

Abstract
Our previous studies have demonstrated that a decrease in arteriolar diameter that causes endothelial deformation elicits the release of nitric oxide (NO). Thus we hypothesized that cardiac contraction, via deformation of coronary vessels, elicits the release of NO and increases in coronary flow. Coronary flow was measured at a constant perfusion pressure of 80 mmHg in Langendorff preparations of rat hearts. Hearts were placed in a sealed chamber surrounded with perfusion solution. The chamber pressure could be increased from 0 to 80 mmHg to generate extra-cardiac compression. To minimize the impact of metabolic vasodilatation and rhythmic changes in shear stress, nonbeating hearts, by perfusing the hearts with a solution containing 20 mM KCl, were used. After extracardiac compression for 10 or 20 s, coronary flow increased significantly, concurrent with an increased release of nitrite into the coronary effluent and increased phosphorylation of endothelial NO synthase in the hearts. Inhibition of NO synthesis eliminated the compression-induced increases in coronary flow. Shear stress-induced dilation could not account for this increased coronary flow. Furthermore, in isolated coronary arterioles, without intraluminal flow, the release of vascular compression elicited a NO-dependent dilation. Thus this study reveals a new mechanism that, via coronary vascular deformation, elicited by cardiac contraction, stimulates the endothelium to release NO, leading to increased coronary perfusion.
Keywords: endothelial deformation, nitric oxide, coronary circulation
We Demonstrated Previously that a brief Compression of Single Isolated mesenteric arterioles elicits an endothelium-dependent, nitric oxide (NO)-mediated dilation. Also, a unidirectional compression of cultured endothelial cells stimulates, time dependently, the release of NO from the cells (26). These studies indicate that vascular deformation coincident with endothelial deformation induced by physical forces stimulates endothelial cells to produce vasoactive substances to regulate vascular tone.
Multiple mechanisms are involved in the regulation of the coronary circulation, among them cardiac metabolism (8), as well as local mechanisms including myogenic constriction (12, 18, 20) and flow-induced dilation (13, 23), all of which have been demonstrated to participate significantly in the control of vascular resistance. It is well known that in the coronary circulation, vessels are rhythmically compressed by contraction of the myocardium. Unlike the physical forces exerted by pulsatile pressure, in which the vessels are subjected to circumferential stretch (1), myocardial contraction provides for circumferential compression of the vessels. Thus studies aimed to evaluate the physiological significance of cardiac contraction in the regulation of the coronary circulation become of considerable interest, because they may implicate this physical force as being a factor in the local regulation of myocardial blood flow.
Given that endothelial deformation caused by changes in vascular diameter elicits the release of NO and that coronary vessels are constantly subjected to rhythmic compression by cardiac contraction, it is legitimate to ask the questions as to whether coronary endothelium is sensitive to the cardiac contraction-induced deformation, and, if so, whether the deformation contributes to the synthesis and release of NO followed by changes in coronary blood flow. Thus we tested the hypothesis that compression of the heart is followed by a NO-mediated increase in coronary flow. The data obtained confirm our hypothesis suggesting, therefore, that cardiac contraction could be considered not only as a functional pump to maintain cardiac output but also as being involved in the direct regulation of coronary blood flow via the activation of coronary endothelial NO synthase (eNOS).
Methods
All protocols were approved by the Institutional Animal Care and Use Committee of New York Medical College and conformed to the present guidelines of the National Institutes of Health and the American Physiological Society for the use and care of laboratory animals.
Isolated hearts
Normal Wistar rats (250 g) were anesthetized with pentobarbital (50 mg/kg ip) and heparinized (500 IU/kg ip). After the thorax was opened, the heart was quickly excised and mounted via the ascending aorta onto a perfusion apparatus (Fig. 1). The heart was perfused retrograde with a nonrecirculating physiological salt solution (PSS) at a constant hydrostatic pressure of 80 mmHg. The PSS contained (in mM) 118 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 11 glucose, 25 NaHCO3, 2 pyruvate, and 0.5 EDTA and was equilibrated with 95% O2-5% CO2 at 37°C and pH 7.4. Coronary flow was measured using a square-wave electromagnetic flowmeter (Carolina Medical Electronics). The pericardium and adherent lung tissue were removed, and the cut ends of the vena cava were ligated. The pulmonary artery was cannulated to obtain the coronary venous effluent. The heart was then placed in a chamber, which was sealed with a lid as illustrated in Fig. 1. In this arrangement, an increase in chamber pressure generates an extracardiac compression. A single compression was accomplished by switching the extracardiac pressure (EP) stopcock from the outlet (zero pressure) to the EP reservoir (Fig. 1). The magnitude of the compression was controlled by the height of the reservoir. The chamber solution was changed periodically.
Fig. 1.

Perfusion system and compression chamber for isolated rat hearts.
A 30-min equilibration period was provided. To eliminate the influence of a change in myocardial metabolism on coronary flow, a nonbeating heart was used, by exchanging PSS with PSS with a total of 20 mM KCl (PSS-KCl), by substituting equimolar sodium with potassium in the buffer. Chamber pressure was then increased from 0 to 80 mmHg for 10 and 20 s, respectively, to generate extracardiac compression. Changes in mean coronary flow and chamber pressure were continuously recorded. Because the increase in chamber pressure to 80 mmHg would not only generate deformation of myocardial microvessels but also would compress large epicardial vessels to stop coronary perfusion, as a control, coronary perfusion pressure (controlled by an inflow stopcock, as illustrated in Fig. 1) was turned off for a period of 10 and 20 s to generate zero coronary flow. Results of changes in coronary flow upon restoring perfusion pressure were compared with those after cardiac compression was released. After inhibition of NO synthesis, which was accomplished by the administration of Nω-nitro-l-arginine methyl ester (l-NAME; 2 × 10−4 M) to nonbeating hearts, protocols of increasing chamber pressure and turning off of inflow pressure were repeated. The function of coronary endothelium and smooth muscle cells in nonbeating hearts was also assessed by bolus injections (into the coronary inflow solution, Fig. 1) of ACh (100 μl, 10−6 M) and sodium nitroprusside (SNP; 30 μl, 10−4 M) before and after l-NAME. To further compare to compression-induced increase in coronary flow, in separate experiments, coronary flow of nonbeating hearts was also measured in response to a 10-s occlusion of coronary outflow (by clamping the catheter of the pulmonary artery). In this instance, transmural pressure of coronary vessels was maintained at a high level and coronary flow was decreased to zero. To eliminate a possible accumulation of interstitial fluid in response to coronary outflow occlusion, all three measurements (inflow and outflow occlusions and extracardiac compression) were performed only once in each heart. All experiments were completed within 3 h.
Measurement of coronary effluent nitrite
Coronary effluents were collected for 1 min in beating and nonbeating hearts before and immediately after cardiac compressions. The amount of NO released in the coronary effluent was assessed via quantification of its decomposition product, nitrite, by a fluorometric assay (17) with a spectrofluorometer (SFM25, Kontron). For quantification of the amount of NO in the sample, standard curves of nitrite (sodium nitrite of 0–640 nM) were constructed using PSS as a vehicle. Background readings were subtracted from sample readings, and final results are expressed as nanomoles per minute.
Western blot analysis
Isolated hearts were perfused via the following three protocols: protocol I, 30 min of PSS (beating hearts, ∼260 beats/min); protocol II, 30 min of PSS-KCl (nonbeating hearts); and protocol III, 30 min of PSS-KCl and then 2 min of 80 mmHg of EP (nonbeating hearts plus compression). After perfusion, hearts were snap frozen in liquid nitrogen and pulverized. Tissue homogenates (200 μg protein) [containing 1% protease inhibitor cocktail (Sigma P8340) and phosphatase inhibitor cocktail (Sigma P5726)] were separated on a SDS-PAGE gel (7.5% acrylamide), transferred to a polyvinylidene difluoride membrane, and probed with primary anti-bodies of phospho-eNOS (p-eNOS; Ser1177, Cell Signaling) and eNOS (BD Transduction Laboratories). Secondary antibodies were conjugated to horseradish peroxidase according to the Amersham ECL-Plus protocol.
Isolated coronary arterioles
The excised heart was pinned to the Silastic bottom of a dissecting dish containing ice-cold MOPS (3 mM)-buffered PSS (MOPS-PSS) at pH 7.4. A myocardial branch of the left anterior descending coronary artery was isolated and cannulated in a perfusion chamber containing MOPS-PSS at 37°C and pH 7.4. The vessel chamber was then sealed air tight with a lid, as described in our previous study (26). Intravascular pressure was maintained constant at 80 mmHg with an inflow pressure-servo system. The vessels were equilibrated for 60 min to generate myogenic tone. Extravascular pressure (EVP; chamber pressure) was then increased from 0 to 75 mmHg for 20 and 60 s, respectively. The methods to generate EVP were similar to those described in Fig. 1. Thereafter, l-NAME (2 × 10−4 M) was administered to inhibit NO synthesis, and the effects of EVP on the diameter of vessels were reassessed. The internal diameter of arterioles was measured continuously with a television scanning device (Living Systems; Burlington, VT).
Statistical analysis
Data are expressed as means ± SE. Statistical analysis was performed using repeated-measures ANOVA, followed by the Tukey-Kramer post hoc test and Student's t-test. Statistical significance was accepted at a level of P < 0.05.
Results
In the presence of 80 mmHg of perfusion pressure, mean coronary flow in PSS-perfused beating hearts and PSS-KCl-perfused nonbeating hearts was 10.9 ± 0.7 and 10.6 ± 0.6 ml/min, respectively. In nonbeating hearts, the administration of ACh and SNP significantly increased coronary flow (Table 1). Inhibition of NO synthesis with l-NAME (2 × 10−4 M) significantly decreased mean coronary flow to 6.7 ± 0.4 ml/min (by 35.6 ± 2.5%) and inhibited ACh-induced dilation without affecting SNP-induced dilation.
Table 1. Effects of ACh and SNP on coronary flow.
| Control | l-NAME (2 × 10−4M) | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Baseline | Response | %Change | Baseline | Response | %Change | |
| ACh | 10.7±0.8 | 13.4±0.9* | 26.3±6.0 | 6.3±0.6 | 4.7±0.8* | −25.8±9.2 |
| SNP | 10.1±0.6 | 13.8±0.6* | 40.7±10.2 | 5.8±0.4 | 10.2±0.9* | 77.2±7.2 |
Values are means ± SE; n = 7 hearts. Mean coronary flow in physiological salt solution-KCl-perfused nonbeating hearts in response to bolus injections of ACh (100 μl of 10−6 M) and sodium nitroprusside (SNP; 30 μl of 10−4 M) are shown. l-NAME, Nω-nitro-L-arginine methyl ester.
P < 0.05.
Perfusion of hearts with PSS-KCl stopped myocardial contraction, resulting in nonpulsatile coronary flow (Fig. 2A) and dilated hearts. A transient increase in coronary flow was seen immediately after PSS was replaced with PSS-KCl, followed by a gradual return (∼2 min) of coronary flow back to the control level. In continuous PSS-KCl-perfusion, a constant coronary flow in nonbeating hearts was maintained for at least 1 h. By switching the perfusion back to PSS, spontaneous contraction of the heart and pulsatile coronary flow were recovered (data not shown). Figure 2B, top, shows a representative recording in which occlusion of coronary perfusion via turning off the inflow stopcock or increasing EP from 0 to 80 mmHg for 10 s elicited increases in mean coronary flow. The magnitude and duration of increases in coronary flow were much greater when the hearts were subjected to an increase in EP than to occlusion. In the presence of l-NAME (Fig. 2B, bottom), the increases in mean coronary flow elicited by occlusion or changes in EP were eliminated, indicating that the responses were mediated by NO.
Fig. 2.
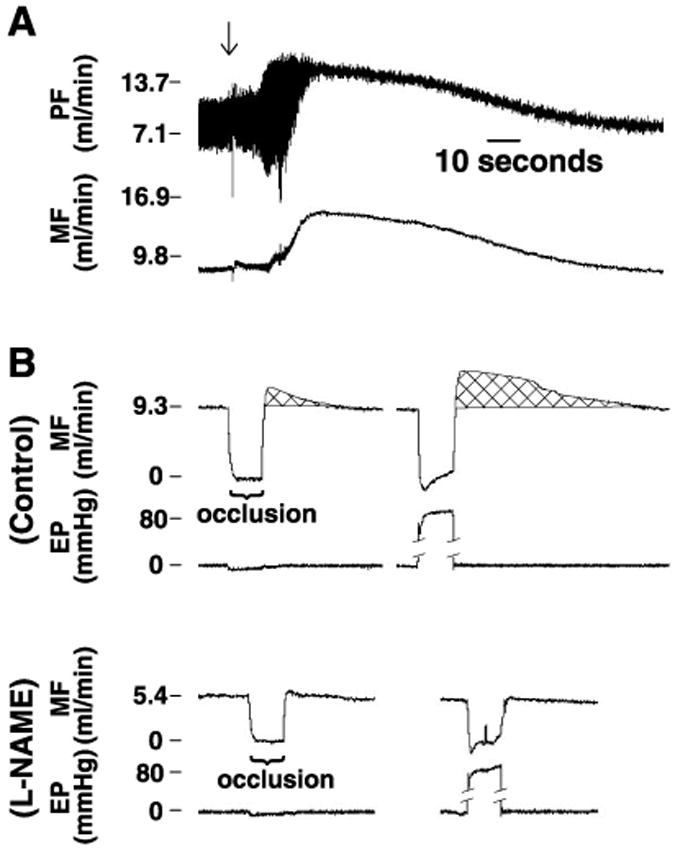
Representative recordings of coronary flow and extracardiac pressure in an isolated and perfused rat heart. A: changes in pulsatile and mean coronary flow (PF and MF, respectively) after the physiological salt solution (PSS) was exchanged with PSS containing a total of 20 mM KCl (PSS-KCl). Arrow indicates the start of perfusion with PSS-KCl. B: changes in mean coronary flow in response to occlusion of coronary perfusion via turning off the inflow stopcock and increasing extracardiac pressure (EP) to 80 mmHg for 10 s in control and after inhibition of nitric oxide (NO) synthesis with Nω-nitro-l-arginine methyl ester (l-NAME; 2 × 10−4 M).
The difference in increases in the integrated flow (flow rate × duration of a response) as a function of change in EP or inflow occlusion is summarized in Fig. 3. In control, both EP and occlusion significantly increased integrated flow. There was no difference in the integrated flow between 10 and 20 s of occlusion. However, the integrated flow was significantly greater in the response to 20 s than to 10 s of EP and was significantly greater in response to either 10 or 20 s of EP than to occlusion. l-NAME abolished EP- or occlusion-induced responses.
Fig. 3.

Increase in integrated flow in response to increases in EP and stoppage of coronary perfusion for 10 and 20 s in control and after inhibition of NO synthesis with l-NAME (2 × 10−4M). Increase in integrated flow, as illustrated by the crosshatched areas in Fig. 2B, was calculated as the difference between the area (flow rate × duration) under a specific response curve and a control area (control flow rate × duration of the response). Data are means ± SE (n = 8–14).
Figure 4 further compares increases in integrated coronary flow-induced by outflow occlusion from those induced by inflow occlusion and extracardiac compression. The magnitude and duration of increases in coronary flow were similar to those induced by inflow occlusion but less than those induced by extracardiac compression (Fig. 4A). Summary data of increases in integrated flow, shown in Fig. 4B, demonstrate that extracardiac compression-induced increase in coronary flow is significantly greater than that induced by outflow occlusion as well as that induced by inflow occlusion.
Fig. 4.
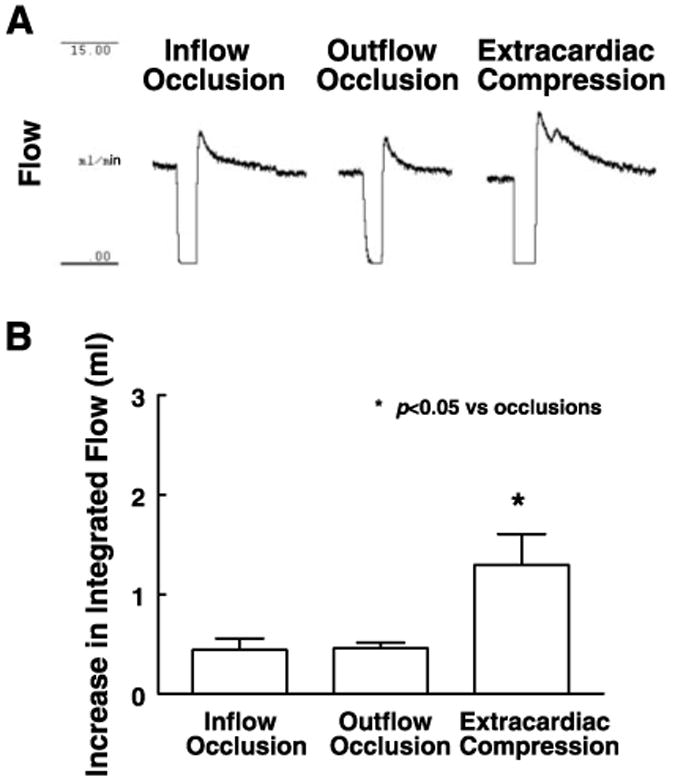
Increase in integrated flow in response to 10 s of inflow and outflow occlusions and extracardiac compression (0–80 mmHg). A: representative recordings. B: summary data of 5 experiments. Data are means ± SE.
Coronary effluent nitrite was measured to assess the activation of NO synthesis. Figure 5 shows that, although there was a significant amount of coronary effluent nitrite in nonbeating hearts, the production of nitrite was ∼10 times higher in beating hearts. In nonbeating hearts, a single extracardiac compression of 80 mmHg for 20 s significantly increased nitrite production approximately fivefold; however, the enhanced production of nitrite after the compression was still significantly less than that in beating hearts.
Fig. 5.

Release of NO, measured as nitrite in the coronary effluent, in beating hearts and in nonbeating hearts before and after 20 s of EP (80 mmHg). Coronary effluent was collected for 1 min in each experimental condition. Average release of nitrite (in nmol/min) was calculated by the total volume collected during 1 min. Each bar represents the average of nine measurements (means ± SE).
Cardiac compression-induced increases in NO production were further assessed by comparing the level of phosphorylation of eNOS in beating, nonbeating, and nonbeating hearts subjected to extracardiac compression. Figure 6A shows that the level of phosphorylation of eNOS is lower in nonbeating than beating hearts. Figure 6A also shows that a single compression increases phosphorylation of eNOS in nonbeating hearts. Summary data of densitometric ratios of p-eNOS to eNOS are shown in Fig. 6B. The results indicate that without cardiac contraction, the level of p-eNOS significantly decreased by 82%. In nonbeating hearts, extracardiac compression increased p-eNOS about fourfold and the level of p-eNOS became 70% (P > 0.05) of that in hearts with spontaneous contraction.
Fig. 6.

A: representative Western blots of phospho-endothelial NO synthase (p-eNOS) and eNOS. B: densitometric ratio of p-eNOS and eNOS in beating (I), nonbeating (II), and nonbeating hearts with extracardiac compression (III). Data are means ± SE; n = 3–5. NS, no statistical difference.
To further distinguish the activation of NO synthesis induced by cardiac compression from that by shear stress, single coronary arterioles were isolated and studied in a no-flow condition. The average passive diameter at 80 mmHg of intravascular pressure was 129 ± 7 μm, and the average basal diameter was 75 ± 2 μm. We found that increases in EVP to 75 mmHg, due to a reduction of transmural pressure, decreased the internal diameter of arterioles to ∼25 μm. After the 20- and 60-s compressions were released, the diameter of arterioles immediately returned to control level and continued to increase further above the control, exhibiting a dilator response. Summary data of these experiments are shown in Fig. 7. The peak changes in diameter and the duration of the dilation were proportional to the rise in EVP, as the area under the dilation curves was significantly increased in response to 20 s of compression and was further significantly increased after 60 s of compression. Inhibition of NO synthesis with l-NAME (2 × 10−4 M), which did not affect the basal tone of the vessels (73 ± 2 μm), eliminated EVP-induced dilations.
Fig. 7.

Vasodilatation of isolated rat coronary arterioles induced by release of extravascular pressure (EVP) in control and after administration of l-NAME (2 × 10−4 M). EVP was increased from 0 to 75 mmHg for 20 and 60 s. Post-EVP dilation is presented as an area that was calculated as the difference between a product of diameter (in μm) and duration (in s) under a specific response curve and a product of the control diameter and the duration of the same response curve. Each bar represents an average of 6 measurements (means ± SE).
Discussion
This study demonstrates that cardiac compression, a physical force that mimics cardiac contraction in vivo, causes a significant increase in coronary flow, concurrent with an enhanced phosphorylation of eNOS and a greater production of nitrite. In addition, extravascular compression of isolated coronary arterioles in a no-flow condition also elicits a NO-dependent dilator response.
The resistance of coronary vessels is mainly regulated by myocardial contraction, myocardial metabolites, and local intrinsic mechanisms, including shear stress-dependent dilation and myogenic constriction. In a beating heart, with each cardiac cycle, due to myocardial compression, the diameter of coronary vessels and the shape of vascular endothelial cells are altered, accompanied by changes in the release of tissue metabolites, shear stress, and intravascular pressure. It is extremely difficult in a beating heart preparation to separate endothelial deformation-dependent regulation of coronary flow from other mechanisms listed above. For this reason, nonbeating hearts were used in the experiments to keep the influence of metabolic effects, shear stress, and intravascular pressure at relatively constant levels. Our data demonstrated that in PSS-KCl-perfused nonbeating hearts, coronary flow was maintained constant, mean coronary flow was comparable to that in beating hearts, and endothelium-dependent and -independent dilator responses were preserved. The effects of cardiac compression and occlusions on coronary flow were assessed immediately after the cessation of myocardial contraction and the achievement of a stable coronary flow to reduce the influence of hyperkalemic PSS on vascular function (9, 29). By placing a simple retrograde perfused nonbeating heart in a sealed chamber, we were able to mimic the effects of cardiac contraction on coronary vessels by increasing the hydraulic pressure of the chamber to compress the whole heart evenly. In this condition, endothelial deformation of coronary vessels is mainly induced by changes in vascular shape and/or a reduction in circumferential length of the vessels due to a decrease in transmural pressure and passive movements of the ventricular wall by cardiac compression. However, unlike vascular compression generated by a beating heart, an increase in chamber pressure will not only compress microvessels in the myocardium but also large epicardial vessels. Moreover, transmission of chamber pressure to intramyocardial vessels may be different depending on the myocardial layers, elastance, and volume. Additionally, to minimize the influence of rhythmic compression on the generation of pulsatile shear stress and intravascular pressure, single compressions with long duration were used in the present study. Nevertheless, our results showed that a single compression of nonbeating hearts increased coronary flow and that the increases were dependent on the activation of NO synthase (Figs. 2 and 3).
The endothelial function of coronary vessels is sensitive to mechanical stimulation. Early studies (14), by comparing the cGMP content of platelets passing through a working or arrested heart, have demonstrated that mechanical stimulation potentiates the release of an endothelium-derived relaxing factor. More recent studies (19), by using a porphyrinic sensor placed in the left ventricular myocardium, have further demonstrated the existence of cyclic and preload-dependent changes in NO release in beating rabbit hearts. Cardiac contraction increases intramyocardial pressure (IMP), which decreases transmural pressure of coronary vessels and hence decreases the diameter of the vessels, causing enhanced shear stress and vascular deformation. It has been shown that in systole, arterioles of the subendocardium decrease their diameter by about 20%, whereas arterioles of the subepicardium decrease their diameter by about 2% (10, 30). It has also been shown that systolic IMP exhibits a transmural gradient oriented from endocardium toward the epicardium (16). Accordingly, NO release was found to be higher in the endocardium than in the myocardium (19). These results clearly indicate a close relationship between the synthesis of NO and mechanical stimulation of cardiac contraction in coronary vessels. In addition to endothelium-dependent mechanisms, an endothelium-independent dilation induced by pulsatile pressure was found in isolated pig coronary resistance vessels (7). The exact mechanism of the dilation was not ascertained, but a decrease in intracellular calcium was clearly not involved in the response (24). Thus both endothelium-dependent and -independent mechanisms are involved in the mechanical stimulation-induced modulation of coronary vessel function. The contribution of the present study is the demonstration of the existence of a cardiac compression-dependent, endothelial deformation-induced release of NO in the regulation of coronary flow.
It has been well known that during myocardial contraction, the diameter of coronary vessels and velocity of blood flow are altered (3, 30), causing a change of shear stress in these vessels, leading to vasodilator responses. Some previous studies have reported that pulsatile flow enhances flow-induced dilation and NO production in the coronary circulation (2, 21). Also, an elevated blood velocity and level of shear stress have been observed in vivo in the coronary circulation of dogs administered vasoconstrictor agents, followed by increases in NO production (25). On the other hand, vascular deformation-induced by myocardial contraction may change the sensitivity of coronary vessels to shear stress, as indicated by the evidence that changes in endothelial shape alter the mechanical connections between transmembrane extracellular matrix receptors, integrins, cytoskeletal filaments, and nuclear scaffolds (6, 15), all of which have been implicated in the mechanical force-induced upregulation of eNOS activity (27, 28, 31). Thus it is important to distinguish vascular deformation-dependent increases in coronary flow from those induced by decreasing pressure or by increasing flow/shear stress. Cardiac compression reduces transmural pressure of coronary vessels, which may cause a myogenically dependent decrease in vascular tone. On the other hand, release of the compression will cause a rapid resumption of coronary flow, which may induce a shear stress-dependent dilation. Two control experiments were performed. First, by turning off the coronary inflow stopcock, coronary flow and transmural pressure of coronary vessels decreased without external compression and inward movement of the ventricular wall as that employed to increase chamber pressure. Then, by turning on the stopcock, resumption of coronary flow and transmural pressure would be similar to that induced by an increase in chamber pressure. Second, a 10-s occlusion of coronary outflow resulted in a decrease in coronary flow without a decrease in transmural pressure and generation of external compression. After release of the occlusion, a similar increase in coronary flow was induced. We found that in either case, a much smaller increase in coronary flow ensued than the one induced by the release of extracardiac compression (Figs. 3 and 4), suggesting that cardiac compression-induced increases in coronary flow, although partially contributed to by flow/shear stress-induced dilation and/or a reduction in myogenic tone, are mainly mediated by a vascular deformation-dependent mechanism. Moreover, the results showing that the increases in coronary flow were sensitive to l-NAME also suggest that the mechanism involved is dependent on endothelial deformation-induced release of NO.
Release of an occlusion of large coronary vessels results in reactive hyperemia (11, 23). The mechanisms of this response involve NO-dependent, myogenic and metabolic dilations of coronary vessels. In the present study, the hearts underwent extracardiac compression caused by an increase in hydraulic pressure in the compression chamber. In this condition, while large coronary vessels on the surface of the heart would be compressed or even occluded, myocardial microvessels would be compressed as well. The control experiments, in which coronary perfusion was stopped by turning off the inflow stopcock, indicate that there is indeed a reactive hyperemic response in these perfused hearts. However, the response was smaller and mediated mainly by NO-dependent mechanisms, contrasted with that obtained in isolated beating hearts and conscious animals (11, 23). These differences are most likely due to the fact that nonbeating hearts, the metabolism of which is low and relatively constant, were used in the present study. In addition, previous studies have demonstrated that crystalloid perfusion and hyperkalemic cardioplegia, which depolarize the membrane of vascular cells, compromise coronary vessel function (9, 29) and may also contribute to the reduced reactive hyperemia in response to the occlusions. In contrast, when the whole heart was compressed, a significantly greater increase in the integrated flow was observed, indicating that the mechanism involved is different from the reactive hyperemia induced by the release of occlusions of large vessels and rather is a response dependent upon the deformation of myocardial microvessels.
To further explore the nature of these responses, we performed separate experiments on isolated coronary arterioles to assess vasodilator responses as a consequence of releasing extravascular compression. Vessels were perfused with MOPS-PSS at 80 mmHg of intravascular pressure to achieve spontaneous tone. We found that after extravascular compression was released, the vessels dilated in a compression duration-dependent, as well as l-NAME-sensitive, manner (Fig. 7). Because the experiments were conducted in a no-flow condition, and also because the volume of intraluminal fluid was small and the shear stress generated by expelling or refilling the vessel was insignificant, compression-induced dilation can be considered to be independent of stimulation by shear stress. Moreover, that inhibition of NO synthesis did not significantly affect the basal diameter of these isolated vessels and that the dilation was sensitive to l-NAME also suggest that the response was not myogenically generated, because NO does not participate significantly in the generation of myogenic responses in the coronary circulation (12). Thus the data shown in these experiments suggest that endothelial deformation induced by vascular compression enhances NO production and causes vasodilatation.
In the previous study, we demonstrated that endothelial deformation induced by vascular compression releases NO and regulates arteriolar tone (26). In the present study, we further demonstrated that in nonbeating hearts, cardiac compression elicited significantly greater phosphorylation of eNOS than that elicited solely by shear stress (Fig. 6). These data, in concurrence with compression-induced increases in coronary flow and nitrite production, provide molecular evidence that couples cardiac compression with the activation of eNOS. Similar to shear stress-stimulated activation of eNOS, cardiac compression induces phosphorylation of eNOS at Ser1177 or Ser1179 positions, indicating a protein kinase-dependent response (22), although direct evidence as to which specific upstream effectors, such as PKC, PKA, or PKB/Akt, or eNOS, are involved is still unavailable. Do shear stress and compression or deformation of endothelial cells interact with each other? Or do they activate two distinct signaling cascades? On the basis of their synergistic effects on the increase in coronary flow (Figs. 2B and 3) and eNOS phosphorylation (Fig. 6), it is likely that each factor activates eNOS in a distinct fashion. Cardiac synthesis of NO is regulated on a beat-to-beat basis in response to ventricular contraction or applied mechanical force (19). There are two phases of increase in NO release during each cardiac cycle: a slow, less potent one in systole and a burst and more potent increase in diastole. Although the specific stimulus responsible for the release of NO was not clarified in the study, we suggest that this dual-phase phenomenon observed in the course of the cardiac cycle could be explained, at least in part, as the consequence of cardiac compression (first phase) and additional shear stress-dependent responses (second phase). Thus by combining our previous findings with the present results, we posit that in addition to shear stress-dependent NO release, cardiac contraction simultaneously triggers a different signal transduction pathway of NO production, involving coronary vascular compression followed by endothelial deformation and activation of eNOS. Regarding the signal transduction pathways responsible for the cardiac contraction-dependent release of NO, they may involve changes in Ca2+ concentration in coronary endothelial cells, endothelial potassium channels, cytoskeleton, caveolae, and free radical species. Different mechanisms in the regulation of responses to a reduction in vessel diameter have already been proposed, although whether these mechanisms contribute to the cardiac contraction-dependent regulation of coronary flow is still an open question. For instance, cell-to-cell communication between smooth muscle and endothelium has been suggested to be essential for the release of endothelial NO elicited by agonist-induced vessel constriction (4), and compression-induced cutaneous vasodilation has been shown to be mediated by an axon reflex (5). It is tempting to speculate that multiple and functionally redundant mechanisms influence the regulation of coronary vessel diameter and consequent coronary perfusion in vivo during cardiac contraction. This conclusion may well form the basis of a significantly greater enhancement of eNOS phosphorylation (Fig. 6) as well as production of nitrite (Fig. 5) in beating than resting hearts that were subjected to extracardiac compression or changes in shear stress.
In conclusion, cardiac compression elicited a NO-mediated increase in coronary flow, which is independent of shear stress- or myogenically mediated mechanisms. Also, an enhanced phosphorylation of eNOS, as a function of cardiac compression, reveals the linkage, at the molecular level, that couples cardiac contraction, as a possible physiological stimulus in vivo, to the activation of eNOS. These novel findings may have pathophysiological significance in that, for example, heart failure, characterized by a compromised cardiac contractility due to a reduced compressive stimulation of coronary endothelium, may result in a reduction of NO release and an impairment in the regulation of the coronary microcirculation, leading to increased oxygen consumption and deterioration of function.
Acknowledgments
Grants This study was supported by National Heart, Lung, and Blood Institute Grants HL-43023, HL-68813, and HL-070653.
References
- 1.Busse R, Fleming I. Pulsatile stretch and shear stress: physical stimuli determining the production of endothelium-derived relaxing factors. J Vasc Res. 1998;35:73–84. doi: 10.1159/000025568. [DOI] [PubMed] [Google Scholar]
- 2.Canty JM, Jr, Schwartz JS. Nitric oxide mediates flow-dependent epicardial coronary vasodilation to changes in pulse frequency but not mean flow in conscious dogs. Circulation. 1994;89:375–384. doi: 10.1161/01.cir.89.1.375. [DOI] [PubMed] [Google Scholar]
- 3.Chilian WM, Marcus ML. Phasic coronary blood flow velocity in intramural and epicardial coronary arteries. Circ Res. 1982;50:775–781. doi: 10.1161/01.res.50.6.775. [DOI] [PubMed] [Google Scholar]
- 4.Dora KA, Doyle MP, Duling BR. Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. Proc Natl Acad Sci USA. 1997;94:6529–6534. doi: 10.1073/pnas.94.12.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fromy B, Abraham P, Saumet JL. Non-nociceptive capsaicin-sensitive nerve terminal stimulation allows for an original vasodilatory reflex in the human skin. Brain Res. 1998;811:166–168. doi: 10.1016/s0006-8993(98)00973-1. [DOI] [PubMed] [Google Scholar]
- 6.Girard PR, Nerem RM. Shear stress modulates endothelial cell morphology and F-actin organization through the regulation of focal adhesion-associated proteins. J Cell Physiol. 1995;163:179–193. doi: 10.1002/jcp.1041630121. [DOI] [PubMed] [Google Scholar]
- 7.Goto M, VanBavel E, Giezeman MJ, Spaan JA. Vasodilatory effect of pulsatile pressure on coronary resistance vessels. Circ Res. 1996;79:1039–1045. doi: 10.1161/01.res.79.5.1039. [DOI] [PubMed] [Google Scholar]
- 8.Guyton AC, Ross JM, Carrier O, Walker JR. Evidence for tissue oxygen demend as the major causing autoregulation. Circ Res. 1964;(I):14–15. I-60–I-68. [PubMed] [Google Scholar]
- 9.He GW, Ge ZD, Yim AP, Yang Q, Zhang RZ. Electrophysiologic and mechanical evidence of superiority of hyperpolarizing versus depolarizing cardioplegia in protection of endothelium-derived hyperpolarizing factor-mediated endothelial function: a study in coronary resistance arteries. J Thorac Cardiovasc Surg. 2004;127:1773–1780. doi: 10.1016/j.jtcvs.2003.09.051. [DOI] [PubMed] [Google Scholar]
- 10.Hiramatsu O, Goto M, Yada T, Kimura A, Chiba Y, Tachibana H, Ogasawara Y, Tsujioka K, Kajiya F. In vivo observations of the intramural arterioles and venules in beating canine hearts. J Physiol. 1998;509:619–628. doi: 10.1111/j.1469-7793.1998.619bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kostic MM, Schrader J. Role of nitric oxide in reactive hyperemia of the guinea pig heart. Circ Res. 1992;70:208–212. doi: 10.1161/01.res.70.1.208. [DOI] [PubMed] [Google Scholar]
- 12.Kuo L, Chilian WM, Davis MJ. Coronary arteriolar myogenic response is independent of endothelium. Circ Res. 1990;66:860–866. doi: 10.1161/01.res.66.3.860. [DOI] [PubMed] [Google Scholar]
- 13.Kuo L, Davis MJ, Chilian WM. Endothelium-dependent, flow-induced dilation of isolated coronary arterioles. Am J Physiol Heart Circ Physiol. 1990;259:H1063–H1070. doi: 10.1152/ajpheart.1990.259.4.H1063. [DOI] [PubMed] [Google Scholar]
- 14.Lamontagne D, Pohl U, Busse R. Mechanical deformation of vessel wall and shear stress determine the basal release of endothelium-derived relaxing factor in the intact rabbit coronary vascular bed. Circ Res. 1992;70:123–130. doi: 10.1161/01.res.70.1.123. [DOI] [PubMed] [Google Scholar]
- 15.Maniotis AJ, Chen CS, Ingber DE. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc Natl Acad Sci USA. 1997;94:849–854. doi: 10.1073/pnas.94.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mihailescu LS, Abel FL. Intramyocardial pressure gradients in working and nonworking isolated cat hearts. Am J Physiol Heart Circ Physiol. 1994;266:H1233–H1241. doi: 10.1152/ajpheart.1994.266.3.H1233. [DOI] [PubMed] [Google Scholar]
- 17.Miles AM, Wink DA, Cook JC, Grisham MB. Determination of nitric oxide using fluorescence spectroscopy. Methods Enzymol. 1996;268:105–120. doi: 10.1016/s0076-6879(96)68013-6. [DOI] [PubMed] [Google Scholar]
- 18.Miller FJ, Jr, Dellsperger KC, Gutterman DD. Myogenic constriction of human coronary arterioles. Am J Physiol Heart Circ Physiol. 1997;273:H257–H264. doi: 10.1152/ajpheart.1997.273.1.H257. [DOI] [PubMed] [Google Scholar]
- 19.Pinsky DJ, Patton S, Mesaros S, Brovkovych V, Kubaszewski E, Grunfeld S, Malinski T. Mechanical transduction of nitric oxide synthesis in the beating heart. Circ Res. 1997;81:372–379. doi: 10.1161/01.res.81.3.372. [DOI] [PubMed] [Google Scholar]
- 20.Rajagopalan S, Dube S, Canty JM., Jr Regulation of coronary diameter by myogenic mechanisms in arterial microvessels greater than 100 μm in diameter. Am J Physiol Heart Circ Physiol. 1995;268:H788–H793. doi: 10.1152/ajpheart.1995.268.2.H788. [DOI] [PubMed] [Google Scholar]
- 21.Recchia FA, Senzaki H, Saeki A, Byrne BJ, Kass DA. Pulse pressure-related changes in coronary flow in vivo are modulated by nitric oxide and adenosine. Circ Res. 1996;79:849–856. doi: 10.1161/01.res.79.4.849. [DOI] [PubMed] [Google Scholar]
- 22.Shaul PW. Regulation of endothelial nitric oxide synthase: location, location, location. Annu Rev Physiol. 2002;64:749–774. doi: 10.1146/annurev.physiol.64.081501.155952. [DOI] [PubMed] [Google Scholar]
- 23.Smith TP, Jr, Canty JM., Jr Modulation of coronary autoregulatory responses by nitric oxide. Evidence for flow-dependent resistance adjustments in conscious dogs. Circ Res. 1993;73:232–240. doi: 10.1161/01.res.73.2.232. [DOI] [PubMed] [Google Scholar]
- 24.Sorop O, Spaan JA, VanBavel E. Pulsation-induced dilation of subendocardial and subepicardial arterioles: effect on vasodilator sensitivity. Am J Physiol Heart Circ Physiol. 2002;282:H311–H319. doi: 10.1152/ajpheart.2002.282.1.H311. [DOI] [PubMed] [Google Scholar]
- 25.Stepp DW, Merkus D, Nishikawa Y, Chilian WM. Nitric oxide limits coronary vasoconstriction by a shear stress-dependent mechanism. Am J Physiol Heart Circ Physiol. 2001;281:H796–H803. doi: 10.1152/ajpheart.2001.281.2.H796. [DOI] [PubMed] [Google Scholar]
- 26.Sun D, Huang A, Recchia FA, Cui Y, Messina EJ, Koller A, Kaley G. Nitric oxide-mediated arteriolar dilation after endothelial deformation. Am J Physiol Heart Circ Physiol. 2001;280:H714–H721. doi: 10.1152/ajpheart.2001.280.2.H714. [DOI] [PubMed] [Google Scholar]
- 27.Sun D, Huang A, Sharma S, Koller A, Kaley G. Endothelial microtubule disruption blocks flow-dependent dilation of arterioles. Am J Physiol Heart Circ Physiol. 2001;280:H2087–H2093. doi: 10.1152/ajpheart.2001.280.5.H2087. [DOI] [PubMed] [Google Scholar]
- 28.Uematsu M, Ohara Y, Navas JP, Nishida K, Murphy TJ, Alexander RW, Nerem RM, Harrison DG. Regulation of endothelial cell nitric oxide synthase mRNA expression by shear stress. Am J Physiol Cell Physiol. 1995;269:C1371–C1378. doi: 10.1152/ajpcell.1995.269.6.C1371. [DOI] [PubMed] [Google Scholar]
- 29.Wang SY, Stamler A, Tofukuji M, Deuson TE, Sellke FW. Effects of blood and crystalloid cardioplegia on adrenergic and myogenic vascular mechanisms. Ann Thorac Surg. 1997;63:41–49. doi: 10.1016/s0003-4975(96)00644-3. [DOI] [PubMed] [Google Scholar]
- 30.Yada T, Hiramatsu O, Kimura A, Goto M, Ogasawara Y, Tsujioka K, Yamamori S, Ohno K, Hosaka H, Kajiya F. In vivo observation of subendocardial microvessels of the beating porcine heart using a needle-probe videomicroscope with a CCD camera. Circ Res. 1993;72:939–946. doi: 10.1161/01.res.72.5.939. [DOI] [PubMed] [Google Scholar]
- 31.Ziegler T, Bouzourene K, Harrison VJ, Brunner HR, Hayoz D. Influence of oscillatory and unidirectional flow environments on the expression of endothelin and nitric oxide synthase in cultured endothelial cells. Arterioscler Thromb Vasc Biol. 1998;18:686–692. doi: 10.1161/01.atv.18.5.686. [DOI] [PubMed] [Google Scholar]