocardium, and in different subcellular compartments within myocytes;
The Ca2+ binding affinity or Kd of the dye can distort the linearity of its response if it is too low or too high relative to Cai transients;
The calibration of the dye's fluorescence to Cai depends on rapid and accurate measurements of its fluorescence in the absence of Ca2+ (Fmin) and when all the dye is bound to Ca2+ (Fmax) by saturating the intracellular space with Ca2+;
The potential cross-talk between the fluorescence of the voltage-sensitive dye and the Ca2+ indicator.
There are often conflicting results in the literature regarding the behavior of these dyes, but they are often difficult to assess because of differences in experimental protocols, which influence the outcome and interpretation of Cai signals measured with organic Ca2+ indicators.
Distribution of Rhod-2 in the Heart
The delivery of dye to the heart by a bolus injection of dye in the coronary vessels can in principle distribute the dye among different cell types in the heart (myocytes as well as other, noncontracting endothelial cells). In addition, the delivery of the dye to various sub-cellular organelles such as the mitochondria, the nuclei, and endoplasmic and sarcoplasmic reticulum can be a potential source of error when attempting to measure cytosolic Cai. Dye accumulation in the mitochondria poses a particularly serious challenge to the interpretation of Ca2+-sensitive signals. There is a general agreement in the literature that free Ca2+ in the matrix of mitochondria (Ca2+m) reaches substantial concentrations (10 to 20 μM), but controversies remain as to whether or not there are beat-to-beat oscillations of Ca2+m in cardiac myocytes (Dedkova and Blatter, 2008). Thus, dye accumulation in the mitochondria not only can yield errors in measurements of diastolic and systolic free cytosolic Ca2+ (Ca2+c) but could also yield spurious measurements on the shape and time course of Ca2+c transients. For these reasons, it is important to carefully assess the subcellular distribution of organic dyes to validate Ca2+c transients in physiological and pathological conditions.
Uptake of Rhod-2 in endothelial cells
One concern is that endothelial cells may take up Ca2+ dyes and give spurious signals. For example, cardiac endothelial cells in rabbit hearts were found to take up considerable amounts of Indo-1AM, which was demonstrated by adding bradykinin, which elevated Cai from endothelial cells but not myocytes (Lorell et al., 1990). Uptake of Indo-1 by endothelial cells might have caused spurious elevations of Cai during the initial phase of ischemia—an effect originating from endothelial cells rather than an increase of Cai in myocytes. In rabbit hearts loaded with Rhod-2AM as described above, the addition of a bradykinin bolus (0.1 nmol) did not change Cai (Del Nido et al., 1998). Differences between the Rhod-2 and Indo-1 results were most likely due to the differences in loading procedures. A bolus addition of dye may be too brief for significant trapping of Rhod-2AM in endothelial cells, whereas Indo-1 (10 μM) was loaded by recirculating 10 μM of dye for 30 to 40 min. It should be noted that the binding of voltage-sensitive dyes (VSDs) to endothelial membranes does not pose a serious problem, because endothelial cells are not electrically active and the binding of VSDs would merely shift the background fluorescence and reduce the fractional fluorescence change per AP.
Possible accumulation of Rhod-2 in mitochondria
Early studies (dihydro-Rhod-2)
A major pitfall regarding intracellular Ca2+ indicator dyes is the potential accumulation of the dye in subcellular organelles (e.g., mitochondria or nucleus), resulting in errors not only in the levels of diastolic and systolic Cai but also in the kinetics of Cai transients. The first report that Rhod-2 might accumulate in the mitochondria of smooth muscle cells dealt with the dihydro form of Rhod-2, which was thought to accumulate in the mitochondria (Mix et al., 1994). When the AM form of a dye is introduced into cells, the dye diffuses freely into the cells and subcellular compartments until it is hydrolyzed by native esterase enzymes and becomes trapped in that particular compartment. Rhod-2AM has a net delocalized positive charge, which, in principle, facilitates its accumulation into mitochondria due to their negative potential (– 120 to – 180 mV). To further improve the selectivity of dye accumulation in the mitochondrial matrix, Rhod-2AM was reduced with sodium borohydride to produce dihydro-Rhod-2AM, which was loaded into hepatocytes for 60 min. The use of dihydro-Rhod-2AM was supposed to enhance mitochondrial loading because it could only exhibit Ca2+-dependent fluorescence changes after it was oxidized, which occurred preferentially in the mitochondria (Mix et al., 1994; Hajnoczky et al., 1995). Hepatocytes were loaded with dihydro-Rhod-2AM (1 to 2 μM) in the presence of 2 mM acetoacetate to stimulate mitochondrial oxidation of the probe, then the hepatocytes were kept in medium for 12 to 16 hr to eliminate dihydro-Rhod-2AM from the cytosol by diffusion and exocytosis (Hajnoczky et al., 1995). Dihydro-Rhod-2AM was thought to provide mitochondrial Ca2+ measurements because mitochondrial uncoupler and the ATPase inhibitor oligomycin altered the Rhod-2 response but not Fura-2 signals that represented cytosolic Ca2+. It is important to note that Fura-2AM was loaded under very different conditions (20 to 30 min in the presence of 100 μM sulfinpyrazone at 37°C), and the two signals were not measured from the same cells. While potentially powerful, the use of redox chemistry to load the mitochondrial matrix with a dye has not been pursued, largely because the dihydro form of the dye may be toxic.
Rhod-2 in mitochondria
The use of Rhod-2AM as a mitochondrial Ca2+ (Ca2+m) indicator has met with mixed results, and the interpretation of Rhod-2-based Ca2+m measurements remains controversial due to pitfalls in the various experimental approaches that have been used for Rhod-2 Ca2+m measurements.
In rat neonatal myocytes (Jou and Sheu, 1994) and rat brain astrocytes (Jou et al., 1996), Rhod-2 Ca2+m measurements were validated by comparing Rhod-2 signals to Ca2+c signals measured with Fura-2AM. However, the reference Ca2+c measured with Fura-2 could not be relied upon to selectively represent Ca2+c since Fura-2 has been known to accumulate extensively in the mitochondria (Blatter and Wier, 1990; Haworth and Redon, 1998).
There has been a tacit assumption that Rhod-2 is the dye of choice to measure Ca2+m because of its delocalized positive charge, which would drive its accumulation in mitochondria with a negative internal potential. Moreover, Ca2+ indicators lacking a positive charge, such as Indo 1-AM, Fura 2AM, or Fluo-3AM, accumulate even better in mitochondria and have been used to monitor Ca2+m (McDonough and Button, 1989; Miyata et al., 1991; Allen et al., 1992; Chacon et al., 1996; Schreur et al., 1996). Thus, the charge distribution on the dye is not a particularly useful predictor of mitochondrial localization.
Another approach to measure Ca2+m measurements was to load the –AM ester form of the dye for ~60 min to allow the dye to diffuse into the mitochondria, then quench cytosolic dye by bathing the cells with Mn2+ (McDonough and Button, 1989; Haworth and Redon, 1998). The remaining Cai signal following Mn2+ was interpreted to reflect changes in Ca2+m (McDonough and Button, 1989). However, it should be emphasized that Mn2+ does not fully quench Rhod-2, and the approach cannot be used to separate Ca2+m form Ca2c (Del Nido et al., 1998).
In rat chromaffin cells, Ca2+m was monitored by loading the cells with Rhod-2AM for 35 to 50 min at 22° to 25°C in DMSO plus 10% Pluronic 147 (Babcock et al., 1997). Ca2+m measured with Rhod-2 was compared to Ca2+c measured with either Indo 1 (100 μM), or Calcium green (10 μM), introduced into the cytosol with a patch pipet. The introduction of a Ca2+ indicator by whole-cell dialysis with a micropipet provided the most convincing approach to measurement of Ca2+c. However, the organelles identified as mitochondria did not have the expected shape and distribution of mitochondria, and the dye's fluorescence appeared as a halo around the presumed mitochondria, with little evidence of dye accumulation in the mitochondrial matrix. Similar measurements of Ca2+m using Rhod-2 in CHO.T cells reported waves of Ca2+ transients from cell to cell and indicated problems of interpretation, because the Rhod-2 was variably but significantly incorporated in nucleoli and other subcellular organelles (Rutter et al., 1996). In rat myocytes loaded with Rhod-2, mitochondrial uncouplers produced a redistribution of Rhod-2 from the mitochondria to cytosol (Duchen et al., 1996).
Put together, these studies highlight the importance of the loading conditions, the need to measure the dye's Kd in various cellular compartments, the complexity of calibration procedures, and the difficulties in loading and trapping Rhod-2 in the mitochondrial matrix.
Distribution of Rhod-2 in isolated guinea pig myocytes
The ability to simultaneously map Vm and Cai from Langendorff-perfused hearts required the validation that Cai transients represented Ca2+c and not spurious effects from Rhod-2 trapped in the mitochondria. To load the cytosolic compartment with Rhod-2 with negligible accumulation of dye in subcellular compartments, several loading conditions were tested. The distribution of Rhod-2 in the heart was found to be highly dependent on the time of dye exposure and on temperature during dye loading. The working strategy was to load intact hearts and isolated myocytes with Rhod-2 at 37°C for ~2 min with a large gradient of Rhod-2AM between the extracellular and intracellular spaces. As the dye diffuses first in the cytosol, it is rapidly hydrolyzed to its nonpermeable form and cannot diffuse further in other compartments. It was important to keep a normal physiological temperature of 37°C (rather than room temperature) to ensure a high enzymatic activity of native cytosolic esterases and a rapid immobilization of the dye in the cytosol before further diffusion could occur. Control of the time of dye exposure limits the permeation of the –AM form of the dye in various compartments while obtaining sufficient dye loading for Ca2+c measurements with high S/N ratio. The distribution of Rhod-2 in isolated guinea pig myocytes was investigated by confocal microscopy and mitochondrial membrane potential dyes. The following is a summary of the methods used.
Isolation of myocytes: Guinea pig cardiomyocytes were isolated as previously described (Mitra and Morad, 1985). Briefly, guinea pigs of either sex (250 to 450 g) were injected intraperitoneally with 1000 U heparin, followed by an injection of Nembutal (300 mg/kg). Once the anesthetic took effect, the heart was removed and placed in a Langendorff perfusion setup. The heart was perfused with an oxygenated HEPES-buffered saline solution (HBSS) containing: 135 mM NaCl, 1 mM MgCl2, 10 mM HEPES, 4 mM KCl, and 0 CaCl2, pH 7.2, at 36 ± 2°C. After 7 min, the perfusate was switched to HBSS containing 1 mg/ml collagenase (type II, Worthington) and 0.1 mg/ml pronase (Sigma). After 8 to 10 min, the enzyme solution was replaced by HBSS containing 0.2 mM CaCl2 for 5 min, then the heart was removed, minced, and triturated. Cells were filtered through a 100 μm nylon mesh and allowed to settle by gravity in a tube containing HBSS with 0.2 mM CaCl2. After 20 min, the supernatant was removed and the cells were resuspended in HBSS with 1.0 mM CaCl2. Myocytes were plated on poly-l-lysine-coated coverslips and were used within 8 hr.
Myocytes on coverslips were incubated with Rhod-2AM (5 μM) for 15 to 20 min either at room temperature (23°C) or in an incubator at 37°C, then washed twice with dye-free solution. Alternatively, myocytes were incubated in HBSS containing 10 nM TMRE (tetramethylrhodamine ethyl ester), a probe that accumulates in the mitochondria as a function of mitochondrial potential. Myocytes loaded with Rhod-2 or incubated in TMRE were examined with a laser scanning confocal microscope (LSCM) to compare the distribution of Rhod-2 with that of TMRE. Fluorescence images taken at λexcitation = 514 nm and λemission >555 nm were obtained at seven longitudinal focal planes to select a plane below the cellular membrane rich in mitochondria. Possible errors due to nonspecific binding of TMRE were evaluated by collapsing the mitochondrial potential with the proton ionophore FCCP, which was shown to collapse the mitochondrial membrane potential and decrease the voltage-dependent accumulation of TMRE in the mitochondria (Loew et al., 1993, 1994). Similarly, possible voltage-dependent accumulation of Rhod-2 in the mitochondria was evaluated by imaging the myocytes before and after the addition of FCCP to collapse the mitochondrial potential. Other nonspecific interactions of Rhod-2 with intracellular components were evaluated by permeabilizing the cell membrane with the detergent digitonin to disperse the freely soluble form of Rhod-2 while retaining residual bound Rhod-2 in the myocytes. To discriminate between mitochondrial versus cytosolic loading of Rhod-2, the localization of TMRE (a selective mitochondrial membrane potential probe) was compared to that of Rhod-2.
Figure 12.17.11 demonstrates that when loaded at 37°C for 5 to 10 min, Rhod-2 was selectively distributed in the cytosol with undetectable levels in mitochondria or other subcellular organelles. In Figure 12.17.1, panel A, a guinea pig ventricular myocyte was bathed in 10 nM TMRE, which exhibited the typical punctate appearance of mitochondrial labels. The addition of FCCP collapsed the mitochondrial membrane potential, resulting in a rapid loss of TMRE from the mitochondrial matrix (panel B). Guinea pig myocytes loaded with Rhod-2 exhibited a relatively homogeneous distribution of fluorescence throughout the cell, which was markedly different from that of TMRE (panel C); the addition of the proton ionophore FCCP abolished the mitochondrial membrane potential (as shown in panel B), but did not alter the Rhod-2 fluorescence distribution (panel D). Even though panels C and D showed that the Rhod-2 fluorescence emanated predominantly from the cytosol, the possibility of Rhod-2 accumulation in the mitochondria was not entirely excluded. For example, when a myocyte was moderately loaded with Rhod-2, the fluorescence was markedly reduced and was less homogeneous (e.g., up to 100-fold gradient of intensity), as in panel E, compared to panel C. In this case, the appearance of fluorescence “bands” could be mistaken for mitochondrial labeling instead of dye exclusion by the myofilaments, which act as dye-diffusion barriers. However, treatment of such myocytes with the detergent digitonin resulted in the complete loss of Rhod-2 fluorescence (100-fold decrease in intensity), and with no residual fluorescence in the cell (panel F). Thus, almost all the dye diffused out of digitonin-treated cells, indicating that little or no dye was trapped in the mitochondria or any other subcellular organelle.
Figure 12.17.11.
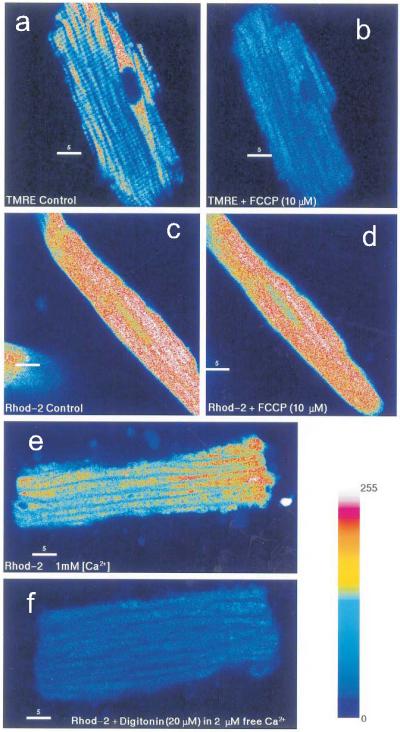
Subcellular distribution of Rhod-2 in guinea pig myocytes. Panels (A) and (B): confocal fluorescence images of a guinea pig ventricular myocytes bathed with TMRE (10 nM) in the absence (A) and presence (B) of 10 μM FCCP) (FCCP is an H+ ionophore which collapses Δψ—membrane potential—across the mitochondria). TMRE was rapidly accumulated in the mitochondria (10 to 15 min), and exhibited the typical punctate pattern associated with mitochondria aligned in myocytes below the surface membrane (Chacon et al., 1996). Panels (C) and (D) illustrate confocal fluorescence images of myocytes loaded with Rhod-2AM (5 μM) at 37°C, resulting in very different dye distribution compared to the punctate pattern of mitochondria shown in panel A. The subsequent addition of FCCP collapsed the mitochondrial potential but had no effect on Rhod-2 fluorescence (panel D). To test the possibility that Rhod-2 remained trapped in mitochondria, myocytes were loaded with Rhod-2AM (2 μM), then a low concentration of detergent was added to the bathing medium to allow cytosolic Rhod-2 to diffuse out of the cells without changing the mitochondrial membrane permeability. In panel (E), Rhod-2 loading produced what could be mistaken for the punctate appearance of mitochondrial stains, but when digitonin (20 μM) was then added to permeabilize the surface membrane (but not the mitochondrial membrane), Rhod-2 diffused out of the myocyte and there was no detectable level of Rhod-2 in the cell (panel F). Note that the cytosol and the mitochondria contained high levels of Ca2+ (2 μM), such that Rhod-2 trapped in the mitochondria and nondiffusible Rhod-2 in the cytosol would fluoresce strongly and would be readily detected in controls. The color-coded bar represents the relative fluorescence intensity. For color figure go to http://www.currentprotocols.com.
Subcellular distribution of Rhod-2 in Langendorff-perfused mouse hearts
The subcellular distribution of Rhod-2 was also examined by confocal microscopy in perfused mouse hearts loaded with Rhod-2 to discriminate between mitochondrial versus cytosolic loading. The perfused heart was mounted horizontally in a chamber with a Sylgard bottom carved in the shape of the heart and a 3-mm-diameter glass window (0.2 mm thick) on the bottom. The chamber was placed on the stage of the inverted microscope such that the objective viewed the left ventricular epicardium. To acquire confocal images, KCl (20 mM) was added to the perfusate to arrest the heart. An argon laser excited the epicardial surface and fluorescence was collected through a confocal aperture by a photomultiplier under manual gain and black-level control. Confocal images were recorded from hearts loaded with Rhod-2, then after perfusion with 20 μM digitonin and 2 μM free Ca2+ to permeabilize cell membranes and release Rhod-2 trapped in the cytosol while retaining Rhod-2 trapped in mitochondria and other subcellular organelles (Baker et al., 2004). Confocal images of perfused mouse hearts loaded with Rhod-2 indicated that the dye was not trapped in mitochondria, and readily diffused out of ventricular cells once permeabilized with digitonin. Thus, reports of mitochondrial dye accumulation may depend more on the loading conditions (temperature and time), metabolic state, and rate of dye esterification than the positive charge of the dye.
Rhod-2 calibration of Cai in Langendorff -perfused hearts
The calibration method for nonratiometric Ca2+ indicators was first established in single cells, and was applied to Langendorff-perfused hearts. A calibration curve for fluorescence (F) versus [Ca2+]i was generated based on the equation: [Ca2+]i = Kd(F – Fmin/Fmax – F), where Fmin and Fmax are the fluorescence intensities at zero and saturating Ca2+, respectively, and Kd is the dissociation constant for Ca2+. In single-cell experiments, Fmax was obtained by bathing the cell in a Ca2+ solution and making the cell Ca2+-permeable with a Ca2+ ionophore (A-23187) or a detergent (digitonin or saponin). Fmin will be taken as zero fluorescence because the dye's quantum yield is negligible in the absence of Ca2+.
Determinations of Fmax and Fmin
The approach used to calibrate the dye in isolated myocytes failed in perfused hearts because of the difficulties in determining Fmax and Fmin rapidly and accurately without causing intrinsic absorption changes of the myocardium due to loss of myoglobin and/or dye. Fmax was measured by saturating cytosolic Rhod-2 with a solution containing the Ca2+ ionophore A-23187 (10 μM), high external Ca2+ (5 mM), dithiodipyridine (DTDP = 100 μM), and diacetylmonoxime (DAM = 15 mM). The most important component of this approach was the use of DTDP, which was shown to trigger Ca2+ release from the sarcoplasmic reticulum through the selective sulfhydryl oxidation of hyper-reactive thiols on skeletal and cardiac ryanodine receptors (Zaidi et al., 1989; Prabhu and Salama, 1990). Caffeine was not effective because of the slow and transient rise of Ca2+c; DAM was important to eliminate cardiac contractures during the saturation of Ca2+c, and A-23187 helped accelerate the rise of Ca2+c. The use of digitonin to permeabilize the cell membranes and produce a massive Ca2+ influx failed to produce accurate measurements of Fmax because the rise of Ca2+c was slow and the myocardium changed its color dramatically and became pale white, most likely due to a significant loss of myoglobin from the intracellular milieu. Thus, it was important to validate Fmax by making sure that the absorption spectrum of the tissue did not significantly change before and after perfusion with the solution used to measure Fmax and to verify that the fluorescence intensity increased at 585 nm with no wavelength shifts. Fmin was measured by perfusing the heart with a solution containing 0 Ca2+, 5 mM EGTA, and A23187 to access the intracellular milieu, chelate free Ca2+, and quench the Rhod-2 fluorescence. In whole hearts, Fmin measurements were very similar to the tissue fluorescence measured before loading the heart with Rhod-2AM, because the fluorescence of the free dye tended to be very weak.
Determination of Kd
The effects of protein binding, viscosity and solution polarity have been shown to alter the Kd of Ca2+ indicator dyes. Hove-Madsen and Bers (1992) measured the Kd of Indo-1 in permeabilized rabbit myocytes and found it to be 4- to 5-fold greater than in vitro. The interaction of Indo-1 with cellular proteins was shown to change the spectral and Ca2+-binding properties of the dye (Hove-Madsen and Bers, 1992). Similar effects of dye-protein interactions were found for Fura-2 and Fluo-3 (Scanlon et al., 1987; Blatter and Wier, 1990; Uto et al., 1991; Loughrey et al., 2003). Kd is typically obtained from in vitro solutions that mimic the intracellular milieu. For Rhod-2, Kd = 570 nM in solution, but in the presence of myoglobin (0.5 mM) to mimic the myocyte homogenate, Kd increased to 710 nM (Del Nido et al., 1998). The higher Kd of Rhod-2 was used to calibrate Ca2+c in Langendorffperfused hearts (Del Nido et al., 1998; Choi and Salama, 2000; Choi et al., 2002; Baker et al., 2004).
Calibration of diastolic and systolic Ca2+c
The calibration procedure described above was applied to measure Ca2+c in guinea pig, rabbit, and mouse hearts (Choi and Salama, 2000; Choi et al., 2002; Baker et al., 2004). Diastolic and systolic Ca2+c were respectively 301 ± 66 and 936 ± 182 nM in guinea pig hearts (Choi and Salama, 2000); 320.04 ± 46.5 and 1.10 ± 46.5 nM in rabbit hearts (Choi et al., 2002); and 257 ± 30 and 743 ± 47 nM in FVB mouse hearts (Baker et al., 2004).
Dual Vm and Cai Measurements in Cultured Myocytes
Dual measurements of Vm and Cai in rat neonate myocytes were reported with RH 237 and Fluo-3AM (Fast and Ideker, 2000) as well as various low- and high-affinity Ca2+ indicator dyes (Fast, 2005). The combination of RH 237 and Fluo-3 used the same excitation for both dyes (500 ± 25 nm) and a 560 ± 60 nm interference filter for Fluo-3 fluorescence and a long-pass (>600 nm) filter to measure the RH237 fluorescence (Fast and Ideker, 2000). The selection of these dyes and these wavelengths compromised the optimum excitation of RH 237; the signals were also noteworthy for the slow recovery of the Fluo-3 Cai signals (>130 msec). The incomplete recovery of Cai signals could be in part due to the use of probenicid (1 mM), which is a blocker of anion transport that reduced the rate of Fluo-3 exocytosis from 50% in 10 min to 50% of loss of dye in 45 min (Fast and Ideker, 2000). The slow kinetics and recovery of Fluo-3 Cai transients lead to a comparison of low- and high-affinity Ca2+ indicators. High-affinity dyes (Fluo-3 and Rhod-2) detected Cai transients that were twice as long as low affinity dyes (Fluo-4FF and Rhod-FF). When electrical shocks were applied early during the AP plateau, low-affinity dyes reported decreases in Cai at sites of both positive and negative Vm, whereas high-affinity dyes reported negligible changes during the AP plateau and large increases in diastolic Cai (Fast, 2005). The lack of Cai calibration or measurements of dye distributions in the myocytes make it difficult to interpret these signals. For example, the Ca2+ dyes were loaded in cultured myocytes with long incubation periods (45 to 60 min), which typically leads to dye accumulation in mitochondria and other subcellular organelles; the cells were bathed in probenicid (1 mM), which can produce toxic effects; the optical recordings relied on the spectral properties of the dyes that were measured in ethanol or aqueous solutions, not when loaded in the cells; an image of dye-loaded myocytes suggested that the dyes were primarily located in mitochondria; and the Kd of these dyes was not measured in a cytosolic environment, but low-affinity dyes (Kd ~ 10 μM) are likely to track Ca2+m and not Ca2c (Fast, 2005).
Table 12.17.4 summarizes the spectral and Ca2+-binding properties of Ca2+ indicator dyes that have been used in dual optical mapping of Vm and Cai.
Table 12.17.4.
Properties of Calcium-Sensitive Dyes
| λex (nm) |
λem (nm) |
Kd (nM) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Fmax/Fmina (Rmax/Rmin) | Ca2+-free | Ca2+-bound | Ca2+-free | Ca2+-bound | In vitrob | In vivo | |||
| Indo-1 | 20-80 | 346 | 330 | 475 | 401 | 230 | 844c | ||
| Fura-2 | 13-25 | 363 | 335 | 512 | 505 | 140 | — | ||
| Fluo-3 | 506 | 526 | 325 | 898d | |||||
| Fluo-4 | >100e | 345 | — | ||||||
| Fluo-4FF | 494 | 516 | 9700 | — | |||||
| Rhod-2 | 14f to >100e | 549 | 552 | No fluorescence | 581 | 570 | 710g | ||
| Rhod-FF | 580 | 19,000 | — | ||||||
*Unless specified otherwise, all data from Handbook: A Guide to Fluorescent Probes and Labeling Technologies, by Molecular Probes/Invitrogen.
Fmax/Fmin for nonratiometric dyes (Fluo and Rhod) and Rmax/Rmin for ratiometric (Indo-1 and Fura-2).
As reported by Molecular Probes, measured at 22°C and in 100 mM KCl, 10 mM MOPS, pH 7.2.
From intact isolated rabbit myocytes. (Bassani et al., 1995).
From intact isolated rabbit myocytes (Loughrey et al., 2003).
In vitro measurement, reported by Molecular Probes.
In vitro measurement with 0.5 mM myoglobin. (Del Nido et al., 1998).
DEALING WITH MOTION ARTIFACTS
By its very design and function, the heart is a construct of muscles whose constituents, myocytes, contract mechanically following the electrical activation and the subsequent elevation of intracellular Ca2+. Without any countermeasures, mechanical contraction will significantly distort optical signals recorded from an optical mapping system. This section will discuss possible countermeasures that can be used individually or in combination to abate motion artifacts sufficiently so as to avoid distortions of the shape and time course of APs and Cai transients.
Mechanical Restraint
This is a basic method that has minimal physiological impact on the heart under observation, but is also the least effective, so it should be considered as a technique to be combined with other more effective methods. Its implementation usually comes in the form of a chamber, within which the heart is immersed in the cardiac eluate (Fig. 12.17.12A). The cannula that comes through the top of the chamber, to which the heart is tied by the aorta, provides one point of restraint. Then, the heart is lightly squeezed between the window in front of the chamber facing the macro lens and the backstop on the opposite side of the window. Care should be exercised in squeezing the heart, since if it is squeezed too much, some of the coronary vessels may get blocked and make the heart ischemic. There can also be two bars or braces on the opposite sides perpendicular to the window and the backstop, further immobilizing the heart, as well as providing a set of metal contacts that can be used to pick up pseudo-ECG signals or deliver electrical stimulation. Those five points of restraint can effectively reduce lateral motions, although intrinsic changes in texture from contraction will still be there (Salama et al., 1987; Efimov et al., 1994). Verification that the chamber reduced motion artifacts without impeding cardiac function was obtained by measuring left ventricular pressure and fluorescence from a heart loaded with an internal fluorescence standard, 5(6)-carboxy-2′, 7′-dichlorofluorescein, with the heart in the chamber, before and after applying pressure against the heart with the optical window (Fig. 12.17.12B; Del Nido et al., 1998). Similarly, excessive loading of the heart with Rhod-2 would be expected to buffer Ca2+c and reduce force generation. The recommended concentrations of Rhod-2 shown in Table 12.17.3 were determined by measuring left ventricular pressure before and after loading the heart with Rhod-2 and finding safe dye concentrations that did not significantly reduce force generation.
Figure 12.17.12.

A cardiac chamber abates motion artifacts with no effect on left ventricular pressure. (A) A guinea pig heart placed in a chamber designed with front, rear and side branches to minimize motion. (B) Simultaneous left ventricular (LV) pressure and fluorescence measurements of motion artifacts (MA) obtained by loading the heart with fluorescein used as a control dye. In plot a, MA are clearly noticeable from the fluorescein control before tightening the chamber around the heart. Tightening the chamber produced a marked reduction of MA with no detectable change in LV pressure.
Elimination of Ca2+ from Tyrode's Solution
Perhaps the simplest method to eliminate motion is to perfuse the heart with Ca2+-free Tyrode's solution for 10 to 20 min, which depletes cytosolic Ca2+ and abolishes contractions (Salama and Morad, 1976). Ca2+-free Tyrode's provides an excellent approach to test candidate dyes as possible voltage-sensitive probes using amphibian hearts. Frog (Rana pipiens) and bullfrog (Rana catesbeiana) hearts can be perfused without direct oxygenation of the buffer, at room temperature; they can be placed in a spectrophotometer to measure voltage-dependent spectral changes of the dyes, and the hearts contain little or no myoglobin, which reduces tissue absorption and absorption changes as a function of oxygen. Ca2+-free Tyrode's makes it possible to characterize the dyes responses to pharmacological and ionic interventions and to measure the sensitivity of the dye to changes in transmembrane potential (ΔF/F). Ca2+-free Tyrode's is typically not suitable for most studies, since intracellular Ca2+ (Cai) is markedly reduced and Cai transients have a direct impact on electrical activity, action potential characteristics, and arrhythmia susceptibility. In addition, some mammalian hearts, like rat hearts, do not fire action potentials in Ca2+ perfusate. Nevertheless, reducing Ca2+ by 50% in the perfusate can be used as part of an overall strategy to reduce motion artifacts in experiments where changes in Cai are not essential to the interpretation of the data.
Electro-Mechanical Uncouplers
These pharmacological agents are known to disrupt a component of the myocyte's contractile apparatus, leading to normal electrical activation devoid of mechanical contraction. Electromechanical uncouplers impart their effects by interacting with the cytoskeleton or sarcomeres of myocytes to alter the normal physiological contraction cycle despite relatively normal action potential and Cai transients. However, one cannot overemphasize their different side effects and variations in efficacy, as well as their sometimes considerable influence on normal action potential and Cai handling, and known species differences (Sellin and McArdle, 1994; Baker et al., 2004).
Diacetyl monoxime (DAM) or 2,3-butanedione monoxime (BDM)
The acronyms DAM and BDM refer to the same compound; it is a noncompetitive inhibitor of myosin II that inhibits myosin ATPase activity that acts by altering protein phosphorylation to prevent elevated Cai from generating mechanical contraction (Li et al., 1985; Gwathmey et al., 1991; Baker et al., 2004). It is effective in the range of 15 to 20 mM, and has been reported to reversibly eliminate mechanical contraction, and for this reason has been used in a large number of studies. However, it is known to significantly alter electrophysiological properties of working myocytes as well as specialized cells of the conduction system, in particular the atrio-ventricular (AV) node. In ventricular myocytes, DAM was reported to alter electrical restitution (APD and refractory period), conduction velocity, and Cai transients (Liu et al., 1993; Baker et al., 2004; Cheng et al., 2004; Kettlewell et al., 2004). DAM is soluble in water and is directly dissolved in Tyrode's solution, to be continuously perfused for the duration of the time period for which the motion should be inhibited. In studies of impulse propagation across the AV node, perfusion with DAM for more than 15 min permanently abolished AV nodal electrical activity and could only be used judiciously for brief periods (10 min), then washed out to insure “normal” AV node functions (Choi and Salama, 2000). In rodent hearts, DAM prolongs the action potential duration, whereas in other mammalian hearts, DAM shortens the action potential duration (Li et al., 1985; Baker et al., 2004).
Cytochalasin D (cyto-D)
Cyto-D is a fungal metabolite that was found to inhibit actin polymerization and is thereby able to significantly reduce or completely eliminate mechanical contraction of the myocardium in a reversible manner (Biermann et al., 1998; Jalife et al., 1998; Wu et al., 1998). Cyto-D is considered superior to DAM in that it has negligible effects on APD and its transmural gradient (Wu et al., 1998), but has been found to introduce significant changes to voltage-dependent Na+ channels (Undrovinas et al., 1995), inward rectifier K+ channels (Mazzanti et al., 1996), and ATP-sensitive K+ channels (Furukawa et al., 1996). Also, it has been reported that cyto-D changes the shape of action potential in mouse hearts from its characteristic sharp one with no plateau phase (APD90 of 40 to 50 msec) to one with a noticeable plateau (APD90 of ~200 msec; Jalife et al., 1998; Baker et al., 2004). Minimal effective concentration range is 5 to 10 μM, diluted from a stock solution dissolved in DMSO (up to 2 mg/ml, or 50 mM), to be continuously perfused with Tyrode's solution.
Blebbistatin
Blebbistatin is a recently discovered EC uncoupling agent that is reported to noncompetitively inhibit only certain types of myosin II with high specificity (Straight et al., 2003; Allingham et al., 2005) and to have significantly fewer side effects on both action potential shapes and Cai transients (Fedorov et al., 2007; Li and Nattel, 2007). It was successfully used in inhibiting motion in intact rabbit hearts, single rat ventricular myocytes (Fedorov et al., 2007), and mouse papillary muscle (Dou et al., 2007). Its minimal effective concentration range is 5 to 10 μM, and, like cyto-D, it should be diluted from a stock solution dissolved in DMSO (soluble up to 100 mM, but usually prepared in 1 mg/ml or 3.4 mM concentration), to be continuously perfused in the Tyrode's solution.
Ratiometry
The basic idea behind this method is to have an extra optical path (or channel) with a different range of emission wavelengths and use this extra data to compensate for motion artifacts from the main channel(s) via post processing. The premise is that the change in fluorescence due to the quantity measured would differ significantly between the two properly chosen wavelengths, or, in the ideal case, be opposite to each other, while the change due to the motion would stay more or less the same. Therefore, proper ratiometric processing of the two images taken simultaneously would be able to significantly reduce motion artifacts, while the functional signal is preserved (Knisley et al., 2000; Kong et al., 2003; Inagaki et al., 2004; Knisley and Pollard, 2005; Brown et al., 2007). Since this is essentially a passive method, i.e., everything happens in the optics and image acquisition part, it can reduce motion artifacts without having any physiological side effects whatsoever. One obvious downside is the necessity of one extra optical path complete with its own dedicated dichroic mirror, emission filter, and camera, making the whole optical mapping setup even more complicated. Another possible downside is the fact that it does not really remove the motion from the heart itself, but rather compensates for it with post processing via extra data, meaning that the site each pixel of the camera is looking at does change with the heart's motion, introducing an uncertainty to the signal's origin, and also that it will be impossible to “completely” remove the motion artifact.
KINETICS OF AP AND Ca2+C TRANSIENTS IN GUINEA PIG HEARTS
Dual mapping of APs and Ca2+c can be used to obtain detailed kinetic measurements of AP and Ca2+c transients. In guinea pig hearts paced at 300 msec cycle length, the mean time to peak for AP and Ca2+c (10% to 90% of upstroke) was 8.2 ± 0.7 and 13.9 ± 1.0 msec, respectively, with a time delay between AP and Ca2+c of 9.4 ± 1.1 msec (Choi and Salama, 2000). The time-to-peak for Ca2+c transients 24.9 ± 3.5 msec (Fig. 12.17.13).
Figure 12.17.13.

Rise time of Ca2+c transients. In a Rhod-2AM-loaded guinea pig heart, the rise time of Ca2+c transients was measured by superimposing the second derivative of the Ca2+c upstroke and the trace; rise time was the time between the maximum and minimum of the second derivative.
The downstroke of Ca2+c transients was dependent on the species, heart rate, and experimental conditions. In mouse hearts with brief APs in the range of 20 to 25 msec, Ca2+c transients had rapid monophasic downstrokes with no plateau phase (see Fig. 12.17.7, panel C and Fig. 12.17.9). In hearts with longer APs, Ca2+c transients exhibited more complex downstrokes which were dependent on the external Ca2+ concentrations. Figure 12.17.14 illustrates Ca2+c transients recorded at 1 mM (A), 0.75 mM (B), 0.5 mM (C), and 0.25 mM (D) free Ca2+ in the perfusate. The shape and time course of Ca2+c transients could not be attributed to cross-talk with the voltage-sensitive dye because the heart was loaded only with Rhod-2AM, and, although the heart was perfused with cytochalasin-D, all motion artifacts could not be excluded.
Figure 12.17.14.

Kinetics of guinea pig Ca2+c transients. Ca2+c transients were measured from a guinea pig heart loaded with Rhod-2AM, placed in the chamber described in Figure 12.17.12, and perfused with cytochalasin-D to abate motion artifacts. The downstroke of Ca2+c transients exhibited complex biphasic recoveries to baseline and decreases in durations that were dependent on Ca2+ in the perfusate: (A), 1; (B), 0.75; (C), 0.5; and (D), 0.25 mM free Ca2+.
FINDINGS AND SIGNIFICANCE
Simultaneous optical mapping of Vm and Cai has made a considerable contribution to our understanding of cardiac arrhythmia mechanisms and the special role of intracellular Ca2+ as a trigger of electrical instabilities. The first reported determination of Vm and Cai was carried out in Langendorff guinea pig hearts, and compared the restitution kinetics of the two parameters (Choi and Salama, 2000). Classic restitution kinetics were studied by applying 10 to 20 control beats at a constant cycle length (S1-S1 = 300 msec), followed by a single premature impulse applied at variable S1-S2 interval. During a normal action potential and Cai recording, the Cai up-stroke was delayed by 10 msec compared to the action potential upstroke, and Cai delay had a tendency to increase with shorter cycle lengths (increasing heart rate), but the increase was not statistically significant. However, at the shortest S1-S2 interval of 180 msec, the Cai to Vm delay became significantly longer at the base than the apex, indicating a spatial heterogeneity of excitation-contraction coupling at the base and the apex of the heart. Abrupt changes in heart rate were found to elicit concordant Vm and Cai alternans, which lasted for 12 heart beats and where every odd and even beat oscillated between large and small Cai transient. Transient concordant alternans were caused by alternans in conduction velocity due to apex-base heterogeneities of refractory periods (Choi and Salama, 2000). In a rabbit model of drug-induced long QT type 2 (LQT2), early afterdepolarizations (EADs) that fire during the long plateau phase of the action potential were elicited by a rise of Cai that preceded the voltage depolarization of the EAD (Choi et al., 2002). Figure 12.17.15 shows examples of normal action potential and Cai transients (A) and Vm and Cai recordings during EADs elicited by perfusing the rabbit heart with E4031 (0.5 μM), a selective inhibitor of the delayed fast-rectifying K+ current, IKr. Phase maps were generated by plotting Cai as a function of Vm, after Vm and Cai traces were normalized between 0 and 1. During a normal action potential (A), the phase plot of Cai versus Vm has a counterclockwise trajectory (see arrows) in the shape of a “boomerang” (Fig. 12.17.15A, bottom plot). In contrast, during EADs, Cai versus Vm plots had trajectories in the opposite, clockwise direction, indicating that Cai elevation preceded the voltage depolarization of EADs (Fig. 12.17.15B, bottom plot). The kinetic analysis of Vm and Cai shows that the spontaneous Ca2+ release from the sarcoplasmic reticulum network precedes the depolarization during an EAD, which in turn progresses to a polymorphic ventricular arrhythmia, called Torsade de Pointes (Choi et al., 2002).
Figure 12.17.15.

Simultaneous Vm and Cai recordings from a Langendorff perfused rabbit heart treated with E4031 (0.5 μM) to induce LQT2. Vm and Cai measured during a normal cardiac beat (A) and during EADs (B) were normalized between 0 and 1 to plot Cai versus Vm and thereby generate phase maps. Phase trajectories were counterclockwise during a normal beat (control A) and clockwise during EADs (EADs B), indicating that during EADs, Cai elevation preceded the voltage depolarization.
A number of pathologies (i.e., heart failure, ischemia/reperfusion, long QT) have been linked to anomalies in Cai handling and spontaneous Ca2+ release that elicits triggered activity, EADs, and arrhythmias (Salama, 2006). The ability to map Vm and Cai simultaneously has changed the focus of the literature on arrhythmia mechanisms from a “voltage-centric” to a “Cai-centric” point of view (Salama, 2006). Yet, recent reports persist in promoting the reactivation of the L-type Ca2+ current, ICa,L, as the driving force for EADs, independent of Ca2+ release from the sarcoplasmic reticulum (Xie et al., 2009). Figure 12.17.16 shows that in rabbit LQT2, Cai oscillations occur during the middle of the action potential plateau, long before voltage oscillations can be detected, which provides compelling evidence that spontaneous Ca2+ release and Cai cycling occur independently of membrane potential changes and reactivation of ICa,L.
Figure 12.17.16.

Vm (white) and Cai (red) were simultaneously recorded from a rabbit heart before (A) and after perfusion with the IKr blocker dofetilide (0.2 μM) for 2 min (B), 5 min (C), 7 min (D), and 10 min (E). The marked Cai instabilities in B and C clearly precede Vm instabilities and demonstrate a loss of voltage control. For the color version of this figure, go to http://www.currentprotocols.com.
Figure 12.17.3.

Dual optical mapping apparatus based on two CMOS from SciMedia. A photograph of a dual mapping system based on two CMOS cameras has general features similar to those shown in Figure 12.17.1, except for the dimensions of the CMOS compared to the PDAs, which require changes in the lenses used to focus the excitation beam and to focus images of the heart on the smaller CMOS sensors (1 × 1 cm2) compared to the larger PDA sensing surface (1.8 × 1.8 cm2).
Footnotes
INTERNET RESOURCES
http://www.dalsa.com/markets/ccd_vs_cmos.asp
Discussion of CCD versus CMOS on Dalsa Corporation Web site.
http://www.olympusmicro.com/primer/techniques/fluorescence/fluorosources.html
Abramowitz, M. and Davidson, M.W. Microscopy Primer: Light Sources.
http://repairfaq.ece.drexel.edu/sam/laserdio.htm
Goldwasser, S.M. 2008. Sam's Laser FAQ: Diode lasers.
http://www.lumileds.com/pdfs/DS34.pdf
Philips/Lumileds. 2004. LUXEON V LED Emitter Datasheet.
LITERATURE CITED
- Allen SP, Stone D, McCormack JG. The loading of fura-2 into mitochondria in the intact perfused rat heart and its use to estimate matrix Ca2+ under various conditions. J. Mol. Cell. Cardiol. 1992;24:765–773. doi: 10.1016/0022-2828(92)93390-6. [DOI] [PubMed] [Google Scholar]
- Allingham JS, Smith R, Rayment I. The structural basis of blebbistatin inhibition and specificity for myosin II. Nat. Struct. Mol. Biol. 2005;12:378–379. doi: 10.1038/nsmb908. [DOI] [PubMed] [Google Scholar]
- Babcock DF, Herrington J, Goodwin PC, Park YB, Hille B. Mitochondrial participation in the intracellular Ca2+ network. J. Cell. Biol. 1997;136:833–844. doi: 10.1083/jcb.136.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker LC, Wolk R, Choi BR, Watkins S, Plan P, Shah A, Salama G. Effects of mechanical uncouplers, diacetyl monoxime, and cytochalasin-D on the electrophysiology of per-fused mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 2004;287:H1771–H1779. doi: 10.1152/ajpheart.00234.2004. [DOI] [PubMed] [Google Scholar]
- Bassani JW, Bassani RA, Bers DM. Calibration of indo-1 and resting intracellular [Ca]i in intact rabbit cardiac myocytes. Biophys. J. 1995;68:1453–1460. doi: 10.1016/S0006-3495(95)80318-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biermann M, Rubart M, Moreno A, Wu J, Josiah-Durant A, Zipes DP. Differential effects of cytochalasin D and 2,3 butane-dione monoxime on isometric twitch force and transmembrane action potential in isolated ventricular muscle: Implications for optical measurements of cardiac repolarization. J. Cardiovasc. Electrophysiol. 1998;9:1348–1357. doi: 10.1111/j.1540-8167.1998.tb00110.x. [DOI] [PubMed] [Google Scholar]
- Blatter LA, Wier WG. Intracellular diffusion, binding, and compartmentalization of the fluorescent calcium indicators indo-1 and fura-2. Biophys. J. 1990;58:1491–1499. doi: 10.1016/S0006-3495(90)82494-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NH, Dobrovolny HM, Gauthier DJ, Wolf PD. A fiber-based ratiometric optical cardiac mapping channel using a diffraction grating and split detector. Biophys. J. 2007;93:254–263. doi: 10.1529/biophysj.106.101154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacon E, Ohata H, Harper IS, Trollinger DR, Herman B, Lemasters JJ. Mitochondrial free calcium transients during excitation-contraction coupling in rabbit cardiac myocytes. FEBS Lett. 1996;382:31–36. doi: 10.1016/0014-5793(96)00138-x. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Li L, Nikolski V, Wallick DW, Efimov IR. Shock-induced arrhythmogenesis is enhanced by 2,3-butanedione monoxime compared with cytochalasin D. Am. J. Physiol. Heart Circ. Physiol. 2004;286:H310–H318. doi: 10.1152/ajpheart.00092.2003. [DOI] [PubMed] [Google Scholar]
- Choi BR, Salama G. Simultaneous maps of optical action potentials and calcium transients in guinea-pig hearts: Mechanisms underlying concordant alternans. J. Physiol. 2000;529:171–188. doi: 10.1111/j.1469-7793.2000.00171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BR, Burton F, Salama G. Cytosolic Ca2+ triggers early afterdepolarizations and Torsade de Pointes in rabbit hearts with type 2 long QT syndrome. J. Physiol. 2002;543:615–631. doi: 10.1113/jphysiol.2002.024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BR, Hatton WJ, Hume JR, Liu T, Salama G. Low osmolarity transforms ventricular fibrillation from complex to highly organized, with a dominant high-frequency source. Heart Rhythm. 2006a;3:1210–1220. doi: 10.1016/j.hrthm.2006.06.026. [DOI] [PubMed] [Google Scholar]
- Choi BR, Liu T, Salama G. Calcium transients modulate action potential repolarizations in ventricular fibrillation. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2006b;1:2264–2267. doi: 10.1109/IEMBS.2006.260059. [DOI] [PubMed] [Google Scholar]
- Choi BR, Jang W, Salama G. Spatially discordant voltage alternans cause wavebreaks in ventricular fibrillation. Heart Rhythm. 2007;4:1057–1068. doi: 10.1016/j.hrthm.2007.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CC, Nguyen BL, Tan AY, Chang PC, Lee HL, Lin FC, Yeh SJ, Fishbein MC, Lin SF, Wu D, Wen MS, Chen PS. Intracellular calcium dynamics and acetylcholine-induced triggered activity in the pulmonary veins of dogs with pacing-induced heart failure. Heart Rhythm. 2008;5:1170–1177. doi: 10.1016/j.hrthm.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen L. Optical monitoring of activity in the nervous system: A brief history. Tsitologiia. 1995;37:1136–1141. [PubMed] [Google Scholar]
- Dedkova EN, Blatter LA. Mitochondrial Ca2+ and the heart. Cell Calcium. 2008;44:77–91. doi: 10.1016/j.ceca.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Del Nido PJ, Glynn P, Buenaventura P, Salama G, Koretsky AP. Fluorescence measurement of calcium transients in perfused rabbit heart using rhod 2. Am. J. Physiol. 1998;274:H728–H741. doi: 10.1152/ajpheart.1998.274.2.H728. [DOI] [PubMed] [Google Scholar]
- Djurisic M, Zochowski M, Wachowiak M, Falk CX, Cohen LB, Zecevic D. Optical monitoring of neural activity using voltage-sensitive dyes. Methods Enzymol. 2003;361:423–451. doi: 10.1016/s0076-6879(03)61022-0. [DOI] [PubMed] [Google Scholar]
- Dou Y, Arlock P, Arner A. Blebbistatin specifically inhibits actin-myosin interaction in mouse cardiac muscle. Am. J. Physiol. Cell. Physiol. 2007;293:C1148–C1153. doi: 10.1152/ajpcell.00551.2006. [DOI] [PubMed] [Google Scholar]
- Duchen MR, Leyssens A, Mojet MH, Peuchen S. Properties of rhod 2 as an indicator of intra-mitochondrial calcium (abstract). J. Physiol. 1996;494:9P. [Google Scholar]
- Efimov IR, Huang DT, Rendt JM, Salama G. Optical mapping of repolarization and refractoriness from intact hearts. Circulation. 1994;90:1469–1480. doi: 10.1161/01.cir.90.3.1469. [DOI] [PubMed] [Google Scholar]
- Efimov IR, Nikolski VP, Salama G. Optical imaging of the heart. Circ. Res. 2004;95:21–33. doi: 10.1161/01.RES.0000130529.18016.35. [DOI] [PubMed] [Google Scholar]
- Entcheva E, Bien H. Macroscopic optical mapping of excitation in cardiac cell networks with ultra-high spatiotemporal resolution. Prog. Biophys. Mol. Biol. 2006;92:232–257. doi: 10.1016/j.pbiomolbio.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Fast VG. Simultaneous optical imaging of membrane potential and intracellular calcium. J. Electrocardiol. 2005;38:107–112. doi: 10.1016/j.jelectrocard.2005.06.023. [DOI] [PubMed] [Google Scholar]
- Fast VG, Ideker RE. Simultaneous optical mapping of transmembrane potential and intracellular calcium in myocyte cultures. J. Cardiovasc. Electrophysiol. 2000;11:547–556. doi: 10.1111/j.1540-8167.2000.tb00008.x. [DOI] [PubMed] [Google Scholar]
- Fedorov VV, Lozinsky IT, Sosunov EA, Anyukhovsky EP, Rosen MR, Balke CW, Efimov IR. Application of blebbistatin as an excitation-contraction uncoupler for electrophysiologic study of rat and rabbit hearts. Heart Rhythm. 2007;4:619–626. doi: 10.1016/j.hrthm.2006.12.047. [DOI] [PubMed] [Google Scholar]
- Fossum ER. Active-pixel sensors challenge CCDs. Laser Focus World. 1993a;29:83–87. [Google Scholar]
- Fossum ER. Active pixel sensors: Are CCDs dinosaurs? Proc. SPIE. 1993b;1900:2–14. [Google Scholar]
- Furukawa T, Yamane T, Terai T, Katayama Y, Hiraoka M. Functional linkage of the cardiac ATP-sensitive K+ channel to the actin cytoskeleton. Pflugers Arch. 1996;431:504–512. doi: 10.1007/BF02191896. [DOI] [PubMed] [Google Scholar]
- Gwathmey JK, Hajjar RJ, Solaro RJ. Contractile deactivation and uncoupling of crossbridges: Effects of 2,3-butanedione monoxime on mammalian myocardium. Circ. Res. 1991;69:1280–1292. doi: 10.1161/01.res.69.5.1280. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Haworth RA, Redon D. Calibration of intracellular Ca transients of isolated adult heart cells labelled with fura-2 by acetoxymethyl ester loading. Cell Calcium. 1998;24:263–273. doi: 10.1016/s0143-4160(98)90050-1. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Kamanu SD, Ono N, Kawase A, Chou CC, Weiss JN, Karagueuzian HS, Lin SF, Chen PS. Calcium transient dynamics and the mechanisms of ventricular vulnerability to single premature electrical stimulation in Langendorff-perfused rabbit ventricles. Heart Rhythm. 2008;5:116–123. doi: 10.1016/j.hrthm.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hove-Madsen L, Bers DM. Indo-1 binding to protein in permeabilized ventricular myocytes alters its spectral and Ca binding properties. Biophys. J. 1992;63:89–97. doi: 10.1016/S0006-3495(92)81597-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki M, Hidaka I, Aiba T, Tatewaki T, Sunagawa K, Sugimachi M. High resolution optical mapping of cardiac action potentials in freely beating rabbit hearts. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2004;5:3578–3580. doi: 10.1109/IEMBS.2004.1404006. [DOI] [PubMed] [Google Scholar]
- Jalife J, Morley GE, Tallini NY, Vaidya D. A fungal metabolite that eliminates motion artifacts. J. Cardiovasc. Electrophysiol. 1998;9:1358–1362. doi: 10.1111/j.1540-8167.1998.tb00111.x. [DOI] [PubMed] [Google Scholar]
- Jou M-J, Sheu S-S. Mitochondrial Ca2+ oscillations in single living cells revealed by rhod-2 and laser confocal microscopy. Biophys. J. 1994;66:A94. [Google Scholar]
- Jou MJ, Peng TI, Sheu SS. Histamine induces oscillations of mitochondrial free Ca2+ concentration in single cultured rat brain astrocytes. J. Physiol. 1996;497:299–308. doi: 10.1113/jphysiol.1996.sp021769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katra RP, Laurita KR. Cellular mechanism of calcium-mediated triggered activity in the heart. Circ. Res. 2005;96:535–542. doi: 10.1161/01.RES.0000159387.00749.3c. [DOI] [PubMed] [Google Scholar]
- Katra RP, Pruvot E, Laurita KR. Intracellular calcium handling heterogeneities in intact guinea pig hearts. Am. J. Physiol. Heart Circ. Physiol. 2004;286:H648–H656. doi: 10.1152/ajpheart.00374.2003. [DOI] [PubMed] [Google Scholar]
- Kettlewell S, Walker NL, Cobbe SM, Burton FL, Smith GL. The electrophysiological and mechanical effects of 2,3-butane-dione monoxime and cytochalasin-D in the Langendorff perfused rabbit heart. Exp. Physiol. 2004;89:163–172. doi: 10.1113/expphysiol.2003.026732. [DOI] [PubMed] [Google Scholar]
- Knisley SB, Justice RK, Kong W, Johnson PL. Ratiometry of transmembrane voltage-sensitive fluorescent dye emission in hearts. Am. J. Physiol. Heart Circ. Physiol. 2000;279:H1421–H1433. doi: 10.1152/ajpheart.2000.279.3.H1421. [DOI] [PubMed] [Google Scholar]
- Knisley SB, Pollard AE. Use of translucent indium tin oxide to measure stimulatory effects of a passive conductor during field stimulation of rabbit hearts. Am. J. Physiol. Heart Circ. Physiol. 2005;289:H1137–H1146. doi: 10.1152/ajpheart.00064.2005. [DOI] [PubMed] [Google Scholar]
- Kong W, Walcott GP, Smith WM, Johnson PL, Knisley SB. Emission ratiometry for simultaneous calcium and action potential measurements with coloaded dyes in rabbit hearts: Reduction of motion and drift. J. Cardiovasc. Electrophysiol. 2003;14:76–82. doi: 10.1046/j.1540-8167.2003.02077.x. [DOI] [PubMed] [Google Scholar]
- Lakireddy V, Baweja P, Syed A, Bub G, Boutjdir M, El-Sherif N. Contrasting effects of ischemia on the kinetics of membrane voltage and intracellular calcium transient underlie electrical alternans. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H400–H407. doi: 10.1152/ajpheart.00502.2004. [DOI] [PubMed] [Google Scholar]
- Lakireddy V, Bub G, Baweja P, Syed A, Boutjdir M, El-Sherif N. The kinetics of spontaneous calcium oscillations and arrhythmogenesis in the in vivo heart during ischemia/reperfusion. Heart Rhythm. 2006;3:58–66. doi: 10.1016/j.hrthm.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Lan DZ, Pollard AE, Knisley SB, Fast VG. Optical mapping of V(m) and Ca(i)(2+) in a model of arrhythmias induced by local catecholamine application in patterned cell cultures. Pflugers Arch. 2007;453:871–877. doi: 10.1007/s00424-006-0162-6. [DOI] [PubMed] [Google Scholar]
- Laude B, de Martino A, Drévillon B, Benattar L, Schwartz L. Full-field optical coherence tomography with thermal light. App. Optics. 2002;41:6637–6645. doi: 10.1364/ao.41.006637. [DOI] [PubMed] [Google Scholar]
- Laurita KR, Rosenbaum DS. Mechanisms and potential therapeutic targets for ventricular arrhythmias associated with impaired cardiac calcium cycling. J. Mol. Cell. Cardiol. 2008;44:31–43. doi: 10.1016/j.yjmcc.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurita KR, Singal A. Mapping action potentials and calcium transients simultaneously from the intact heart. Am. J. Physiol. Heart Circ. Physiol. 2001;280:H2053–H2060. doi: 10.1152/ajpheart.2001.280.5.H2053. [DOI] [PubMed] [Google Scholar]
- Li D, Nattel S. Pharmacological elimination of motion artifacts during optical imaging of cardiac tissues: Is blebbistatin the answer? Heart Rhythm. 2007;4:627–628. doi: 10.1016/j.hrthm.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Li T, Sperelakis N, Teneick RE, Solaro RJ. Effects of diacetyl monoxime on cardiac excitation-contraction coupling. J. Pharmacol. Exp. Ther. 1985;232:688–695. [PubMed] [Google Scholar]
- Liu Y, Cabo C, Salomonsz R, Delmar M, Davidenko J, Jalife J. Effects of diacetyl monoxime on the electrical properties of sheep and guinea pig ventricular muscle. Cardiovasc. Res. 1993;27:1991–1997. doi: 10.1093/cvr/27.11.1991. [DOI] [PubMed] [Google Scholar]
- Loew LM, Carrington W, Tuft RA, Fay FS. Physiological cytosolic Ca2+ transients evoke concurrent mitochondrial depolarizations. Proc. Natl. Acad. Sci. U.S.A. 1994;91:12579–12583. doi: 10.1073/pnas.91.26.12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loew LM, Tuft RA, Carrington W, Fay FS. Imaging in five dimensions: Time-dependent membrane potentials in individual mitochondria. Biophys. J. 1993;65:2396–2407. doi: 10.1016/S0006-3495(93)81318-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London B, Baker LC, Lee JS, Shusterman V, Choi BR, Kubota T, McTiernan CF, Feldman AM, Salama G. Calcium-dependent arrhythmias in transgenic mice with heart failure. Am. J. Physiol. Heart Circ. Physiol. 2003;284:H431–H441. doi: 10.1152/ajpheart.00431.2002. [DOI] [PubMed] [Google Scholar]
- London B, Baker LC, Petkova-Kirova P, Nerbonne JM, Choi BR, Salama G. Dispersion of repolarization and refractoriness are determinants of arrhythmia pheno-type in transgenic mice with long QT. J. Physiol. 2007;578:115–129. doi: 10.1113/jphysiol.2006.122622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorell BH, Apstein CS, Cunningham MJ, Schoen FJ, Weinberg EO, Peeters GA, Barry WH. Contribution of endothelial cells to calcium-dependent fluorescence transients in rabbit hearts loaded with indo 1. Circ. Res. 1990;67:415–425. doi: 10.1161/01.res.67.2.415. [DOI] [PubMed] [Google Scholar]
- Loughrey CM, MacEachern KE, Cooper J, Smith GL. Measurement of the dissociation constant of Fluo-3 for Ca2+ in isolated rabbit cardiomyocytes using Ca2+ wave characteristics. Cell Calcium. 2003;34:1–9. doi: 10.1016/s0143-4160(03)00012-5. [DOI] [PubMed] [Google Scholar]
- Mazzanti M, Assandri R, Ferroni A, DiFrancesco D. Cytoskeletal control of rectification and expression of four substates in cardiac inward rectifier K+ channels. FASEB J. 1996;10:357–361. doi: 10.1096/fasebj.10.2.8641571. [DOI] [PubMed] [Google Scholar]
- McDonough PM, Button DC. Measurement of cytoplasmic calcium concentration in cell suspensions: Correction for extracellular Fura-2 through use of Mn2+ and probenecid. Cell Calcium. 1989;10:171–180. doi: 10.1016/0143-4160(89)90071-7. [DOI] [PubMed] [Google Scholar]
- Mendis S, Kemeny SE, Fossum ER. CMOS active pixel image sensor. IEEE Trans. Electron. Devices. 1994;41:452–453. [Google Scholar]
- Mendis SK, Pain B, Nixon RH, Fossum ER. Low-light-level image sensor with on-chip signal processing. Proc. SPIE. 1993;1952:23–33. [Google Scholar]
- Minta A, Kao JP, Tsien RY. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J. Biol. Chem. 1989;264:8171–8178. [PubMed] [Google Scholar]
- Mitra R, Morad M. A uniform enzymatic method for dissociation of myocytes from hearts and stomachs of vertebrates. Am. J. Physiol. 1985;249:H1056–H1060. doi: 10.1152/ajpheart.1985.249.5.H1056. [DOI] [PubMed] [Google Scholar]
- Mix TCH, Drummond RM, Tuft RA, Fay FS. Mitochondria in smooth muscle cells sequester Ca2+ following stimulation of cell contraction. Biophys. J. 1994;66:A97. [Google Scholar]
- Miyata H, Silverman HS, Sollott SJ, Lakatta EG, Stern MD, Hansford RG. Measurement of mitochondrial free Ca2+ concentration in living single rat cardiac myocytes. Am. J. Physiol. 1991;261:H1123–H1134. doi: 10.1152/ajpheart.1991.261.4.H1123. [DOI] [PubMed] [Google Scholar]
- Prabhu SD, Salama G. Reactive disulfide compounds induce Ca2+ release from cardiac sarcoplasmic reticulum. Arch. Biochem. Biophys. 1990;282:275–283. doi: 10.1016/0003-9861(90)90117-h. [DOI] [PubMed] [Google Scholar]
- Roell W, Lewalter T, Sasse P, Tallini YN, Choi BR, Breitbach M, Doran R, Becher UM, Hwang SM, Bostani T, von Maltzahn J, Hofmann A, Reining S, Eiberger B, Gabris B, Pfeifer A, Welz A, Willecke K, Salama G, Schrickel JW, Kotlikoff MI, Fleischmann BK. Engraftment of connexin 43-expressing cells prevents post-infarct arrhythmia. Nature. 2007;450:819–824. doi: 10.1038/nature06321. [DOI] [PubMed] [Google Scholar]
- Rutter GA, Burnett P, Rizzuto R, Brini M, Murgia M, Pozzan T, Tavaré JM, Denton RM. Subcellular imaging of intramitochondrial Ca2+ with recombinant targeted aequorin: Significance for the regulation of pyruvate dehydrogenase activity. Proc. Natl. Acad. Sci. U.S.A. 1996;93:5489–5494. doi: 10.1073/pnas.93.11.5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba S, Mathier MA, Mehdi H, Liu T, Choi BR, London B, Salama G. Dual-dye optical mapping after myocardial infarction: Does the site of ventricular stimulation alter the properties of electrical propagation? J. Cardiovasc. Electrophysiol. 2008;19:197–202. doi: 10.1111/j.1540-8167.2007.00998.x. [DOI] [PubMed] [Google Scholar]
- Salama G. Arrhythmia genesis: Aberrations of voltage or Ca2+ cycling? Heart Rhythm. 2006;31:67–70. doi: 10.1016/j.hrthm.2005.10.025. [DOI] [PubMed] [Google Scholar]
- Salama G, Choi BR. Imaging ventricular fibrillation. J. Electrocardiol. 2007;40:S56–S61. doi: 10.1016/j.jelectrocard.2007.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama G, Choi BR, Azour G, Lavasani M, Tumbev V, Salzberg BM, Patrick MJ, Ernst LA, Waggoner AS. Properties of new, long-wavelength, voltage-sensitive dyes in the heart. J. Membr. Biol. 2005;208:125–140. doi: 10.1007/s00232-005-0826-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama G, Lombardi R, Elson J. Maps of optical action potentials and NADH fluorescence in intact working hearts. Am. J. Physiol. 1987;252:H384–H394. doi: 10.1152/ajpheart.1987.252.2.H384. [DOI] [PubMed] [Google Scholar]
- Salama G, Morad M. Merocyanine 540 as an optical probe of transmembrane electrical activity in the heart. Science. 1976;191:485–487. doi: 10.1126/science.191.4226.485. [DOI] [PubMed] [Google Scholar]
- Scanlon M, Williams DA, Fay FS. A Ca2+-insensitive form of fura-2 associated with polymorphonuclear leukocytes: Assessment and accurate Ca2+ measurement. J. Biol. Chem. 1987;262:6308–6312. [PubMed] [Google Scholar]
- Schreur JH, Figueredo VM, Miyamae M, Shames DM, Baker AJ, Camacho SA. Cytosolic and mitochondrial [Ca2+] in whole hearts using indo-1 acetoxymethyl ester: Effects of high extracellular Ca2+. Biophys. J. 1996;70:2571–2580. doi: 10.1016/S0006-3495(96)79828-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellin LC, McArdle JJ. Multiple effects of 2,3-butanedione monoxime. Pharmacol. Toxicol. 1994;74:305–313. doi: 10.1111/j.1600-0773.1994.tb01365.x. [DOI] [PubMed] [Google Scholar]
- Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Dissecting temporal and spatial control of cytokinesis with a myosin II inhibitor. Science. 2003;299:1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- Tallini YN, Brekke JF, Shui B, Doran R, Hwang SM, Nakai J, Salama G, Segal SS, Kotlikoff MI. Propagated endothelial Ca2+ waves and arteriolar dilation in vivo: Measurements in Cx40BAC GCaMP2 transgenic mice. Circ. Res. 2007;101:1300–1309. doi: 10.1161/CIRCRESAHA.107.149484. [DOI] [PubMed] [Google Scholar]
- Tallini YN, Ohkura M, Choi BR, Ji G, Imoto K, Doran R, Lee J, Plan P, Wilson J, Xin HB, Sanbe A, Gulick J, Mathai J, Robbins J, Salama G, Nakai J, Kotlikoff MI. Imaging cellular signals in the heart in vivo: Cardiac expression of the high-signal Ca2+ indicator GCaMP2. Proc. Natl. Acad. Sci. U.S.A. 2006;103:4753–4758. doi: 10.1073/pnas.0509378103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RY. New calcium indicators and buffers with high selectivity against magnesium and protons: Design, synthesis, and properties of prototype structures. Biochemistry. 1980;19:2396–2404. doi: 10.1021/bi00552a018. [DOI] [PubMed] [Google Scholar]
- Undrovinas AI, Shander GS, Makielski JC. Cytoskeleton modulates gating of voltage-dependent sodium channel in heart. Am. J. Physiol. 1995;269:H203–H214. doi: 10.1152/ajpheart.1995.269.1.H203. [DOI] [PubMed] [Google Scholar]
- Uto A, Arai H, Ogawa Y. Reassessment of fura-2 and the ratio method for determination of intracellular Ca2+ concentrations. Cell Calcium. 1991;12:29–37. doi: 10.1016/0143-4160(91)90082-p. [DOI] [PubMed] [Google Scholar]
- Wu J, Biermann M, Rubart M, Zipes DP. Cytochalasin D as excitation-contraction uncoupler for optically mapping action potentials in wedges of ventricular myocardium. J. Cardiovasc. Electrophysiol. 1998;9:1336–1347. doi: 10.1111/j.1540-8167.1998.tb00109.x. [DOI] [PubMed] [Google Scholar]
- Xie L-H, Chen F, Karagueuzian HS, Weiss JN. Oxidative stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ. Res. 2009;104:79–86. doi: 10.1161/CIRCRESAHA.108.183475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi NF, Lagenaur CF, Abramson JJ, Pessah I, Salama G. Reactive disulfides trigger Ca2+ release from sarcoplasmic reticulum via an oxidation reaction. J. Biol. Chem. 1989;264:21725–21736. [PubMed] [Google Scholar]
- Zecevic D, Djurisic M, Cohen LB, Antic S, Wachowiak M, Falk CX, Zochowski MR. Imaging nervous system activity with voltage-sensitive dyes. Curr. Protoc. Neurosci. 2003;23:6, 17, 1–6, 17, 29. doi: 10.1002/0471142301.ns0617s23. [DOI] [PubMed] [Google Scholar]
- Zimmermann H. Integrated Silicon Opto-electronics. Springer-Verlag, Berlin. 2000 [Google Scholar]