

Abstract
In 2010, a new recessive cohesinopathy disorder, designated Warsaw breakage syndrome (WABS), was described. The individual with WABS displayed microcephaly, pre- and postnatal growth retardation, and abnormal skin pigmentation. Cytogenetic analysis revealed mitomycin C (MMC)-induced chromosomal breakage; however, an additional sister chromatid cohesion defect was also observed. WABS is genetically linked to bi-allelic mutations in the ChlR1/DDX11 gene which encodes a protein of the conserved family of Iron–Sulfur (Fe–S) cluster DNA helicases. Mutations in the budding yeast ortholog of ChlR1, known as Chl1, were known to cause sister chromatid cohesion defects, indicating a conserved function of the gene. In 2012, three affected siblings were identified with similar symptoms to the original WABS case, and found to have a homozygous mutation in the conserved Fe–S domain of ChlR1, confirming the genetic linkage. Significantly, the clinically relevant mutations perturbed ChlR1 DNA unwinding activity. In addition to its genetic importance in human disease, ChlR1 is implicated in papillomavirus genome maintenance and cancer. Although its precise functions in genome homeostasis are still not well understood, ongoing molecular studies of ChlR1 suggest the helicase plays a critically important role in cellular replication and/or DNA repair.
Keywords: ChlR1, DDX11, Warsaw breakage syndrome, Cohesinopathy, Helicase, Genomic instability, Genetic disease
Clinical and cellular features of Warsaw breakage syndrome
Warsaw breakage syndrome is genetically linked to mutations in ChlR1 (DDX11) that encodes a member of the clinically relevant Iron–Sulfur (Fe–S) DNA helicase family
In 2010, Johan P. de Winter and colleagues [1] reported the first case of Warsaw breakage syndrome (WABS), an individual from Warsaw, Poland, with several congenital abnormalities that included severe pre- and postnatal growth retardation, microcephaly, facial dysmorphy, deafness, ventricular septal defects, mild intellectual disability (ID), and abnormal skin pigmentation. Mitomycin C (MMC)-induced chromosomal breakage was elevated in lymphocytes derived from the affected individual as well as EBV-immortalized B lymphoblasts, suggesting a potential diagnosis of Fanconi anemia (FA), a rare genetic disorder characterized by progressive bone marrow failure, congenital abnormalities, and cancer. However, cytogenetic analysis also showed additional chromosomal abnormalities in untreated lymphocyte cultures from the affected individual that included defective sister chromatid cohesion. The sister chromatid cohesion defects are caused by heterochromatin repulsion resulting in chromosomes with a ‘railroad’ appearance (RR), consistent with centromeric cohesion defects and total premature chromatid separation (PCS). Chromosomal breakage and cohesion defects including PCS were further elevated in WABS lymphocyte cultures that had been exposed to MMC or the topoisomerase I inhibitor camptothecin (CPT). These observed cohesion defects were similar to those typically observed in lymphoblasts from individuals with the rare genetic disorder Roberts syndrome (RBS) characterized by skeletal deformities but not FA, suggesting that the affected individual had a distinct clinical phenotype with combined FA- and RBS-like cytogenetic features. A search for candidate genes revealed that the individual with the new genetic disorder WABS possessed compound heterozygous mutations in DDX11 (MIM# 601150; NM_0306533.3), constituted by a splice mutation in intron 22 in the maternal allele (IVS22 + 2T) that resulted in deletion of 10 base pairs (bp) in exon 22 and a 3-bp in-frame deletion in exon 26 (c2689–2691 del) of the paternal allele resulting in an unstable protein (Fig. 1). The ChlR1 gene, also known as DDX11, resides on human chromosome 12p11 and encodes an ortholog of the yeast Chl1, a Superfamily 2 (SF2) ATP-dependent DEAH-box DNA helicase [2, 3]. Subsequent biochemical studies of a recombinant ChlR1 protein harboring the clinically relevant 3-bp deletion (c2689–2691 del) characterized by the loss of a single amino acid (lysine) at the C-terminus of the protein (ChlR1-K897del) demonstrated that the mutation inactivated its catalytic ATPase and helicase function [4]. In order to avoid misdiagnosis of FA and the cohesinopathies RBS and WABS, a recommendation was made that a cytogenetic analysis for spontaneous PCS and related chromosomal cohesion defects be included with tests for chromosomal breakage [5].
Fig. 1.

ChlR1 belongs to a family of sequence-related Iron–Sulfur (Fe–S) cluster DNA helicases. ChlR1 contains a conserved ATPase/helicase core domain characterized by the eight conserved helicase motifs (purple) and Fe–S cluster domain (yellow). ChlR1 belongs to a family of human DNA helicases (XPD, FANCJ, RTEL1) that are implicated in genetic diseases and cancer. The positions of the mutations genetically linked to WABS are shown above the Homo sapiens (Hs) ChlR1 depiction. Below the human Fe–S helicases are ChlR1 homologs from Saccharomyces pombe (Sp), Saccharomyces cerevisiae (Sc), Caenorhabditis elegans (Ce), and Mus musculus (Mmus), followed by the sequence-related Fe–S helicases MmusRTEL1 and CeDOG-1. The sequence length and percent similarity and identity with the ChlR1 ATPase/helicase domain for each of the Fe–S helicases is shown on the right. The BRCA1 and MLH1 interaction sites, as well as the BLM interaction domain are shown in FANCJ. The PCNA interaction motif (PIP) in ScChl1 and RTEL1, as well as RING finger domain (RING) in RTEL1 is shown
ChlR1 shares sequence homology in the ATPase motor core domain with the human SF2 DNA helicases FANCJ (MIM# 609054), ERCC2 (XPD; MIM# 126340), and RTEL1 (MIM# 608833) (Fig. 1). A peculiar structural feature of the family of DNA helicases to which ChlR1 belongs is the presence of a conserved Iron–Sulfur (Fe–S) motif between helicase motifs IA and II [6]. Structural and biochemical studies of Fe–S DNA helicases have helped to determine the functional importance of the Fe–S motif for DNA binding and interaction of the helicase with the junction of single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA), critical for its mechanism of catalytic disruption of the DNA double helix to unwind the complementary Watson–Crick strands (for review, see [7]). The genetic linkage of several chromosomal instability disorders with distinct clinical symptoms to other genes of the Fe–S cluster helicase family emphasizes the clinical importance of the Fe–S DNA helicases (Table 1). Bi-allelic mutations in XPD are responsible for Xeroderma pigmentosum (XP), XP combined with Cockayne syndrome (CS), trichothiodystrophy (TTD), or cerebro-oculo-facial-skeletal (COFS) syndrome [8]. XPD is implicated in nucleotide excision repair (NER) and initiation of RNA polymerase II transcription [9]. Bi-allelic FANCJ mutations are linked to FA Complementation Group J [10–12], and mono-allelic FANCJ mutations predispose individuals to breast or ovarian cancer [13–15]. FANCJ interacts with the tumor suppressor BRCA1 [13] (Fig. 1) and plays a role in the replication stress response [16, 17] and interstrand cross-link (ICL) repair, the latter mediated by its interaction with the mismatch repair factor MLH1 [18] (Fig. 1). Bi-allelic RTEL1 mutations are linked to Dyskeratosis congenita (DC) and Hoyeraal-Hreidarsson syndrome (HHS) [19–24]. RTEL1 is involved in the maintenance of telomere length, presumably by its helicase activity on unusual DNA structures associated with the chromosome end [25]. Recently, homozygous recessive mutations genetically linked to HHS with severe immunodeficiency were mapped to a highly conserved residue in the RING finger domain responsible for interaction with E2 ubiquitin-conjugating enzymes [20] (Fig. 1). In addition to the RING domain, RTEL1 [26, 27] as well as Saccharomyces cerevisiae (Sc) Chl1 [28] contain a PCNA interaction motif (PIP); however, binding of ScChl1 to PCNA via the PIP box was not confirmed experimentally. Thus, DDX11 belongs to a high profile family of Fe–S cluster DNA helicases important for the maintenance of genomic stability. Furthermore, ChlR1 has conserved its biological functions throughout evolution, as evidenced by the aberrant organismal and cellular phenotypes associated with mutations in genes encoding proteins sharing sequence homology with ChlR1 in yeast, nematode, and mouse (discussed below) (Fig. 1).
Table 1.
Clinical features of genetic disorders with mutations in sequence-related ChlR1 helicases
| Gene | Disease | Prominent clinical symptoms | Ref. |
|---|---|---|---|
| ChlR1 | WABS | Pre- and postnatal growth failure, microcephaly, deafness, facial dysmorphy, ventricular septal defects, intellectual disability | [1, 32] |
| XPD | XP | Severe photosensitivity, hyperpigmentation, skin cancer | [8] |
| XP/CS | Photosensitivity, neurological defects | ||
| TTD | Brittle hair and nail ichtyosis, postnatal growth failure, impaired sexual development | ||
| COFS | Progressive neurological degeneration, calcifications, cataracts, microcornea, optic atrophy, joint contractures, growth failure | ||
| FANCJ | FA | Congenital abnormalities, progressive bone marrow failure, cancer | [10–12] |
| Breast and ovarian cancera | [13–15] | ||
| RTEL-1 | DC | Progressive bone marrow failure, nail dysplasia, oral leukoplakia, abnormal skin pigmentation, cancer, pulmonary fibrosis, liver disease | [19–24] |
| HHS | Severe form of DC characterized by cerebellar hypoplasia, and may present with severe immunodeficiency and enteropathy | ||
| Gliomab | [92–94] |
aHeterozygotes
bPolymorphisms
In addition to the Fe–S cluster helicases, mutations in genes encoding DNA helicases outside the Fe–S cluster family are implicated in human diseases and play important roles in DNA repair and maintenance of genomic stability [29]. For example, SF2 helicases of the highly conserved RecQ family are required for genomic stability and several are implicated in genetic diseases characterized by premature aging and/or cancer. Interestingly, studies of a Recql4 mutant mouse model for Type II Rothmund–Thomson syndrome revealed that cells from the mutant mice displayed elevated frequencies of premature centromere separation and aneuploidy, suggesting a role of the RecQl4 helicase in sister chromatid cohesion [30]. In the greater scope, Skibbens et al. [31] suggest an interesting hypothesis that cohesinopathy disorders are more common than previously thought and share an underlying transcription dysregulation. It is of great interest to ascertain the importance and role of cohesion defects in cancer predisposition and developmental disorders such as RBS or Cornelia de Lange syndrome as well as helicase disorders characterized by accelerated aging.
Two years after the initial report of the genetic linkage between ChlR1 and WABS, a second paper was published in 2012 describing three affected siblings in a Lebanese consanguineous family with novel bi-allelic homozygous mutations in ChlR1 [c.788G > A (p.R263Q)] resulting in the replacement of a conserved lysine with a glutamine in the Fe–S domain [32] (Fig. 1). These individuals showed many of the symptoms associated with WABS, including ID growth retardation, microcephaly, facial dysmorphism, deafness (in two of the siblings), and cardiac malformations (in one of the siblings). The Lebanese individuals with WABS were distinguished from the Polish individual with WABS by their more severe ID and absence of skin pigmentation abnormality. Metaphase cells from MMC-treated lymphocytes of two affected siblings (that were tested) showed elevated chromosome breakage and cohesion defects (RR, PCS), consistent with the observations made for the first individual clinically diagnosed with WABS. Biochemical analysis of the ChlR1-R263Q recombinant protein showed that the mutation impaired ChlR1 helicase activity by perturbing its DNA binding and DNA-dependent ATPase activity. However, it should be mentioned that the recombinant ChlR1 protein with the R263Q mutation retained partial catalytic activity at higher protein concentrations [32], whereas the ChlR1-K897del mutation completely inactivated DNA helicase activity [4]. The ChlR1-R263Q mutation resides at the site of a highly conserved arginine residue in the Fe–S domain found in a number of SF2 helicases including FANCJ, XPD, and RTEL [33]. A mutation of the homologous arginine residue in XPD (R112H) causes the developmental disorder TTD, loss of XPD helicase activity, defective NER, and reduction in TFIIH [34, 35]. Thus, the pathogenic effects of a missense mutation of the homologous amino acid confirm the clinical and molecular relevance of the conserved Fe–S domain in ChlR1 and XPD helicases. As more mutations in helicase disorders are discovered, it will be insightful to characterize the molecular and cellular phenotypes of genetic variants in order to correlate genotype–phenotypes relationships that may influence the heterogeneity of clinical symptoms.
Cellular phenotypes of ChlR1-deficient human cells
The first study to evaluate the consequence of ChlR1 deficiency in human cells demonstrated that depletion of ChlR1 by RNA interference (RNAi) caused mitotic failure with chromosome segregation defects and abnormal sister chromatid cohesion [36], consistent with the observations reported in the clinical descriptions of WABS [1, 32]; furthermore, lymphoblasts from an individual affected by WABS were sensitive to the replication stress-inducing agents MMC or CPT as measured by growth inhibition assays [1]. Transfection of normal ChlR1 cDNA into the mutant cells corrected the chromosomal instability and sensitivity to CPT or MMC, confirming the genetic defect. Primary skin fibroblasts from the affected individual were also sensitive to MMC, but not X-rays or UV light as measured by survival assays [1]. Thus, ChlR1-deficient cells were sensitive to agents that introduce DNA damage which impedes replication but not irradiation treatments that elicit NER or repair of single-strand breaks (SSB) or DSBs induced by X-rays.
Independent studies from the Noguchi laboratory confirmed that RNAi depletion of ChlR1 in HeLa cells caused cohesion defects [37]. In more recent work, they investigated the importance of ChlR1 in replication recovery from DNA damage. It was found that ChlR1 depletion caused increased sensitivity and DNA damage accumulation in HeLa cells exposed to the DNA cross-linking agent cisplatin [38], a result that was consistent with earlier MMC survival studies [1]. It was also observed that ChlR1-depleted cells pre-treated with hydroxyurea followed by exposure to cisplatin were delayed in their ability to replicate compared to control short hairpin (sh)RNA cells, suggesting a role of ChlR1 in replication fork recovery [38]. Because the homologous recombination (HR) pathway plays a primary role downstream in the correction of ICLs, the effect of ChlR1 deficiency on the persistence of an inducible I-PpoI DSB at genomic DNA target sites was evaluated by measuring Chk2 phosphorylation, a signal for the ATM-dependent checkpoint. The percent damaged DNA in ChlR1-depleted cells was greater than control shRNA-treated cells from 2–8 h post-induction of I-PpoI relocalization to the nucleus. The observation that ChlR1-depleted cells treated with the radiomimetic agent bleomycin accumulated a greater level of 53BP1 foci, a marker of DSBs, compared to control shRNA cells also suggested a role of ChlR1 in DSB repair. Further studies are need to explain the apparent discrepancy of these findings with the earlier observation that primary skin fibroblasts from the individual with WABS were not sensitive to X-rays [1], a treatment that would be expected to induce DSBs. The biological endpoints and cell types are different in the two studies, and previous observations that DNA damage protein markers accumulate at chromatin foci in the absence of DNA damage may be relevant [39]. Accumulation of double-strand breaks in DDX11-deficient cells should be evaluated by independent assays such as a neutral comet or pulse field gel analysis.
Conserved roles of ChlR1 homologs in model genetic organisms
Saccharomyces cerevesiae and S. pombe Chl-1
Genetic analysis of the Saccharomyces cerevisiae homolog Chl1 provided the first evidence for a role in sister chromatid cohesion [3, 40]. chl1 was demonstrated to genetically interact with two genes (CTF7/ECO1 and CTF18/CHL12) that function in sister chromatid cohesion [3]. Ctf7/Eco1 acetyltransferase is responsible for modifying chromatin-bound cohesins, thereby playing an integral regulatory role in tethering sister chromatid pairs [41]. The specialized replication factor C (Ctf18/Dcc1/Ctf8) and DNA polymerase-associated Ctf4 are required to maintain sister chromatid cohesion in S. pombe cells arrested for long periods during mitosis [42]. chl1 served as a high copy suppressor of ctf18Δ spore lethality, and the expression of CHL1 was determined to be required for chromatid disjunction during meiosis II [42]. A synthetic lethal relationship was found to exist between S. pombe chl1Δ and ctf18Δ [43]. Consistent with the interaction of Chl1 with replication proteins [42, 44], expression of chl1 + rescued the sensitivity of swi1Δ, a mutant of the replication fork protection complex, to agents that induce replication stress [43]. In more recent work, the Skibbens laboratory provided experimental evidence that S. cerevisiae Ctf7/Eco1 and Chl1 are associated with FEN-1, the nuclease responsible for Okazaki fragment processing of the newly replicated lagging strand [45]. They proposed a model that temporally couples cohesion establishment to lagging strand processing. Altogether, these findings establish that Chl1 interacts with the replication machinery and suggest that the helicase is likely to act during lagging strand synthesis/maturation to insure normal sister chromatid cohesion and chromosome segregation. Very recently, Chl1 was implicated in chromatin recruitment of various factors (Scc2, cohesin) that are necessary for the establishment of sister chromatid cohesion [46, 47]. A replication-coupled deposition model was proposed in which Chl1 helicase regulates cohesion enrichment during S phase [47]. This model challenges the idea that cohesion rings are preassembled prior to passage of the replication fork. We anticipate that Chl1 (or its mammalian homolog ChlR1) unwinds DNA replication or repair intermediates (5′ flaps, D-loops, or alternate DNA structures, e.g., G-quadruplexes) during critical quality control steps to enable smooth deposition of cohesion factors onto stable genomic duplex DNA (see below).
Mus musculus ChlR1
In mice, genes encoding four Fe–S DNA helicases are known to exist: XPD, FANCJ, RTEL, and ChlR1 [6]. XPD and ChlR1 lack the extensive C-terminal region found in FANCJ and RTEL1, and only FANCJ among the Fe–S cluster DNA helicases contains the conserved BRCA1 binding domain [13]. In 2007, the Lahti laboratory created a mutant mouse harboring a directed homozygous deletion of the Walker A box (motif I) in ChlR1 responsible for ATP hydrolysis [48]. Loss of ChlR1 in mouse resulted in embryonic lethality. It was determined that the aneuploidy apparent in ChlR1−/− embryos occurred as a consequence of defective sister chromatid cohesion and placental malformation. Aneuploid cells in chlr1-null mouse embryos were characterized by partial loss of sister chromatid cohesion as well as elevated G2/M cell cycle arrest and apoptosis. More recently, a mouse N-ethyl-N-nitrosurea (ENU)-induced mutation (cetus) resulting in a nonconservative change of an amino acid in motif V of the helicase core domain in ChlR1 was generated and characterized by Cota and Garcia–Garcia [49]. Embryonic lethality and placental defects of the cetus ChlR1 mutant mouse were similar to those of the mouse from the Lahti laboratory. In addition, the latter study identified embryonic phenotypes in the cetus ChlR1 mutant mouse including loss of somatic mesoderm, morphological defects in the neural tube and heart, and extensive apoptosis from early gastrula stages [49].
Caenorhabditis elegans CHL-1
A boot-strapped phylogenetic tree analysis of human ChlR1 with other organisms revealed the existence of four iron–sulfur helicases from Caenorhabditis elegans, namely XPD, DOG-1, RTEL-1, and CHL-1 [50]. CHL-1 showed the greatest sequence identity and similarity with Chl1 in yeast and ChlR1 in humans. This led members of the Rose laboratory to investigate the genetic importance of the chl-1 gene in C. elegans. The introduction of a homozygous deletion within the helicase core domain of chl-1 resulted in sterile worms characterized by germline abnormalities, abnormal karyotypes, and reduced proliferation of D motor neurons and seam cells indicative of somatic cell loss [50]. Chung et al. [50] investigated a potential role of CHL-1 in the metabolism of G-rich tracts in the C. elegans genome, prompted by previous work showing that disruption of the sequence-related dog-1 gene in C. elegans resulted in a peculiar form of genomic instability characterized by deletions upstream of guanine-rich DNA [51]. Chromosomal instability as measured by PCR analysis to detect deletions originating from a polyguanine tract at the vab-1 locus was not observed in chl-1 mutants; however, elevated guanine tract deletions were observed in chl-1 dog-1 double mutants, leading the authors to conclude that CHL-1 preserves G-tract instability that occurs in the absence of DOG-1 [50]. However, the role of CHL-1 to help cells deal with G-tract instability in the absence of DOG-1 is not unique because a similar relationship exists between DOG-1 and proteins implicated in HR repair and translesion synthesis [52].
Worms harboring the dog-1 mutation alongside a mutation [him-1 (e879)] in a cohesin ortholog of SMC1 also displayed elevated guanine tract instability [50]. However, it remains unclear if the chromosomal instability observed in worms defective in CHL-1 and DOG-1 is attributed to a role of the helicase in resolving G-tract-associated DNA structures during replication or repair, and if the molecular basis for this defect is distinct from or related to that observed in worms that are mutated in both the SMC1 cohesin ortholog and DOG-1.
Enzymatic functions of ChlR1
DNA substrate specificity of ChlR1 helicase
Helicases use the energy of nucleoside triphosphate hydrolysis (typically ATP) to fuel the disruption of hydrogen bonds between base pairs of complementary DNA, RNA, or DNA–RNA double helical molecules [53]. They are also involved in the disruption of other forms of structured nucleic acids (e.g., G4 or four-stranded Holliday Junctions). DNA helicases are important in virtually all aspects of cellular DNA metabolism with important roles in replication fork progression, DNA repair, recombination, chromosome segregation, and telomere maintenance. They are also important in transcription or replication initiation by locally unwinding the DNA duplex near promoter regions or origins, respectively. Moreover, helicase deficiencies are linked to genetic disorders and cancer [29]. Characterizing the catalytic functions and substrate specificity of ChlR1 helicase (Table 2) will be useful for understanding its molecular mechanism(s) and potential involvement in cellular pathways of nucleic acid metabolism.
Table 2.
DNA substrate specificity of ChlR1
| DNA substrates unwound by ChlR1 | Ref. |
|---|---|
| Partial duplex with 5′ ssDNA tail | [2, 54] |
| Forked duplex with 5′ and 3′ ssDNA tails | [4] |
| 5′ flap substrate | [4] |
| Bi-molecular anti-parallel G4 | [4, 67] |
| Tetra-molecular parallel G4a | [4, 67] |
| Three-stranded D-loop | [4] |
| DNA substrates not unwound by ChlR1 | Ref. |
|---|---|
| Partial duplex with 3′ ssDNA tail | [4, 54] |
| 3′ flap | [54] |
| Synthetic replication fork | [4] |
| Blunt duplex | [4] |
| Unimolecular G4 | [67] |
| Holliday Junction | [4] |
See text for details
aRelatively low but detectable ChlR1 helicase activity
Initial studies from the Lahti [2] and Hurwitz [54] laboratories showed that recombinant human ChlR1 protein expressed and purified from insect cells or human cells, respectively, behaved like other Fe–S DNA helicases as a DNA-dependent ATPase and unwound duplex DNA with a 5′ to 3′ directionality in which a 5′ single-stranded loading tail was required for helicase loading. Although the structural or biochemical basis for the 5′ to 3′ directionality of ChlR1 helicase is unknown, analysis of the sequence-related XPD helicase has provided insight into some basic features of how the Fe–S cluster helicases achieve unwinding directionality. Kuper et al. determined the co-crystal structure of Thermoplasma acidophilium (Ta) XPD in complex with a short DNA fragment, which defined the binding polarity of the Fe–S helicase to the DNA molecule [55]. Concomitantly, the Spies laboratory biochemically analyzed the nature of the interaction of TaXPD and associated mutants with its DNA substrate, leading them to conclude that TaXPD bound single-stranded DNA (ssDNA) with a defined polarity, and that ligand-induced conformational changes in the motor domain of TaXPD determined its directionality for DNA translocation [56]. Presumably, ChlR1 would behave the same as XPD in terms of its biochemical properties and DNA substrate specificity; however, some differences among the Fe–S cluster DNA helicases exist (see below). Furthermore, the assembly state of ChlR1 is unknown, whereas biochemical studies demonstrated that the optimal assembly state of FANCJ for catalytic activity is a dimer [57]. TaXPD is believed to exist as a monomer in solution [58] and to unwind double-stranded DNA (dsDNA) as a monomer in 1-bp steps [59], whereas eukaryotic XPD is a stable component of the multi-subunit general transcription factor TF IIH complex that also contains the XPB helicase and is implicated in RNA polymerase II transcription initiation and NER [9]. In the future, it will be important to carefully examine the ChlR1 assembly state which may influence its substrate specificity or catalytic efficiency.
Our laboratory examined the DNA substrate specificity of recombinant ChlR1 expressed and purified from human cells, and determined that a minimal 5′ ssDNA tail length of ~15 nucleotides (nt) is required for the helicase to efficiently unwind adjacent duplex [4]. ChlR1 preferentially unwound a forked duplex DNA substrate flanked by noncomplementary 5′ and 3′ ssDNA arms. A 3′ ssDNA tail of just 5 nt significantly improved ChlR1 unwinding of the forked duplex substrate, and a 10-nt 3′ ssDNA tail was found to be optimal. Thus, only a minimal segment of 3′ ssDNA flanking the duplex dramatically enhances the ability of DDX11 to effectively unwind the adjacent duplex also flanked by a 5′ tail. Cellular studies and biochemical protein interactions supporting a role of ChlR1 in replication or DNA repair [54] (discussed below) led us to test ChlR1 helicase activity on model DNA substrates resembling key intermediates of these pathways. ChlR1 efficiently unwound a three-stranded 5′ flap structure which represents an intermediate of lagging strand synthesis and DNA repair pathways such as base excision repair. In contrast to its robust helicase activity on the 5′ flap structure, ChlR1 poorly unwound either a 3′ flap or synthetic replication fork with duplex leading and lagging strand arms, a finding that is consistent with its requirement for a 5′ ssDNA tail for loading. ChlR1 was also inactive on a four-stranded Holliday Junction substrate, a model DNA structure for a late intermediate of HR, but was able to unwind a D-loop substrate with an invading 3′ ssDNA considered an early HR intermediate. Although there is currently no cellular evidence for a direct role of ChlR1 in HR repair, it is conceivable that ChlR1 helicase activity on a D-loop substrate may be physiologically important. ChlR1 may act as an anti-recombinase to prevent untimely or imperfect base-pairing between DNA sequences, an activity proposed for the sequence-related RTEL helicase [25]. Alternatively, unwinding the invading strand of a D-loop by a helicase may be important for the promotion of synthesis-dependent strand annealing or capturing the end of a second DSB [60]. Another possibility is that ChlR1 may unwind a telomeric sub-structure known as a t-loop consisting of a telomeric 3′ ssDNA overhang that loops around and hybridizes to a complementary strand of the adjacent double-stranded telomeric sequence to create a D-loop. An involvement of ChlR1 in telomere metabolism has been suggested based on observations that ChlR1−/− mouse embryos displayed reduced chromatin density at their telomeres [61], and ChlR1-depleted human melanoma cells showed a significantly reduced length of their chromosomes compared to control-siRNA-transfected melanoma cells [62]. Although a direct role of ChlR1 in telomere metabolism is unknown, it is plausible that ChlR1 and other DNA helicases may help to either create DNA structures required for efficient telomere capping by shelterin proteins or smooth telomeric DNA synthesis by the replication machinery.
ChlR1 resolves a class of G-quadruplex DNA structures
In recent years, G-quadruplex (G4) DNA structures have acquired considerable interest because of their predicted abundance in the human genome and their potential impact in many areas of nucleic acid metabolism, including DNA replication, transcription, and telomere stability [63, 64]. Mounting evidence suggests that G4 DNA poses a highly relevant source of genomic instability, and that specialized DNA helicases help cells deal with these and other aberrant DNA structures by catalytically resolving them in an ATP-dependent manner. Based on previous evidence that the sequence-related C. elegans DOG-1 [51] and human FANCJ helicases [65, 66] played a role in the resolution of G-rich sequences prone to form G-quadruplexes, it was plausible that ChlR1 might have a function in G4 DNA metabolism. We determined that ChlR1 could efficiently unwind a two-stranded anti-parallel G4 substrate (G2′), and, to a significantly lesser extent, a four-stranded parallel G4 DNA substrate [4]; however, ChlR1 completely failed to unwind a unimolecular G4 DNA substrate [67]. The bi-molecular G4 substrate more efficiently unwound by ChlR1 possessed two 5′ ssDNA tails which could theoretically permit two DDX11 helicases to load onto the substrate, provided there are not steric constraints. ChlR1 bound the G2′ substrate with greater affinity compared to the four-stranded G4 substrate, which probably contributes to its substrate preference [4]. The preferential binding of ChlR1 to the two-stranded G4 substrate was confirmed by sequestration assays in which ChlR1 preincubated with G2′ was significantly more resistant to challenge by a dT45 competitor compared to the four-stranded G4 [4]. Although it is not well understood, the precise role of ChlR1 or other DNA helicases in G4 metabolism, and the growing interest in the stability, recognition, and functions of distinct G4 topologies, will fuel new research directions and discoveries.
In contrast to ChlR1, the sequence-related TaXPD helicase failed to unwind all the topologies of G4 tested (uni-, bi, tetra-molecular) whereas FANCJ helicase unwound all forms of G4 DNA including a unimolecular G-quadruplex [67]. Moreover, FANCJ-depleted U-2 OS osteosarcoma cells exposed to the G4 binding ligand telomestatin (TMS) displayed elevated γ-H2AX foci, a marker for DNA damage, whereas ChlR1-depleted U-2 OS cells did not [67]. In this same study, ChlR1 depletion caused U-2 OS cells to be sensitive to a DNA cross-linking agent, a result consistent with other studies [1, 38]. These findings suggest a specialization among Fe–S cluster helicases to resolve G4 DNA, with FANCJ having a more prominent role, consistent with its robust activity on the entropically favored unimolecular G4 substrate. Nonetheless, ChlR1 or other G4-resolving helicases (e.g., BLM [68]) may unwind G quartets that stabilize G-rich DNA junctions between strands that form during recombination between sister chromatids or during replication to produce daughter chromosomes. Alternatively, ChlR1 may unwind G4 DNA in specialized situations such as the maturation of Okazaki fragments created during lagging strand synthesis. This idea would be consistent with the interaction of ChlR1 with replication proteins including the DNA processing nuclease Flap Endonuclease 1 (FEN-1) [54] (see below). A reasonable hypothesis based on the observation that C. elegans CHL-1 preserves G-tract instability that occurs in the absence of the FANCJ homolog DOG-1 [50] is that human ChlR1 and FANCJ share partially redundant roles in G4 metabolism with FANCJ being the more prominent player. Jones and Rose proposed an interesting hypothesis to explain why DOG-1 may have dual roles in G4 DNA metabolism and resistance to DNA cross-linking agents [69]. They suggested that G-rich structures (e.g., G4) that accumulated in dog-1 mutants may be susceptible to covalent linkage upon exposure to a DNA cross-linking agent. A similar scenario may be relevant for human FANCJ and ChlR1, given that cells deficient in either of these helicases are sensitive to DNA cross-linking agents. A potentially informative experiment would be to test for a synergistic effect between low doses of a DNA cross-linking agent and a G4-interactive compound. One would hypothesize that, if G4 stabilization predisposes genomic DNA to cross-linking, then cells deficient in a G4 resolving helicase (e.g., ChlR1, FANCJ, or PIF1) would be hypersensitive to the combined exposure to a G4 binder and DNA cross-linker.
Genetic studies that explore the potential functional redundancy of ChlR1 with other relevant Fe–S helicases (FANCJ, RTEL) in model vertebrate organisms may provide a useful platform. In chicken DT40 cells, a combined genetic defect in FANCJ and FANCC resulted in an additive effect on cisplatin sensitivity, suggesting a role of FANCJ outside the classical FA pathway [70]. To explain this, Hiom proposed a hypothesis related to that of Jones and Rose [69], in which guanines nested within a G4 structure reside in close proximity and can be preferentially cross-linked to one another [71]. Therefore, FANCJ may not only operate in the FA pathway but also function in G4 metabolism to facilitate the repair of the damaged sequence. In light of these ideas, the potential importance of ChlR1 to resolve two-stranded G4 DNA for the preservation of sister chromatid cohesion or fidelity of DNA repair should be carefully examined in model genetic organisms and cell-based models. A useful tool for this purpose may be a biologically active G4 ligand that specifically binds G2′ DNA. Synthetic chemistry is a prominent area in G-quadruplex research, and it seems likely that small molecules tailored to bind G4 DNA structures with defined topologies are likely to be forthcoming [72]. Indeed, we recently determined that two structurally related bisquinolinium compounds potently inhibited FANCJ helicase activity on unimolecular G4 by 100- to 1,000-fold greater than two- or four-stranded G4 [67]. The development of antibodies (e.g., [73, 74]) or molecular beacons that specifically recognize certain G-quadruplex architectures should help to elucidate their biological significance and characterize the cellular functions of proteins that bind or resolve G4 DNA. In support of this approach, we recently demonstrated enrichment of G4 DNA in FANCJ-deficient cells using a G4-specific antibody [73]. G4-specific antibodies may be useful to investigate the potential role of ChlR1 in G4 metabolism.
Disruption of protein-DNA complexes by ChlR1
ChlR1 is also able to use its motor ATPase function to disrupt protein–DNA interactions modeled by the high affinity interaction of streptavidin homo-tetramer bound to a biotinylated single-strand DNA oligonucleotide [4]. The efficiency of streptavidin displacement by ChlR1 was dependent on the length of the 5′ ssDNA adjacent to the position of covalently attached biotin bound by streptavidin. The length dependence for streptavidin displacement by ChlR1 reflects similar observations made for its helicase activity on duplex DNA molecules flanked by a 5′ ssDNA tail, suggesting that increasing the tail length may enable multiple helicase molecules to load or an optimal helicase assembly state. Indeed, the Fe–S helicase FANCJ unwinds duplex DNA most efficiently as a dimer [57]; however, the influence of ChlR1 oligomerization on its catalytic functions has not been reported. In future, it will be important to determine if ChlR1, like FANCJ [75], can efficiently disrupt endogenous protein-DNA complexes such as the Rad51 nucleoprotein filament that plays a critical role during strand invasion into a recipient duplex in HR repair of DSBs. Alternatively, ChlR1 may facilitate sister chromatid cohesion by helping to preserve replication fork integrity and progression via its ability to strip proteins bound to duplex DNA. The ability of ChlR1 (or other helicases) to displace proteins bound to duplex DNA may be regulated by interactions with their protein partners. Smooth replication fork progression, facilitated by accessory helicases such as ChlR1, more than likely plays a role in sister chromatid cohesion.
To ascertain the relative importance of ATPase-driven ChlR1 protein displacement versus helicase activity, it would be highly informative to characterize a ChlR1 separation-of-function mutant which retains the ability to strip proteins bound to DNA, but not helicase activity, or vice versa. To our knowledge, such a separation-of-function mutant has not been identified for any helicase protein; however, the discovery of new and higher resolution structures of DNA repair proteins, including the Fe–S cluster helicases, is likely to lead to the creation of site-directed helicase mutants with novel properties that will be useful for genetic rescue studies. Accordingly, we characterized a clinically relevant FA Complementation Group J mutation corresponding to a FANCJ–A349P amino acid substitution in the conserved Fe–S domain that effectively uncoupled DNA translocation from helicase activity or disruption of protein–DNA complexes [76], suggesting that more subtle alterations to helicase proteins can have deleterious effects both in vitro and in vivo. Clearly, elucidation of the catalytic functions and molecular mechanism(s) of action of ChlR1 in cell biology will require more work.
Roles and pathways of ChlR1 to preserve chromosomal integrity and genome homeostasis
ChlR1 protein interactions
Although still in its early stage, protein interaction studies have begun to provide a better picture of the cellular pathways in which human ChlR1 protein participates (Table 3). Evidence from yeast implicated a role of Chl1 in the establishment of sister chromatid cohesion; therefore, Parish et al. [36] investigated whether human ChlR1 was associated with proteins of the cohesion complex. Using an antibody directed against the purified recombinant ChlR1 protein, they performed co-immunoprecipitation experiments and found that endogenously expressed ChlR1 was associated with the cohesion subunits Scc1, Smc1, and Smc3. These initial studies of human ChlR1 were highly significant and substantiated by the discovery that RNAi depletion of ChlR1 resulted in defective mitotic progression and subsequent mitotic failure, and a cohesion abnormality in which the associated sister chromatid pairs were significantly further apart at the centromeric region, indicative of a sister chromatid cohesion defect [36].
Table 3.
Protein interactions of ChlR1
| ChlR1 partner | Physical interaction | Direct functional interaction | Ref. |
|---|---|---|---|
| Cohesin | Yes | ND | [36] |
| RPA | ND | RPA stimulates ChlR1 helicase activity | [54] |
| Ctf18-RFC | Yes | Ctf18-RFC stimulates ChlR1 helicase activity | [54] |
| PCNA | Yes | ND | [54] |
| FEN-1 | Yes | ChlR1 stimulates FEN-1 5′ flap cleavage | [54] |
| Timeless–Tipin | Yes | ND | [37] |
See text for details
ND not determined
The Hurwitz laboratory reported that RNAi depletion of ChlR1 or the structure-specific nuclease FEN-1 implicated in replication and DNA repair resulted in defective sister chromatid cohesion in human cells [54]. ChlR1 helicase activity was found to be stimulated by the cohesion establishment factor (Ctf18-replication factor C; RFC) [54]. Furthermore, ChlR1 was observed to directly bind Ctf18-RFC, proliferating cell nuclear antigen (PCNA), a well-known processivity clamp for DNA replication machinery, and FEN-1 that is responsible for cleaving 5′ ssDNA flaps during Okazaki fragment maturation in cellular DNA replication [54]. Collectively, these studies helped to build a case that human ChlR1, like its yeast homolog Chl1 [45], facilitates sister chromatid cohesion by directly participating in lagging strand processing.
In vitro experiments with purified recombinant proteins demonstrated that ChlR1 helicase activity could be stimulated by the ssDNA binding protein, Replication Protein A (RPA), to unwind partial duplex DNA substrates as long as 500 bp in a specific manner because the E. coli ssDNA binding protein (ESSB) failed to enhance ChlR1 helicase activity [54]. It is tempting to speculate that RPA stimulates ChlR1 activity by physically binding to the helicase and enhancing its processivity in a manner analogous to that shown for the interaction of RPA70 subunit (RPA1) with the Werner syndrome helicase (WRN) [77]. However, there is no report of a direct physical interaction between RPA and ChlR1. Nonetheless, the interaction of RPA with ChlR1 is likely to be relevant as shown for the interaction of RPA with human RecQ helicases [78], as well as the sequence-related human Fe–S cluster DNA helicase FANCJ [79].
In addition to proteins that help to insure replication fork processivity and/or process DNA structures associated with the replication fork, checkpoint proteins facilitate signaling to insure coordination between cell cycle progression, DNA repair, and fork stabilization. Among the checkpoint proteins are an evolutionarily conserved protein pair known as Timeless and Tipin. The Timeless–Tipin protein complex plays an important but not fully understood role in the intra-S-phase checkpoint when replication forks stall, and helps to restore fork progression under conditions of replication stress. The Noguchi laboratory investigated the role of Timeless–Tipin in replication fork stabilization and sister chromatid cohesion, and found that, in human cells, Timeless–Tipin is not only associated with cohesion sub-units (Smc1, Smc3, SA1) known to be important for sister chromatid cohesion but also with ChlR1 [37]. They made two important observations regarding the interaction of Timeless–Tipin and ChlR1 from cell-based experiments: (1) RNAi-depletion of Timeless resulted in reduction of ChlR1 protein levels, suggesting that Timeless helps to stabilize ChlR1 or maintain its association with chromatin where ChlR1 is more stable; and (2) exogenous overexpression of ChlR1 reduced cohesion defects caused by RNAi-depletion of Timeless or its associated partner Tipin, suggesting that ChlR1 cooperates with Timeless–Tipin to maintain proper sister chromatid cohesion. It was surmised that the stabilizing effect of Timeless–Tipin on replication forks may allow ChlR1 to maintain its proper association with chromatin, whereby ChlR1-catalyzed DNA unwinding and protein interactions with factors that process lagging strand intermediates enable newly replicated sister chromatids to undergo appropriate cohesion.
Mammalian ChlR1 maintains proper heterochromatin organization
Mammalian ChlR1 is implicated in heterochromatin organization, which maintains chromosome regions in a condensed state and transcriptionally silenced [61]. The ATPase-driven functions of ChlR1 helicase activity and catalytic disruption of protein–DNA complexes are likely to be important for ChlR1’s role in heterochromatin formation and function; however, experimental evidence is lacking. What has been observed is that ChlR1-depleted human cells as well as mouse cells isolated from ChlR1−/− embryos display dispersed localization of constitutive heterochromatin and disrupted centromere clustering [61]. In addition, ChlR1−/− embryos showed decreased DNA methylation within major satellite repeats and reduced chromatin density at the telomeres [61]. The cumulative evidence suggests that the involvement of ChlR1 in heterochromatin organization is likely to influence transcription and may also affect sister chromatid cohesion. Based on experimental evidence in yeast, the Skibbens laboratory proposed that Chl1-mediated cohesion establishment is temporally coupled to lagging strand processing [45]. The post-fork cohesion establishment model is analogous to how histone protein complexes are deposited onto new sister chromatids and become subsequently modified to confer epigenetic status. Therefore, the involvement of ChlR1 in heterochromatin, and its interaction with FEN-1 to insure efficient lagging strand processing [54], will likely influence cohesion as well as epigenetic events that affect programmed transcription. The placental defect associated with ChlR1 null embryos [48] probably reflects an abnormal heterochromatin state. Defective heterochromatinization may underlie the diverse array of developmental defects and phenotypic characteristics for individuals with WABS. From a larger perspective, the distinct developmental defects of cohesinopathy disorders may in part reflect dysregulated expression of genes encoding cohesins and regulatory proteins [80].
Model to explain the potential role of ChlR1 in sister chromatid cohesion
Based on the current evidence, we suggest a working model to explain the role of ChlR1 in sister chromatid cohesion. This model relies heavily on the described genetic interactions and physical associations of the yeast Chl-1 homolog with proteins important for sister chromatid cohesion and DNA replication. Moreover, the recently described physical and functional interactions of ChlR1 with human DNA replication proteins and the potential involvement of the ChlR1 motor ATPase to unwind DNA structures that affect heterochromatin state further support its role in chromosomal stability maintenance. ChlR1 may help to coordinate unwinding of hairpins that arise during strand displacement synthesis of the lagging strand or more complex DNA structures (e.g., G2′ quadruplexes) that may form during replication or recombination (Fig. 2). In addition, interaction of ChlR1 with FEN-1 and accessory replication proteins (RFC, PCNA) may help to enable timely processing of lagging strand intermediates that arise during DNA replication. The proposed role of ChlR1 during Okazaki fragment processing would provide the opportunity for efficient cohesin-mediated sister chromatid tethering during the complex process of chromatinization, resulting in efficacious sister chromatid cohesion. It is also conceivable that the catalytic functions and protein interactions of ChlR1 operate not only during semi-conservative DNA replication but also during DNA repair synthesis or recombinational repair. Further studies are required to test these ideas, which will hopefully yield a better understanding of how ChlR1 operates. Moreover, it seems probable that ChlR1 may share its duties with other proteins that help process aberrant DNA structures, because genetic deficiencies in proteins responsible for flap processing and other relevant DNA metabolic processes result in cohesion defects.
Fig. 2.
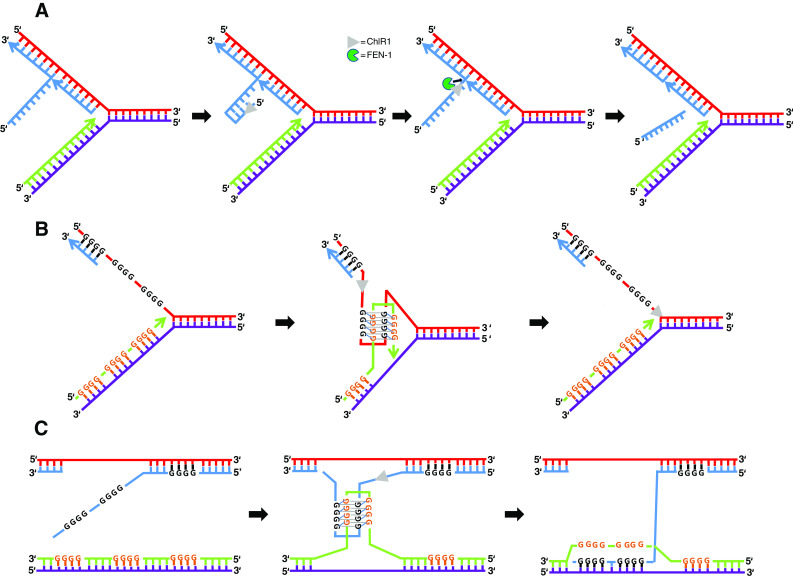
Model depicting the role of ChlR1 in processing of DNA structures during cellular DNA replication that would affect sister chromatid cohesion. a Efficient maturation of Okazaki fragments is believed to be important for sister chromatid cohesion. 5′ flaps created by strand displacement synthesis can form hairpins that are refractory to cleavage by FEN-1. ChlR1 5′ to 3′ helicase activity may unwind the hairpin structure, enabling FEN-1 to track along the single-strand and perform cleavage at the junction. ChlR1 interacts with FEN-1 and stimulates it 5′ flap cleavage, resulting in a ligatable nick that can be sealed by ligase. b, c ChlR1 resolution of a G-quadruplex structure that arises during replication of parental DNA (b) or recombination between sister chromatids (c) may suppress chromosomal instability. ChlR1 efficiently unwinds a specialized form of G-quadruplex DNA in which anti-parallel strands form a G-tetrad stack (G2′). The ability of ChlR1 to resolve G2′ quadruplexes may help replication to proceed (b) or untangle sister chromatids to allow appropriate HR repair (c)
Previously, we discussed the scenario that acetyltransferases such as p300 or the human orthologs Esco1 and Eso2, the latter mutated in the cohesinopathy RBS [81], regulate the functions of enzymes (nucleases, helicases) involved in the processing of DNA replication intermediates [82]. It will be informative to explore the possibility that ChlR1 is a target for posttranslational modification (e.g., acetylation) that regulates its catalytic functions or protein interactions which play a role in sister chromatid cohesion. Conceivably, the cellular defects underlying cohesinopathy disorders like WABS may be attributed to inefficient coupling of lagging strand synthesis to the process of sister chromatid cohesion, leading to replication fork destabilization and chromosomal instability.
Novel roles of ChlR1 in viral genome maintenance and cancer
Role of ChlR1 in papillomavirus genome maintenance
The papilloma virus life cycle is highly coordinated with the differentiation of the host epithelium, using the DNA damage response of the host cell as a highly potent signaling mechanism to replicate its viral genome [83]. Papillomaviruses vegetatively amplify their genomes in differentiated cells by a proposed mechanism in which HR-dependent replication exploits the host cell DNA damage response. Two virally encoded proteins, E1 and E2, bind to defined sites within the viral origin of replication. E1 protein is a DNA helicase, whereas E2 protein is a transcriptional regulator that helps to load E1 onto the origin of replication. E1 is dispensable during the maintenance phase of papillomavirus replication, but E2 is essential for the maintenance of the viral genome. Understanding how the papillomaviral proteins E1 and E2 coordinate with host cell replication, recombination, and DNA repair proteins to enable papillomavirus recombination-dependent replication should be informative for efforts to devise strategies to combat human papilloma virus (HPV) infection.
ChlR1 interacts with the papillomavirus E2 protein in vitro and in vivo, and is required for HPV genome maintenance and segregation [84]. E2 and DDX11 colocalize at early stages of mitosis [84], and ChlR1 enables E2 to load onto mitotic chromosomes during DNA replication [85]. A single amino acid substitution in E2 (W130R) that abrogates E2 protein binding to ChlR1 prevents its association with mitotic chromosomes and episomal maintenance of the virus in cell culture [84]. Parish et al. found that E2 remains associated with the chromosomes following ChlR1 removal, suggesting that ChlR1 may act during replication to facilitate the association of viral genomes onto chromosomes by a kinetochore-independent mechanism. It is unclear whether ChlR1 catalytic function is required for it to enable E2 to load onto chromosomes during DNA replication, but it seems likely. If so, then suppression of HPV infection may be achieved therapeutically by targeted ChlR1 depletion via RNA interference or by a small molecule DDX11 helicase inhibitor. The involvement of ChlR1 and other DNA repair proteins in HPV replication may have other clinical implications. For example, individuals with the genetic disorder FA display an increased susceptibility to head-and-neck squamous cell carcinomas [86]. Although somewhat controversial, research suggests that human papillomavirus is elevated in some cases of FA deficiency frequently associated with squamous cell carcinoma susceptibility [87]. It is possible that viral DNA may integrate into the host genome by an aberrant pathway when the FA pathway is dysfunctional. Understanding the role of ChlR1 or other host cellular proteins in HPV infection may help to identify suitable targets to combat carcinogenesis.
JunB regulates ChlR1 expression leading to premature sister chromatid separation
Characterizing how cancer cells or normal cells regulate the expression of certain DNA helicases may be insightful for understanding the upstream events and mechanisms whereby chromosomal anomalies arise during carcinogenesis. For example, it was recently determined that JunB, a component of the Activator Protein-1 transcription complex, accumulates during the G2/M phase of the cell cycle and causes transcriptional repression of ChlR1, leading to premature sister chromatid separation [88]. Such genetic infidelity during mitosis may contribute to enhanced cell proliferation in certain cancers such as anaplastic large cell lymphoma (ALCL), an aggressive form of non-Hodgkin lymphoma promoted by JunB. These observations are interesting in light of the extensive chromosomal abnormalities displayed in ALCL [89]. Further studies should explore ChlR1 and other helicases as predictive biomarkers for personalized cancer therapy.
ChlR1 is required for advanced melanoma cell survival
To identify genes that are differentially regulated with progression from early to advanced melanoma, the Becker laboratory profiled tissues by whole genome expression analysis and determined that ChlR1 is significantly up-regulated [62]. This led them to extensively examine ChlR1 expression patterns in primary and metastatic melanomas, which revealed an eight-fold increase in ChlR1 up-regulation in first stage invasive melanoma cells compared to noninvasive melanoma cells in situ. Moreover, expression of ChlR1 is high in advanced melanoma. RNAi depletion of ChlR1 in human melanoma cells resulted in a rapid change in their morphology, and a loss of cell-to-cell contact within 24 h. The ChlR1-depleted melanoma cells acquired a tightly condensed chromosome appearance and an increased number of chromosomes with only partially closed or open/separated arms, indicative of a chromosome segregation defect, as well as shorter average chromosome lengths. Further analysis revealed that ChlR1-depleted melanoma cells showed reduced proliferation and elevated apoptosis. Based on their findings, the authors proposed that targeted inhibition of ChlR1 may represent an approach for treating advanced melanoma that is resistant to more conventional radiometric or chemotherapy strategies.
Future perspective for ChlR1 research
Understanding the molecular and cellular functions of ChlR1 helicase should help to elucidate the underlying basis for the pathology of the very rare chromosomal instability disorder, Warsaw breakage syndrome. An improved knowledge of ChlR1 cellular pathways will be informative for the clinical diagnosis of the disorder, which partially overlaps with other rare genetic diseases including RBS and FA. It is important that clinicians and basic scientists who specialize in the study of rare chromosomal instability disorders communicate regularly with each other and the affected individuals and their families. Studying a rare genetic disorder like WABS presents an excellent opportunity to understand the molecular-genetic basis for a disease and its relationship to a biological process like sister chromatid cohesion which offers a window for comprehending other related but distinct disorders. There is still much to learn about the underlying basis for sister chromatid cohesion defects in clinical disorders, and ChlR1 may provide a clue. It seems likely that a role of the DNA helicase in replication is involved; however, perturbations to other processes including transcription probably contribute to phenotypes of cohesinopathy disorders like WABS. Further exploration of ChlR1 and other host proteins in papillomavirus genome maintenance may yield insights with clinical significance.
It is probable that ChlR1, like a number of DNA helicases, is differentially regulated in transformed or neoplastic cells and tissues in addition to melanoma (for review, see [29]). An existing challenge in the field is to understand why an increasing number of helicases are elicited to help cells deal with replication stress or DNA damage in cancer cells, and whether they are tailored to specific functions or tissue types. Indeed, early studies demonstrated that keratinocyte growth factor strongly induced the ChlR1 gene [90]. The latest work from the Becker laboratory [62] suggests that ChlR1 may be a useful target for battling melanoma, but it is unclear what the consequence(s) of targeted ChlR1 inhibition would be for normal cells, or if other cancer cell types also rely on elevated ChlR1 expression for proliferation. As significantly, what is the functional consequence of up-regulated expression of ChlR1 (or other DNA helicases) in the tumor microenvironment that may not be evident in experimental tissue culture or mouse model settings? While it is evident that depletion of certain helicases adversely affects growth or proliferation of human cancer cells grown in culture and increases their sensitivity or radiation or chemotherapy drugs [29], it is unclear how effective targeting a helicase for RNAi depletion or small molecule inhibition would be in a clinical setting. Animal models should be useful. One concern is that cancer cells treated with a DNA-damaging agent evoke a complex network of response pathways involving numerous proteins acting separately or in protein complexes at one or multiple steps of a given pathway. For example, an expanding list of DNA helicases, including ChlR1, are implicated in the cellular response to DNA cross-linking agents, such as MMC, or CPT, that are frequently used to combat leukemia and other cancers [91]. Devising a strategy to enhance the potency of chemotherapy drug regime by targeted helicase inhibition requires a molecular dissection of how uniquely or redundantly these helicases perform in the cross-link response and at what levels. Nonetheless, the prominence of DNA helicases like ChlR1 in human diseases and mechanisms that confer an elevated DNA damage response in cancer continues to prompt their attention for further experimental investigation.
Acknowledgments
This work is funded in part by the National Institutes of Health (NIH) and the National Institute on Aging (NIA) (R.M.B.), and by the Saskatchewan Health Research Foundation (SHRF) and the Natural Sciences and Engineering Research Council of Canada (NSERC) (Y.W.). We thank Drs. H. Vallabhaneni and J. Yin (NIA–NIH) for critically reading the manuscript. We wish to dedicate this paper to the memory of Dr. Johan P. de Winter, an outstanding scientist who led efforts in understanding the molecular-genetic basis of Warsaw breakage syndrome, Fanconi anemia, and other genetic disorders.
References
- 1.van der Lelij P, Chrzanowska KH, Godthelp BC, Rooimans MA, Oostra AB, Stumm M, Zdzienicka MZ, Joenje H, De Winter JP. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am J Hum Genet. 2010;86:262–266. doi: 10.1016/j.ajhg.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirota Y, Lahti JM. Characterization of the enzymatic activity of hChlR1, a novel human DNA helicase. Nucleic Acids Res. 2000;28:917–924. doi: 10.1093/nar/28.4.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skibbens RV. Chl1p, a DNA helicase-like protein in budding yeast, functions in sister-chromatid cohesion. Genetics. 2004;166:33–42. doi: 10.1534/genetics.166.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu Y, Sommers JA, Khan I, De Winter JP, Brosh RM., Jr Biochemical characterization of Warsaw breakage syndrome helicase. J Biol Chem. 2012;287:1007–1021. doi: 10.1074/jbc.M111.276022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Lelij P, Oostra A.B, Rooimans M.A, Joenje H, De Winter J.P (2010) Diagnostic overlap between Fanconi Anemia and the cohesinopathies: Roberts syndrome and Warsaw breakage syndrome. Anemia, Article ID 565268 [DOI] [PMC free article] [PubMed]
- 6.Rudolf J, Makrantoni V, Ingledew WJ, Stark MJ, White MF. The DNA repair helicases XPD and FancJ have essential Iron–Sulfur domains. Mol Cell. 2006;23:801–808. doi: 10.1016/j.molcel.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 7.Wu Y, Brosh RM., Jr DNA helicase and helicase-nuclease enzymes with a conserved iron–sulfur cluster. Nucleic Acids Res. 2012;40:4247–4260. doi: 10.1093/nar/gks039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Digiovanna JJ, Kraemer KH. Shining a light on Xeroderma pigmentosum. J Invest Dermatol. 2012;132:785–796. doi: 10.1038/jid.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Egly JM, Coin F. A history of TFIIH: two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst) 2011;10:714–721. doi: 10.1016/j.dnarep.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 10.Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, Arwert F, Mathew CG, Zdzienicka MZ, Hiom K, De Winter JP, Joenje H. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group. J Nat Genet. 2005;37:934–935. doi: 10.1038/ng1625. [DOI] [PubMed] [Google Scholar]
- 11.Levran O, Attwooll C, Henry RT, Milton KL, Neveling K, Rio P, Batish SD, Kalb R, Velleuer E, Barral S, Ott J, Petrini J, Schindler D, Hanenberg H, Auerbach AD. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet. 2005;37:931–933. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- 12.Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreasse PR, Cantor SB. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell. 2005;8:255–265. doi: 10.1016/j.ccr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 13.Cantor SB, Bell DW, Ganesan S, Kas EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA, Livingston DM. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105:149–160. doi: 10.1016/S0092-8674(01)00304-X. [DOI] [PubMed] [Google Scholar]
- 14.Rafnar T, Gudbjartsson DF, Sulem P, Jonasdottir A, Sigurdsson A, Jonasdottir A, Besenbacher S, Lundin P, Stacey SN, Gudmundsson J, et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet. 2011;43:1104–1107. doi: 10.1038/ng.955. [DOI] [PubMed] [Google Scholar]
- 15.Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006;38:1239–1241. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 16.Greenberg RA, Sobhia B, Pathania S, Cantor SB, Nakatani Y, Livingston DM. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006;20:34–46. doi: 10.1101/gad.1381306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suhasini AN, Rawtani NA, Wu Y, Sommers JA, Sharma S, Mosedale G, North PS, Cantor SB, Hickson ID, Brosh RM., Jr Interaction between the helicases genetically linked to Fanconi anemia group J and Bloom’s syndrome. EMBO J. 2011;30:692–705. doi: 10.1038/emboj.2010.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng M, Litman R, Xie J, Sharma S, Brosh RM, Jr, Cantor SB. The FANCJ/MutLalpha interaction is required for correction of the cross-link response in FA-J cells. EMBO J. 2007;26:3238–3249. doi: 10.1038/sj.emboj.7601754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ballew BJ, Yeager M, Jacobs K, Giri N, Boland J, Burdett L, Alter BP, Savage SA. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita. Hum Genet. 2013;132:473–480. doi: 10.1007/s00439-013-1265-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ballew BJ, Joseph V, De S, Sarek G, Vannier JB, Stracker T, Schrader KA, Small TN, O’Reilly R, Manschreck C, et al. A recessive founder mutation in Regulator of Telomere Elongation Helicase 1, RTEL1, underlies severe immunodeficiency and features of Hoyeraal Hreidarsson Syndrome. PLoS Genet. 2013;9:e1003695. doi: 10.1371/journal.pgen.1003695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng Z, Glousker G, Molczan A, Fox AJ, Lamm N, Dheekollu J, Weizman OE, Schertzer M, Wang Z, Vladimirova O, et al. Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal-Hreidarsson syndrome. Proc Natl Acad Sci USA. 2013;110:E3408–E3416. doi: 10.1073/pnas.1300600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le GT, Jullien L, Touzot F, Schertzer M, Gaillard L, Perderiset M, Carpentier W, Nitschke P, Picard C, Couillault G, et al. Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum Mol Genet. 2013;22:3239–3249. doi: 10.1093/hmg/ddt178. [DOI] [PubMed] [Google Scholar]
- 23.Lee J. Telomere shortening by mutations in the RTEL1 helicase cause severe form of Dyskeratosis Congenita, Hoyerall-Hreidarsson syndrome. Clin Genet. 2013;84:210. doi: 10.1111/cge.12175. [DOI] [PubMed] [Google Scholar]
- 24.Walne AJ, Vulliamy T, Kirwan M, Plagnol V, Dokal I. Constitutional mutations in RTEL1 cause severe Dyskeratosis congenita. Am J Hum Genet. 2013;92:448–453. doi: 10.1016/j.ajhg.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell. 2012;149:795–806. doi: 10.1016/j.cell.2012.03.030. [DOI] [PubMed] [Google Scholar]
- 26.Ding H, Schertzer M, Wu X, Gertsenstein M, Selig S, Kammori M, Pourvali R, Poon S, Vulto I, Chavez E, Tam PP, Nagy A, Lansdorp PM. Regulation of murine telomere length by Rtel: an essential gene encoding a helicase-like protein. Cell. 2004;117:873–886. doi: 10.1016/j.cell.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 27.Vannier JB, Sandhu S, Petalcorin MI, Wu X, Nabi Z, Ding H, Boulton SJ. RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science. 2013;342:239–242. doi: 10.1126/science.1241779. [DOI] [PubMed] [Google Scholar]
- 28.Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–679. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 29.Brosh RM., Jr DNA helicases involved in DNA repair and their roles in cancer. Nat Rev Cancer. 2013;13:542–558. doi: 10.1038/nrc3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mann MB, Hodges CA, Barnes E, Vogel H, Hassold TJ, Luo G. Defective sister-chromatid cohesion, aneuploidy and cancer predisposition in a mouse model of type II Rothmund-Thomson syndrome. Hum Mol Genet. 2005;14:813–825. doi: 10.1093/hmg/ddi075. [DOI] [PubMed] [Google Scholar]
- 31.Skibbens RV, Colquhoun JM, Green MJ, Molnar CA, Sin DN, Sullivan BJ, Tanzosh EE. Cohesinopathies of a feather flock together. PLoS Genet. 2013;9:e1004036. doi: 10.1371/journal.pgen.1004036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capo-chichi J-M, Bharti SK, Sommers JA, Yammine T, Chouery E, Patry L, Rouleau GA, Samuels ME, Hamdan FF, Michaud JL, Brosh RM, Jr, Megarbae A, Kibar Z. Identification and biochemical characterization of a novel mutation in DDX11 causing Warsaw breakage syndrome. Human Mutat. 2012;334:103–107. doi: 10.1002/humu.22226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suhasini AN, Brosh RM., Jr Disease-causing missense mutations in human DNA helicase disorders. Mutat Res. 2012;752:138–152. doi: 10.1016/j.mrrev.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Botta E, Nardo T, Lehmann AR, Egly JM, Pedrini AM, Stefanini M. Reduced level of the repair/transcription factor TFIIH in trichothiodystrophy. Hum Mol Genet. 2002;11:2919–2928. doi: 10.1093/hmg/11.23.2919. [DOI] [PubMed] [Google Scholar]
- 35.Dubaele S, De Proietti SL, Bienstock RJ, Keriel A, Stefanini M, Van HB, Egly JM. Basal transcription defect discriminates between Xeroderma pigmentosum and Trichothiodystrophy in XPD patients. Mol Cell. 2003;11:1635–1646. doi: 10.1016/S1097-2765(03)00182-5. [DOI] [PubMed] [Google Scholar]
- 36.Parish JL, Rosa J, Wang X, Lahti JM, Doxsey SJ, Androphy EJ. The DNA helicase ChlR1 is required for sister chromatid cohesion in mammalian cells. J Cell Sci. 2006;119:4857–4865. doi: 10.1242/jcs.03262. [DOI] [PubMed] [Google Scholar]
- 37.Leman AR, Noguchi C, Lee CY, Noguchi E. Human timeless and tipin stabilize replication forks and facilitate sister-chromatid cohesion. J Cell Sci. 2010;123:660–670. doi: 10.1242/jcs.057984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shah N, Inoue A, Woo LS, Beishline K, Lahti JM, Noguchi E. Roles of ChlR1 DNA helicase in replication recovery from DNA damage. Exp Cell Res. 2013;319:2244–2253. doi: 10.1016/j.yexcr.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soutoglou E, Misteli T. Activation of the cellular DNA damage response in the absence of DNA lesions. Science. 2008;320:1507–1510. doi: 10.1126/science.1159051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holloway L. CHL1 is a nuclear protein with an essential ATP binding site that exhibits a size-dependent effect on chromosome segregation. Nucleic Acids Res. 2000;28:3056–3064. doi: 10.1093/nar/28.16.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rudra S, Skibbens RV. Cohesin codes—interpreting chromatin architecture and the many facets of cohesin function. J Cell Sci. 2013;126:31–41. doi: 10.1242/jcs.116566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petronczki M, Chwalla B, Siomos MF, Yokobayashi S, Helmhart W, Deutschbauer AM, Davis RW, Watanabe Y, Nasmyth K. Sister-chromatid cohesion mediated by the alternative RF-CCtf18/Dcc1/Ctf8, the helicase Chl1 and the polymerase-alpha-associated protein Ctf4 is essential for chromatid disjunction during meiosis II. J Cell Sci. 2004;117:3547–3559. doi: 10.1242/jcs.01231. [DOI] [PubMed] [Google Scholar]
- 43.Ansbach AB, Noguchi C, Klansek IW, Heidlebaugh M, Nakamura TM, Noguchi E. RFCCtf18 and the Swi1-Swi3 complex function in separate and redundant pathways required for the stabilization of replication forks to facilitate sister chromatid cohesion in Schizosaccharomyces pombe. Mol Biol Cell. 2008;19:595–607. doi: 10.1091/mbc.E07-06-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mayer ML, Pot I, Chang M, Xu H, Aneliunas V, Kwok T, Newitt R, Aebersold R, Boone C, Brown GW, Hieter P. Identification of protein complexes required for efficient sister chromatid cohesion. Mol Biol Cell. 2004;15:1736–1745. doi: 10.1091/mbc.E03-08-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rudra S, Skibbens RV. Sister chromatid cohesion establishment occurs in concert with lagging strand synthesis. Cell Cycle. 2012;11:2114–2121. doi: 10.4161/cc.20547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borges V, Smith DJ, Whitehouse I, Uhlmann F. An Eco1-independent sister chromatid cohesion establishment pathway in S. cerevisiae. Chromosoma. 2013;122:121–134. doi: 10.1007/s00412-013-0396-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rudra S, Skibbens RV. Chl1 DNA helicase regulates Scc2 deposition specifically during DNA-replication in Saccharomyces cerevisiae . PLoS ONE. 2013;8:e75435. doi: 10.1371/journal.pone.0075435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Inoue A, Li T, Roby SK, Valentine MB, Inoue M, Boyd K, Kidd VJ, Lahti JM. Loss of ChlR1 helicase in mouse causes lethality due to the accumulation of aneuploid cells generated by cohesion defects and placental malformation. Cell Cycle. 2007;6:1646–1654. doi: 10.4161/cc.6.13.4411. [DOI] [PubMed] [Google Scholar]
- 49.Cota CD, Garcia-Garcia MJ. The ENU-induced cetus mutation reveals an essential role of the DNA helicase DDX11 for mesoderm development during early mouse embryogenesis. Dev Dyn. 2012;241:1249–1259. doi: 10.1002/dvdy.23810. [DOI] [PubMed] [Google Scholar]
- 50.Chung G, O’Neil NJ, Rose AM. CHL-1 provides an essential function affecting cell proliferation and chromosome stability in Caenorhabditis elegans . DNA Repair (Amst) 2011;10:1174–1182. doi: 10.1016/j.dnarep.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 51.Cheung I, Schertzer M, Rose A, Lansdorp PM. Disruption of dog-1 in Caenorhabditis elegans triggers deletions upstream of guanine-rich DNA. Nat Genet. 2002;31:405–409. doi: 10.1038/ng928. [DOI] [PubMed] [Google Scholar]
- 52.Youds JL, O’Neil NJ, Rose AM. Homologous recombination is required for genome stability in the absence of DOG-1 in Caenorhabditis elegans. Genetics. 2006;173:697–708. doi: 10.1534/genetics.106.056879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lohman TM, Bjornson KP. Mechanisms of helicase-catalyzed DNA unwinding. Annu Rev Biochem. 1996;65:169–214. doi: 10.1146/annurev.bi.65.070196.001125. [DOI] [PubMed] [Google Scholar]
- 54.Farina A, Shin JH, Kim DH, Bermudez VP, Kelman Z, Seo YS, Hurwitz J. Studies with the human cohesin establishment factor, ChlR1. Association of ChlR1 with Ctf18-RFC and Fen1. J Biol Chem. 2008;283:20925–20936. doi: 10.1074/jbc.M802696200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuper J, Wolski SC, Michels G, Kisker C. Functional and structural studies of the nucleotide excision repair helicase XPD suggest a polarity for DNA translocation. EMBO J. 2012;31:494–502. doi: 10.1038/emboj.2011.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pugh RA, Wu CG, Spies M. Regulation of translocation polarity by helicase domain 1 in SF2B helicases. EMBO J. 2011;31:503–514. doi: 10.1038/emboj.2011.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu Y, Sommers JA, Loiland JA, Kitao H, Kuper J, Kisker C, Brosh RM. The Q motif of FANCJ DNA helicase regulates its dimerization, DNA binding, and DNA repair function. J Biol Chem. 2012;287:21699–21716. doi: 10.1074/jbc.M112.351338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pugh RA, Honda M, Leesley H, Thomas A, Lin Y, Nilges MJ, Cann IK, Spies M. The iron-containing domain is essential in Rad3 helicases for coupling of ATP hydrolysis to DNA translocation and for targeting the helicase to the single-stranded DNA-double-stranded DNA junction. J Biol Chem. 2008;283:1732–1743. doi: 10.1074/jbc.M707064200. [DOI] [PubMed] [Google Scholar]
- 59.Qi Z, Pugh RA, Spies M, Chemla YR. Sequence-dependent base pair stepping dynamics in XPD helicase unwinding. Elife. 2013;2:e00334. doi: 10.7554/eLife.00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 61.Inoue A, Hyle J, Lechner MS, Lahti JM. Mammalian ChlR1 has a role in heterochromatin organization. Exp Cell Res. 2011;317:2522–2535. doi: 10.1016/j.yexcr.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bhattacharya C, Wang X, Becker D. The DEAD/DEAH box helicase, DDX11, is essential for the survival of advanced melanomas. Mol Cancer. 2012;11:82. doi: 10.1186/1476-4598-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bochman ML, Paeschke K, Zakian VA. DNA secondary structures: stability and function of G-quadruplex structures. Nat Rev Genet. 2012;13:770–780. doi: 10.1038/nrg3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu Y, Brosh RM., Jr G-quadruplex nucleic acids and human disease. FEBS J. 2010;277:3470–3488. doi: 10.1111/j.1742-4658.2010.07760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.London TB, Barber LJ, Mosedale G, Kelly GP, Balasubramanian S, Hickson ID, Boulton SJ, Hiom K. FANCJ is a structure-specific DNA helicase associated with the maintenance of genomic G/C tracts. J Biol Chem. 2008;283:36132–36139. doi: 10.1074/jbc.M808152200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu Y, Shin-Ya K, Brosh RM., Jr FANCJ helicase defective in Fanconia Anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol Cell Biol. 2008;28:4116–4128. doi: 10.1128/MCB.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bharti SK, Sommers JA, George F, Kuper J, Hamon F, Shin-Ya K, Teulade-Fichou MP, Kisker C, Brosh RM., Jr Specialization among iron–sulfur cluster helicases to resolve G-quadruplex DNA structures that threaten genomic stability. J Biol Chem. 2013;288:28217–28229. doi: 10.1074/jbc.M113.496463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun H, Karow JK, Hickson ID, Maizels N. The Bloom’s syndrome helicase unwinds G4 DNA. J Biol Chem. 1998;273:27587–27592. doi: 10.1074/jbc.273.42.27587. [DOI] [PubMed] [Google Scholar]
- 69.Jones M, Rose A. A DOG’s view of Fanconi Anemia: insights from C. elegans. Anemia. 2012;2012:323721. doi: 10.1155/2012/323721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bridge WL, Vandenberg CJ, Franklin RJ, Hiom K. The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat Genet. 2005;37:953–957. doi: 10.1038/ng1627. [DOI] [PubMed] [Google Scholar]
- 71.Hiom K. FANCJ: solving problems in DNA replication. DNA Repair (Amst) 2010;9:250–256. doi: 10.1016/j.dnarep.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 72.Garner TP, Williams HE, Gluszyk KI, Roe S, Oldham NJ, Stevens MF, Moses JE, Searle MS. Selectivity of small molecule ligands for parallel and anti-parallel DNA G-quadruplex structures. Org Biomol Chem. 2009;7:4194–4200. doi: 10.1039/b910505k. [DOI] [PubMed] [Google Scholar]
- 73.Henderson A, Wu Y, Huang YC, Chavez EA, Platt J, Johnson FB, Brosh RM, Jr, Sen D, Lansdorp PM. Detection of G-quadruplex DNA in mammalian cells. PMID: Nucleic Acids Res; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lam EY, Beraldi D, Tannahill D, Balasubramanian S. G-quadruplex structures are stable and detectable in human genomic DNA. Nat Commun. 2013;4:1796. doi: 10.1038/ncomms2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sommers JA, Rawtani N, Gupta R, Bugreev DV, Mazin AV, Cantor SB, Brosh RM., Jr FANCJ uses its motor ATPase to disrupt protein-DNA complexes, unwind triplexes, and inhibit rad51 strand exchange. J Biol Chem. 2009;284:7505–7517. doi: 10.1074/jbc.M809019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu Y, Sommers JA, Suhasini AN, Leonard T, Deakyne JS, Mazin AV, Shin-Ya K, Kitao H, Brosh RM., Jr Fanconi anemia Group J mutation abolishes its DNA repair function by uncoupling DNA translocation from helicase activity or disruption of protein-DNA complexes. Blood. 2010;116:3780–3791. doi: 10.1182/blood-2009-11-256016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Doherty KM, Sommers JA, Gray MD, Lee JW, von Kobbe C, Thoma NH, Kureekattil RP, Kenny MK, Brosh RM., Jr Physical and functional mapping of the RPA interaction domain of the Werner and Bloom syndrome helicases. J Biol Chem. 2005;280:29494–29505. doi: 10.1074/jbc.M500653200. [DOI] [PubMed] [Google Scholar]
- 78.Sharma S, Doherty KM, Brosh RM. Mechanisms of RecQ helicases in pathways of DNA metabolism and maintenance of genomic stability. Biochem J. 2006;398:319–337. doi: 10.1042/BJ20060450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gupta R, Sharma S, Sommers JA, Kenny MK, Cantor SB, Brosh RM., Jr FANCJ (BACH1) helicase forms DNA damage inducible foci with replication protein A and interacts physically and functionally with the single-stranded DNA-binding protein. Blood. 2007;110:2390–2398. doi: 10.1182/blood-2006-11-057273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Horsfield JA, Print CG, Monnich M. Diverse developmental disorders from the one ring: distinct molecular pathways underlie the cohesinopathies. Front Genet. 2012;3:171. doi: 10.3389/fgene.2012.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vega H, Waisfisz Q, Gordillo M, Sakai N, Yanagihara I, Yamada M, van GD, Kayserili H, Xu C, Ozono K, et al. Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat Genet. 2005;37:468–470. doi: 10.1038/ng1548. [DOI] [PubMed] [Google Scholar]
- 82.Bharti SK, Banerjee T, Brosh RM., Jr Setting the stage for cohesion establishment by the replication fork. Cell Cycle. 2012;11:2228. doi: 10.4161/cc.20962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sakakibara N, Chen D, McBride AA. Papillomaviruses use recombination-dependent replication to vegetatively amplify their genomes in differentiated cells. PLoS Pathog. 2013;9:e1003321. doi: 10.1371/journal.ppat.1003321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Parish JL, Bean AM, Park RB, Androphy EJ. ChlR1 is required for loading papillomavirus E2 onto mitotic chromosomes and viral genome maintenance. Mol Cell. 2006;24:867–876. doi: 10.1016/j.molcel.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 85.Feeney KM, Saade A, Okrasa K, Parish JL. In vivo analysis of the cell cycle dependent association of the bovine papillomavirus E2 protein and ChlR1. Virology. 2011;414:1–9. doi: 10.1016/j.virol.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 86.Romick-Rosendale LE, Lui VW, Grandis JR, Wells SI. The Fanconi anemia pathway: repairing the link between DNA damage and squamous cell carcinoma. Mutat Res. 2013;743–744:78–88. doi: 10.1016/j.mrfmmm.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Park JW, Pitot HC, Strati K, Spardy N, Duensing S, Grompe M, Lambert PF. Deficiencies in the Fanconi anemia DNA damage response pathway increase sensitivity to HPV-associated head and neck cancer. Cancer Res. 2010;70:9959–9968. doi: 10.1158/0008-5472.CAN-10-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Perez-Benavente B, Garcia JL, Rodriguez MS, Pineda-Lucena A, Piechaczyk M, de Font MJ, Farras R. GSK3-SCF(FBXW7) targets JunB for degradation in G2 to preserve chromatid cohesion before anaphase. Oncogene. 2013;32:2189–2199. doi: 10.1038/onc.2012.235. [DOI] [PubMed] [Google Scholar]
- 89.Mussolin L, Pillon M, Bonato P, Leszl A, Franceschetto G, Di MA, d’Amore ES, Sainati L, Rosolen A. Cytogenetic analysis of pediatric anaplastic large cell lymphoma. Pediatr Blood Cancer. 2010;55:446–451. doi: 10.1002/pbc.22550. [DOI] [PubMed] [Google Scholar]
- 90.Frank S, Werner S. The human homologue of the yeast CHL1 gene is a novel keratinocyte growth factor-regulated gene. J Biol Chem. 1996;271:24337–24340. doi: 10.1074/jbc.271.40.24337. [DOI] [PubMed] [Google Scholar]
- 91.Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011;11:467–480. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Egan KM, Thompson RC, Nabors LB, Olson JJ, Brat DJ, Larocca RV, Brem S, Moots PL, Madden MH, Browning JE, Ann CY. Cancer susceptibility variants and the risk of adult glioma in a US case-control study. J Neurooncol. 2011;104:535–542. doi: 10.1007/s11060-010-0506-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shete S, Hosking FJ, Robertson LB, Dobbins SE, Sanson M, Malmer B, Simon M, Marie Y, Boisselier B, Delattre JY, et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet. 2009;41:899–904. doi: 10.1038/ng.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wrensch M, Jenkins RB, Chang JS, Yeh RF, Xiao Y, Decker PA, Ballman KV, Berger M, Buckner JC, Chang S, et al. Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat Genet. 2009;41:905–908. doi: 10.1038/ng.408. [DOI] [PMC free article] [PubMed] [Google Scholar]