

Abstract
Hypoxia induces the stabilization and transcriptional activation of the hypoxia-inducible factor 1α (HIF-1α) protein, the regulatory member of the HIF-1 complex. The molecular mechanisms that are responsible for oxygen sensing and the downstream pathways utilized by the hypoxic signal are still poorly understood. One hypothesis for oxygen sensing has postulated that reactive oxygen species generated at mitochondrial complex III are the initiators of the hypoxic signal. Here we find that mitochondrial DNA-less (ρo) cells have a normal response to hypoxia, measured at the level of HIF-1α protein stabilization, nuclear translocation, and its transcriptional activation activity. Furthermore, overexpression of catalase, either in the mitochondria or in the cytosol, fails to modify the hypoxia response indicating that hydrogen peroxide is not a signaling molecule in the hypoxic signaling cascade that culminates with HIF-1 activation.
In multicellular organisms, oxygen, as the final acceptor of electrons in the respiratory chain, is an absolute requirement for life. Hypoxia, the decrease in oxygen supply to tissues, determines a series of metabolic and systemic adaptations that tend to overcome the detrimental effect of the lack of oxygen. Central to this adaptation is the regulation of hypoxia-responsive genes that control multiple cellular and systemic functions (reviewed in Ref. 1). Most of these genes are regulated by a common mechanism of transcriptional activation that involves the hypoxia-inducible factor 1 (HIF-1)1 complex (2). The HIF-1 complex is composed of two subunits. HIF-1α and HIF-1β, members of the helix loop helix-PAS family of transcription factors. The function of the HIF-1 complex is primarily regulated by the abundance of the HIF-1α subunit. Although HIF-1β is constitutively expressed in normoxic cells, the HIF-1α subunit is only detectable in hypoxic cells. Under normoxic conditions, the HIF-1α protein is ubiquitinated and rapidly degraded by the proteasomal system (3, 4). The ubiquitination process depends on its interaction with the von Hippel Lindau protein (VHL) that acts as a ubiquitin protein ligase (5–7). Hypoxia, transition metals, and iron chelators inhibit this degradation process and allow for HIF-1α accumulation and formation of the transcriptionally active complex. Significantly, HIF-1α responses are very rapid and occur at levels of hypoxia that are well within the physiological range (8). Furthermore, the induction of HIF-1α during hypoxia is practically instantaneous, with protein HIF-1α detected in the nucleus as early as 2 min after exposure to low oxygen (9).
Despite the advances in the area of gene regulation by hypoxia, the molecular mechanisms that are responsible for oxygen sensing and the downstream pathways utilized by the hypoxic signal are still poorly understood. In biological systems, oxygen reactivity is usually associated with metals, most notably iron, as it intervenes in two types of reactions in the following ways: (a) as an acceptor of electrons in redox reactions, or (b) in liganding to heme groups. In the latter case, as it happens in its interactions with hemoglobin, there is no transfer of electrons. This last property suggested to Goldberg et al. (10) that oxygen sensing in vertebrates may utilize a rapidly turning over heme-containing protein, such that oxygen-dependent changes in conformation would initiate the hypoxia response. However, inhibitors of heme synthesis had no significant affect on hypoxia response (11, 12). Other models for oxygen sensing are based on electron transfer or redox reactions. In redox reactions, oxygen acts as the acceptor of electrons and may result in generation of reactive oxygen species (ROS). Based on the stimulatory effect of iron chelators on HIF-1α activation, Srinivas et al. (11) proposed that iron may be interacting directly, or through an intermediate iron-binding protein, with HIF-1α by inducing localized oxidative reactions that would act as a signal for degradation. However, no such protein has yet been found (11). Acker and colleagues (13, 14) have postulated that a low output NADPH oxidase, similar to the one present in neutrophils, could act as an oxygen sensor. In this model, hydrogen peroxide would be continuously generated by the oxidase in an oxygen-dependent manner and would have a continuous negative tonic effect on HIF-1α survival. During hypoxia, the decrease in peroxide production would result in HIF-1α accumulation. However, evidence against this model is the finding that diphenylene iodonium (DPI), an NADPH oxidase inhibitor, decreases rather than stimulates the hypoxia response (15). Chandel et al. (16) have proposed another redox model of oxygen sensing based on the production of ROS by the mitochondria. These investigators have reported that during hypoxia there is an increase in superoxide production at the level of complex III of the respiratory electron transfer chain. The increase in ROS production would be proportional to the degree of hypoxia and would be, following dismutation to hydrogen peroxide, the starting point of the hypoxic signal. Experimental support for their model is the unresponsiveness to hypoxia of cells lacking mitochondrial DNA, ρo cells. Furthermore, blocking of electron transfer at complex I by rotenone resulted in inhibition of the hypoxia response. However, other studies had found that inhibitors of cytochrome c oxidase, like cyanide and azide, had no effect on the hypoxia response and suggested that the mitochondria was not involved in oxygen sensing. The results by Chandel et al. (16) prompted us to re-evaluate the role of mitochondria in HIF-1α activation. Our results indicate that the integrity of the mitochondrial respiratory chain is not necessary for hypoxia response. Furthermore, they show that hydrogen peroxide is not an intermediary molecule involved in oxygen sensing.
MATERIALS AND METHODS
Cell Lines and Culture Conditions
HeLa cells were obtained from ATCC and grown in high glucose DMEM (Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum, penicillin, and streptomycin. HepG2 cells and its derivative catalase overexpressing cells Mc5 (mitochondrial localization) and C33 (cytoplasmic localization) were obtained from Dr. Cederbaum (Mt. Sinai School of Medicine, New York, NY) and grown in minimal essential medium (Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT), penicillin (100 units/ml), and streptomycin (100 µg/ml) (17). Mitochondrial DNA-less (ρo cells) were prepared by prolonged exposure to ethidium bromide as described by King and Attardi (18) and grown in DMEM supplemented with 5% or 15% fetal bovine serum, penicillin, streptomycin, sodium pyruvate (1 mM), and uridine (50 µg/ml). Cells were maintained at saturation humidity at 37 °C in 5% CO2, 95% air. For hypoxic stimulation, the cultures were flushed in a modular incubator (Billups-Rothenburg, Del Mar, CA) with a gas mixture of 0.5% O2, 5% CO2, and nitrogen was balanced. Cells were transfected with LipofectAMINE Plus (Life Technologies, Inc.) as described previously (19). Expression vectors used in the functional Luciferase assays have been described previously (19).
Nuclear Extracts, Electrophoretic Mobility Shift Assay, and Immunoblot Analysis
Nuclear extracts were prepared from normal or treated cells as described previously (19). Electrophoretic mobility shift assay was performed by incubating 10 µg of nuclear extract with 32P-labeled double stranded oligonucleotide probe from the human HIF-1 Epo enhancer as described (4). For immunoblot analysis, equal amounts of nuclear extracts (10 –20 µg of protein) were fractionated by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes prior to detection using a stabilized alkaline phosphatase system (Promega, Madison, WI) (19). Monoclonal anti-HIF-1α antibody was purchased from Transduction Laboratories (Lexington, KY), and alkaline phosphatase-conjugated anti-mouse secondary antibody was purchased from Southern Biotechnology (Birmingham, AL).
Indirect Immunofluorescence Microscopy
Cells growing on glass slides were fixed with 4% paraformaldehyde in PBS (pH 8.0) for 10 min and then washed with PBS three times. Subsequently, they were permeabilized with 0.5% Triton X-100 in PBS for 5 min and rinsed again three times with PBS. After blocking nonspecific binding with 10% fetal calf serum in PBS for 30 min, the cells were incubated with a monoclonal anti-HIF-1α antibody (Novus Biologicals, Littleton, CO). Primary antibody was detected by incubation with an fluorescein isothiocyanate-conjugated rabbit anti-mouse antibody (Southern Biotechnology, Birmingham, AL) at room temperature for 30 min. Following extensive washing in PBS and mounting in Miowol solution (Calbiochem), the cells were visualized and photographed utilizing a fluorescence microscope(Nikon).
RNA Isolation and Ribonuclease Protection Assays
Total RNA was isolated with the Trizol reagent (Life Technologies, Inc.) according to the manufacturer’s instructions. RNase protection assays was performed utilizing a 32P-labeled single stranded vascular endothelial growth factor (VEGF) probe as described previously (20).
RESULTS
Activation of HIF-1α by Hypoxia in ρo Cells
Mitochondria are the center of oxidative phosphorylation, a process by which ATP is formed as a result of the transfer of electrons from NADH or FADH2 to O2 by a series of electron carriers. The mammalian mitochondrial DNA encodes thirteen proteins, all subunits of the respiratory chain enzymes, including critical catalytic subunits for complex I (NADH dehydrogenase), complex III (bcl complex), complex IV (cytochrome c oxidase), and ATP synthetase. Elimination of mitochondrial DNA from cells by prolonged ethidium bromide treatment results in the absence of respiratory chain electron transfer and mitochondrial ATP production by oxidative phosphorylation (18). Cells lacking mitochondrial DNA can survive, however, when provided with external sources of pyrimidines and pyruvate (18). We utilized two independently generated ρo cell lines, one derived from the human osteosarcoma cells (143B206) (18) and one from a human fibroblast cell line (GM 701) (21). The ρo cells and the corresponding parental cell lines were subjected to hypoxia (0.5% O2) for 4–6 h, and the nuclear extracts were analyzed for HIF-1α expression by Western blots and for HIF-1 complex formation by electrophoretic mobility shift analysis. As shown in Fig. 1, a and b, exposure to hypoxia and desferrioxamine results in increased expression of HIF-1α protein in both ρo cell lines, their parental wild types, and HeLa cells. Similarly, hypoxia, desferrioxamine, and cobalt induced the formation of the HIF-1 DNA binding complexes, as measured by gel shift assays (Fig. 1c). Furthermore, rotenone, an inhibitor of mitochondrial complex I, had no effect on the hypoxia responses (not shown).
Fig. 1. Expression of HIF-1a in ρo cells.
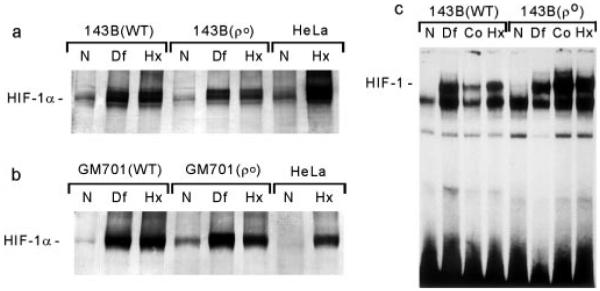
Parental (WT) and ρo cells were exposed to hypoxia (Hx; 0.5% O2), desferrioxamine (Df; 130 µM), or cobalt (Co; 100 µM) for 4–6 h, and nuclear extracts were analyzed by Western blots (a) and (b) or electrophoretic mobility shift assays (c). N, normoxia.
The activation of HIF-1α expression by hypoxia in ρo cells was confirmed by immunofluorescence and functional studies. Fig. 2a shows the induction of nuclear fluorescence, detected with an anti-HIF-1α monoclonal antibody, in parental 143B (wild type) and ρo 143B206 cells exposed to hypoxia (2 and 5) or treated with desferrioxamine (3 and 6). To test whether the transcriptional activation of HIF-1α by hypoxia was conserved in ρo cells, we utilized an expression vector containing the transcriptionally active C-terminal domain of HIF-1α protein (amino acids 740 to 826) fused to the DNA-binding domain of the yeast transactivator GAL-4 (19). Cotransfection of this plasmid with a luciferase reporter containing a GAL-4 DNA-binding site showed that hypoxia induced the transactivational activity of HIF-1α, both in ρo and wild type cells (Fig. 2b). Furthermore, confirmation of the hypoxia responsiveness of ρo cells was obtained by the hypoxic activation of the VEGF gene as shown in the RNase protection assay in Fig. 2c.
Fig. 2. Functional activity of HIF-1α in ρo cells.
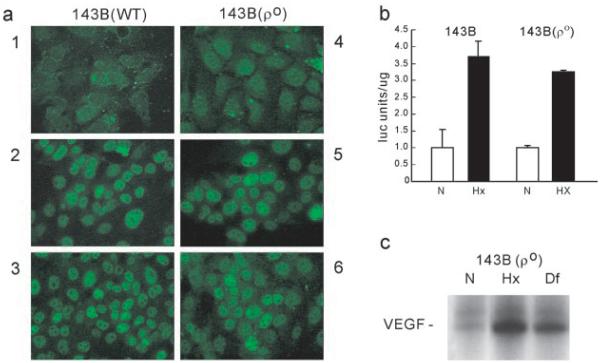
a, nuclear translocation. Wild type (WT) and ρo cells were maintained in normoxia (1 and 4) or exposed to hypoxia (2 and 5) or desferrioxamine (3 and 6) for 2–4 h and immunostained with an anti HIF-1α monoclonal antibody. b, transcriptional activity. Wild type and ρo cells were transfected with a GAL-4-HIF-1α fusion (amino acids 740–826) and a GAL-4-DNA-binding domain luciferase expression plasmid and maintained in normoxia (N) or exposed to hypoxia (Hx). Results are expressed as the mean of arbitrary luciferase units per µg of protein ± S.D. c, RNase protection assay for Northern blot VEGF expression in ρo cells exposed to hypoxia (Hx) or desferrioxamine (Df).
Effect of Overexpression of Catalase on HIF-1α Expression
Most of the currently proposed redox models for oxygen sensing suggest that H2O2 is the intermediary signal molecule in the hypoxia response. To evaluate the role of hydrogen peroxide in oxygen sensing, we utilized cell lines that overexpress the enzyme catalase in either the mitochondrial or cytosolic compartments. These cell lines were produced by introducing a catalase cDNA or a catalase cDNA with a superoxide dismutase mitochondrial leader sequence into a human hepatoma cell line (HepG2 cells). Published results (17) have shown that these cell lines are more resistant to H2O2-, menadione-, or antimycin A-induced apoptosis as compared with cells transfected with the vector alone. After exposure to hypoxia and desferrioxamine of control and catalase-overexpressing cells, the induction of HIF-1α was measured by Western blots and electrophoretic motility shift assays. Fig. 3a shows that overexpression of catalase, either in the cytosol (C33 cells) or in the mitochondria (Mc5 cells), does not affect the response to hypoxia or change the basal expression of HIF-1α protein. Similar results were obtained using the gel shift assay (not shown). The effect of catalase overexpression on the transcriptional activity of HIF-1α was evaluated by using the GAL-4-HIF-1α fusion system, as described above. Fig. 3b indicates that overexpression of catalase does not affect the transcriptional response to hypoxia (Fig. 3d).
Fig. 3. Hypoxia responses in catalase overexpressing cells.
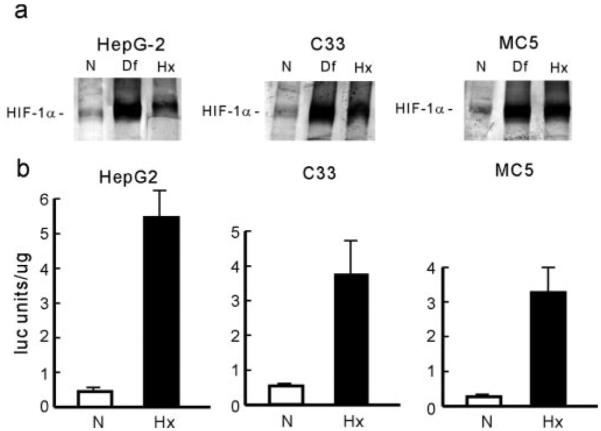
a, expression of HIF-1α. Control HepG2 cells or cells overexpressing catalase in the cytosol (C33) or in the mitochondria (MC5) were exposed to normoxia (N), desferrioxamine (Df), or hypoxia (Hx) for 2–4 h, and nuclear extracts were subjected to Western blot analysis. b, HIF-1α transcriptional activity. Control and catalase overexpressing cells were transfected, as described in Fig. 2, and exposed to normoxia (N) or hypoxia (Hx) for 4 h. Results are expressed as arbitrary luciferase units per µg of protein ± S.D.
DISCUSSION
Early work showing that cyanide and sodium azide do not affect the hypoxia response suggested that the mitochondrial respiratory chain was not involved in oxygen sensing (22). However, Chandel et al. (16) have proposed that an increase in superoxide production at the level of mitochondrial complex III (quinol-cytochrome c oxidoreductase) is the initial event that triggers the hypoxia response (16). Superoxide is produced during normal mitochondrial function, and it is rapidly converted to H2O2, a readily diffusible molecule (23). A compelling argument for their hypothesis was the finding that ρo cells, which lack mitochondrial DNA, have an impaired response to hypoxia. In the work presented here, we have utilized two independent human ρo cell lines created by prolonged exposure to low doses of ethidium bromide. This treatment results in inhibition of mitochondrial DNA replication without affecting the nuclear DNA (18). As mitochondrial DNA encodes essential subunits of the respiratory chain and ATP synthetase, ρo cells have no mitochondrial electron transfer and are completely dependent on glycolysis for the generation of ATP. Furthermore, these cells depend on exogenous sources of pyrimidines (pyrimidine auxotrophy). The need for pyrimidine is explained by the fact that the enzyme dihydrooratate dehydrogenase is located in the mitochondrial inner membrane and requires mitochondrial electron transfer for normal function. In the presence of pyruvate and pyrimidines, these cell lines can maintain almost normal cell growth rates. One of the cell lines (143B206) was derived from an osteosarcoma cell line and the other (701–2c) from an SV-40 T-antigen-transformed fibroblast cell line (18, 21). Oxygen consumption in the ρo cells was found to be less than 5% of the control cells, and no cytochrome c oxidase activity was detected (24). The ρo status of both cell lines was confirmed by their inability to grow in pyruvate- and pyrimidine-deficient media and by DNA hybridization studies showing no traces of mitochondrial DNA. Our results with the ρo cells indicate that an active mitochondrial respiratory electron transport chain is not an absolute requirement for the activation of HIF-1 protein in response to hypoxia. Furthermore, we find that in both cell lines the functional activity of the HIF-1 complex, its nuclear translocation, and its hypoxia-induced transactivation activity are preserved, as well. The differences between our results and those of Chandel et al. (16) are unclear and may derive from differences in the cell lines or the procedures used to establish the ρo cells. It should be noted that in the preparation of ρo cells, Chandel et al. (16) utilized a selection that included growth in the presence of rotenone and antimycin. It is not known whether this selection step may affect the properties of the cells.
Electron transfer reactions (redox reactions) appear to be an important component of the mechanisms of oxygen sensing. Acker (13) and Fandrey et al. (14) have postulated that a low output NADPH oxidase, similar to the one present in granulocytes, may initiate the hypoxic response. In their model, the continuous production of superoxide by this NADPH oxidase would act as an active negative signal to induce HIF-1α degradation. During hypoxia, the decrease in hydrogen peroxide would result in increased HIF-1α stability. However, Gleadle et al. (15) observed that DPI, a flavoprotein inhibitor whose targets include NADPH oxidases, decreases, rather than enhances, the hypoxia response (15). Dismutation of superoxide, either spontaneously or through superoxide dismutase, results in the production of hydrogen peroxide. This compound is quite soluble and diffusible and has been postulated as a likely intermediate of the hypoxic signal. Reaction of H2O2 with iron (Fenton reaction) results in the generation of hydroxyradicals, which are highly reactive molecules. To test the role of H2O2 as a signal molecule, we utilized cell lines that overexpress the enzyme catalase either in the cytosol or in the mitochondria. These cell lines have been shown to decompose H2O2 very effectively and to provide resistance to the apoptotic effects of menadione or antimycin (17). Overexpression of catalase, either in the cytosol or in the mitochondria, did not affect the normoxic levels of HIF-1α protein or its response to hypoxic stimulation. These results cast doubt on the role of H2O2 as a signal molecule in the hypoxic response, either as a negative signal, as postulated by Acker (13) and Fandrey et al. (14), or as a positive signal, as postulated by Chandel et al. (16).
The strong stimulatory effect of the iron (III) chelator desferrioxamine in HIF-1α activation suggests that an iron-containing protein(s) is most likely involved in oxygen sensing. In general, iron (III)-chelating agents are not active against hemecontaining enzymes or iron-sulfur cluster proteins, although they do interfere with enzymes containing mono-iron or bi-iron centers coordinated to oxygen ligands, including amino acids hydroxylases, lipoxygenases, and ribonucleotide reductase (25). As shown by the studies presented here, the putative oxygen sensor is independent of the mitochondrial respiratory chain activity, and it does not utilize H2O2 as a signal molecule.
Acknowledgments
We thank Dr. Cederbaum for generously providing the catalase overexpressing cells. We acknowledge the support of and thank our secretary, Rosemarie Silvano, and our graphic illustrator, Drew Likens.
Footnotes
This work was supported in part by Grants 0060194U (to V. S.) and 9950122N (to J. C.) from the American Heart Association and by National Institutes of Health Grant 1RO1 CA89212-02 (to J. C.).
- HIF
- hypoxia-inducible factor
- DPI
- diphenylene iodonium
- ROS
- reactive oxygen species
- DMEM
- Dulbecco’s modified Eagle’s medium
- PBS
- phosphate-buffered saline
- VEGF
- vascular endothelial growth factor
REFERENCES
- 1.Wenger RH. J. Exp. Biol. 2000;203:1253–1263. doi: 10.1242/jeb.203.8.1253. [DOI] [PubMed] [Google Scholar]
- 2.Wang GL, Jiang B-H, Rue EA, Semenza GL. Proc. Natl. Acad. Sci. U. S. A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang LE, Gu J, Schau M, Bunn HE. Proc. Natl. Acad. Sci. U. S. A. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salceda S, Caro J. J. Biol. Chem. 1997;272:22642–22647. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- 5.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe SC, Vaux EC. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 6.Tanimoto K, Makino Y, Pereira T, Poellinger L. EMBO J. 2000;19:4298–4309. doi: 10.1093/emboj/19.16.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Nat. Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 8.Jiang B-H, Semenza GL, Bauer C, Marti HH. Am. J. Physiol. 1996;271:C1172–C1180. doi: 10.1152/ajpcell.1996.271.4.C1172. [DOI] [PubMed] [Google Scholar]
- 9.Jewell UR, Kvietikova I, Scheid A, Bauer C, Wenger RH, Gassman M. FASEB. 2001;15:1312–1314. [PubMed] [Google Scholar]
- 10.Goldberg MA, Dunning SP, Bunn HF. Science. 1988;242:1412–1415. doi: 10.1126/science.2849206. [DOI] [PubMed] [Google Scholar]
- 11.Srinivas V, Zhu X, Salceda S, Nakamura R, Caro J. J. Biol. Chem. 1998;273:18019–18022. doi: 10.1074/jbc.273.29.18019. correction 274, 1180, 1999. [DOI] [PubMed] [Google Scholar]
- 12.Horiguchi H, Bunn HF. Biochim. Biophys. Acta. 2000;1495:231–236. doi: 10.1016/s0167-4889(99)00169-x. [DOI] [PubMed] [Google Scholar]
- 13.Acker H. Ann. N. Y. Acad. Sci. 1994;718:1–10. doi: 10.1111/j.1749-6632.1994.tb55698.x. [DOI] [PubMed] [Google Scholar]
- 14.Fandrey J, Frere S, Jelkmann W. Biochem. J. 1994;303:507–510. doi: 10.1042/bj3030507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gleadle JM, Ebert BL, Ratcliffe PJ. Eur. J. Biochem. 1995;234:92–99. doi: 10.1111/j.1432-1033.1995.092_c.x. [DOI] [PubMed] [Google Scholar]
- 16.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. J. Biol. Chem. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 17.Bai J, Rodriguez AM, Melendez JA, Cederbaum AI. J. Biol. Chem. 1999;274:26217–26224. doi: 10.1074/jbc.274.37.26217. [DOI] [PubMed] [Google Scholar]
- 18.King MP, Attardi G. Methods Enzymol. 1996;264:304–313. doi: 10.1016/s0076-6879(96)64029-4. [DOI] [PubMed] [Google Scholar]
- 19.Srinivas V, Zhang L-P, Zhu X-H, Caro J. Biochem. Biophy. Res. Commnun. 1999;260:557–561. doi: 10.1006/bbrc.1999.0878. [DOI] [PubMed] [Google Scholar]
- 20.Minchenko A, Bauer T, Salceda S, Caro J. Lab. Invest. 1994;71:374–379. [PubMed] [Google Scholar]
- 21.Jacobson MD, Burne JF, King MP, Miyashita T, Reed JC, Raff MC. Nature. 1993;361:365–369. doi: 10.1038/361365a0. [DOI] [PubMed] [Google Scholar]
- 22.Necas E, Thorling EB. Am. J. Physiol. 1972;222:1187–1190. doi: 10.1152/ajplegacy.1972.222.5.1187. [DOI] [PubMed] [Google Scholar]
- 23.Han D, Williams E, Cadenas E. Biochem. J. 2001;353:411–416. doi: 10.1042/0264-6021:3530411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.King MP, Attardi G. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 25.Liu ZD, Lockwood M, Rose S, Theobald AE, Hider RC. Biochem. Pharmacol. 2001;61:285–290. doi: 10.1016/s0006-2952(00)00551-7. [DOI] [PubMed] [Google Scholar]