

Abstract
Background
The mechanisms underlying the association between diabetes and coronary artery disease (CAD) risk are unclear. We aimed to assess this association by studying genetic variants that have been shown to associate with type 2 diabetes (T2DM). If the association between diabetes and CAD is causal, we expected to observe an association of these variants with CAD as well.
Methods and Results
We studied all genetic variants currently known to be associated with T2DM at a genome-wide significant level (p<5*10−8) in CARDIoGRAM, a genome-wide data-set of CAD including 22,233 CAD cases and 64,762 controls. Out of the 44 published T2DM SNPs 10 were significantly associated with CAD in CARDIoGRAM (OR>1, p<0.05), more than expected by chance (p=5.0*10−5). Considering all 44 SNPs, the average CAD risk observed per individual T2DM risk allele was 1.0076 (95% confidence interval (CI), 0.9973–1.0180). Such average risk increase was significantly lower than the increase expected based on i) the published effects of the SNPs on T2DM risk and ii) the effect of T2DM on CAD risk as observed in the Framingham Heart Study, which suggested a risk of 1.067 per allele (p=7.2*10−10 vs. the observed effect). Studying two risk scores based on risk alleles of the diabetes SNPs, one score using individual level data in 9856 subjects, and the second score on average effects of reported beta-coefficients from the entire CARDIoGRAM data-set, we again observed a significant - yet smaller than expected - association with CAD.
Conclusions
Our data indicate that an association between type 2 diabetes related SNPs and CAD exists. However, the effects on CAD risk appear to be by far lower than what would be expected based on the effects of risk alleles on T2DM and the effect of T2DM on CAD in the epidemiological setting.
Keywords: genome-wide association, cardiovascular disease, coronary artery disease, SNP, type-2 diabetes
Introduction
Type 2 diabetes mellitus (T2DM) and coronary artery disease (CAD) are strongly associated conditions.(1) As a consequence, guidelines suggest that all patients with T2DM should be screened for CAD and, vice versa, all patients with CAD should be screened for T2DM.(2) Although diabetics carry a 2- and 3-fold higher probability to present with incident CAD (2;3) there is an ongoing debate whether T2DM is a causal factor. Interestingly, prevalent CAD also associated with an increased probability of incident T2DM.(4) This may be explained by the fact that even impaired glucose tolerance, or prediabetes, increases the risk of CAD resulting in clinically evident coronary events even before T2DM can be diagnosed.(5) However, the variable order in which the two diseases manifest raises doubt whether the association between T2DM and CAD is confounded rather than causal. Indeed, hypothetically T2DM and CAD could both be precipitated in parallel by common preceding factors such as a sedentary life style, an inadequate diet, obesity or others.(6) Observational studies, whether cross-sectional or longitudinal, are not able to fully exclude these and other confounders in the complex etiology of both T2DM and CAD.(7) Here we utilized genetic information to further study the commonly held view that the observed association between T2DM and CAD is due to a causal relationship.
Specifically, we tested if single nucleotide polymorphisms (SNPs) that affect T2DM risk also associate with the risk of an individual developing CAD. Such risk increase, if present, would be expected to be independent of exogenous environmental factors, since these confounders are expected to be distributed evenly in the respective genotype groups.(8) Thus, if T2DM SNPs were also found to be associated with increased CAD risk, this analysis based on genetic information would support the evidence of T2DM as a causal risk factor for CAD.
A series of large-scale genomewide association studies (GWAS) have reported that 44 SNPs are associated with T2DM in Caucasians with a genomewide level of significance (p<5×10−8).(9–16) In the present analysis, we assessed whether the respective T2DM associated risk alleles also associate with an increased risk for CAD. We used data from the CARDIoGRAM Consortium, which has meta-analyzed genomewide data from 14 studies, including 22,233 cases with CAD and 64,762 controls.(17;18) We compared the quantitative effects of T2DM SNPs on CAD risk as observed in CARDIoGRAM with those that could be expected based on the reported effects of these SNPs on diabetes risk (in the published literature) and the effect of diabetes on CAD risk in the Framingham Heart Study.
Methods
SNP selection
We systematically searched the literature including NHGRI GWAS Catalog (http://www.genome.gov/gwastudies/; access date: 09/15/2012) for SNPs with genomewide significant (p<5*10−8) associations with T2DM in Caucasians by using the terms “genomewide, GWAS, type 2 diabetes”(9–16). For loci reported to be associated with T2DM, we searched for the respective SNPs in the CARDIoGRAM database.
If the SNPs did not pass quality control in CARDIoGRAM we identified proxy-SNPs using the SNAP (SNP Annotation and Proxy Search)-tool by searching with a r2-threshold of 0.8 and a distance limit of 500 BPs.(19) The identified proxy-SNPs were then tested for association with CAD in CARDIoGRAM.
Study samples
CARDIoGRAM consortium
Details about the CARDIoGRAM Consortium have been reported elsewhere.(18) In brief, this consortium combined genomewide association data on CAD from several studies and consortia: CHARGE (Cohorts for Heart and Aging Research in Genomic Epidemiology)(20); CADomics (18); ADVANCE (Atherosclerotic Disease VAscular functioN and genetiC Epidemiology study) (21); deCODE CAD study (22); LURIC (Ludwigshafen Risk and Cardiovascular Health Study) (23)/AtheroRemo 1 and 2; MIGen (Myocardial Infarction Genetics Consortium) (24); MedStar (25); Ottawa Heart Genomics Study (OHGS) (26); PennCATH (25); the Wellcome Trust Case Control Consortium (WTCCC) (27;28); and the German Myocardial Infarction Family Studies (GerMIFS) I, II, and III (KORA).(28–30) A detailed description of probands (Cases/Controls) in the participating studies is presented in the supplementary data (Supplementary table 1).
German MI Family Study (GerMIFS) I and II and WTCCC
Individual level data from the German MI Family Studies (GerMIFS) I and II and from WTCCC (28–30), which both participated in the CARDIoGRAM Consortium, were used to generate a genetic risk score as detailed below. All CAD cases were characterized by i) premature myocardial infarction (before the age of 60 years) and ii) >1 first-degree relative with an MI/CAD before the age of 70 years (in most cases a sibling) within in the GerMIFS I and II. CAD was defined as having documented coronary bypass surgery or percutaneous coronary intervention (PCI).(27–31) All patients recruited, were also characterized as having survived with CAD for long enough to be diagnosed, recruited and studied. CAD within the WTCCC study was defined as having a validated myocardial infarction, a history of PCI, coronary bypass surgery or angina with a positive noninvasive testing before the age of 66. Conversely, the control group was defined as having no known CAD up to the age at which they were recruited and studied.
Framingham Heart Study
The Framingham Study is a large prospective cohort study of the determinants of cardiovascular disease, that includes several thousand participants, from three generations.(32–34) Pooled data from the Original Cohort and from the Offspring Cohort were used to quantify the magnitude of the association between type 2 diabetes and CAD (n=7872), as detailed below (see Expected effects of type 2 diabetes-SNPs on CAD).
Statistical methods
Observed association of SNPs and CAD in CARDIoGRAM
The statistical analysis plan for the meta-analyses of the CARDIoGRAM Consortium has been described elsewhere in detail.(18) Within each participating cohort, the associations between SNPs and CAD were tested using a logistic regression model, adjusting for age, sex and potential population stratification and assuming an additive genetic model. Based on the cohort-specific effect estimates and their standard errors, a meta-analysis was performed, using random effects or fixed effects models, as appropriate. Observed associations between T2DM-SNPs and CAD were expressed as odds ratios (ORs) and their 95% Wald confidence intervals (CI).
We assessed whether T2DM risk alleles displayed a positive association with CAD (OR>1) and determined the nominal significance of this effect. The risk allele (whether minor or major) was defined as the allele that was previously associated with a higher incidence or prevalence of T2DM.(9–16) Associations that were judged to have study-wide statistical significance for CAD, were identified after Bonferroni adjustment for multiple testing.
SNPs previously associated with a risk factor other than T2DM (e.g. lipid traits, hypertension) were considered as pleiotropic SNPs. We excluded 2 SNPs at KLF14 (rs972283) and GCKR (rs780094) from further analysis because the expected association with CAD presumed to be mediated via diabetes was smaller or similar to that which might be attributable to mediation via another risk factor (HDL cholesterol, please see below). Such assessment was based on the “expected” effects of diabetes risk (see below) and the respective risk conferred by the other risk factor. Specifically, if the SNP associated effect size on CAD risk for the non-DM risk factor was larger than 50% of the effect size hypothetically associated via diabetes, that SNP was excluded (n=2 SNPs). In order to adjust for a small remaining effect of a SNP on another trait (with a hypothetical effect size on CAD smaller than 50% of that hypothetically mediated via diabetes), we subtracted from the effect observed for that SNP on CAD risk an effect that potentially could have been caused by the other risk trait (n=3 SNPs). An example is given in the supplement. This calculation implies that respective risk traits (e.g. lipids) and diabetes affect CAD risk in an independent fashion. SNPs being not in strong linkage disequilibrium (r2 < 0.2) were treated as independent signals for each of the traits.
Also, we added calculations without any exclusions of pleiotropic SNPs nor any adjustments for other risk factors (see below).
We also studied the proportion of SNPs, which were positively (OR>1) associated with CAD. Based on the null hypothesis of no shared association with CAD it is expected that this number would be approximately 50% (i.e. the proportion of risk alleles with an OR>1 purely by chance). This hypothesis was tested using an exact binomial test.
Weighted risk score
Based on individual-level data from 4,030 CAD cases and 5,826 controls of the German MI Family studies I and II and WTCCC CAD consortium (27–30), a weighted risk score was calculated as follows: for each subject, the number of risk alleles of each SNP was counted (range 0–2), and then multiplied with its effect on expected CAD risk (see below for calculation of expected CAD effects). This product (number of risk alleles x expected betaSNP→CAD) was summed over all SNPs within each participant. Individual missing genotypes were imputed by the most frequent genotype at the respective SNP. We compared the empirical distributions of the genetic risk scores in cases, as compared to controls, and quantified the strength of association by deriving odds ratios for each quintile of the risk score, using the bottom quintile as the reference. To clarify if a linear trend of increasing odds ratios from the bottom to the top quintile can be observed, we performed a Cochran-Armitage test of trend.
The German MI Family studies I and II and the WTCCC CAD consortium offer a comprehensive opportunity to study the association between T2DM associated SNPs and CAD based on individual-level data with respect to other circumventing traditional risk factors like HDL- and LDL cholesterol or high blood pressure (please see below). Another eligibility criterium for the selected studies is the statistical power of the available datasets: 4,030 CAD cases and 5,826 controls allow detecting even small effects of SNPs on disease manifestations.
A second risk score analysis summarized data of the respective SNPs in the broader CARDIoGRAM combined cohorts. Specifically, the risk score was weighted using the average effects (reported betas) for the SNPs. These aggregate effects were derived from a study of 22,233 cases and 64,762 controls. Details of such risk score calculation are described in the supplementary data and have been reported previously.(35)
Expected effects of type 2 diabetes-SNPs on CAD
For each T2DM SNP we estimated the quantitative effect that it might have on the risk of CAD as mediated via the associated effect on diabetes. The “expected” diabetes-mediated effect of each T2DM-SNP on CAD was calculated based on 1) the mean effect sizes estimated for each SNP associated with T2DM, as previously described in the literature (9–16) and 2) the mean effect size of a prior diagnosis of T2DM on the future risk of CAD in the Framingham Heart Study. Whenever T2DM SNPs have been reported in more than one paper, we took the effect estimate from the largest (meta-) analysis. The effect of T2DM on the odds of CAD was assessed in 7,872 Framingham participants (mean age 47 year, 52% women) over a 10-year time period using a logistic regression model that adjusted for age and sex. In the Framingham Heart Study CAD was defined as one of the following conditions: MI, coronary insufficiency, angina, or coronary death before age 75, consistent with previous publications.(36)
Comparison of observed and expected SNP-effects on CAD
We aimed to compare the observed effects on CAD in CARDIoGRAM to the calculated effects based on I) the reported SNP effects on T2DM and II) the effects of T2DM on CAD derived from the Framingham Heart Study. Furthermore, we calculated the average observed and estimated effects of T2DM risk alleles on CAD across all SNPs. We tested the null hypothesis that the average observed effect of T2DM-SNPs on CAD was associated with an odds ratio greater than 1.0 using paired t-tests.
Results
Proportion of T2DM in CARDIoGRAM
For assessment of the proportion of T2DM cases in CARDIoGRAM, individual data from 8,093 CAD cases and 9,951 controls were available within the respective cohorts (17;18). We found 1,544 (19.1%) CAD cases and 789 controls (7.9%) to be affected by T2DM (p<0.05). Most risk alleles previously identified for T2DM displayed a higher prevalence in the T2DM patients studied in CARDIoGRAM as compared to CAD patients/controls without T2DM (Supplementary Table 2).
Individual SNP analysis
From the literature, we identified 48 SNPs associated with T2DM at a genomewide significant level (p<5*10−8). Two SNPs (rs4457053 (ZBED3), rs5945326 (DUSP9)) were excluded because of poor genotyping quality in CARDIoGRAM and no proxy SNP in linkage disequilibrium (r2>0.8 within 500 bp). Further two SNPs, KLF14 (rs972283) and GCKR (rs780094), have shown prominent effect sizes on both T2DM and lipid traits. Since we were not able to relate the risk of CAD conferred by these two SNPs to the two intermediate phenotypes, i.e. diabetes and HDL, we excluded both from further analysis in the main paper. An analysis including these SNPs gave similar results and is shown in the supplement. Thus, in the main paper 44 T2DM-SNPs were tested for association with CAD. Out of these, 3 SNPs (MC4R (rs12970134), CILP2 (rs10401969) and LOC64673/IRS (rs2943651)) have been reported to associate weakly with other CAD risk factors (e.g. HDL, LDL). We adjusted for these reported effects on the second risk factor other than diabetes following the criteria given in the method section.
Ten out of 44 tested T2DM SNPs were nominally significantly (p<0.05) associated with an increase of CAD in CARDIoGRAM (Figure 1). This proportion is much higher than expected based on an alpha level of 0.05 (10 of 44 = 23%, p=5.0*10−5). Only one SNP at the LOC64673/IRS1 was associated with CAD with study-wide statistical significance as determined by adjusting for multiple testing (p=3.4*10−5, the Bonferroni adjusted p value threshold was 0.0011. Overall, 29 out of 44 SNPs displayed odds ratios for CAD greater than 1, a proportion (65.9%) that is higher than would be expected only by chance (p=0.024).
Figure 1.
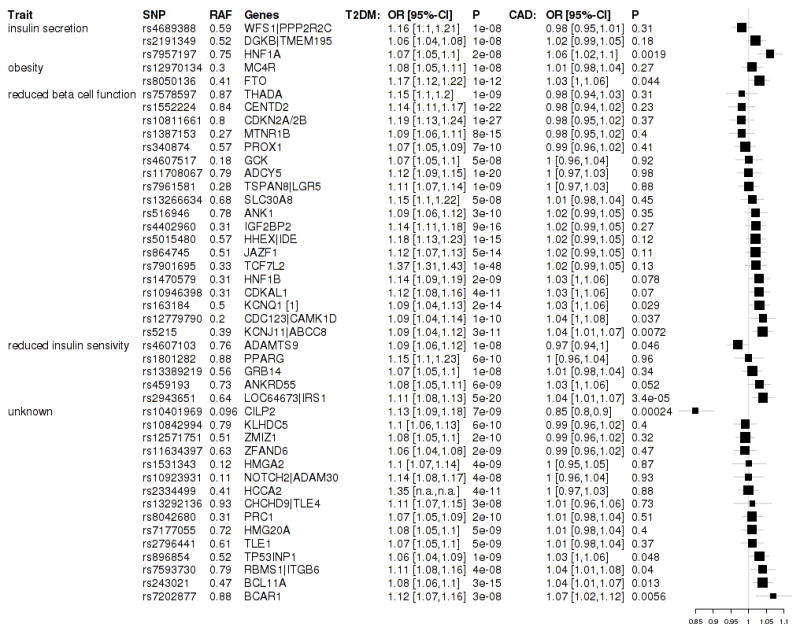
Genes and respective SNPs (rs numbers) are listed. We grouped SNPs according to the presumed pathophysiological mechanisms leading to DM.(16,38,41) RAF denotes risk allele frequency and OR T2D the change in diabetes risk associated with the T2D risk allele. p CAD displays the p-values for association of CAD within CARDIoGRAM. Forest plots display the associations of type 2 diabetes associated SNPs with CAD in CARDIoGRAM. Boxes represent the odds ratios (ORs) and whiskers 95% confidence intervals. Size of boxes proportional to inverse variance of ORs.
The average increase of CAD risk per T2DM risk allele was 1.0076 (95% confidence interval (CI), 0.9973–1.018). When we grouped the SNPs according to the pathophysiological mechanisms that are thought to lead to T2DM (Figure 1), we observed no evidence that any of these interacted with the association with CAD.
We carried out two sensitivity analyses. Firstly exclusion of all SNPs with the potential of secondary effects by risk factors other than diabetes (Supplementary Figure 1 and 2) and secondly, adjustment for all SNPs including the two that we had excluded for profound effects on other risk factors (Supplementary Figure 3 and 4). As the results were not markedly altered by these measures we felt that the principle conclusions are not affected by known pleiotropy of diabetes SNPs.
Comparison of the expected and observed risk
In the Framingham Heart Study, a prior diagnosis of T2DM conferred 80% increased odds of developing overt CAD over 10 years of assessment, as compared to individuals that were initially free of a diagnosis of T2DM at study baseline.(37)
On average, the SNPs studied here were previously reported to be associated with an increase in the risk of T2DM of 11.7% per risk allele (9–16). Based on i) this mean effect of a SNP on T2DM risk (9–16) and ii) the effect of T2DM on CAD (as determined in the Framingham Heart Study (37)), we assessed the ‘expected’ effect size on CAD for each T2DM-SNP. Such expected effect on CAD was calculated to be, on average, 1.067 per risk allele, which significantly contrasted with the effect that was observed in CARDIoGRAM (1.0076) (Figure 2). Thus, the mean observed effect of SNPs on CAD risk was significantly smaller than the mean expected effect on CAD risk (p=7.16*10−10).
Figure 2.
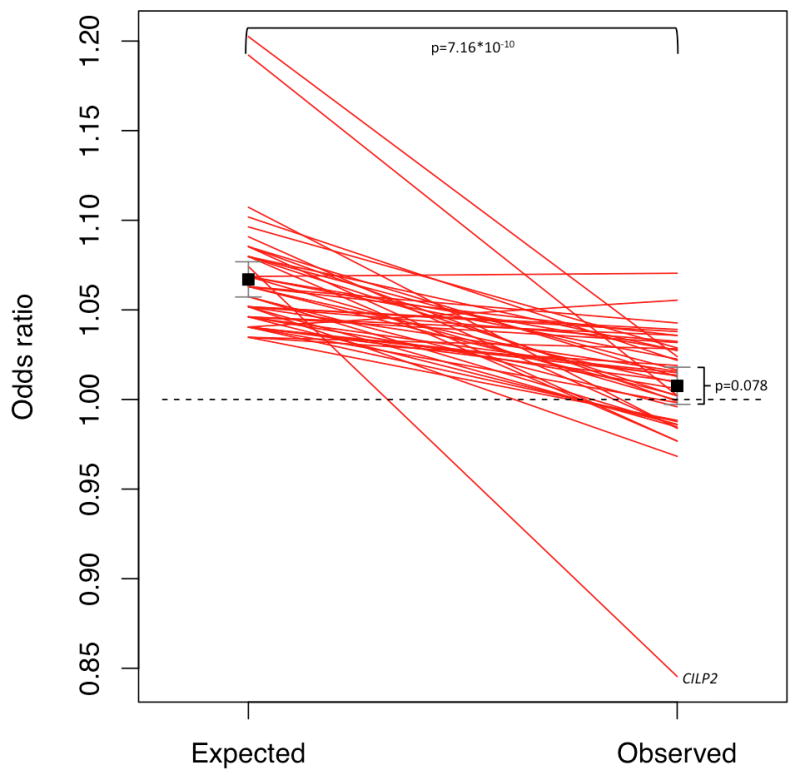
Plot displaying the expected and observed effect sizes for the association of T2DM associated SNPs with CAD. The expected effects were calculated from the effects of SNPs on T2DM (in GWAS analysis) and the impact of T2DM on CAD risk (in the Framingham Heart Study). The observed effect was taken from the CARDIoGRAM meta-analysis on CAD GWAS studies.
On top of the line chart the p-value for the difference between the expected and observed effects in CARDIoGRAM is shown.
On the right the p-value of the ORs of T2DM SNPs in the CARDIoGRAM sample is shown. After removal of SNPs with potential pleiotropic effects the respective p value observed in CARDIOGRAM was 0.001 (as shown in Supplementary Figure 2).
Risk score
In the German MI Family Studies I & II and the WTCCC study, we assessed the aggregate effect of T2DM-SNPs on CAD risk by calculating a weighted genetic risk score for each individual.
Compared to individuals with the lowest weight of risk alleles (i.e. lowest quintile of the risk score distribution), those in the highest quintile had 11% (95% CI, −2–26%) higher odds for CAD (Figure 3). Furthermore, the Cochran-Armitage trend test indicates a linear increase in risk from bottom to top quintile (p=0.04).
Figure 3.
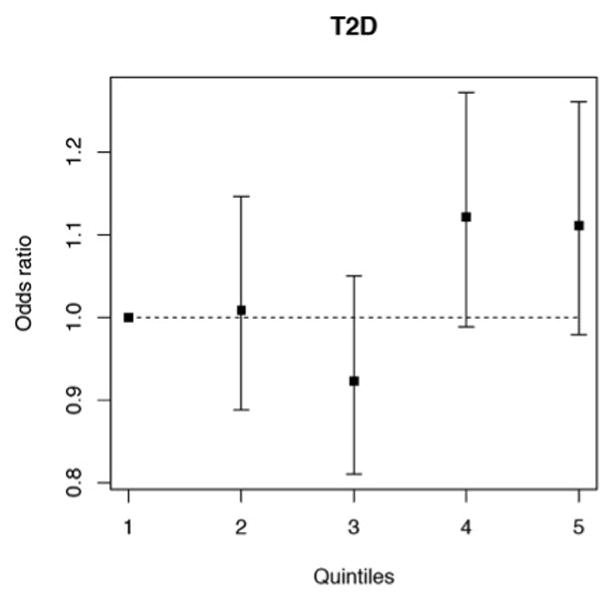
Association of a weighted genetic risk score with CAD (based on individual-level data of the German MI Family studies I and II and WTCCC CAD consortium). Odds ratios for the association with CAD risk for individuals divided into quintiles (1.–5.) by their individual weight of type 2 diabetes risk alleles, using the 1st quintile as a reference. The Cochran-Armitage trend test indicates a linear increase in risk from bottom to top quintile (p=0.04).
Moreover, we correlated the diabetes SNP score with other CAD risk factors (Supplementary Table 3) in the KORA study.(31) As a positive control, we observed a highly significant association of this score with incident diabetes. However, we found no association between the genetic diabetes risk score and other traditional risk factors like HDL cholesterol, LDL cholesterol, triglycerides, smoking and arterial hypertension. The diabetes SNP score was also significantly associated with BMI > 30, reflecting the close interrelation between diabetes and obesity, which has been underlined by a recent meta-analysis where BMI associated SNPs have been also found to increase the risk of type 2 diabetes.(4) In addition to these cross-sectional analyses, we prospectively related the diabetes SNP score to incident CAD (91 events) during 13.8 years of follow-up in 3064 individuals of the KORA study. Each 1-SD increment in the score was associated with a 17.9% increased hazards for CAD, which is similar to the association of the diabetes SNP-score and CAD risk in the CARDIoGRAM GWAS analysis. However, the association in the much smaller KORA study failed to reach statistical significance (Supplementary Table 4).
We performed a second risk score analysis based on summary statistic data from CARDIoGRAM. The respective associations with CAD in this much larger data sample showed a significant association with CAD: the p-value for this second risk score was 5.8*10−5 (OR 1.083, 95% CI, 1.042–1.126) per 1-SD increase in the genetic risk score. In order to display the results of this score we grouped the SNPs into quintiles according to their individual T2DM risk. We plotted these groups against the effects on CAD risk and observed the strongest effect on CAD risk in the group representing SNPs with an intermediate risk for T2DM (Supplementary Figure 5). In this analysis we had no clear indication of a correlation between CAD risk and the number and effect sizes of diabetes SNPs.
Discussion
Studying more than 80,000 individuals we observed that several SNPs known to associate with T2DM also increase the risk for CAD by a small margin. Indeed, of the 44 T2DM risk alleles tested about 2/3 displayed an OR for CAD>1, and 10 SNPs displayed association with CAD at a nominal significance level of 5%. Accordingly, individuals being in the top quintile of a genetic risk score based on individual number and weight of T2DM risk alleles had a slightly higher risk for CAD as compared to individuals in the bottom quintile of this score. In contrast, we found no convincing correlation between the number and weight of T2DM risk alleles and CAD risk in calculations based on summary statistic from the CARDIoGRAM consortium. Moreover, the average increase of CAD risk of diabetes risk alleles was by far smaller than expected. Thus, while T2DM SNPs appear to have some impact on CAD risk in general, we found no clear proportional risk increase depending on the published effects sizes on T2DM associated with these SNPs.
In contrast to observational studies on the association between T2DM and CAD risk or randomised, controlled interventional trials, which might also sufficiently address causality, our analysis is not based on blood glucose levels or other measures to diagnose T2DM; rather we used risk alleles that have been previously shown to increase the risk of diabetes manifestation as surrogates. The genomewide data set of CARDIoGRAM allowed us to include almost all SNPs previously identified to affect T2DM with genome-wide significance. Thereby, we were able to include nearly the full genetic information currently available for T2DM in this analysis on CAD risk. In this respect, our data extend previous molecular-genetic studies that tested association of T2DM SNPs with CAD.(10;38)
Vice versa, only a small fraction of genetic variants associated with CAD in GWAS showed a relation to traditional risk factors and none of the CAD-SNPs were associated with glycemic traits.(17;39;40)
A caveat of our approach is that the T2DM SNPs affect blood glucose levels and thus the risk of T2DM by different functional mechanisms. Thus, these SNPs may also affect a number of other (intermediate) phenotypes. We therefore grouped the T2DM-SNPs according to their putative pathways leading to T2DM (e.g. reduced beta-cell mass, beta-cell dysfunction, others).(38;41) However, we found a similar proportion of SNPs associated with CAD in each of these subgroups. This finding substantiates the notion that SNPs irrespective of the mechanisms affecting T2DM risk are responsible for the observed association with CAD and thus diabetes by itself can be considered causative. We also excluded 2 SNPs with known profound effects on other risk factors and conducted two further analyses (please see supplementary data) with and without SNPs with the potential of pleiotropic effects and found consistent results. Nevertheless, we cannot definitively rule out that residual pleiotropic effects have influenced our findings.
A limitation of our study is that the CARDIoGRAM database does not allow testing within a single sample whether the diabetes risk alleles are indeed associated with a higher prevalence of T2DM and therefore a higher risk of CAD in these patients. However, such prospectively studied cohort must involve ≫ 100.000 subjects studied for 10–20 years, given the incidence of diabetes and CAD and the SNPs currently known to affect disease risk. Also, type 2 diabetes might be underdiagnosed within our control group since the diagnosis of diabetes is often challenging and diabetes can be subclinical for a long time before presenting as a clinical burden. The genetic information employed in our study comes from large-scale meta-analyses of genomewide association studies for diabetes and CAD involving each more than 80,000 subjects. Despite a few limitations, these datasets are highly reliable in terms of associations between SNPs and disease (either diabetes or CAD). Furthermore, the evidence pointing at diabetes as a risk factor for CAD has been derived from the Framingham Heart Study, which displays probably one of the best characterized epidemiological studies. In addition, several other epidemiological studies assessed comparable risk estimates for the association of T2DM and CAD.(42–45) Based on this quality of data, we felt that the CARDIoGRAM dataset provides an adequate genotypic and phenotypic tool (with respect to coronary disease) to investigate the association between diabetes associated SNPs and CAD even if information about incident disease manifestation and control of diabetes is not available to us in this specific GWAS meta-analysis. Hence, an additional analysis (e.g. after exclusion of all patients with known diabetes) was not possible to run.
Interestingly, the effects on CAD risk that we observed for T2DM SNPs were smaller than expected given i) their increase in the probability to develop T2DM and ii) the subsequent risk of persons with T2DM to develop CAD as observed in the Framingham study. This is in contrast to SNPs primarily shown to associate with LDL cholesterol or blood pressure, which associated with CAD risk even more strongly than what was expected given their effects on respective risk factors (Supplementary Figure 6).(46;47) There are several possible explanations for this observation. First, epidemiological studies may in fact overestimate the effect of T2DM on CAD risk. In other words, insufficient adjustment of confounders, e.g. dietary and lifestyle factors that contribute to the manifestation of both diseases, may inflate the association observed between diabetes and CAD in observational studies. This point is emphasized by recent findings from clinical trials which disappointed in that aggressive blood glucose lowering failed to further decrease coronary events.(48) Second, the effect sizes of T2DM SNPs regarding the chance to develop T2DM may be in fact smaller than reported. Such overestimation (winners curse) has been found for a number of molecular-genetic associations discovered by GWAS.(49) It may also result in an overestimation of the expected effect of the diabetes SNP score on CAD risk in our mendelian randomisation approach, albeit similar studies on LDL cholesterol, triglycerides or blood pressure revealed that the observed effects were even larger than the expected effects of the SNPs on CAD risk. (Supplementary Figure 6). Hence, the effects estimated based on GWAS data might differ from those being observed in the general population.
Third, the consecutive timing of developing T2DM and CAD was not captured in our investigation on a large group of CAD patients, which was somewhat selected for early onset of the disease. Specifically, as compared to other quantitative risk factors, e.g. genetically mediated high blood pressure or LDL-cholesterol levels, T2DM often manifests in the late adulthood and only then starts to affect the risk of CAD. Such patients may be underrepresented in CARDIoGRAM. Fourth, several other factors, e.g. medication and life style changes may reduce the risk of cardiovascular events once the diagnosis of diabetes is made. Finally, it is possible that some of the T2DM SNPs display pleiotropic effects on other confounding factors and thereby reducing CAD risk. An example for this phenomenon is the CILP2 SNP, which increases the risk of T2DM but appears to decrease the risk of CAD. However, it is not conceivable that almost all SNPs display such beneficial effects on CAD risk in parallel to increasing T2DM susceptibility. Despite all these limitations it is remarkably that T2DM SNPs, i.e. the genetically mediated diabetes risk, conferred a much smaller increase in CAD risk than expected from observational epidemiological studies. It is conceivable that such mendelian randomization like approach is also suitable for other complex phenotypes (e.g. like inflammatory diseases) to elucidate the nature of association. Taken together our data suggest a moderate association of T2DM SNPs with CAD consistent with T2DM being a weak but causal risk factor for CAD. The finding that the risk increase mediated by the SNPs was much smaller than expected from epidemiological data on the risk factor diabetes mellitus is in line with the notion that both T2DM and CAD may be precipitated in parallel by yet unknown confounding risk factors. Finally, our findings add to the complex genetic foundation of CAD risk.
Supplementary Material
Acknowledgments
Supported by the EU-funded Integrated Projects Cardiogenics (LSHM-CT-2006-037593), CVgenes@target and ENGAGE as well as the BMBF-funded German National Genome Network (NGFN-Plus) Project Atherogenomics (FKZ: 01GS0831) and e:AtheroSysMed, with participation in the German Excellence Center of Cardiovascular Research (DZHK), partner site Munich Heart Alliance and the Leducq consortium CADgenomics. WL has in part been supported by the BMBF-funded (federal ministry for education and research) project GANI_MED (03IS2061A). Information regarding the CARDIoGRAM members, acknowledgements, funding sources and disclosures are detailed in the online supplement.
Source(s) of Funding: Information regarding the sources of funding for CARDIoGRAM is detailed in the online supplement.
Footnotes
Authors contributions:
HJ wrote the manuscript and researched data. CL researched data and edited the manuscript. CPN and NJS undertook analysis of the WTCCC data. WL and NJS contributed to discussion and reviewed/edited the manuscript. MJP, CPN, SK, GMP, BFV, MPR, TLA, EB, CH, RL, RP, RR, UT, AP, CC, RR, JRT, IRK, RSV, JE and NJS reviewed and edited the manuscript. HS designed the study, reviewed/edited the manuscript and is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Conflict(s) of Interest/Disclosure(s): None specific to this article. Information regarding disclosures for CARDIoGRAM is detailed in the online supplement.
References
- 1.Franco OH, Steyerberg EW, Hu FB, Mackenbach J, Nusselder W. Associations of diabetes mellitus with total life expectancy and life expectancy with and without cardiovascular disease. Arch Intern Med. 2007;11:1145–1151. doi: 10.1001/archinte.167.11.1145. [DOI] [PubMed] [Google Scholar]
- 2.Rydén L, Standl E, Bartnik M, Van den Berghe G, Betteridge J, de Boer M-J, et al. Guidelines on diabetes, pre-diabetes, and cardiovascular diseases: executive summary. The Task Force on Diabetes and Cardiovascular Diseases of the European Society of Cardiology (ESC) and of the European Association for the Study of Diabetes (EASD) Eur Heart J. 2007;1:88–136. doi: 10.1093/eurheartj/ehl260. [DOI] [PubMed] [Google Scholar]
- 3.Preis SR, Pencina MJ, Hwang S-J, D’Agostino RB, Sr, Savage PJ, et al. Trends in cardiovascular disease risk factors in individuals with and without diabetes mellitus in the Framingham Heart Study. Circulation. 2009;3:212–220. doi: 10.1161/CIRCULATIONAHA.108.846519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doerr R, Hoffmann U, Otter W, Heinemann L, Hunger-Battefeld W, Kulzer B, et al. Oral glucose tolerance test and HbAc for diagnosis of diabetes in patients undergoing coronary angiography the Silent Diabetes Study. Diabetologia. 2011;11:2923–2930. doi: 10.1007/s00125-011-2253-y. [DOI] [PubMed] [Google Scholar]
- 5.DeFronzo RA, Abdul-Ghani M. Assessment and treatment of cardiovascular risk in prediabetes: impaired glucose tolerance and impaired fasting glucose. Am J Cardiol. 2011;108:3B–24B. doi: 10.1016/j.amjcard.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 6.Laakso M, Kuusisto J. Epidemiological evidence for the association of hyperglycaemia and atherosclerotic vascular disease in non-insulin-dependent diabetes mellitus. Ann Med. 1996;5:415–418. doi: 10.3109/07853899608999101. [DOI] [PubMed] [Google Scholar]
- 7.Nathan DM, Cleary PA, Backlund J-YC, Genuth SM, Lachin JM, Orchard TJ, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;25:2643–2653. doi: 10.1056/NEJMoa052187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lawlor DA, Harbord RM, Sterne JAC, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;8:1133–1163. doi: 10.1002/sim.3034. [DOI] [PubMed] [Google Scholar]
- 9.Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;2:105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;7:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;5829:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rung J, Cauchi S, Albrechtsen A, Shen L, Rocheleau G, Cavalcanti-Proença C, et al. Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat Genet. 2009;10:1110–1115. doi: 10.1038/ng.443. [DOI] [PubMed] [Google Scholar]
- 13.Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;5:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thorleifsson G, Walters GB, Gudbjartsson DF, Steinthorsdottir V, Sulem P, Helgadottir A, et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet. 2009;1:18–24. doi: 10.1038/ng.274. [DOI] [PubMed] [Google Scholar]
- 15.Willer CJ, Speliotes EK, Loos RJF, Li S, Lindgren CM, Heid IM, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;1:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.the DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium. Morris AP, Voight BF, Teslovich TM, Ferreira T, Segre AV, Steinthorsdottir V, et al. Large scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;9:981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;4:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Preuss M, König IR, Thompson JR, Erdmann J, Absher D, Assimes TL, et al. Design of the Coronary ARtery DIsease Genome-Wide Replication And Meta-Analysis (CARDIoGRAM) Study: A Genome-wide association meta-analysis involving more than 22 000 cases and 60 000 controls. Circ Cardiovasc Genet. 2010;5:475–483. doi: 10.1161/CIRCGENETICS.109.899443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O’Donnell CJ, de Bakker PIW. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Psaty BM, O’Donnell CJ, Gudnason V, Lunetta KL, Folsom AR, Rotter JI, et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;1:73–80. doi: 10.1161/CIRCGENETICS.108.829747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Assimes TL, Knowles JW, Basu A, Iribarren C, Southwick A, Tang H, et al. Susceptibility locus for clinical and subclinical coronary artery disease at chromosome 9p21 in the multi-ethnic ADVANCE study. Hum Mol Genet. 2008;15:2320–2328. doi: 10.1093/hmg/ddn132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;5830:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 23.Winkelmann BR, März W, Boehm BO, Zotz R, Hager J, Hellstern P, et al. Rationale and design of the LURIC study--a resource for functional genomics, pharmacogenomics and long-term prognosis of cardiovascular disease. Pharmacogenomics. 2001;1:S1–73. doi: 10.1517/14622416.2.1.S1. [DOI] [PubMed] [Google Scholar]
- 24.Kathiresan S, Voight BF, Purcell S, Musunuru K, Ardissino D, Mannucci PM, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;3:334–341. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lehrke M, Millington SC, Lefterova M, Cumaranatunge RG, Szapary P, Wilensky R, et al. CXCL16 is a marker of inflammation, atherosclerosis, and acute coronary syndromes in humans. J Am Coll Cardiol. 2007;4:442–449. doi: 10.1016/j.jacc.2006.09.034. [DOI] [PubMed] [Google Scholar]
- 26.McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;5830:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;5:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Genome-wide association study of 14,000 cases of seven common diseases and 3, 000 shared controls. Nature. 2007;7145:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Erdmann J, Willenborg C, Nahrstaedt J, Preuss M, König IR, Baumert J, et al. Genome-wide association study identifies a new locus for coronary artery disease on chromosome 10p11.23. Eur Heart J. 2011;2:158–168. doi: 10.1093/eurheartj/ehq405. [DOI] [PubMed] [Google Scholar]
- 30.Erdmann J, Grosshennig A, Braund PS, König IR, Hengstenberg C, Hall AS, et al. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet. 2009;3:280–282. doi: 10.1038/ng.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wichmann H-E, Gieger C, Illig T. KORA-gen--resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen. 2005;67:S26–30. doi: 10.1055/s-2005-858226. [DOI] [PubMed] [Google Scholar]
- 32.Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families. The Framingham offspring study. Am J Epidemiol. 1979;3:281–290. doi: 10.1093/oxfordjournals.aje.a112813. [DOI] [PubMed] [Google Scholar]
- 33.Dawber TR, Kannel WB, Revotskie N, Stokes J, 3rd, Kagan A, Gordon T. Some factors associated with the development of coronary heart disease: six years’ follow-up experience in the Framingham study. Am J Public Health Nations Health. 1959;49:1349–1356. doi: 10.2105/ajph.49.10.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Splansky GL, Corey D, Yang Q, Atwood LD, Cupples LA, Benjamin EJ, et al. The Third Generation Cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: design, recruitment, and initial examination. Am J Epidemiol. 2007;11:1328–1335. doi: 10.1093/aje/kwm021. [DOI] [PubMed] [Google Scholar]
- 35.International Consortium for Blood Pressure Genome-Wide Association Studies. Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;7367:103–109. doi: 10.1038/nature10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Agostino RB, Sr, Vasan RS, Pencina MJ, Wolf PA, Cobain M, Massaro JM, et al. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation. 2008;6:743–753. doi: 10.1161/CIRCULATIONAHA.107.699579. [DOI] [PubMed] [Google Scholar]
- 37.Pencina MJ, D’Agostino RB, Sr, Larson MG, Massaro JM, Vasan RS. Predicting the 30-year risk of cardiovascular disease: the framingham heart study. Circulation. 2009;24:3078–84. doi: 10.1161/CIRCULATIONAHA.108.816694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grarup N, Sparsø T, Hansen T. Physiologic characterization of type 2 diabetes-related loci. Curr Diab Rep. 2010;6:485–497. doi: 10.1007/s11892-010-0154-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.CARDIoGRAMplusC4D Consortium. Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;1:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schunkert H, Erdmann J, Samani NJ. Genetics of myocardial infarction: a progress report. Eur Heart J. 2010;8:918–925. doi: 10.1093/eurheartj/ehq038. [DOI] [PubMed] [Google Scholar]
- 41.McCarthy MI. Genomics, type 2 diabetes, and obesity. N Engl J Med. 2010;24:2339–2350. doi: 10.1056/NEJMra0906948. [DOI] [PubMed] [Google Scholar]
- 42.Pan W, Cedres LB, Liu K, Dyer A, Schoen- berger JA, Shekelle RB, et al. Relationship of clinical diabetes and asymptomatic hyperglycemia to risk of coronary heart disease mortality in men and women. Am J Epidemiol. 1986;3:504–516. doi: 10.1093/oxfordjournals.aje.a114266. [DOI] [PubMed] [Google Scholar]
- 43.Kleinman JC, Donahue RP, Harris MI, Finucane FF, Madans JH, Brock DB. Mortality among diabetics in a national sample. Am J Epidemiol. 1988;2:389–401. doi: 10.1093/oxfordjournals.aje.a114979. [DOI] [PubMed] [Google Scholar]
- 44.Folsom AR, Szklo M, Stevens J, Liao F, Smith R, Eckfeldt JH. A prospective study of coronary heart disease in relation to fast- ing insulin, glucose, and diabetes: the Ath- erosclerosis Risk in Communities (ARIC) Study. Diabetes Care. 1997;6:935–942. doi: 10.2337/diacare.20.6.935. [DOI] [PubMed] [Google Scholar]
- 45.Fraser GE, Strahan TM, Sabaté J, Beeson WL, Kissinger D. Effects of traditional coronary risk factors on rates of incident coronary events in a low-risk population: the Adventist Health Study. Circulation. 1992;2:406–413. doi: 10.1161/01.cir.86.2.406. [DOI] [PubMed] [Google Scholar]
- 46.Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;9841:572–80. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lieb W, Jansen H, Loley C, Pencina MJ, Nelson CP, Newton-Cheh C, et al. Genetic predisposition to higher blood pressure increases coronary artery disease risk. Hypertension. 2013;5:995–1001. doi: 10.1161/HYPERTENSIONAHA.111.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riddle MC. Effects of intensive glucose lowering in the management of patients with type 2 diabetes mellitus in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Circulation. 2010;8:844–846. doi: 10.1161/CIRCULATIONAHA.110.960138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garner C. Upward bias in odds ratio estimates from genome-wide association studies. Genet Epidemiol. 2007;4:288–95. doi: 10.1002/gepi.20209. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.