

Abstract
Peptide therapeutics is a promising field for emerging anti-cancer agents. Benefits include the ease and rapid synthesis of peptides and capacity for modifications. An existing and vast knowledge base of protein structure and function can be exploited for novel peptide design. Current research focuses on developing peptides that can (1) serve as tumor targeting moieties and (2) permeabilize membranes with cytotoxic consequences. A survey of recent findings reveals significant trends. Amphiphilic peptides with clusters of hydrophobic and cationic residues are features of anti-microbial peptides that confer the ability to eradicate microbes and show considerable anti-cancer toxicity. Peptides that assemble and form pores can disrupt cell or organelle membranes and cause apoptotic or necrotic death. Cell permeable and tumor-homing peptides can carry biologically active cargo to tumors or tumor vasculature. The challenge lies in developing the clinical application of therapeutic peptides. Improving delivery to tumors, minimizing non-specific toxic effects and discerning pharmacokinetic properties are high among the needs to produce a powerful therapeutic peptide for cancer treatment.
Keywords: Tumor-targeting, membrane, anti-microbial, Bcl-2 family, apoptosis, necrosis, cell-penetrating, mitochondria, cytotoxicity, vasculature
1. INTRODUCTION
Cancer is not a single disease but a group of diseases characterized by the deregulated growth of abnormal cells. Driving this uncontrolled growth are a series of mutations that cause aberrant expression of gene products essential for regulating proliferation, survival, and growth activities of cells. Hence, cancer results from defects in the most basic biological operations of cells: the ability to respond to growth signals, engage cell death programs to eliminate unnecessary, excess or damaged cells, and the formation of new blood vessels and ability to invade tissue. The challenge to clinicians and researchers seeking effective therapeutic approaches is to eliminate cancerous cells while sparing the normal, healthy tissue. While significant progress has been made in recent years, most current treatments for cancers involve surgery or chemo-, radiation, and hormone therapies that have changed little in the past decade. As an example, conventional chemotherapeutics (i.e.: DNA alkylating agents), which target proliferating cancer cells, also damage healthy growing cells but may fail to eliminate quiescent or non-proliferating cancer cells [1, 2]. Moreover, the development of drug resistance is associated with current treatments, and may be due to abnormalities in drug transporters or detoxifying enzymes that affect the interaction between the drug and its target. Defects in DNA repair mechanisms and the apoptotic or cell death pathway can also lead to the development of cancer drug resistance.
Even if initial therapies are successful, the risk of cancer recurrence remains a problem for patients. In an effort to find new cancer drugs, the evolution of small molecules as drugs has driven research endeavors; but problems with unforeseen off-target effects have created the need for alternative approaches. In this review, we will focus on a novel class of anti-cancer agents that are based on peptides. These small molecular weight molecules can be modeled after endogenous proteins, are readily synthesized in a cost efficient manner and are amenable to modifications. Peptides have applications in both diagnostics and cancer treatments, providing specificity for tumor tissues, decreased chances of developing resistance, and low toxicity. While a number of reviews have described in detail the use of individual types of peptides (i.e. antimicrobial, cell penetrating or tumor targeting), in this review, we will survey the general advancements made in the discovery and application of therapeutic peptides for cancer treatment with the intent to demonstrate both the benefits and challenges of this promising biomedical research direction.
1.1. Properties of Peptides
Peptides that could be developed into therapies to treat cancer can be organized into three major groups as shown in Fig. (1). In the first group are those peptides, usually naturally occurring or derived from a known protein, which have inherent membrane binding capacity, can form pores or disrupt membranes, and through this mode of action are cytotoxic (Fig. 1A). Anti-microbial peptides (AMPs), like cecropins and magainins, are part of this first group as are “pore-forming” peptides derived from the Bcl-2 family of apoptosis mediators. In the second group are cell penetrating peptides (CPPs), such as the trans-activating transcriptional activator (Tat) from HIV (human immunodeficiency virus), which function like a “Trojan horse” and internalize tethered cargo (Fig. 1B). The problems associated with most CPPs, like Tat, are related to the absence of tumor cell specificity, requiring modifications or fusion with a tumor-specific ligand for optimal anti-cancer effectiveness. The third group is comprised of tumor targeting peptides (TTPs) that are specific for tumor-related surface markers, such as membrane receptors, and can be used to deliver cytotoxic cargo (i.e. drugs or a cytotoxic peptide) specifically to tumor tissue or vasculature (Fig. 1C). Examples of these are peptides with RGD (arginine/glycine/aspartic acid) or NGR (asparagine/glycine/arginine) motifs that bind to integrins or cell surface associated molecules or receptors frequently overexpressed on tumor cells or tumor-associated blood vessels. CPPs may directly penetrate membranes or like TTPs employ a biologically active mechanism like endocytosis for internalization of cargo. To produce the ideal therapeutic peptide, one would need to integrate the essential characteristics from each of the peptides in these groups: tumor targeting, cell membrane penetrating and death-inducing.
Fig. 1. Classes of Therapeutic Peptides.

Current therapeutic peptides can be classified into three major divisions. A) Anti-microbial peptides (i.e. magainins or cecropins) or pore forming peptides (i.e. derived from the Bcl-2 family of apoptosis mediators) are targeted to characteristic features of certain membranes and typically incorporate into the membrane to form pores, affecting both membrane stability and membrane potential. B) Cell penetrating peptides, for example the Tat peptide derived from HIV, are usually arginine rich and inert to the cell. These may enter cells directly through membranes (as shown), or use an endocytic mechanism (C) to incorporate within the cell, and are often tethered to another more active compound or peptide, making this a “Trojan horse” class. C) Targeting peptides, such as RGD or NGR peptides, are specific for a plasma membrane component overexpressed by the tumor vasculature. These peptides are typically used to target a secondary chemotherapy agent or a biological preferentially to tumors.
Many peptides that have inherent cytotoxicity against cancer cells (i.e. those in group 1, Fig. 1A) possess a cationic, amphiphilic structure that allows them to target the negatively charged membranes of cancer cells (and bacteria), but not normal mammalian cells (see below), and may have pore-forming activity. These peptides, which have an affinity for anionic phospholipid head groups, exert their cytotoxic effects by docking in target membranes and causing depolarization [3,4]. The mechanism of action underlying depolarization, in turn, is related to the ability of cationic, membrane-active peptides to form toroidal pores [3, 4]. In a toroidal pore, as compared to a “barrel stave” pore, the peptides and lipids assemble into a type of organized, supra-molecular structure, causing curvature of the membrane to form a pore through which ions or small molecules can pass (Fig. 2). An alternative mode to explain membrane disruption by peptides is the “carpet” model; peptides accumulate parallel to the membrane surface and destabilize the membrane, causing permeabilization. Other structural features can contribute to the lethality of a peptide. For example, the position of a tryptophan residue, adjacent to the cationic region of a short amphipathic peptide and facilitated by the electrostatic interaction, aids in the insertion and subsequent cytotoxic activity of the peptide [5]. Such amphipathic bioactive peptides can also be used to drive “self-assembly” to form higher order peptide structures that can either be targeted directly to the membrane or can be guided through the membrane to attack intracellular targets upon assembly [6].
Fig. 2. Schematic of pore types.
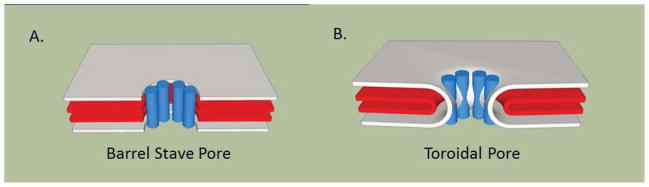
Models of the (A) barrel-stave and (B) toroidal pores that can be formed by anti-microbial or pore-forming peptides are shown. Models of pores were generated using SketchUp (Google). The layers represent the outer and inner membranes with the cylinders indicating the peptides.
1.2. Properties of Tumors
1.2.1. Membranes of Cancer Cells
To be useful as anti-cancer agents, cytotoxic peptides like AMPs (Fig. 1A) need to target and damage the membranes of cancer cells, sparing non-cancer cells. Important in this regard is that fact that cancer cells have significant differences in their cell membranes that favor the targeting of such cytotoxic peptides. In non-cancer cells, the total membrane charge is more neutral due to the presence of zwitterionic phospholipids like phosphatidylcholine (PC) and sphingomyelin, with phosphatidylserine (PS) and phosphotidylethanolamine (PE) located in the inner leaflet of plasma membrane [7]. Cancer cells, in contrast, lose this membrane symmetry and expose anionic PS on the outer leaflet of the plasma membrane, increasing the overall negative charge [8]. Exposure of PS is linked to the metastatic phenotype [9] and has been documented to occur with multiple cancer tissue types (reviewed in [10]). Another source for increased negative charges on the membrane of cancerous cells is the sialic acid residues linked to glycolipids and glycoproteins like mucins. Mucin 1 (Muc 1) is highly expressed in most breast carcinomas as well as in ovarian, pancreatic, and lung cancers as examples [11]. Proteoglycans with highly negatively charged side chains, heparin sulfate and chondroitin sulfate, can also contribute to changing the charge on the surface of cancer cell membranes. A number of cancers with altered proteoglycan expression and sulfonation have been documented [12, 13]. In addition to increased negative charges on the cell membranes of cancer cells, changes in membrane fluidity are associated with some cancers [14, 15] and may be linked to higher cholesterol levels [16]. Another difference is the increased surface area due to more microvilli on cancer cells compared to non-cancer cells [17]. This collective evidence indicates that significant differences between the cell membranes of cancer cells and non-cancer cells exists that could support the specific targeting and cytotoxic action of a peptide therapeutic.
1.2.2. Tumor Vasculature
Solid tumors pose a particular challenge to therapeutics because drugs introduced through blood vessels can only penetrate a few cells deep into tumor tissues [18]. Reasons may include poor blood flow through tumor vessels [19] and high interstitial pressure within in tumors that may stem from aberrant lymphatic flow [19, 20]. However, the problems of getting therapeutics into tumors may be offset by tumor leakiness caused by rapidly growing and abnormal tumor neovasculature. This alteration in the fluid dynamics of tumors causes cellular metabolites to accumulate in tumors at higher concentrations than in normal tissue [21]. Termed enhanced permeability and retention (EPR), these effects can differ from tumor to tumor, posing a significant variable in treatment effectiveness. Besides being uneven and leaky, tumor blood vessels express surface and extracellular matrix (ECM) proteins that are different from normal blood vessels. Thus, an alternative treatment approach to improve delivery of anti-cancer drugs to tumors is to direct treatments to tumor blood vessels by using tumor-targeting peptides (Fig. 1C) that home to tumor vasculature.
The expression of some of these proteins is associated with angiogenesis. Examples are the overexpression of certain types of αβ integrins [22]. Since these integrins are uniquely overexpressed, they could serve as sites for the targeting peptides to home to tumor vasculature. The ECM also has markers that could help direct peptides to tumors, such as the expression of an alternative spliced form of fibronectin, which is linked to tumor vessels [23]. Peptides can also be used to target fibrin-fibronectin complexes in the walls or stroma of tumor blood vessels [24, 25]. Collagen expression is another potential target for tumor-specific peptides. However, angiogenesis normally occurs in the context of inflammation as well as in regenerating tissues. Consequently, targeting angiogenic markers on tumor vasculature could also led to collateral damage of tissues undergoing repair or responding to infections. The need for TTPs with more focused recognition for molecules specific to tumor vasculature is evident.
2. PORE-FORMING AND CYTOTOXIC PEPTIDES
2.1. Anti-Microbial Peptides
Anti-microbial peptides (AMPs) (also called host defense peptides or cationic antimicrobial peptides) are low molecular weight molecules (~10–40 amino acids) with a cationic amphipathic structure that enables them to interact with anionic lipid membranes. Naturally occurring AMPs are part of the protective innate immune response to microbes in many species that include mammals, amphibians and insects. Because of their mode of action, in which the AMPs target membranes, AMPs could be used as alternative cancer therapeutics. In AMP databases, more than 100 AMPs have been characterized as having potential anti-tumor activity [26]. Lipid membranes are the major target of most AMPs; therefore, the development of drug resistance is less likely to occur since damaging cytotoxicity can take place within minutes of peptide introduction [27]. AMPs can induce death in cancer cells through two general mechanisms: apoptosis or necrosis. A necrotic death pathway could be the result of AMPs targeting negatively charged molecules on the cell membrane of cancerous cells, leading to cell lysis, while apoptosis could be the consequence of mitochondrial membrane disruption. AMP-mediated membrane permeabilization may occur through various mechanisms, such “barrel stave” or “carpet” models of membrane perturbation or the formation of organized channels or “toroidal” pores (Fig. 2). AMPs could also have non-membrane activities, such as activation of immune mechanisms that result in anti-tumor effects [28]. The anti-cancer activity of specific classes of AMPs is described below. The first two examples highlight well-studied AMPs and their anti-cancer effects, while the second two exemplify the current trends for discovery of new AMPs with cytotoxicity against cancerous cells.
2.1.1. Magainins
Magainins, derived from the skin of the African clawed frog, Xenopus laevis, were among the first AMPs tested for anti-cancer activity [29]. Early work showed that magainin 2 formed an amphiphilic α-helical structure (Fig. 3A) that porated membranes [30] and improved the survival of animals bearing tumors [31]. Local treatment with magainin 2 in a xenograft model of tumors in nude mice, led to the ablation of the tumors [32]. Multiple lines of evidence support the cytotoxic effects of magainin 2 in melanoma, breast and lung cancers. As example, magainin 2 was highly effective inhibiting the proliferation of bladder cancer cell lines with a range of IC50 values of 31–135μM (BrdU assay), while comparable IC50 values for normal fibroblast cells lines were undetermined [33]. Additional studies demonstrated that the anti-cancer effects of magainins were selective. Lower concentrations of magainins were found to be toxic to cancer cells but not lymphocytes or fibroblasts, and magainins were resistant to serum proteolysis [34,32]. Conjugation of magainin 2 to a tumor-homing peptide, bombesin, exemplified the potential uses of the anti-tumor effects of the conjugate peptide (MG2B) [35]. Bombesin alone was not cytotoxic, while MG2B had an IC50 range of 10–15 μM in breast cancer and melanoma cell lines that was at about 10 times lower than unconjugated magainin 2 and 6–10 lower than the IC50 for normal fibroblasts [35]. The utility of magainin 2 as an anti-cancer agent is bolstered by the fact that it’s mechanism of action makes it a poor candidate for development of resistance [36].
Fig. 3. Solved structures of peptides used in pre-clinical and clinical trials.
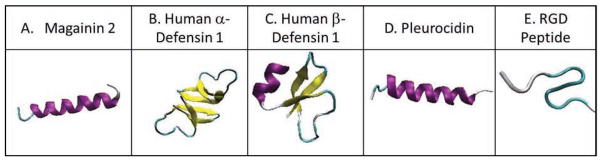
(A) The structure of the Magainin 2 α-helical peptide is shown and was derived from pdb ID of 2MAG. (B) Human α-defensin 1, a mostly α-sheet peptide has a pdb ID of 3LO9. (C) Human β-defensin 1 peptide has a pdb ID of 2NLS. (D) Pleurocidin structure is from pdb ID 1Z64. (E) The RGD peptide is based on pdb ID 1FUL.
2.1.2. Cecropins
Cecropin A and B belong to the cecropin-family of AMPs isolated from the hemolymph of the giant silk moth Hyalophora cecropia and were later found in mammals. The basic structure of both cecropins A and B consists of two α helices, bearing an amphipathic N-terminus and a hydrophobic C-terminus, with cytotoxic anti-cancer activity associated with the features of the N-terminus [37,38]. Both cecropins A and B can lyse cancer cells at concentrations below that which damage normal cells [39,40]. Cecropin B and analogs displayed IC50 values that ranged from 3–17 μM against different cancer cell lines, like HL-60, K-562 or Jurkat, while IC50 values for 3T3 fibroblasts were >50 μM [39]. Cecropin B also showed in vitro killing activity against multi-drug resistance (MDR) human breast and ovarian cancer cell lines and also in vivo anti-cancer effects in mice with colon adenocarcinomas [41]. The cecropin-family of antimicrobial peptides have selective and cytotoxic efficacy by targeting nonpolar lipid cell membranes and forming ion-permeable channels as shown in non-muscle invasive bladder cancer [42]. In the case of bladder cancer cells, average IC50 values for cell viability ranged between 185 and 251 μg/ml, doses that were much lower than killed fibroblasts [42]. A synergistic lethal effect was observed when cecropin A was combined with 5-fluoroouracil in the treatment of human leukemia cells [40].
2.1.3. Defensins
Defensins belong to one of the subgroups of AMPs with variations of the β-sheet structure (Figs. 3B, 3C). Defensins are small (3.5–4 kD), cysteine-rich peptides that are found in mammals, insects, plants, and fungi. Humans produce α and β defensins as part of an effective innate immune response to pathogens. Alpha-defensins (HNP1, HNP2, HNP3) can be found in the granules of neutrophils or intestinal Paneth cells (HD5, HD6) whereas Beta-defensins are mainly produced in the mucosal environment. Permeabilization of membranes by HNPs is accomplished by their ability to form channels in lipid bilayers [43]. Several studies found defensins in fluids and cells associated with malignancies [44]. In colorectal cancer, levels of HNP1–3 were increased in tumor tissues compared to normal tissues [45]. Such work suggested that defensins could be used as biomarkers for early detection of cancer. As therapeutic tools, the cytotoxic activity of HNPs1–3 has been reported for human tumor cells, with 100 μg/ml HNP1 showing significant cytotoxicity against oral squamous cell carcinoma [46]. Other anti-cancer effects documented for HNPs include the ability to cause DNA damage and interfere with neovascularization of tumors [47,48]. One problem that emerges is that HNP-mediated cytotoxicity is not specific to tumor cells, but can also cause the lysis of normal leukocytes or epithelial cell, and is inhibited by serum [49, 50]. To address this problem, novel variants of defensins have been developed. Coprisin is an insect defensin-like peptide from which a synthetic peptide (cationic 9-mer dimer), called CopA3, was synthesized [51]. CopA3 displayed antimicrobial activity and inhibited the growth of pancreatic (IC50 of 61.7μM) and hepatocellular (IC50 of 67.8μM) cancer cells. This is an example of a synthetic peptide based on a naturally occurring substance that exhibits inhibitory activities against microbes and has potential use as a novel anti-cancer agent.
2.1.4. Pleurocidin
Pleurocidin was originally isolated from the winter flounder, Pleuronectes americanus [52] which express high levels of bioactive peptides most likely as result of their heavy reliance on innate immune responses. Similar to other AMP’s[52], pleurocidin possess as a signal sequence at the N-terminus and an acidic peptide sequence at the C-terminus, which may promote secretion while protecting the host and enabling pore formation. Such conserved sequences are shared among other AMPs [53]. Using the sequences from conversed regions of pleurocidin, new pleurocidin-like cationic AMPs were discovered that had inhibitory activity [54]. Two of these, NRC-03 and NRC-07, have also exhibited activity against breast cancer cells [55]. Examination of the sequence of NRC-03 indicated an unstructured cationic amino terminus and an α-helical segment linked by two glycine residues (Fig. 3D). The sequence of NRC-07 suggested that it could also form an α-helix. Subsequent evaluation of the tumoricidal action revealed that both NRC-03 and NRC-07 could kill breast cancer cells and enhance the toxicity of chemotherapeutic drugs such as cisplatin or docetaxel [55]. At 50μM, NRC-03 and NRC-07 displayed about 75–94% cytotoxicity against breast cancer cell lines [55]. Evaluation of cells treated with NRC-03 and NRC-07 showed that these AMPs caused cell death by binding to negatively charged molecules on the cell membrane. Death could also ensue upon loss of mitochondrial membrane integrity. In this manner, both AMPs could inhibit the growth of breast cancer cells in a murine xenograft model [55]. Interestingly, the pleurocidin-family of cationic AMPs tends to exhibit cytotoxicity in slow-growing cancer cells that overexpress P-glycoprotein [55].
2.2. BCL-2 Family Peptides and BH3 Mimetics
Mitochondria are central to the apoptotic process. The intermembrane space of mitochondria holds numerous proteins that are effectors of cell death, the classic example being cytochrome c. Control over the release of apoptosis-inducing factors, such as cytochrome C, from mitochondria is governed by the Bcl-2 family of proteins. The Bcl-2 family is comprised of two groups: anti-apoptotic proteins like Bcl-2, Bcl-XL and Mcl-1, and pro-apoptotic proteins like Bax, Bak, Bid, Bim or Bad [56]. The latter group is subdivided into the multi-domain effectors of cell death, like Bax or Bak, and the BH3-only proteins, like Bad, Bim or Bid, that regulate the activity of anti- or pro-apoptotic members [57]. The mechanism of action for the Bcl-2 family proteins depends on their ability to associate with organelles, specifically the mitochondria or ER, and elicit effects that lead to the release of apoptotic factors. Characterizing this activity at a molecular level has been challenging, since Bcl-2 proteins may adopt distinct conformations whether in soluble or membrane-bound form. However, similarity of structure with pore forming proteins like colicin or toxins [58–60] suggested that the Bcl-2 proteins likely have pore-forming activity. This permeabilizing effect has been demonstrated with Bax [61] and Bid (with Bax) [62] and led to the testing of minimal forms of Bax or Bid for poration activity (see below). Because of their importance in regulating apoptotic activity, Bcl-2 proteins are frequently mutated in cancerous cells in order to deregulate cell survival [63–65]. Indeed, Bcl-2 itself is one of the first examples of an oncogene, underscoring the role of this family of proteins in oncogenesis and tumor formation [66]. The release of apoptosis inducing factors from mitochondria depends on the balance of Bcl-2 proteins, with cell fate determined by over expression of one group over the other. For the therapeutic purpose of inducing cancer cell death, efforts are being directed at developing agents that either perturb the balance between pro- and anti-apoptotic Bcl-2 proteins or directly disrupt mitochondrial membrane integrity. The Bax-related pore forming peptides and the BH3-related peptides and mimetics are prominent examples of these efforts.
2.2.1. Bax-Related Pore-Forming Peptides
Bax is formed by nine α-helices of which the α5, α6 and α9 helices have the features of membrane binding domains. Peptides based on the α5 and α6 helices of Bax caused permeabilization of artificial lipid vesicles, resulting in the release of sequestered contents [67, 68]. Biophysical measurements indicated that the α5 or α6 helix of Bax could form a toroidal-like pore structure (Fig. 2) in lipid vesicles, particularly involving lysine/arginine residues in the helix-turn-helix region [68]. The lipidic pore forming properties of these helices are a point of similarity with AMPs [69]. Such studies provided insights on how the full-length Bax protein could permeabilize mitochondrial membranes and induce apoptosis. As a result, there is interest in producing minimally active forms of Bax to induce apoptosis for therapeutic applications. In this regard, GFP-tagged versions of the α5 and α6 helices of Bax induced apoptosis and, when fused to a cell-penetrating motif, caused cell death (LC50 ~15 μM) and the regression of murine mammary adenocarcinomas [70].
2.2.2. BH3-Related Peptides and Mimetics
Overexpression of anti-apoptotic proteins like Bcl-2 or Mcl-1 tips the balance in favor of cell survival and in part underlies the development of resistance to conventional therapeutics or chemotherapy [71, 72]. It follows that interfering with the ability of Bcl-2 anti-apoptotic proteins to interact and inhibit pro-apoptotic proteins, like Bax, could restore apoptosis in cancer cells. This is the premise behind the use of BH3 peptides and the development of BH3 mimetics. A peptide containing the BH3 domain from Bax caused apoptosis in treated cells, but this effect was due to interference with the interaction between Bax and Bcl-XL and not the direct ability of the peptide to activate Bax or induce its translocation to the mitochondria [73]. Such data indicated that molecules based on the BH3 domains of Bcl-2 proteins could promote apoptosis of cancer cells by blocking the association between pro-apoptotic multi-domain proteins and anti-apoptotic counterparts. However, such peptides had poor cellular permeability and issues with solubility and stability as well as the ability to target and permeabilize cancer cells. To address this, a Bid BH3 peptide was modified using a chemical staple to stabilize its α-helical conformation [74]. The stapled Bid BH3 peptide was able to activate Bax [75], was resistant to protease degradation and, by maintaining its α-helical form, could penetrate cell membranes and induce apoptosis as was shown in vitro and in vivo with leukemia cells [74].
Success with modified BH3 peptides and the presence of a deep groove within the structure of Bcl-2 proteins, like Bcl-XL [76], led to the development of small molecules or BH3 mimetics. This is an example of understanding a protein’s function and how peptides derived affect the protein’s activity can lead to pharmacologically active agents that mimic the peptide’s action. Three different classes of BH3 mimetics have had significant success in killing cancer cells in vitro and in pre-clinical mouse models and are undergoing clinical investigation in human cancer. The first of these, navitoclax/ABT-737, are molecular mimetics of the BH3-only protein, Bad, that displayed cytotoxicity in in vitro (IC50 <10 nM), and in vivo (75 mg/kg/d) tumor models [77, 78]. These compounds were selective against Bcl-2 and Bcl-XL and less so for Mcl-1 [79,80], although Bim could play a role in this latter effect. Obatoclax had less affinity for Bcl-2 and more for Mcl-1 [81], while AT-101 had modest affinity for Bcl-2, Bcl-XL and Mcl-1 and could disrupt the interactions between Bcl-XL and Bax or Bad [81]. While the affinity for Bcl-2 anti-apoptotic proteins is a feature of these compounds (more so for Obatolcax and AT-101), they also have significant anti-tumor effects that are independent of their intended BH3-mimetic properties [82]. These “off target” effects may be as relevant and critical as their intended Bcl-2/Bcl-XL/Mcl-1 inhibiting activities. In clinical trials, navitoclax demonstrated toxicity against selected hematological malignancies [83, 84].
3. CELL-PENETRATING AND TUMOR-TARGETING PEPTIDES
The anti-cancer efficacy of a drug or compound is limited by its capacity to target, penetrate membranes and localize within tumors. The ability to target cancerous tissues, and concentrate the desired therapeutic agent within those tissues, can be achieved via tumor-specific markers, specifically those found on tumors or tumor vasculature. Identification and characterization of tumor markers are the foundation of knowledge from which tumor-targeting peptides (TTPs) (also known as homing peptides) are being developed. As examples, the screening of phage-displayed peptide libraries and antibody based screens has successfully identified a number of TTPs that function in vivo and bind to molecules that are accessible and expressed on tumor vasculature. Most TTPs that enable delivery of therapeutic agents to tumors can be divided in two basic types. TTPs in the first group are linked to cell-penetrating peptides (CPPs) which allow the cargo to be transported through the cell membrane. On their own, these CPPs are not specific; specificity can be added by union with a TTP. TTPs that can bind directly to a tumor marker and, as a result, internalize cargo comprise the second group. Among the first CPPs discovered was HIV-Tat [85,86]. Attaching the Tat sequence (GRKKRRQRRRPPQ) could enable the internalization of cargo from the surrounding media by numerous cell types [87]. Peptides with inherent tumor targeting capacity are those with RGD (arginine/glycine/aspartic acid) or NGR (asparagine/glycine/arginine) sequences [88]. Effective use of Tat-like and RGD-like sequences to deliver cargo to tumors resulted in the development of numerous hybrid anti-cancer compounds in which the TTP (for example) is linked with a drug or cytotoxic peptide. In the following sections, we will review the various applications of CPPs, the RGD/NGR-delivery system and, using the synthetic, AMP-like cytotoxic peptide, (KLAKLAK)2, discuss the efficacy of newly emerging methods to introduce peptides into cancer cells.
3.1. Tat and pH-Sensitive Peptides
CPPs that deliver cargo to cells can range from small molecules (<1kDA) to large microparticles and can be conjugated to their cargo or interact in a manner that allows cellular delivery of a drug, vector or liposome. Of the CPPs extensively studied are the HIV-Tat peptide, penetratin (Antp), transportan and its variant TP10, and repeating units of arginine, R8 and R9. Common features of these peptides are cationic residues which can, in some instances, be separated by hydrophobic residues, leading to the formation of amphiphilic helices. The ability to penetrate membranes is equated with hydrophobicity and some CPPs can be extremely hydrophobic, for example the PFDYLI peptide [89]. However, extensive study of CPPs has yet to conclusively demonstrate the mechanisms by which these gain entry into cells, be it directly through the plasma membrane (Fig. 1B) or through an endocytic pathway that is clathrin-dependent or independent (Fig. 1C). Determining how CPPs and their cargo enter cells and escape endosomes or lysosomes to reach the cytosol (or nucleus) remains a technically challenging endeavor. This is complicated by the observation that small changes in experimental procedures or the sequence of the CPP can have profound effects on biological outcomes. Moreover, the effective extracellular dose range of CPPs can be over 10μM, indicating that endocytic delivery (and escape) may lead to inefficient cytosolic accumulation. The major challenge for the therapeutic use of most CPPs, like Tat or R8, remains the complete lack of tumor cell specificity. As example, an intraperitoneal injection of Tat fused to β-galactosidase resulted in recovery from kidneys, liver, lungs among other tissues, including the brain [90]. There is a need to find ways to home CPPs to tumor tissues. Recent studies have exploited the distinct physiology of the tumor microenvironment, such acidic pH, in order to produce novel CPPs with increased specificity for cancerous cells. Examples of how Tat-like sequence can be adapted for optimal tumor targeting and the development of pH sensitive CPPs are discussed.
3.1.1. Tat
Numerous studies have demonstrated the utility of Tat fusions to enable cell penetration of proteins, peptides or oligonucleotides [91]. Pre-clinical cancer studies, however, are limited by the lack of tumor specificity. One example was the conjugation of Tat to a peptide that activates p53, which was used to treat mice with implanted human cancerous tissue with positive results [92]. More recently, a peptide (with the Met docking sequence) that blocks the interaction between hepatocyte growth factor (HGF) and its receptor, Met, was delivered using either Tat or Antp. At concentrations of 10–25 μM Tat-Met or Antp-Met peptides inhibited HGF-mediated growth of cells and higher doses resulted in cytostatic activity [93]. Generation of a hybrid compound in which Tat was linked to chitosan/doxorubicin displayed IC50 values in the nanomolar range, using CT26 adenocarcinoma cells, which was two-fold less than chitosan/doxorubicin alone [94]. These studies show the potential anti-cancer use of Tat and similar CPPs as well as the need for modifications that enable tumor targeting and concentration of toxic cargo. One approach being tested is to hide the cationic CPP during transit to the tumor tissue and then expose the membrane penetrating activity of the CPP at the tumor site. Examples of this are pH-sensitive Tat-modified pegylated (PEG) liposomes or “Smart” compounds [95]. Steric hindrance provided by PEG shields the Tat peptide until the acidic tumor environment results in cleavage of the linker to which PEG was attached [96]. The efficacy of this system was shown in a liposomal formulation incorporating Doxorubicin (Doxil), with LC50 values demonstrating cytotoxicity in MCF-7 cells at 2 μg/ml compared to 38 μg/ml for Doxil alone [97].
3.1.2. pHLIP
A number of pathological conditions like cancer result in an acidic extracellular environment [98]. Tumor acidity is therefore a useful parameter for developing CPPs. A novel peptide was discovered, called pH (low)-dependent Insertion Peptide (pHLIP), that is derived from the bacteriorhodopsin C helix [99]. pHLIP can be found in three states: (1) water-soluble at neutral or pH >7, (2) on the surface of a lipid bilayer at neutral pH, and (3) inserted across the bilayers as a monomeric alpha-helix at pH < 6 [100, 101]. Hence, folding and membrane insertion of pHLIP occur when the pH drops from neutral (~7.4) to acidic (7.0–6.5 or less) [100]. The energy released from this conformational change of pHLIP can be used to move cargo across membranes [101]. As a result, pHLIP is an pH-targeting peptide shown to accumulate in acidic tissues such as tumors [102] and deliver cargo such as dyes, peptides and toxins [101]. Using a pHLIP-NIR (near infrared) dye construct, pHLIP was shown to accumulate and persist in tumor tissue at levels five times greater than other tissues like the kidneys [103]. Hence, pHLIP is a new type of CPP that exploits the inherent acidity of tumors and can transport cytotoxic cargo under conditions in which conventional anti-cancer drugs may be ineffective.
3.2. RGD and NGR-Containing Peptides
Operating from the premise that endothelial cells in tumor blood vessels have distinct protein markers not found in normal blood vessels, a phage peptide library was used to identify peptides that would “home” to tumor vasculature [88]. Two peptides with tumor-targeting sequences were discovered, one containing the RGD motif (Fig. 3E) and another with the NGR motif. Biochemical studies revealed that RGD is found in the integrin-interaction site of many ECM proteins. Hence RGD recognizes a class of integrins associated with tumors, specifically αvβ3 and αvβ5 integrins, which are overexpressed in angiogenic blood vessels [22, 104, 105]. NGR peptides recognize and bind to aminopeptidase N (also known as CD13) that is overexpressed by endothelial cells of many tumors [88,106] and could lead to improved tumor selectivity. More recently, NGR-containing peptides were found to convert to isoDGR by asparagine deamidation, resulting in αvβ3 integrin ligands that could mediate endothelial cell functions and tumor growth [107].
Both RGD and NGR peptides have been used to target a number of compounds to tumors and are being tested in different phases of human clinical trials (Table 1). An RGD peptide (GSSSGRGDSPA) coupled to a PEG (poly (ethylene glycol)) modified stearic acid-grafted chitosam micelle increased the efficacy of uptake in tumor types that overexpress integrins. This allowed for the shuttling of cargo (doxorubicin) into tumor cells [108]. By using c(RGDyK), a cyclic RGD peptide, the targeted delivery and uptake of micellar-like nanoparticles containing hydrophobic chemotherapeutic agents to cancerous cells overexpressing integrins was achieved [109]. The NGR targeting sequence was initially linked to doxorubicin and resulted in tumor regression in a murine xenograft model [88]. More recently, NGR was coupled to platinum anti-cancer drugs to enhance their targeting and incorporation within tumors [110]. This sequence homed specifically to solid tumors and, more strictly, to the endothelium of angiogenic blood vessels, and delivered a liposomal doxorubicin compound to treat orthotopic neuroblastoma xenografts in mice [111, 112]. The NGR peptide fused with a 15kDa actin fragment induced apoptosis in vitro by integrating into the cytoskeleton [113]. NGR and/or STR-R4 peptides, attached to the distal ends of PEG on liposomes, enhanced the uptake of the liposomes in CD13 positive cells [114]. Supported by such findings, cilengitide, the salt of a cyclized RGD-based pentapeptide, was made (EMD 121974, Merck). Cilengitide is undergoing testing in phase I, II and III clinical trials for non-small cell lung cancer, glioma, head and neck cancer, and prostate cancer to name a few (Table 1). Use in glioblastoma patients in phase II trials resulted in a six-month progression free survival rate of 12–15% [115, 116]. Another promising clinical approach is the delivery of the cytokine TNF (tumor necrosis factor) via RGD or NGR peptides. For NGR-hTNF, there are a number of on-going phase II–III clinical trials for ovarian, lung, colon and other cancers (Table 1). Using NGR-hTNF in a cohort of patients with hepatocellular carcinoma, minimal to no toxicity was observed and median survival rates of 8.9 months (average is 6 months) was achieved [117].
Table 1.
Representative List of Pre-Clinical and Clinical Trials of Therapeutic Peptides
| PEPTIDE NAME | THERAPEUTIC USE | CLINICAL DEVELOPMENT PHASE | REFERENCES/CLINICAL TRIAL SITE |
|---|---|---|---|
|
| |||
|
Group I Peptides
| |||
| Cecropin A and B | Cancer: | ||
| Leukemia | Pre-Clinical | Hui et al., 2002 | |
| Bladder | Pre-Clinical | Suttmann et al., 2008 | |
|
| |||
| Pleurocidin | Cancer: breast | Pre-Clinical | Hilchie et al., 2011 |
|
| |||
| Magainin2 | Cancer: | ||
| Bladder | Pre-Clinical | Lehmann et al., 2006 | |
| Anti-microbial: | |||
| Diabetic foot ulcers | Phase III | MarcoChem Corporation | |
|
| |||
| β-defensin | Anti-microbial: | Eastern Virginia Medical School/Merck | |
| Inflammatory Biomarker | Phase IV | ||
|
| |||
| α5-α6 Bax peptide | Cancer: apoptosis | Pre-Clinical | Valero et al, 2010 |
|
| |||
| BH3 domain | Cancer: apoptosis | Pre-Clinical | Walensky et al, 2004 |
|
| |||
| KLAKLAK | Cancer: glioblastoma | Pre-Clinical | Agemy et al, 2011 |
|
| |||
|
Group II Peptides
| |||
| Tat | Cancer | Pre-Clinical | |
| HIV: vaccine | Phase I, II | Istituto Superiore di Sanita | |
|
| |||
|
Group III Peptides
| |||
| RGD (Cilengitide, Delta 24-RGD, Delta 24-RGD 4C, RGD-K5) | Cancer: | ||
| Brain | Phase I | M.D. Anderson and others | |
| Phases II, | Erasmus Medical Center and others | ||
| Ovarian | Phase I | University of Alabama at Birmingham | |
| Head and Neck | Phase I and II | Merck, Chang Gung Memorial Hospital | |
| Prostate | Phase II | University of Michigan Cancer Center | |
| Lung | Phase I and II | Merck, University Hospital Mannheim | |
| Melanoma | Phase II | M.D. Anderson | |
|
| |||
| NGR (NGR-hTNF) | Cancer: | ||
| Ovarian | Phase II | MolMed S.p.A. | |
| Mesothelioma | Phase II, III | MolMed S.p.A. | |
| Lung | Phase II | MolMed S.p.A. | |
| Sarcoma | Phase II | MolMed S.p.A. | |
| Colon | Phase II | MolMed S.p.A. | |
| Hepatic | Phase II | MolMed S.p.A. | |
The next generation of RGD peptides is being designed to improve the penetration of cargo into tumors. Permeation of drugs into tumor tissue is a problem given that blood flow through tumor vessels may be abnormal and tumors could have high interstitial pressures [18, 19]. Novel RGD peptides have been adapted with a tissue penetration motif, R/KXXR/K, which must be exposed for activity – called the C end rule (CendR). An RGD peptide containing a CendR motif, iRGD, was developed that binds to integrins and then upon proteolytic cleavage, produces the CendR motif, which enables binding to neuropilin-1 (NRP-1), triggering tissue penetration [118, 119]. A potential use for iRGD is increasing the amount of drugs that penetrates tumors. Combining iRGD with drugs like Paclitaxel (Taxol) or Doxorubicin allowed increased accumulation of the drugs in tumor tissues, indicating its possible use in combination therapies [119].
3.3. Targeting the KLA Peptide to Tumors
The amphipathic α-helix is a common motif found in AMPs as well as other biologically active peptides. The ability to permeabilize membranes and result in cytotoxicity is a common activity for many of these peptides, including most AMPs being developed for anti-cancer applications. Using this information, synthetic peptides based on the selectivity of AMPs for membranes were produced to replicate these amphipathic features [120]. Of these, the most successful is (KLAKLAK)2 (KLA peptide), which displayed significant anti-microbial activity at 1% the concentrations needed to kill mammalian cells [120]. Because of its selectivity for anionic membranes of prokaryotes, the KLA peptide is poorly permeable to eukaryotic plasma membranes and needs to be fused to CPPs and/or TTPs to promote cellular uptake and tumor targeting. Once inside the cell, the KLA peptide will disrupt mitochondria. Fusing the KLA peptide with a tumor-homing motif like RGD (RGD-4C) resulted in the apoptotic cell death of cancer cells [121]. Another approach, while less tumor specific, included fusing cationic sequences to the KLA peptide to promote plasma membrane insertion. When coupled to a cationic peptide transduction domain (PTD-5), the KLA peptide became a potent inducer of apoptosis [122]. Attachment of a cell penetrating domain (R7 or RRRRRRR) to the KLA peptide increased cellular uptake and efficient apoptosis in tumor cell lines and human xenografts [123]. The IC50 of R7-KLA ranged from 3–25 μM in different cancer cell lines, while KLA alone had IC50 values >100 μM [123]. For specific tumor targeting, a prostate-specific membrane antigen (PSMA) targeting peptide (PTP) was conjugated to the KLA peptide and induced dose-dependent death of human prostate cancer cell lines by apoptotic (~10μM PTP-KLA) and oncotic/necrotic (~25 μM PTP-KLA) mechanisms. The PTP-KLA fusion peptide exploited the expression of the PSMA on prostate cancer cells for targeted delivery of the KLA peptide [124]. Along the same line, fusing the NH2-terminus of TCTP (human translationally controlled tumor protein) to the KLA peptide resulted in TCTP-KLA which displayed an IC50 between 7–10 μM and caused regression of lung carcinomas implanted in mice [125]. These examples show the potential use of TTPs fused to a cytotoxic peptide, which on its own has no selective tumor-homing capacity, to produce a highly efficacious, tumor cell killing complex.
Direct modifications to the KLA peptide or encapsulation in nanoparticles are being explored to develop the cytotoxic and tumor-targeting activity of the peptide, while reducing potential toxic “off-target” effects. The addition of 3-triphenylphosphonium cations (TPPs) improved the ability of the KLA peptide to target to mitochondria and induce apoptosis (i.e. 30μM caused 60% mortality of HeLa cells), increasing the biological activity of the peptide [126]. The formation of a pH-responsive nanocomplex consisting of GCS-g-DMA (glycol chitosan graft with 2,3-dimethylmaleic acid) and KLA peptide resulted in release of the KLA peptide in response to increasing acidic pH, and the accumulation of the KLA peptide in murine tumors [127]. In order to produce a nanosystem to target tumor vasculature, the KLA peptide was fused to a TTP (CGKRK peptide) and multivalent presentation achieved using iron oxide nanoparticles. The complex targeted mitochondria and induced apoptosis, causing significant tumor regression in a murine model of glioblastoma [128].
4. CONCLUSIONS AND CHALLENGES
The application of peptide therapeutics in the treatment of cancer is not a new field but one that has resurged in the recent years with the discovery of new tumor-targeting markers and naturally occurring or synthetic cytotoxic peptides. Features of tumors that could facilitate the targeting and uptake of peptides include the increased negative charge of the plasma membrane of tumor cells and the leakiness of the tumor vasculature. Taking advantage of tumor-specific membrane differences, AMPs are proving to be effective anti-cancer agents that cause cell lysis and induce apoptosis, with a lower risk of developing resistance. Similarly, pore-forming cytotoxic peptides based on transmembrane sequences from apoptosis-inducing Bcl-2 proteins or BH3 peptides/mimetics, which interfere with the binding of anti- and pro-apoptotic Bcl-2 proteins, can promote mitochondrial-mediated apoptosis of tumor cells, specifically those overexpressing pro-survival proteins. However, neither AMPs nor Bcl-2 family-derived peptides can directly target tumors via tumor-specific markers. Such targeting is mediated by TTPs that in most part do not have inherent cytotoxic activity but can deliver drugs or cytotoxic peptides directly to tumors. Among the most effective TTPs are those bearing RGD or NGR motifs that bind to receptors overexpressed on endothelial cells of tumor vasculature. Modifications of these TTPs can elicit changes in the tumor environment to promote the accumulation of higher concentrations of anti-cancer agents or drugs within tumors. The combinations of TTPs with cytotoxic peptides or drugs in nanoparticles or as part of a self-assembled complex could produce the “magic bullet” that specifically targets and eradicates tumor cells.
The challenges to produce efficient and specific tumor-eradicating peptides not toxic to normal tissue are severalfold. AMPs or cytotoxic “pore-forming” peptides (like those based on the Bcl-2 family) are promising anti-cancer agents that can be effective against tumor cells at low concentrations non-lethal to normal tissue. However, AMPs or “pore-forming” peptides have no selective tumor targeting capacity. While, AMPs were initially thought to “punch holes” in membranes, the realization is that a more complex mechanism of action is involved. Whether this mechanism is based on structural properties, the formation of a channel or pore, or just thinning of the membrane has yet to be understood. Other issues that need to be addressed include determining potential toxicity or “off-target” effects, improving stability (in serum) or bioavailability, assessing pharmacokinetic properties and ensuring delivery to tumor cells. Delivery is among the most important concerns. The need to improve selectivity of AMPs or pore-forming peptides for cancer cells may require a better understanding of the structure-activity relationship of these molecules. The fusion of cytotoxic peptides with TTPs is a logical direction that has proven effective, reducing concentrations needed to achieve cytotoxic IC50 values. To produce the next generation of TTPs that bind more specifically to target surface molecules on tumor cells or tumor vasculature requires new discovery tools. In this regard, the screening of phage-displayed peptide libraries has already proved productive with the identification of the RGD and NGR motifs. Other approaches include the bioinformatics-driven design of a peptide library based on the solved crystal structures of proteins in which virtual docking of peptides is incorporated and new ligands for receptor targets can be identified. The ultimate goal, however, is to bring the use of peptide therapeutics into the clinic. As shown in Table 1, most cytotoxic peptides, like the antimicrobial peptides, are in pre-clinical stages of testing and have yet to enter human clinical trials for cancer. The critical and validating research findings that result from testing in vitro cancer cell lines and in vivo mouse tumor models are just the initial steps leading to the translational studies that will, if successful, positively impact upon the survival rates of cancer patients.
Acknowledgments
This work was supported in part by an NIH/NIGMS grant GM083324 (Khaled).
ABBREVIATIONS
- AMP
Anti-microbial peptides
- Antp
Antenapedia
- Bcl-2
B-cell Lymphoma 2
- BH
Bcl-2 homology
- CCP
Cell penetrating peptide
- CendR
C end rule
- CopA3
Coprisin analog peptide A3
- ECM
Extracellular matrix
- EPR
Enhanced permeability and retention
- ER
Endoplasmic reticulum
- GCS-g-DMA
Glycol chitosan graft with 2,3-dimethylmaleic acid
- HD
Human alpha defensin
- HNP
Human Neutrophil Peptide
- Muc 1
Mucin 1
- NGR
Asparagine/Glycine/Arginine
- NRP-1
Neuropilin-1
- PC
phosphatidycholine
- PE
Phosphatidylethanolamine
- PEG
Poly (ethylene glycol)
- pHLIP
pH (low)-dependent Insertion Peptide
- PS
Phosphatiydserine
- PSMA
Prostate-specific membrane antigen
- PTP
PSMA targeting peptide
- PTD-5
Peptide transduction domain
- RGD
Arginine/Glycine/Aspartic acid
- Tat
Trans-activating transcriptional activator
- TCTP
Human translationally controlled tumor protein
- TNF
Tumor necrosis factor
- TPP
3-triphenylphosphonium
- TTP
Tumor targeting peptide
Footnotes
CONFLICT OF INTEREST
None declared.
References
- 1.Kalyanaraman B, Joseph J, Kalivendi S, Wang S, Konorev E, Kotamraju S. Doxorubicin-induced apoptosis: implications in cardiotoxicity. Mol Cell Biochem. 2002;234–235:119–124. [PubMed] [Google Scholar]
- 2.Naumov GN, Townson JL, MacDonald IC, Wilson SM, Bramwell VH, Groom AC, Chambers AF. Ineffectiveness of doxorubicin treatment on solitary dormant mammary carcinoma cells or late-developing metastases. Breast Cancer Res Treat. 2003;82:199–206. doi: 10.1023/B:BREA.0000004377.12288.3c. [DOI] [PubMed] [Google Scholar]
- 3.Fuertes G, Gimenez D, Esteban-Martin S, Garcia-Saez A, Sanchez O, Salgado J. Role of membrane lipids for the activity of pore forming peptides and proteins. Adv Exp Med Biol. 2010;677:31–55. doi: 10.1007/978-1-4419-6327-7_4. [DOI] [PubMed] [Google Scholar]
- 4.Fuertes G, Gimenez D, Esteban-Martin S, Sanchez-Munoz OL, Salgado J. A lipocentric view of peptide-induced pores. Eur Biophys J. 2011;40:399–415. doi: 10.1007/s00249-011-0693-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rekdal O, Haug BE, Kalaaji M, Hunter HN, Lindin I, Israelsson I, Solstad T, Yang N, Brandl M, Mantzilas D, Vogel HJ. Relative spatial positions of tryptophan and cationic residues in helical membrane-active peptides determine their cytotoxicity. J Biol Chem. 2012;287:233–244. doi: 10.1074/jbc.M111.279281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Branco MC, Sigano DM, Schneider JP. Materials from peptide assembly: towards the treatment of cancer and transmittable disease. Curr Opin Chem Biol. 2011;15:427–434. doi: 10.1016/j.cbpa.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bevers EM, Comfurius P, Zwaal RF. Regulatory mechanisms in maintenance and modulation of transmembrane lipid asymmetry: pathophysiological implications. Lupus. 1996;5:480–487. doi: 10.1177/096120339600500531. [DOI] [PubMed] [Google Scholar]
- 8.Utsugi T, Schroit AJ, Connor J, Bucana CD, Fidler IJ. Elevated expression of phosphatidylserine in the outer membrane leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res. 1991;51:3062–3066. [PubMed] [Google Scholar]
- 9.Riedl S, Rinner B, Asslaber M, Schaider H, Walzer S, Novak A, Lohner K, Zweytick D. In search of a novel target - phosphatidylserine exposed by non-apoptotic tumor cells and metastases of malignancies with poor treatment efficacy. Biochim Biophys Acta. 2011;1808:2638–2645. doi: 10.1016/j.bbamem.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riedl S, Zweytick D, Lohner K. Membrane-active host defense peptides--challenges and perspectives for the development of novel anticancer drugs. Chem Phys Lipids. 2011;164:766–781. doi: 10.1016/j.chemphyslip.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bafna S, Kaur S, Batra SK. Membrane-bound mucins: the mechanistic basis for alterations in the growth and survival of cancer cells. Oncogene. 2010;29:2893–2904. doi: 10.1038/onc.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fadnes B, Rekdal O, Uhlin-Hansen L. The anticancer activity of lytic peptides is inhibited by heparan sulfate on the surface of the tumor cells. BMC Cancer. 2009;9:183. doi: 10.1186/1471-2407-9-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.viel-Ronen S, Lau SK, Pintilie M, Lau D, Liu N, Tsao MS, Jothy S. Glypican-3 is overexpressed in lung squamous cell carcinoma, but not in adenocarcinoma. Mod Pathol. 2008;21:817–825. doi: 10.1038/modpathol.2008.37. [DOI] [PubMed] [Google Scholar]
- 14.Sherbet GV. Membrane fluidity and cancer metastasis. Exp Cell Biol. 1989;57:198–205. doi: 10.1159/000163526. [DOI] [PubMed] [Google Scholar]
- 15.Sok M, Sentjurc M, Schara M, Stare J, Rott T. Cell membrane fluidity and prognosis of lung cancer. Ann Thorac Surg. 2002;73:1567–1571. doi: 10.1016/s0003-4975(02)03458-6. [DOI] [PubMed] [Google Scholar]
- 16.May GL, Wright LC, Dyne M, Mackinnon WB, Fox RM, Mountford CE. Plasma membrane lipid composition of vinblastine sensitive and resistant human leukaemic lymphoblasts. Int J Cancer. 1988;42:728–733. doi: 10.1002/ijc.2910420517. [DOI] [PubMed] [Google Scholar]
- 17.Zwaal RF, Schroit AJ. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood. 1997;89:1121–1132. [PubMed] [Google Scholar]
- 18.Hambley TW, Hait WN. Is anticancer drug development heading in the right direction? Cancer Res. 2009;69:1259–1262. doi: 10.1158/0008-5472.CAN-08-3786. [DOI] [PubMed] [Google Scholar]
- 19.Jain RK. Transport of molecules, particles, and cells in solid tumors. Annu Rev Biomed Eng. 1999;1:241–263. doi: 10.1146/annurev.bioeng.1.1.241. [DOI] [PubMed] [Google Scholar]
- 20.Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure - an obstacle in cancer therapy. Nat Rev Cancer. 2004;4:806–813. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- 21.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 22.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264:569–571. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 23.Nilsson F, Kosmehl H, Zardi L, Neri D. Targeted delivery of tissue factor to the ED-B domain of fibronectin, a marker of angiogenesis, mediates the infarction of solid tumors in mice. Cancer Res. 2001;61:711–716. [PubMed] [Google Scholar]
- 24.Pilch J, Brown DM, Komatsu M, Jarvinen TA, Yang M, Peters D, Hoffman RM, Ruoslahti E. Peptides selected for binding to clotted plasma accumulate in tumor stroma and wounds. Proc Natl Acad Sci U S A. 2006;103:2800–2804. doi: 10.1073/pnas.0511219103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ye F, Wu X, Jeong EK, Jia Z, Yang T, Parker D, Lu ZR. A peptide targeted contrast agent specific to fibrin-fibronectin complexes for cancer molecular imaging with MRI. Bioconjug Chem. 2008;19:2300–2303. doi: 10.1021/bc800211r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Z, Wang G. APD: the Antimicrobial Peptide Database. Nucleic Acids Res. 2004;32:D590–D592. doi: 10.1093/nar/gkh025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papo N, Shai Y. Host defense peptides as new weapons in cancer treatment. Cell Mol Life Sci. 2005;62:784–790. doi: 10.1007/s00018-005-4560-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chernysh S, Kim SI, Bekker G, Pleskach VA, Filatova NA, Anikin VB, Platonov VG, Bulet P. Antiviral and antitumor peptides from insects. Proc Natl Acad Sci U S A. 2002;99:12628–12632. doi: 10.1073/pnas.192301899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zasloff M. Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc Natl Acad Sci U S A. 1987;84:5449–5453. doi: 10.1073/pnas.84.15.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cruciani RA, Barker JL, Zasloff M, Chen HC, Colamonici O. Antibiotic magainins exert cytolytic activity against transformed cell lines through channel formation. Proc Natl Acad Sci U S A. 1991;88:3792–3796. doi: 10.1073/pnas.88.9.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baker MA, Maloy WL, Zasloff M, Jacob LS. Anticancer efficacy of Magainin2 and analogue peptides. Cancer Res. 1993;53:3052–3057. [PubMed] [Google Scholar]
- 32.Soballe PW, Maloy WL, Myrga ML, Jacob LS, Herlyn M. Experimental local therapy of human melanoma with lytic magainin peptides. Int J Cancer. 1995;60:280–284. doi: 10.1002/ijc.2910600225. [DOI] [PubMed] [Google Scholar]
- 33.Lehmann J, Retz M, Sidhu SS, Suttmann H, Sell M, Paulsen F, Harder J, Unteregger G, Stockle M. Antitumor activity of the antimicrobial peptide magainin II against bladder cancer cell lines. Eur Urol. 2006;50:141–147. doi: 10.1016/j.eururo.2005.12.043. [DOI] [PubMed] [Google Scholar]
- 34.Matsuzaki K, Sugishita K, Harada M, Fujii N, Miyajima K. Interactions of an antimicrobial peptide, magainin 2: with outer and inner membranes of Gram-negative bacteria. Biochim Biophys Acta. 1997;1327:119–130. doi: 10.1016/s0005-2736(97)00051-5. [DOI] [PubMed] [Google Scholar]
- 35.Liu S, Yang H, Wan L, Cai HW, Li SF, Li YP, Cheng JQ, Lu XF. Enhancement of cytotoxicity of antimicrobial peptide magainin II in tumor cells by bombesin-targeted delivery. Acta Pharmacol Sin. 2011;32:79–88. doi: 10.1038/aps.2010.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lincke CR, van der Bliek AM, Schuurhuis GJ, van d V, Smit JJ, Borst P. Multidrug resistance phenotype of human BRO melanoma cells transfected with a wild-type human mdr1 complementary DNA. Cancer Res. 1990;50:1779–1785. [PubMed] [Google Scholar]
- 37.Srisailam S, Kumar TK, Arunkumar AI, Leung KW, Yu C, Chen HM. Crumpled structure of the custom hydrophobic lytic peptide cecropin B3. Eur J Biochem. 2001;268:4278–4284. doi: 10.1046/j.1432-1327.2001.02345.x. [DOI] [PubMed] [Google Scholar]
- 38.Hoskin DW, Ramamoorthy A. Studies on anticancer activities of antimicrobial peptides. Biochim Biophys Acta. 2008;1778:357–375. doi: 10.1016/j.bbamem.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen HM, Wang W, Smith D, Chan SC. Effects of the anti-bacterial peptide cecropin B and its analogs, cecropins B-1 and B-2, on liposomes, bacteria, and cancer cells. Biochim Biophys Acta. 1997;1336:171–179. doi: 10.1016/s0304-4165(97)00024-x. [DOI] [PubMed] [Google Scholar]
- 40.Hui L, Leung K, Chen HM. The combined effects of antibacterial peptide cecropin A and anti-cancer agents on leukemia cells. Anticancer Res. 2002;22:2811–2816. [PubMed] [Google Scholar]
- 41.Moore AJ, Devine DA, Bibby MC. Preliminary experimental anticancer activity of cecropins. Pept Res. 1994;7:265–269. [PubMed] [Google Scholar]
- 42.Suttmann H, Retz M, Paulsen F, Harder J, Zwergel U, Kamradt J, Wullich B, Unteregger G, Stockle M, Lehmann J. Antimicrobial peptides of the Cecropin-family show potent antitumor activity against bladder cancer cells. BMC Urol. 2008;8:5. doi: 10.1186/1471-2490-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kagan BL, Selsted ME, Ganz T, Lehrer RI. Antimicrobial defensin peptides form voltage-dependent ion-permeable channels in planar lipid bilayer membranes. Proc Natl Acad Sci U S A. 1990;87:210–214. doi: 10.1073/pnas.87.1.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Droin N, Hendra JB, Ducoroy P, Solary E. Human defensins as cancer biomarkers and antitumour molecules. J Proteomics. 2009;72:918–927. doi: 10.1016/j.jprot.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 45.Melle C, Ernst G, Schimmel B, Bleul A, Thieme H, Kaufmann R, Mothes H, Settmacher U, Claussen U, Halbhuber KJ, Von EF. Discovery and identification of alpha-defensins as low abundant, tumor-derived serum markers in colorectal cancer. Gastroenterology. 2005;129:66–73. doi: 10.1053/j.gastro.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 46.McKeown ST, Lundy FT, Nelson J, Lockhart D, Irwin CR, Cowan CG, Marley JJ. The cytotoxic effects of human neutrophil peptide-1 (HNP1) and lactoferrin on oral squamous cell carcinoma (OSCC) in vitro. Oral Oncol. 2006;42:685–690. doi: 10.1016/j.oraloncology.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 47.Gera JF, Lichtenstein A. Human neutrophil peptide defensins induce single strand DNA breaks in target cells. Cell Immunol. 1991;138:108–120. doi: 10.1016/0008-8749(91)90136-y. [DOI] [PubMed] [Google Scholar]
- 48.Chavakis T, Cines DB, Rhee JS, Liang OD, Schubert U, Hammes HP, Higazi AA, Nawroth PP, Preissner KT, Bdeir K. Regulation of neovascularization by human neutrophil peptides (alpha-defensins): a link between inflammation and angiogenesis. FASEB J. 2004;18:1306–1308. doi: 10.1096/fj.03-1009fje. [DOI] [PubMed] [Google Scholar]
- 49.Lichtenstein A, Ganz T, Selsted ME, Lehrer RI. In vitro tumor cell cytolysis mediated by peptide defensins of human and rabbit granulocytes. Blood. 1986;68:1407–1410. [PubMed] [Google Scholar]
- 50.Nishimura M, Abiko Y, Kurashige Y, Takeshima M, Yamazaki M, Kusano K, Saitoh M, Nakashima K, Inoue T, Kaku T. Effect of defensin peptides on eukaryotic cells: primary epithelial cells, fibroblasts and squamous cell carcinoma cell lines. J Dermatol Sci. 2004;36:87–95. doi: 10.1016/j.jdermsci.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 51.Kim IW, Kim SJ, Kwon YN, Yun EY, Ahn MY, Kang DC, Hwang JS. Effects of the synthetic coprisin analog peptide, CopA3 in pathogenic microorganisms and mammalian cancer cells. J Microbiol Biotechnol. 2012;22:156–158. doi: 10.4014/jmb.1109.09014. [DOI] [PubMed] [Google Scholar]
- 52.Cole AM, Weis P, Diamond G. Isolation and characterization of pleurocidin, an antimicrobial peptide in the skin secretions of winter flounder. J Biol Chem. 1997;272:12008–12013. doi: 10.1074/jbc.272.18.12008. [DOI] [PubMed] [Google Scholar]
- 53.Yoshida K, Mukai Y, Niidome T, Takashi C, Tokunaga Y, Hatakeyama T, Aoyagi H. Interaction of pleurocidin and its analogs with phospholipid membrane and their antibacterial activity. J Pept Res. 2001;57:119–126. doi: 10.1034/j.1399-3011.2001.00802.x. [DOI] [PubMed] [Google Scholar]
- 54.Patrzykat A, Gallant JW, Seo JK, Pytyck J, Douglas SE. Novel antimicrobial peptides derived from flatfish genes. Antimicrob Agents Chemother. 2003;47:2464–2470. doi: 10.1128/AAC.47.8.2464-2470.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hilchie AL, Doucette CD, Pinto DM, Patrzykat A, Douglas S, Hoskin DW. Pleurocidin-family cationic antimicrobial peptides are cytolytic for breast carcinoma cells and prevent growth of tumor xenografts. Breast Cancer Res. 2011;13:R102. doi: 10.1186/bcr3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leber B, Lin J, Andrews DW. Still embedded together binding to membranes regulates Bcl-2 protein interactions. Oncogene. 2010;29:5221–5230. doi: 10.1038/onc.2010.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shamas-Din A, Brahmbhatt H, Leber B, Andrews DW. BH3-only proteins: Orchestrators of apoptosis. Biochim Biophys Acta. 2011;1813:508–520. doi: 10.1016/j.bbamcr.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 58.Chou JJ, Li H, Salvesen GS, Yuan J, Wagner G. Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell. 1999;96:615–624. doi: 10.1016/s0092-8674(00)80572-3. [DOI] [PubMed] [Google Scholar]
- 59.Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong SL, Ng SL, Fesik SW. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- 60.Suzuki M, Youle RJ, Tjandra N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell. 2000;103:645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 61.Saito M, Korsmeyer SJ, Schlesinger PH. BAX-dependent transport of cytochrome c reconstituted in pure liposomes. Nat Cell Biol. 2000;2:553–555. doi: 10.1038/35019596. [DOI] [PubMed] [Google Scholar]
- 62.Epand RF, Martinou JC, Fornallaz-Mulhauser M, Hughes DW, Epand RM. The apoptotic protein tBid promotes leakage by altering membrane curvature. J Biol Chem. 2002;277:32632–32639. doi: 10.1074/jbc.M202396200. [DOI] [PubMed] [Google Scholar]
- 63.Higashiyama M, Doi O, Kodama K, Yokouchi H, Tateishi R. High prevalence of bcl-2 oncoprotein expression in small cell lung cancer. Anticancer Res. 1995;15:503–505. [PubMed] [Google Scholar]
- 64.Olopade OI, Adeyanju MO, Safa AR, Hagos F, Mick R, Thompson CB, Recant WM. Overexpression of BCL-x protein in primary breast cancer is associated with high tumor grade and nodal metastases. Cancer J Sci Am. 1997;3:230–237. [PubMed] [Google Scholar]
- 65.Dosaka-Akita H, Katabami M, Hommura H, Fujioka Y, Katoh H, Kawakami Y. Bcl-2 expression in non-small cell lung cancers: higher frequency of expression in squamous cell carcinomas with earlier pT status. Oncology. 1999;56:259–264. doi: 10.1159/000011974. [DOI] [PubMed] [Google Scholar]
- 66.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c- myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 67.Garcia-Saez AJ, Coraiola M, Dalla SM, Mingarro I, Menestrina G, Salgado J. Peptides derived from apoptotic Bax and Bid reproduce the poration activity of the parent full-length proteins. Biophys J. 2005;88:3976–3990. doi: 10.1529/biophysj.104.058008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garcia-Saez AJ, Coraiola M, Serra MD, Mingarro I, Muller P, Salgado J. Peptides corresponding to helices 5 and 6 of Bax can independently form large lipid pores. FEBS J. 2006;273:971–981. doi: 10.1111/j.1742-4658.2006.05123.x. [DOI] [PubMed] [Google Scholar]
- 69.Qian S, Wang W, Yang L, Huang HW. Structure of transmembrane pore induced by Bax-derived peptide: evidence for lipidic pores. Proc Natl Acad Sci U S A. 2008;105:17379–17383. doi: 10.1073/pnas.0807764105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Valero JG, Sancey L, Kucharczak J, Guillemin Y, Gimenez D, Prudent J, Gillet G, Salgado J, Coll JL, Aouacheria A. Bax-derived membrane-active peptides act as potent and direct inducers of apoptosis in cancer cells. J Cell Sci. 2011;124:556–564. doi: 10.1242/jcs.076745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miyashita T, Reed JC. Bcl-2 oncoprotein blocks chemotherapy-induced apoptosis in a human leukemia cell line. Blood. 1993;81:151–157. [PubMed] [Google Scholar]
- 72.Schmitt CA, Rosenthal CT, Lowe SW. Genetic analysis of chemoresistance in primary murine lymphomas. Nat Med. 2000;6:1029–1035. doi: 10.1038/79542. [DOI] [PubMed] [Google Scholar]
- 73.Moreau C, Cartron PF, Hunt A, Meflah K, Green DR, Evan G, Vallette FM, Juin P. Minimal BH3 peptides promote cell death by antagonizing anti-apoptotic proteins. J Biol Chem. 2003;278:19426–19435. doi: 10.1074/jbc.M209472200. [DOI] [PubMed] [Google Scholar]
- 74.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, Korsmeyer SJ. A stapled BID BH3 helix directly binds and activates BAX. Mol Cell. 2006;24:199–210. doi: 10.1016/j.molcel.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 76.Petros AM, Medek A, Nettesheim DG, Kim DH, Yoon HS, Swift K, Matayoshi ED, Oltersdorf T, Fesik SW. Solution structure of the antiapoptotic protein bcl-2. Proc Natl Acad Sci U S A. 2001;98:3012–3017. doi: 10.1073/pnas.041619798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DC. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mason KD, Vandenberg CJ, Scott CL, Wei AH, Cory S, Huang DC, Roberts AW. In vivo efficacy of the Bcl-2 antagonist ABT-737 against aggressive Myc-driven lymphomas. Proc Natl Acad Sci U S A. 2008;105:17961–17966. doi: 10.1073/pnas.0809957105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115:3304–3313. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi YX, Sneed T, Verhaegen M, Soengas M, Ruvolo VR, McQueen T, Schober WD, Watt JC, Jiffar T, Ling X, Marini FC, Harris D, Dietrich M, Estrov Z, McCubrey J, May WS, Reed JC, Andreeff M. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 81.Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7:989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 82.Khaw SL, Huang DC, Roberts AW. Overcoming blocks in apoptosis with BH3-mimetic therapy in haematological malignancies. Pathology. 2011;43:525–535. doi: 10.1097/PAT.0b013e32834b1b34. [DOI] [PubMed] [Google Scholar]
- 83.Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, Carney DA, He SZ, Huang DC, Xiong H, Cui Y, Busman TA, McKeegan EM, Krivoshik AP, Enschede SH, Humerickhouse R. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30:488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wilson WH, O’Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, Tulpule A, Dunleavy K, Xiong H, Chiu YL, Cui Y, Busman T, Elmore SW, Rosenberg SH, Krivoshik AP, Enschede SH, Humerickhouse RA. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11:1149–1159. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Green M, Loewenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell. 1988;55:1179–1188. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]
- 86.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 87.Torchilin VP. Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv Drug Deliv Rev. 2008;60:548–558. doi: 10.1016/j.addr.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 88.Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279:377–380. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- 89.Rhee M, Davis P. Mechanism of uptake of C105Y, a novel cell-penetrating peptide. J Biol Chem. 2006;281:1233–1240. doi: 10.1074/jbc.M509813200. [DOI] [PubMed] [Google Scholar]
- 90.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 91.Sawant R, Torchilin V. Intracellular delivery of nanoparticles with CPPs. Methods Mol Biol. 2011;683:431–451. doi: 10.1007/978-1-60761-919-2_31. [DOI] [PubMed] [Google Scholar]
- 92.Snyder EL, Meade BR, Saenz CC, Dowdy SF. Treatment of terminal peritoneal carcinomatosis by a transducible p53-activating peptide. PLoS Biol. 2004;2:E36. doi: 10.1371/journal.pbio.0020036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cantelmo AR, Cammarota R, Noonan DM, Focaccetti C, Comoglio PM, Prat M, Albini A. Cell delivery of Met docking site peptides inhibit angiogenesis and vascular tumor growth. Oncogene. 2010;29:5286–5298. doi: 10.1038/onc.2010.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee JY, Choi YS, Suh JS, Kwon YM, Yang VC, Lee SJ, Chung CP, Park YJ. Cell-penetrating chitosan/doxorubicin/TAT conjugates for efficient cancer therapy. Int J Cancer. 2011;128:2470–2480. doi: 10.1002/ijc.25578. [DOI] [PubMed] [Google Scholar]
- 95.Sawant RM, Hurley JP, Salmaso S, Kale A, Tolcheva E, Levchenko TS, Torchilin VP. “SMART” drug delivery systems: double-targeted pH-responsive pharmaceutical nanocarriers. Bioconjug Chem. 2006;17:943–949. doi: 10.1021/bc060080h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kale AA, Torchilin VP. “Smart” drug carriers: PEGylated TATp-modified pH-sensitive liposomes. J Liposome Res. 2007;17:197–203. doi: 10.1080/08982100701525035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Koren E, Apte A, Jani A, Torchilin VP. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J Control Release. 2011 doi: 10.1016/j.jconrel.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McCarty MF, Whitaker J. Manipulating tumor acidification as a cancer treatment strategy. Altern Med Rev. 2010;15:264–272. [PubMed] [Google Scholar]
- 99.Hunt JF, Rath P, Rothschild KJ, Engelman DM. Spontaneous, pH-dependent membrane insertion of a transbilayer alpha-helix. Biochemistry. 1997;36:15177–15192. doi: 10.1021/bi970147b. [DOI] [PubMed] [Google Scholar]
- 100.Reshetnyak YK, Andreev OA, Segala M, Markin VS, Engelman DM. Energetics of peptide (pHLIP) binding to and folding across a lipid bilayer membrane. Proc Natl Acad Sci U S A. 2008;105:15340–15345. doi: 10.1073/pnas.0804746105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Andreev OA, Engelman DM, Reshetnyak YK. pH-sensitive membrane peptides (pHLIPs) as a novel class of delivery agents. Mol Membr Biol. 2010;27:341–352. doi: 10.3109/09687688.2010.509285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Andreev OA, Engelman DM, Reshetnyak YK. Targeting acidic diseased tissue: New technology based on use of the pH (Low) Insertion Peptide (pHLIP) Chim Oggi. 2009;27:34–37. [PMC free article] [PubMed] [Google Scholar]
- 103.Andreev OA, Dupuy AD, Segala M, Sandugu S, Serra DA, Chichester CO, Engelman DM, Reshetnyak YK. Mechanism and uses of a membrane peptide that targets tumors and other acidic tissues in vivo. Proc Natl Acad Sci U S A. 2007;104:7893–7898. doi: 10.1073/pnas.0702439104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Elayadi AN, Samli KN, Prudkin L, Liu YH, Bian A, Xie XJ, Wistuba II, Roth JA, McGuire MJ, Brown KC. A peptide selected by biopanning identifies the integrin alphavbeta6 as a prognostic biomarker for nonsmall cell lung cancer. Cancer Res. 2007;67:5889–5895. doi: 10.1158/0008-5472.CAN-07-0245. [DOI] [PubMed] [Google Scholar]
- 105.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pasqualini R, Koivunen E, Kain R, Lahdenranta J, Sakamoto M, Stryhn A, Ashmun RA, Shapiro LH, Arap W, Ruoslahti E. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000;60:722–727. [PMC free article] [PubMed] [Google Scholar]
- 107.Corti A, Curnis F, Arap W, Pasqualini R. The neovasculature homing motif NGR: more than meets the eye. Blood. 2008;112:2628–2635. doi: 10.1182/blood-2008-04-150862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cai LL, Liu P, Li X, Huang X, Ye YQ, Chen FY, Yuan H, Hu FQ, Du YZ. RGD peptide-mediated chitosan-based polymeric micelles targeting delivery for integrin-overexpressing tumor cells. Int J Nanomedicine. 2011;6:3499–3508. doi: 10.2147/IJN.S26670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jiang X, Sha X, Xin H, Chen L, Gao X, Wang X, Law K, Gu J, Chen Y, Jiang Y, Ren X, Ren Q, Fang X. Self-aggregated pegylated poly (trimethylene carbonate) nanoparticles decorated with c(RGDyK) peptide for targeted paclitaxel delivery to integrin-rich tumors. Biomaterials. 2011;32:9457–9469. doi: 10.1016/j.biomaterials.2011.08.055. [DOI] [PubMed] [Google Scholar]
- 110.Ndinguri MW, Solipuram R, Gambrell RP, Aggarwal S, Hammer RP. Peptide targeting of platinum anti-cancer drugs. Bioconjug Chem. 2009;20:1869–1878. doi: 10.1021/bc900065r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pastorino F, Brignole C, Marimpietri D, Cilli M, Gambini C, Ribatti D, Longhi R, Allen TM, Corti A, Ponzoni M. Vascular damage and anti-angiogenic effects of tumor vessel-targeted liposomal chemotherapy. Cancer Res. 2003;63:7400–7409. [PubMed] [Google Scholar]
- 112.Pastorino F, Brignole C, Di PD, Nico B, Pezzolo A, Marimpietri D, Pagnan G, Piccardi F, Cilli M, Longhi R, Ribatti D, Corti A, Allen TM, Ponzoni M. Targeting liposomal chemotherapy via both tumor cell-specific and tumor vasculature-specific ligands potentiates therapeutic efficacy. Cancer Res. 2006;66:10073–10082. doi: 10.1158/0008-5472.CAN-06-2117. [DOI] [PubMed] [Google Scholar]
- 113.Lei H, Cao P, Miao G, Lin Z, Diao Z. Expression and functional characterization of tumor-targeted fusion protein composed of NGR peptide and 15-kDa actin fragment. Appl Biochem Biotechnol. 2010;162:988–995. doi: 10.1007/s12010-009-8901-8. [DOI] [PubMed] [Google Scholar]
- 114.Takara K, Hatakeyama H, Ohga N, Hida K, Harashima H. Design of a dual-ligand system using a specific ligand and cell penetrating peptide, resulting in a synergistic effect on selectivity and cellular uptake. Int J Pharm. 2010;396:143–148. doi: 10.1016/j.ijpharm.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 115.Gilbert MR, Kuhn J, Lamborn KR, Lieberman F, Wen PY, Mehta M, Cloughesy T, Lassman AB, Deangelis LM, Chang S, Prados M. Cilengitide in patients with recurrent glioblastoma: the results of NABTC 03–02, a phase II trial with measures of treatment delivery. J Neurooncol. 2012;106:147–153. doi: 10.1007/s11060-011-0650-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Reardon DA, Fink KL, Mikkelsen T, Cloughesy TF, O’Neill A, Plotkin S, Glantz M, Ravin P, Raizer JJ, Rich KM, Schiff D, Shapiro WR, Burdette-Radoux S, Dropcho EJ, Wittemer SM, Nippgen J, Picard M, Nabors LB. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J Clin Oncol. 2008;26:5610–5617. doi: 10.1200/JCO.2008.16.7510. [DOI] [PubMed] [Google Scholar]
- 117.Santoro A, Pressiani T, Citterio G, Rossoni G, Donadoni G, Pozzi F, Rimassa L, Personeni N, Bozzarelli S, Rossoni G, Colombi S, De Braud FG, Caligaris-Cappio F, Lambiase A, Bordignon C. Activity and safety of NGR-hTNF, a selective vascular-targeting agent, in previously treated patients with advanced hepatocellular carcinoma. Br J Cancer. 2010;103:837–844. doi: 10.1038/sj.bjc.6605858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, Hanahan D, Mattrey RF, Ruoslahti E. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16:510–520. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, Ruoslahti E. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science. 2010;328:1031–1035. doi: 10.1126/science.1183057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Javadpour MM, Juban MM, Lo WC, Bishop SM, Alberty JB, Cowell SM, Becker CL, McLaughlin ML. De novo antimicrobial peptides with low mammalian cell toxicity. J Med Chem. 1996;39:3107–3113. doi: 10.1021/jm9509410. [DOI] [PubMed] [Google Scholar]
- 121.Ellerby HM, Arap W, Ellerby LM, Kain R, Andrusiak R, Rio GD, Krajewski S, Lombardo CR, Rao R, Ruoslahti E, Bredesen DE, Pasqualini R. Anti-cancer activity of targeted pro-apoptotic peptides. Nat Med. 1999;5:1032–1038. doi: 10.1038/12469. [DOI] [PubMed] [Google Scholar]
- 122.Mai JC, Mi Z, Kim SH, Ng B, Robbins PD. A proapoptotic peptide for the treatment of solid tumors. Cancer Res. 2001;61:7709–7712. [PubMed] [Google Scholar]
- 123.Law B, Quinti L, Choi Y, Weissleder R, Tung CH. A mitochondrial targeted fusion peptide exhibits remarkable cytotoxicity. Mol Cancer Ther. 2006;5:1944–1949. doi: 10.1158/1535-7163.MCT-05-0509. [DOI] [PubMed] [Google Scholar]
- 124.Rege K, Patel SJ, Megeed Z, Yarmush ML. Amphipathic peptide-based fusion peptides and immunoconjugates for the targeted ablation of prostate cancer cells. Cancer Res. 2007;67:6368–6375. doi: 10.1158/0008-5472.CAN-06-3658. [DOI] [PubMed] [Google Scholar]
- 125.Kim HY, Kim S, Youn H, Chung JK, Shin DH, Lee K. The cell penetrating ability of the proapoptotic peptide, KLAKLAKKLAKLAK fused to the N-terminal protein transduction domain of translationally controlled tumor protein, MIIYRDLISH. Biomaterials. 2011;32:5262–5268. doi: 10.1016/j.biomaterials.2011.03.074. [DOI] [PubMed] [Google Scholar]
- 126.Kolevzon N, Kuflik U, Shmuel M, Benhamron S, Ringel I, Yavin E. Multiple triphenylphosphonium cations as a platform for the delivery of a pro-apoptotic peptide. Pharm Res. 2011;28:2780–2789. doi: 10.1007/s11095-011-0494-6. [DOI] [PubMed] [Google Scholar]
- 127.Lee BR, Oh KT, Oh YT, Baik HJ, Park SY, Youn YS, Lee ES. A novel pH-responsive polysaccharidic ionic complex for proapoptotic D-(KLAKLAK)2 peptide delivery. Chem Commun (Camb) 2011;47:3852–3854. doi: 10.1039/c0cc03590d. [DOI] [PubMed] [Google Scholar]
- 128.Agemy L, Friedmann-Morvinski D, Kotamraju VR, Roth L, Sugahara KN, Girard OM, Mattrey RF, Verma IM, Ruoslahti E. Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma. Proc Natl Acad Sci U S A. 2011;108:17450–17455. doi: 10.1073/pnas.1114518108. [DOI] [PMC free article] [PubMed] [Google Scholar]