

Abstract
We studied the roles of estrogen receptors (ER) and aromatase in the mediation of flow-induced dilation (FID) in isolated arteries of male ERα-knockout (ERα-KO) and wild-type (WT) mice. FID was comparable between gracilis arteries of WT and ERα-KO mice. In WT arteries, inhibition of NO and prostaglandins eliminated FID. In ERα-KO arteries, Nω-nitro-l-arginine methyl ester (l-NAME) inhibited FID by ∼26%, whereas indomethacin inhibited dilations by ∼50%. The remaining portion of the dilation was abolished by additional administration of 6-(2-proparglyoxyphenyl)hexanoic acid (PPOH) or iberiotoxin, inhibitors of epoxyeicosatrienoic acid (EET) synthesis and large-conductance potassium channels, respectively. By using an electrophysiological technique, we found that, in the presence of 10 dyne/cm2 shear stress, perfusate passing through donor vessels isolated from gracilis muscle of ERα-KO mice subjected to l-NAME and indomethacin elicited smooth muscle hyperpolarization and a dilator response of endothelium-denuded detector vessels. These responses were prevented by the presence of iberiotoxin in detector or PPOH in donor vessels. Gas chromatography-mass spectrometry (GC-MS) analysis indicated a significant increase in arterial production of EETs in ERα-KO compared with WT mice. Western blot analysis showed a significantly reduced endothelial nitric oxide synthase expression but enhanced expressions of aromatase and ERβ in ERα-KO arteries. Treatment of ERα-KO arteries with specific aromatase short-interfering RNA for 72 h, knocked down the aromatase mRNA and protein associated with elimination of EET-mediation of FID. Thus, FID in male ERα-KO arteries is maintained via an endothelium-derived hyperpolarizing factor/EET-mediated mechanism compensating for reduced NO mediation due, at least in part, to estrogen aromatized from testosterone.
Keywords: estradiol, testosterone, receptors, nitric oxide, endothelium-derived hyperpolarizing factor
The presence of steroid hormone receptors in vasculature has been recognized for some time (21) and the effect of activation of vascular estrogen receptors (ERs) on genomic and nongenomic vascular responses in both sexes has been amply investigated (6). A normal ER function is required for the cardiovascular development and function in both sexes (17, 37).
Recently, considerable attention has been directed to the issue of how cardiovascular tissues in males respond to the exposure of estrogen. A key enzyme responsible for conversion of androgen to estrogen in males is aromatase, a member of the cytochrome P-450 (CYP) superfamily of enzymes. In males, multiple tissues are involved in the aromatization of testosterone to form estradiol, including the testes, liver, muscle, skin, and adipose tissue (1), as well as vascular smooth muscle and endothelial cells (7, 30). Increasing evidence has been provided that estrogen has beneficial effects on the male cardiovascular system of humans and animals (22). Recent studies highlight the significance of aromatase-derived estrogen in the regulation of endothelial function in the male vasculature. A reduction of endogenous estrogen via aromatase inhibition significantly decreases flow-induced dilation of brachial arteries in healthy young men (19). In male aromatase-knockout (aromatase-KO) mice, which are incapable of synthesizing estrogen, acetylcholine-stimulated release of nitric oxide (NO) is significantly decreased compared with wild-type (WT) controls (18). Thus, aromatase has been defined as an estrogen-producing enzyme in vasculature (7) and has been shown to be essential for the maintenance of normal endothelial function in male vessels (33). Estrogens generated in the male vasculature initiate regulatory functions via activating ERs that are located in both smooth muscle and endothelial cells (26, 34). The role of ERα in the estrogen-dependent potentiation of NO synthesis has been well established (8, 29). Men lacking functional ERα were reported to have early coronary arterial calcification, as well as impairment of endothelial function (31, 32). Specifically, the variation in the gene for ERα, such as with the CC genotype at c.454-397CC or c.454-397T>C was demonstrated to be associated with an increased risk of myocardial infarction in men (28).
Given that estrogen is necessary for the maintenance of endothelial function in the male vasculature and that ERs are required for the mediation of vascular responses, it is legitimate to hypothesize that the release of NO from vessels in response to shear stress is reduced in ERα-KO mice. Also, it remains of interest to seek an answer to the question of whether and by what mechanisms estrogen exerts a beneficial effect on endothelial mechanotransduction to shear stress in the absence of its specific receptor, a target which has been shown to be necessary in estrogen-induced NO production. Thus, our studies were conducted on isolated and cannulated gracilis muscle and mesenteric arteries of male ERα-KO and WT mice. Shear stress-induced arterial dilations and the mediators responsible for the responses were assessed. The data obtained validate our hypothesis that an attenuated NO-mediated portion of flow-induced responses caused by ERα deficiency is compensated for by endothelium-derived hyperpolarizing factor(s) (EDHF), identified as epoxyeicosatrienoic acids (EETs), which are metabolites of arachidonic acid via CYP leading to a normal vasodilator response to shear stress.
Methods
ERα-KO and WT (C57BL/BNT) 12-14 wk-old male mice were purchased from Taconic Farms. All protocols were approved by the Institutional Animal Care and Use Committee of New York Medical College and conform to the current National Institutes of Health and American Physiological Society guidelines for the care and use of laboratory animals.
Isolated Arteries
Mice were killed by inhalation of 100% CO2. Experiments were conducted on arteries isolated from either gracilis muscle or mesentery because of the identical features of their flow-induced dilations, as well as the mediators responsible for the responses (10, 11, 13, 14, 36). The first-order gracilis muscle arteries were isolated for experiments of flow-induced dilation. In experiments of membrane potential measurement, donor vessels were isolated from first-order gracilis muscle arteries of ERα-KO and WT mice. Second-order mesenteric arteries of the corresponding animals were used as detector vessels. The endothelium of the detector vessel was removed by injection of air into the vessel lumen as described previously (10, 11). An RNA interference study was performed on second-order mesenteric arteries of ERα-KO mice because they have adequate length with fewer branches. Also, a sufficient number of vessels can be obtained from the mesentery for RT-PCR and Western blot analysis after transfection with specific short-interfering RNA (siRNA).
Flow/Shear Stress-Induced Dilation
Changes in diameter of arteries in response to increases in perfusate flow (from 0 to 10 μl/min in 2 μl/min steps) were studied at 80 mmHg of perfusion pressure.
First, roles of NO and prostaglandins (PGs) in flow-induced dilation were assessed by exposure of vessels to Nω-nitro-l-arginine methyl ester (l-NAME; 3 × 10−4M) and indomethacin (INDO; 10−5M), inhibitors of NO synthase (NOS) and cyclooxygenase (COX), respectively, after control flow-diameter curves were obtained.
Second, the role of metabolites of CYP/epoxygenase in the mediation of the l-NAME/INDO-resistant portion of flow-induced dilation was assessed by using 6-(2-proparglyoxyphenyl)hexanoic acid (PPOH; 5 × 10−5M), an inhibitor of CYP/epoxygenase (10) responsible for metabolizing arachidonic acid to EETs.
Finally, contribution of potassium channels to PPOH-sensitive flow-induced dilations was evaluated by administration of iberiotoxin (IBTX; 2 × 10−7M), a blocker of large-conductance Ca2+-dependent K+-channels (BKCa).
Assessment of Smooth Muscle Membrane Potential of Detector Vessels
Similar to what was described previously (11), the membrane potential of an endothelium-denuded mesenteric artery (detector vessel) in response to the perfusate that had flowed through a 10 dyne/cm2-stimulated gracilis artery (donor vessel) was recorded. Briefly, a donor vessel was cannulated in chamber A and an endothelium-denuded detector vessel was cannulated in chamber B that was connected serially by a micropipette (∼2 μl volume) to the outflow site of chamber A. Thus, when shear stress was initiated by perfusion of the donor vessel, the perfusate flowed through the detector vessel in which changes in membrane potential and diameter were simultaneously recorded. Intraluminal pressure of the two vessels was maintained at 80 mmHg. The membrane potential was recorded with an electrometer (model IE-210; Warner). Tip resistance of the glass electrode was ∼40–70 MΩ. The electrode was positioned via a micromanipulator (Narishige) and further advanced into a smooth muscle cell of the vessel with an oil hydrostatic micromanipulator (TrentWells). The output of the electrometer and the video calipers were connected to a data acquisition system (model DI-700; Dataq Instruments). A successful intracellular recording was confirmed by the criteria described previously (11).
CYP/Epoxygenase Activity in Skeletal Muscle Arteries
First-order gracilis muscle arteries including their distal branches (∼50–70 μg protein/per sample) isolated from six WT and six ERα-KO mice were incubated in the presence of NADPH (10−3M), INDO (3 × 10−5M), l-NAME (10−4M), arachidonic acid (3 × 10−5M), and dibromododecynyl-methylsulfimide (3 × 10−5M), an inhibitor of ω-hydroxylase, at 37°C for 1 h.
Purification of EETs
A mixture of EET-d8 (8,9-, 11,12-, and 14,15-EET; 4.5 ng) was added to each sample as internal standards. The samples were extracted twice and were purified by RP-HPLC. The fractions were evaporated to dryness and derivatized for gas chromatography-mass spectrometry (GC-MS) analysis.
Derivatization and mass spectrometric analyses
The method of sample derivatization and quantification by negative chemical ionization GC/MS was similar to those described previously (10, 13). The endogenous EETs were identified (ion m/z 319) by comparison of GC retention times with authentic D8-EETs (m/z 327) standards and quantitated by calculating the ratio of abundance.
RNA Interference Study
Vessel culture perfusion system
All perfusion chambers, tubing, reservoirs, and connectors were autoclaved prior to use. The perfusion system was placed in a vertical cell-culture hood (EdgeCARD, Sanford ME) to maintain a sterile environment. The system consists of four 1-ml perfusion chambers that provides an identical experimental environment for four single vessels treated with different agents. The intravascular pressure of the vessels was maintained by four separate pressure reservoirs. The height of the reservoir was precisely controlled. Intraluminal flow was generated by a linear syringe pump coupled with an in-line pressure transducer to monitor the inflow pressure. The outflow pressure (the height of the reservoir) was adjusted accordingly to maintain intravascular pressure constant. The flow rate was adjusted within the submicroliters-per-minute range. The diameter of vessels was measured by a microscope television image shearing system and recorded in a computer. The feasibility of vessel culture systems has been proven by our previous studies (12), and additionally, our preliminary studies further demonstrated constant flow-induced dilations and release of NO in vessels that had been incubated for 7 days.
RNA interference study
The efficiency and specificity for siRNA transfection in isolated vessels have been proven by our preliminary studies by using Hs/Mm-MAPK1 control (positive control) and non-silencing control siRNA labeled with Alexa Fluor 488 (negative control). After transfection of MAPK1 siRNA (5 nM) for 6 h, arterial MAPK1 mRNA was knocked down by ∼70% and by ∼80% after 48 h, whereas the gene expression in time course control vessels (transfected with nonsilencing siRNA for 48 h) was maintained. Also, a successful uptake of siRNA by endothelial cells was confirmed by transfection of vessels with Alexa Fluor 488-labeled siRNA. The RNA interference human/mouse starter kit, as well as the primers, was purchased from Qiagen.
In the present study, four second-order mesenteric arteries isolated from male ERα-KO mice were cannulated at 80 mmHg of intravascular pressure in perfusion chambers. The vessels were superfused with DMEM with 1% antibiotic antimycotic solution without serum. After a 1-h equilibration period, shear stress (10 dyne/cm2)-induced dilation was recorded at 80 mmHg perfusion pressure in the presence of l-NAME (3 × 10−4M) and INDO (10−5M). After that, two vessels were transfected with aromatase siRNA (Mm_Cyp19a1_1_HP siRNA; Qiagen). The siRNA was mixed initially with 3 μl HiPerFect transfection reagent (Qiagen) per 100 μl DMEM at room temperature for 10 min. The mixture was further diluted 1:5 with DMEM to a final concentration of 25 nM siRNA. The siRNA mixture was then administered intra- and extraluminally to the cannulated vessels at 37°C for 4 h without flow. The other two vessels were incubated with transfection reagent without siRNA for the same period of time. After that, the vessels were washed with DMEM and further incubated at 50 mmHg of intravascular pressure with a constant 2 μl/min perfusate flow and in the presence of 5 × 10−10M testosterone for 72 h. Shear stress-induced dilation (in the presence of l-NAME and INDO) was then reassessed at 80 mmHg perfusion pressure. The time course control vessels (incubated with transfection reagent without siRNA), which maintained dilations to shear stress, were then subjected to PPOH or IBTX for 45 min followed by repeating the shear stress-induced responses. The vessels were collected at the end of experiments to determine aromatase mRNA and protein by real time RT-PCR and Western blot analysis, respectively.
Quantitative Real-Time RT-PCR
Total RNA of single vessels was purified using a mini-RNA isolation kit (Zymo Research, Orange, CA). Reverse transcription was performed using 0.5 μg RNA and Superscript II (Invitrogen) as per manufacturer's instructions and was done in duplicate with 10% of the RT product used for PCR amplification in the presence of SYBR Green. Increased fluorescence was determined in real time using a Stratagene M×3000P. Aromatase primers were purchased from Qiagen (Mm_Cyp19a1_1_SG) and the expression of aromatase was normalized to GAPDH.
Western Blot Analysis
Single vessels were homogenized in 1× Laemmli buffer for 1 min, incubated in ice for 30 min, and sonicated twice in ice-cold water with 1 min each and a 5-min interval, and then boiled for 5 min. After a brief centrifugation, samples were loaded on a 10% SDS-PAGE gel and transferred to a PVDF membrane. Membranes were probed with primary antibodies of endothelial NOS (eNOS; 1:1,000; BD Transduction), ERβ (1:500, Affinity Bioreagent), or aromatase (1:1,000, BioVision) overnight at 4°C. Secondary antibodies were conjugated to horseradish peroxidase according to the Amersham ECL-Plus protocol. The exposed film was developed in a Kodak X-Omat developer. Image acquisition and density of specific bands on the film were obtained by an imaging system (Alpha Innotech). Specific bands from arterioles were normalized to GAPDH or β-actin.
Calculations and Statistics
Changes in diameter in responses to increases in flow in each vessel were normalized to its passive diameter. Statistical significance was calculated by repeated-measures ANOVA followed by Tukey/Kramer multiple-comparison test. Data are presented as means ± SE. The number of mice is represented by n. A group t-test was also used in comparison of two independent groups, such as data of GC/MS and molecular studies, etc. Significance level was taken at P < 0.05.
Results
The characteristics of isolated arteries from male WT (n = 9–12) and ERα-KO (n = 8–16) mice are summarized in Table 1. Active diameter of arteries of ERα-KO mice obtained in the presence of 80 mmHg intravascular pressure was significantly smaller than in WT mice. In the same conditions but in Ca2+-free solution, the passive diameter of arteries of each group was comparable. As a result, the basal tone expressed as a percentage of passive diameters was increased in arteries of ERα-KO compared with WT mice (P = 0.087). Inhibition of NO, prostaglandins, and EETs with l-NAME, INDO, and PPOH did not affect basal tone of either group of vessels, but inhibition of potassium channels with IBTX significantly enhanced basal tone in arteries of ERα-KO mice.
Table 1. Characteristics of isolated arteries of mice.
| Wild Type | ERα-KO | |
|---|---|---|
| Active diameter, μm | 90.2±3.5 | 82.1±2.2* |
| Passive diameter, μm | 137.1±3.0 | 131.1±2.8 |
| Basal tone (%passive diameter) in the control | 65.9±3.1 | 62.7±1.2 |
| Basal tone with l-NAME | 65.3±1.8 | 64.0±2.2 |
| Basal tone with INDO | 67.6±1.2 | 63.6±1.5 |
| Basal tone with l-NAME + INDO | 65.3±2.0 | 64.1±1.7 |
| Basal tone with l-NAME + INDO + PPOH | 63.5±1.8 | |
| Basal tone with l-NAME + INDO + IBTX | 56.0±2.4# | |
| Basal tone of detecting vessels | 70.1±1.4 | 67.0±1.3 |
| Resting membrane potential of detector vessels, mV | −26.8±3.5 | −25.3±1.7 |
Values are mean ± SE. ERα-KO, estrogen receptor-α knockout; l-NAME, Nω-nitro-l-arginine methyl ester; INDO, indomethacin; PPOH, 6-(2-propar-glyoxyphenyl)hexanoic acid.
Significant difference from WT (P = 0.03).
Significant difference from ERα-KO control (P = 0.04).
Figure 1 demonstrates the changes in diameter of arteries of ERα-KO and WT mice in response to step increases in flow in the control condition (Fig. 1A) and in the presence of different inhibitors (Fig. 1, B–D). In the control condition, the magnitude of flow-induced dilation was comparable between the two groups, but there was a parallel shift between the two curves due to a difference in basal tone. Flow-induced dilations were also assessed in the presence of inhibitors for NOS (l-NAME), COX (INDO), and CYP/epoxygenase (PPOH), respectively, to identify the mediators responsible for the responses. Similar to our previous findings in arteries of WT mice, (Fig. 1B), l-NAME or INDO alone inhibited flow-induced dilation by ∼50%. Combined administration of both inhibitors abolished the responses, indicating that NO and PGs participate equally in their mediation. In arteries of ERα-KO mice (Fig. 1C), l-NAME had a significantly lesser inhibitory effect (∼26%) on the dilation than in those of WT mice. INDO inhibited dilations by ∼50%, and the remaining portion of dilation was eliminated by an EET synthase inhibitor, PPOH (Fig. 1D), suggesting that EETs are the mediator responsible for the l-NAME/INDO resistant portion of the responses. In separate experiments (summarized in Fig. 1D), IBTX was used to evaluate the role of potassium channels in the EET-mediated flow-induced dilation of ERα-KO arteries. IBTX inhibited the dilation as did PPOH, indicating that EETs activate BKCa of smooth muscle of arteries to initiate vasodilation.
Fig. 1.

Normalized (A) and absolute (B–D) changes in diameter of gracilis muscle arteries of male ERα-knockout (ERα-KO; n = 16) and wild-type (WT; n = 12) mice in response to increases in perfusate flow in the control condition and in the presence of Nω-nitro-l-arginine methyl ester (l-NAME; 5 × 10−4M), indomethacin (INDO; 10−5M), 6-(2-proparglyoxyphenyl)hexanoic acid (PPOH; 5 × 10−5M), and IBTX (2 × 10−7M), respectively. PD, passive diameter. *Significant difference between the 2 curves. #Significant difference from other curves in C.
Figure 2 provides electrophysiological evidence for the EET-dependent hyperpolarization of vascular smooth muscle cells and vasodilation in response to shear stress by an EDHF bioassay. Changes in diameter and membrane potential of detector vessels in response to the perfusate flowing through l-NAME/INDO-treated donor vessels stimulated by shear stress were recorded to examine whether the EETs that are released from ERα-KO donor vessels would not only dilate, but also hyperpolarize detector vessels. The resting membrane potential of detector vessels of ERα-KO mice was comparable to that of WT mice (Fig. 2A and Table 1). In the presence of 10 dyne/cm2 shear stress, the perfusate passing through donor vessels elicited smooth muscle hyperpolarization of detector vessels by −12.00 ± 2.2 mV (Fig. 2B) associated with increases in diameter by 13.2 ± 1.4 μm (Fig. 2C), suggesting that the mediators released from shear stress-stimulated donor vessels hyperpolarize and dilate detector vessels. The hyperpolarization and dilation were eliminated when the donor vessels were exposed to PPOH or the detector vessels to IBTX, suggesting that it is EETs released from donor vessels that activate BKCa channels on detector vessels. However, applying the same level of shear stress to l-NAME/INDO-treated donor vessels isolated from WT mice did not significantly affect the resting membrane potential (−26.8 ± 3.5 vs. −25.0 ± 2.4 mV) or basal diameter (87.3 ± 3.4 vs. 87.0 ± 3.2 μm) of detector vessels.
Fig. 2.

A: original tracing of changes in smooth muscle membrane potential (mV) and diameter (μm) of a detector vessel (mesenteric artery) in response to the perfusate from a donor vessel (gracilis arteries) isolated from male ERα-KO mice, stimulated by 10 dyne/cm2 shear stress. B and C: summarized data of changes in smooth muscle membrane potential (B) and diameter (C) of detector vessels of ERα-KO (n = 5) and WT mice (n = 5) in the presence and absence of IBTX or PPOH. *Significant difference from WT control; #Significant difference from ERα-KO control.
Western blot analysis in Fig. 3 shows a significant reduction of eNOS expression (Fig. 3A) together with an upregulation of ERβ expression (Fig. 3C) in isolated arteries of ERα-KO compared with those of WT mice. Moreover, there is a significant increase in aromatase protein expression in vessels of ERα-KO mice (Fig. 3B). These data illustrate that both the downregulation of NO synthesis and upregulation of ERβ could be the consequence of a deficiency of ERα. This, on the other hand, potentiates vascular CYP activity via a reduced inhibitory effect of NO on CYP and an increased aromatized estradiol to initiate an ERβ-mediated signaling cascade. Indeed, biological evidence of a significantly greater production of EETs in arteries of ERα-KO than in those of WT mice by using GC-MS analysis is shown in Fig. 4, which is indicative of an upregulation of EET synthase or CYP activity. Figure 5 shows the role of aromatase in the mediation of EET-dependent, shear stress-induced dilation in ERα-KO arteries by transfection of vessels with aromatase siRNA. After inhibition of NO and prostaglandin synthesis with l-NAME and INDO, shear stress (10 dyne/cm2)-induced changes in diameter were recorded in arteries of both control and siRNA groups (day 1 in Fig. 5A). After incubation of the vessels with and without aromatase siRNA for 72 h (day 4), shear stress-induced dilation was eliminated in siRNA-treated vessels but was maintained in control vessels and, furthermore, was abolished by PPOH or IBTX, indicating that EET-mediated flow-induced dilation of male ERα-KO arteries is aromatase dependent. RT-PCR (Fig. 5B) and Western blot analyses (Fig. 5C) provide further evidence for the specific knockdown of aromatase mRNA and protein expression in siRNA-transfected vessels, whereas aromatase mRNA and protein were maintained in control vessels.
Fig. 3.
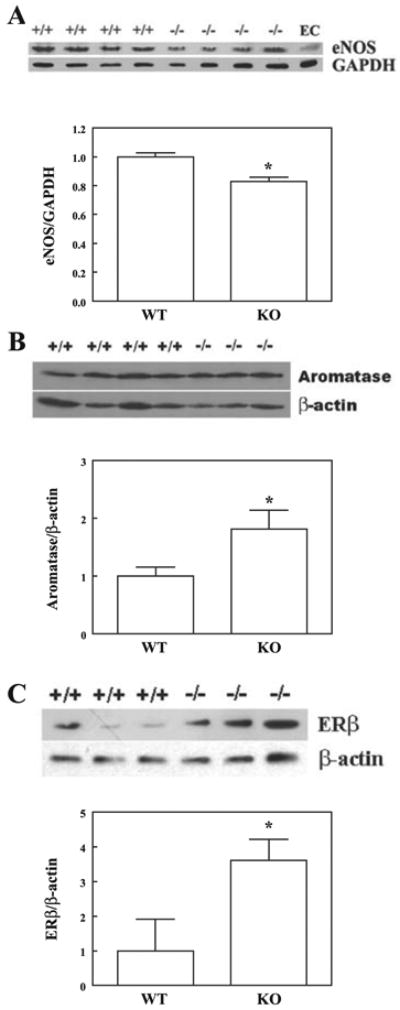
Western blot analysis of endothelial nitric oxide synthase (eNOS; A; n = 2 blots), aromatase (B; n = 2 blots), and ERβ (C; n = 2 blots) in isolated arteries of male ERα-KO and WT mice. *Significant difference from WT.
Fig. 4.

Quantitation of epoxyeicosatrieoic acids (EETs) by GC-MS analysis in gracilis muscle arteries of male ERα-KO (n = 6) and WT (n = 6) mice. *Significant difference from WT.
Fig. 5.
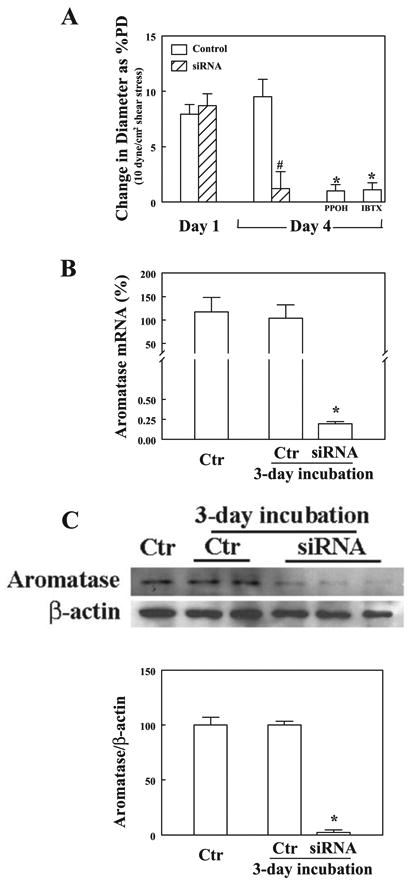
A: changes in diameter of mesenteric arteries of male ERα-KO mice in response to initial shear stress of 10 dyne/cm2 before (day 1) and after transfection with and without aromatase short-interfering RNA (siRNA; 25 nM) for 72 h (day 4), followed by treatment of control (Ctr) vessels with PPOH or IBTX (n = 6 in each group). *Significant difference from control; #significant difference from day 1. B: aromatase mRNA expression in mesenteric arteries of male ERα-KO mice before and after transfection with and without aromatase siRNA for 72 h (n = 5). *Significant difference from controls. C: original tracing and summarized data showing aromatase protein expression in mesenteric arteries of male ERα-KO mice before and after transfection with and without aromatase siRNA for 72 h (n = 2 blots). *Significant difference from controls.
Discussion
This is the first study to investigate effects of an ERα deficiency on arterial regulation of shear stress in male mice. We demonstrated that flow/shear stress-induced dilation in male ERα-KO arteries is well maintained via an EDHF/EET-mediated mechanism compensating for an impaired NO-mediated response as a function of estrogen aromatized from testosterone.
EET/EDHF Mediates Flow-Induced Dilations in Arteries of Male ERα-KO Mice
ERα is necessary for both genomic and nongenomic enhancement of NO synthesis in the vasculature of both sexes (9). This was also proven by our present findings showing that in arteries of ERα-deficient mice, the NO-mediated portion of flow-induced dilation was significantly attenuated (Fig. 1C) as a consequence of reduced eNOS expression (Fig. 3A). We also found that eNOS phosphorylation was not significantly different in vessels of ERα-KO and WT mice, as indicated by a comparable ratio of phospho-eNOS vs. eNOS in the two groups (data not shown). Indeed, in response to ERα deficiency, the vascular production of NO is significantly impaired in both humans and animals (4, 31, 32). On the other hand, a role for estrogen in the regulation of vascular endothelial function in males has also been proven since the beneficial effect of testosterone on the vasculature is dependent upon the presence of aromatase (18, 19, 22). For example, castration of male LDL receptor KO mice hastens the development of atherosclerosis compared with intact mice, while inhibition of aromatase in intact mice reverses the protective effect of testosterone to the level observed in castrated animals (24), indicating that testosterone can prevent the development of atherosclerosis by its conversion to estradiol by aromatase. Since our previous studies demonstrated the beneficial effect of estrogen on augmenting NO-mediated flow/shear stress-induced dilations in gracilis muscle arteries of mice and rats (9, 12) we originally assumed that ERα-KO arteries would have reduced flow-induced dilation due to a reduction of NO release. However, the results obtained were contrary to our expectation in that ERα-KO and WT mice have a comparable magnitude of flow-induced dilation (Fig. 1, A-C).
Subsequent studies were focused on the issue as to what is the mechanism(s) responsible for the preserved endothelial regulation of shear stress and whether estrogen evokes beneficial effects via an ERα-independent pathway in male ERα-KO arteries. We found, as observed previously (10, 36), that in arteries of WT mice, NO and PGs participate equally in the mediation of flow-induced dilation (Fig. 1B). In arteries of ERα-KO mice, however, l-NAME had a lesser effect on flow-induced dilation than in those of WT controls (∼26% vs. 50%). Additional INDO maintained its inhibitory effect (by ∼50%), leaving a portion (∼24%) of the dilation that is resistant to l-NAME and INDO (Fig. 1, C and D). By using a specific inhibitor of CYP/epoxygenase, the nature of the mediator was characterized as EETs. Also, the specific target of EETs was identified as KCa channels on smooth muscle since EDHF bioassay studies show that in the presence of 10 dyne/cm2 shear stress in donor vessels increasing hyperpolarization and vasodilation of detector vessels were elicited in response to the perfusate flowing through donor vessels that had been treated with l-NAME and INDO. This could be prevented by blocking KCa channels in detector vessels or by inhibiting EET synthesis in donor vessels (Fig. 2). Thus, it is EETs that mediate the l-NAME/INDO-resistant portion of flow-induced dilation in arteries of male ERα-KO mice. These results strongly support our conclusions that EETs are the EDHF in arteries of rats and mice (10, 11, 13, 14).
Mechanism Responsible for the Female Phenotypic Flow-Induced Responses in Male ERα-KO Mice
Our previous studies demonstrated that in NO deficiency, EDHF/EET is responsible for the mediation of endothelium-dependent, flow/shear stress-induced dilation of arteries from female but not from arteries of male mice and rats, a response that is purely estrogen- and ER-dependent (10, 11, 13, 36) and therefore has been characterized as a female phenotypic response Thus, the present study showing an EET/EDHF-mediated flow/shear stress-induced dilation in male ERα-KO mice seems to challenge our previous conclusions. With respect to the puzzling emergence of a female phenotypic flow-induced response in male mice, two issues need to be borne in mind, namely the relationship between NO and EDHF/EETs and the role of estrogen in the response.
In physiological conditions, the contribution of EDHF to the regulation of vascular tone is compromised by the presence of NO as indicated by the fact that shear stress-stimulated release of EETs from arteries is only observed when NO synthesis is absent or impaired (9-11, 13, 36), suggesting a negative correlation between NO and CYP/EDHF. Indeed, as we demonstrated in ERα-KO mice, protein expression of eNOS in arteries were significantly reduced (Fig. 3A), whereas arterial production of EETs was significantly increased (Fig. 4). These data are consistent with our functional results showing a switch from NO-mediation to the mediation by EETs (Fig. 1, C and D), and provide molecular and biochemical evidence that forms the basis of EET-mediated dilation to shear stress in compensation for a reduced ERα-related release of NO. Although CYP-mediation of flow-induced dilation in arteries/arterioles is a genomic response, as indicated by the fact that the response was prevented by transcriptional inhibitors (13), the specific CYP gene(s) responsible for the upregulation of EET synthesis in microvessels has not yet been identified. The identification of the specific gene(s) screened from CYP global genes (more than 500 genes categorized in 78 families) will require microarray analysis.
The mechanism responsible for the compensatory upregulation of EETs/EDHF-mediated dilator pathway elicited by shear stress has been demonstrated to be an estrogen-dependent genomic response, since ovariectomy eliminated and estrogen replacement restored the response (13, 14). In response to ERα deficiency, changes in ERβ receptor expression and aromatase activity needed to be addressed. Previous studies reported upregulation of ERβ during the process of vascular injury (20). In the mediation of estrogen-dependent prevention of vascular diseases, ERα and ERβ are able to complement one another (15,16), and both mRNAs are upregulated by shear stress (23). Similar to ERα, ERβ binds specifically to estrogen-responsive elements (ERE) and activates ERE-containing promoters in response to estrogen (25). We presumed therefore, that a compensatory upregulation of ERβ in response to ERα deficiency is responsible for the estrogen-dependent transcriptional upregulation of EET synthesis in male ERα-KO mice. This hypothesis is supported by findings showing significant increases in protein expression of ERβ (Fig. 3C) in ERα-KO vessels. Importantly, arterial expression of aromatase protein was also increased (Fig. 3B) in ERα-KO mice, which provides molecular evidence of increases in vascular estradiol concentrations leading to an enhanced EET synthesis (Fig. 4) in vessels of ERα-KO mice.
Regarding the specific role of estrogen in the responses, there is no significant change in circulating estradiol levels in male ERα-KO mice although there are unusually large increases in serum estradiol in female ERα-KO mice (2, 3, 27). However, there is increasing evidence that plasma estrogen levels may be a poor predictor of effective cellular estrogen concentrations, especially in the vasculature (35). In males, estrogens independent of plasma levels are produced in significant quantities by local tissues through aromatization of testosterone (5, 7). In cultured vascular smooth muscle cells that express aromatase mRNA, exogenous administration of testosterone elicited a significant production of estradiol (7). Thus, in the present study, the significantly increased concentrations of circulating testosterone in male ERα-KO mice (27) provide an extra amount of substrate to be aromatized, especially in the additional presence of an upregulated aromatase expression (Fig. 3B), to increase the local concentration of estradiol that, in turn, in a paracrine manner can further upregulate vascular CYP and EET synthesis (Fig. 4) (13). To confirm our conclusions and evaluate the specific effect of aromatase on the estrogen-dependent, EET-mediated dilator response to shear stress in vessels of male ERα-KO mice, an siRNA was used to knockdown the aromatase gene in the vessels. In our previous studies, we have confirmed that EETs are the mediator of the dilation in response to shear stress in mesenteric, as well as gracilis arteries, when NO and PGs are absent (11). As an estrogen-producing enzyme in the vasculature, aromatase expression in ERα-KO vessels was upregulated. We hypothesized, therefore, that increased vascular estrogen aromatized from testosterone is responsible for this phenotypically female response in male ERα-KO vessels. If this hypothesis is correct, the EET-mediated flow-induced dilation should be eliminated by knockdown of aromatase in the vessels. Thus, we designed an RNA interference study, aimed to confirm whether the EET-mediated flow-induced response is aromatase dependent. EET-mediated, shear stress-induced responses in these l-NAME/INDO treated-vessels were assessed before and after silencing the gene for aromatase. In the present study, a period of 72-h transfection was chosen, based on the necessity for protein degradation. We found that aromatase siRNA specifically knocked down the gene (Fig. 5B) and protein expression (Fig. 5C) in the vessels, which was associated with the elimination of EET-mediated dilator response to shear stress (Fig. 5A). On the other hand, the maintained shear stress-induced dilation in time-course control vessels was eliminated by an inhibitor of EET synthase and a BKCa channel blocker, indicating that it is EETs that mediate shear stress-induced dilation/hyperpolarization in an aromatase-dependent manner. Thus, our results suggest that the cardiovascular protective effects of estrogen are not only of physiological importance in females, but also in males, since the male cardiovascular system is an important source, as well as a target, of estrogen.
In summary, our study provides evidence for the upregulation of aromatase and ERβ expressions in vessels of male ERα-KO mice. We interpret our findings to mean that in male ERα-KO mice the high plasma testosterone and enhanced vascular activity of aromatase increase significantly the vascular concentration of estradiol, a female hormone that is necessary for EET/EDHF-mediated flow/shear stress-induced dilation of arteries, when NO synthesis is impaired. In addition, in view of the questions raised as to whether the maintenance of normal vascular tone by estrogen in males requires ERs and whether estrogen's effect on vascular tone in males is mediated by NO or some other autacoid (22), our studies, by using male ERα-KO mice, confirmed an association between estrogens and EDHF/EETs. Thus, a specific pathway is revealed by which aromatase-derived estrogen through ERβ regulates arterial endothelial function via upregulation of EET synthesis in the male vasculature to compensate for the impairment of NO synthesis.
Acknowledgments
Grants: This study was supported by National Heart, Lung, and Blood Institute Grants HL-070653, HL-68813, and HL-43023.
References
- 1.Bhatnagar AS, Muller P, Schenkel L, Trunet PF, Beh I, Schieweck K. Inhibition of estrogen biosynthesis and its consequences on gonadotrophin secretion in the male. J Steroid Biochem Mol Biol. 1992;41:437–443. doi: 10.1016/0960-0760(92)90369-t. [DOI] [PubMed] [Google Scholar]
- 2.Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 3.Curtis HS, Couse JF, Korach KS. Estrogen receptor transcription and transactivation: estrogen receptor knockout mice: what their phenotypes reveal about mechanisms of estrogen action. Breast Cancer Res. 2000;2:345–352. doi: 10.1186/bcr79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Darblade B, Pendaries C, Krust A, Dupont S, Fouque MJ, Rami J, Chambon P, Bayard F, Arnal JF. Estradiol alters nitric oxide production in the mouse aorta through the α-, but not β-, estrogen receptor. Circ Res. 2002;90:413–419. doi: 10.1161/hh0402.105096. [DOI] [PubMed] [Google Scholar]
- 5.Diano S, Horvath TL, Mor G, Register T, Adams M, Harada N, Naftolin F. Aromatase and estrogen receptor immunoreactivity in the coronary arteries of monkeys and human subjects. Menopause. 1999;6:21–28. [PubMed] [Google Scholar]
- 6.Edwards DP. Regulation of signal transduction pathways by estrogen and progesterone. Annu Rev Physiol. 2005;67:335–376. doi: 10.1146/annurev.physiol.67.040403.120151. [DOI] [PubMed] [Google Scholar]
- 7.Harada N, Sasano H, Murakami H, Ohkuma T, Nagura H, Takagi Y. Localized expression of aromatase in human vascular tissues. Circ Res. 1999;84:1285–1291. doi: 10.1161/01.res.84.11.1285. [DOI] [PubMed] [Google Scholar]
- 8.Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, Sessa WC, Bender JR. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ Res. 2000;87:677–682. doi: 10.1161/01.res.87.8.677. [DOI] [PubMed] [Google Scholar]
- 9.Huang A, Kaley G. Gender-specific regulation of cardiovascular function: estrogen as key player. Microcirculation. 2004;11:9–38. doi: 10.1080/10739680490266162. [DOI] [PubMed] [Google Scholar]
- 10.Huang A, Sun D, Carroll MA, Jiang H, Smith CJ, Connetta JA, Falck JR, Shesely EG, Koller A, Kaley G. EDHF mediates flow-induced dilation in skeletal muscle arterioles of female eNOS-KO mice. Am J Physiol Heart Circ Physiol. 2001;280:H2462–H2469. doi: 10.1152/ajpheart.2001.280.6.H2462. [DOI] [PubMed] [Google Scholar]
- 11.Huang A, Sun D, Jacobson A, Carroll MA, Falck JR, Kaley G. Epoxyeicosatrienoic acids are released to mediate shear stress-dependent hyperpolarization of arteriolar smooth muscle. Circ Res. 2005;96:376–383. doi: 10.1161/01.RES.0000155332.17783.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang A, Sun D, Koller A, Kaley G. 17β-estradiol restores endothelial nitric oxide release to shear stress in arterioles of male hypertensive rats. Circulation. 2000;101:94–100. doi: 10.1161/01.cir.101.1.94. [DOI] [PubMed] [Google Scholar]
- 13.Huang A, Sun D, Wu Z, Yan C, Carroll MA, Jiang H, Falck JR, Kaley G. Estrogen elicits cytochrome P450-mediated flow-induced dilation of arterioles in NO deficiency: role of PI3K-Akt phosphorylation in genomic regulation. Circ Res. 2004;94:245–252. doi: 10.1161/01.RES.0000111525.96232.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang A, Wu Y, Sun D, Koller A, Kaley G. Effect of estrogen on flow-induced dilation in NO deficiency: role of prostaglandins and EDHF. J Appl Physiol. 2001;91:2561–2566. doi: 10.1152/jappl.2001.91.6.2561. [DOI] [PubMed] [Google Scholar]
- 15.Iafrati MD, Karas RH, Aronovitz M, Kim S, Sullivan TR, Jr, Lubahn DB, O'Donnell TF, Jr, Korach KS, Mendelsohn ME. Estrogen inhibits the vascular injury response in estrogen receptor α-deficient mice. Nat Med. 1997;3:545–548. doi: 10.1038/nm0597-545. [DOI] [PubMed] [Google Scholar]
- 16.Karas RH, Hodgin JB, Kwoun M, Krege JH, Aronovitz M, Mackey W, Gustafsson JA, Korach KS, Smithies O, Mendelsohn ME. Estrogen inhibits the vascular injury response in estrogen receptor β-deficient female mice. Proc Natl Acad Sci USA. 1999;96:15133–15136. doi: 10.1073/pnas.96.26.15133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karas RH, Schulten H, Pare G, Aronovitz MJ, Ohlsson C, Gustafsson JA, Mendelsohn ME. Effects of estrogen on the vascular injury response in estrogen receptor α, β (double) knockout mice. Circ Res. 2001;89:534–539. doi: 10.1161/hh1801.097239. [DOI] [PubMed] [Google Scholar]
- 18.Kimura M, Sudhir K, Jones M, Simpson E, Jefferis AM, Chin-Dusting JP. Impaired acetylcholine-induced release of nitric oxide in the aorta of male aromatase-knockout mice: regulation of nitric oxide production by endogenous sex hormones in males. Circ Res. 2003;93:1267–1271. doi: 10.1161/01.RES.0000103172.98986.25. [DOI] [PubMed] [Google Scholar]
- 19.Lew R, Komesaroff P, Williams M, Dawood T, Sudhir K. Endogenous estrogens influence endothelial function in young men. Circ Res. 2003;93:1127–1133. doi: 10.1161/01.RES.0000103633.57225.BC. [DOI] [PubMed] [Google Scholar]
- 20.Lindner V, Kim SK, Karas RH, Kuiper GG, Gustafsson JA, Mendelsohn ME. Increased expression of estrogen receptor-β mRNA in male blood vessels after vascular injury. Circ Res. 1998;83:224–229. doi: 10.1161/01.res.83.2.224. [DOI] [PubMed] [Google Scholar]
- 21.McGill HC, Jr, Sheridan PJ. Nuclear uptake of sex steroid hormones in the cardiovascular system of the baboon. Circ Res. 1981;48:238–244. doi: 10.1161/01.res.48.2.238. [DOI] [PubMed] [Google Scholar]
- 22.Mendelsohn ME, Rosano GM. Hormonal regulation of normal vascular tone in males. Circ Res. 2003;93:1142–1145. doi: 10.1161/01.RES.0000108694.68635.1C. [DOI] [PubMed] [Google Scholar]
- 23.Meyer MR, Haas E, Barton M. Gender differences of cardiovascular disease: new perspectives for estrogen receptor signaling. Hypertension. 2006;47:1019–1026. doi: 10.1161/01.HYP.0000223064.62762.0b. [DOI] [PubMed] [Google Scholar]
- 24.Nathan L, Shi W, Dinh H, Mukherjee TK, Wang X, Lusis AJ, Chaudhuri G. Testosterone inhibits early atherogenesis by conversion to estradiol: critical role of aromatase. Proc Natl Acad Sci USA. 2001;98:3589–3593. doi: 10.1073/pnas.051003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pettersson K, Gustafsson JA. Role of estrogen receptor β in estrogen action. Annu Rev Physiol. 2001;63:165–192. doi: 10.1146/annurev.physiol.63.1.165. [DOI] [PubMed] [Google Scholar]
- 26.Register TC, Adams MR. Coronary artery and cultured aortic smooth muscle cells express mRNA for both the classical estrogen receptor and the newly described estrogen receptor β. J Steroid Biochem Mol Biol. 1998;64:187–191. doi: 10.1016/s0960-0760(97)00155-6. [DOI] [PubMed] [Google Scholar]
- 27.Rissman EF, Wersinger SR, Taylor JA, Lubahn DB. Estrogen receptor function as revealed by knockout studies: neuroendocrine and behavioral aspects. Horm Behav. 1997;31:232–243. doi: 10.1006/hbeh.1997.1390. [DOI] [PubMed] [Google Scholar]
- 28.Shearman AM, Cooper JA, Kotwinski PJ, Miller GJ, Humphries SE, Ardlie KG, Jordan B, Irenze K, Lunetta KL, Schuit SC, Uitterlinden AG, Pols HA, Demissie S, Cupples LA, Mendelsohn ME, Levy D, Housman DE. Estrogen receptor α gene variation is associated with risk of myocardial infarction in more than seven thousand men from five cohorts. Circ Res. 2006;98:590–592. doi: 10.1161/01.RES.0000210578.62102.a6. [DOI] [PubMed] [Google Scholar]
- 29.Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–541. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simpson ER, Clyne C, Rubin G, Boon WC, Robertson K, Britt K, Speed C, Jones M. Aromatase-a brief overview. Annu Rev Physiol. 2002;64:93–127. doi: 10.1146/annurev.physiol.64.081601.142703. [DOI] [PubMed] [Google Scholar]
- 31.Sudhir K, Chou TM, Chatterjee K, Smith EP, Williams TC, Kane JP, Malloy MJ, Korach KS, Rubanyi GM. Premature coronary artery disease associated with a disruptive mutation in the estrogen receptor gene in a man. Circulation. 1997;96:3774–3777. doi: 10.1161/01.cir.96.10.3774. [DOI] [PubMed] [Google Scholar]
- 32.Sudhir K, Chou TM, Messina LM, Hutchison SJ, Korach KS, Chatterjee K, Rubanyi GM. Endothelial dysfunction in a man with disruptive mutation in oestrogen-receptor gene. Lancet. 1997;349:1146–1147. doi: 10.1016/S0140-6736(05)63022-X. [DOI] [PubMed] [Google Scholar]
- 33.Sudhir K, Komesaroff PA. Clinical review 110: cardiovascular actions of estrogens in men. J Clin Endocrinol Metab. 1999;84:3411–3415. doi: 10.1210/jcem.84.10.5954. [DOI] [PubMed] [Google Scholar]
- 34.Venkov CD, Rankin AB, Vaughan DE. Identification of authentic estrogen receptor in cultured endothelial cells. A potential mechanism for steroid hormone regulation of endothelial function. Circulation. 1996;94:727–733. doi: 10.1161/01.cir.94.4.727. [DOI] [PubMed] [Google Scholar]
- 35.White RE. Estrogen and vascular function. Vascul Pharmacol. 2002;38:73–80. doi: 10.1016/s0306-3623(02)00129-5. [DOI] [PubMed] [Google Scholar]
- 36.Wu Y, Huang A, Sun D, Falck JR, Koller A, Kaley G. Gender-specific compensation for the lack of NO in the mediation of flow-induced arteriolar dilation. Am J Physiol Heart Circ Physiol. 2001;280:H2456–H2461. doi: 10.1152/ajpheart.2001.280.6.H2456. [DOI] [PubMed] [Google Scholar]
- 37.Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, Hodgin J, Shaul PW, Thoren P, Smithies O, Gustafsson JA, Mendelsohn ME. Abnormal vascular function and hypertension in mice deficient in estrogen receptor β. Science. 2002;295:505–508. doi: 10.1126/science.1065250. [DOI] [PubMed] [Google Scholar]