

We report on the formation of conducting polymer nanoparticles (CPNs), stabilized by a collagen mimetic peptide (CMP)-polymer amphiphile. CPNs ranging from ∼15-40 nm were readily accessible by modifying amphiphile concentration. Surface presentation of CMPs on CPN precluded intra-/inter-particle trimerization, while preserving their ability to target collagen without pre-activation.
Biomedical imaging technologies are poised to provide insights regarding cellular communication and function by precisely monitoring events at the molecular, cellular, and tissue levels. Since their first use in bioimaging, quantum dots (QDs) have received considerable attention as bioanalytical tools for their unique photophysical properties. Nanoparticles of semiconducting polymers, also referred to as conjugated polymer nanoparticles (CPNs), have emerged as non-cytotoxic alternatives to QDs.1-5 Aside from excellent photostability, CPNs exhibit high fluorescence under one- and two-photon excitation, fast emission rates, and high fluorescence quantum yield.6
CPNs are produced by direct polymerization from microemulsion,7 or by nanoprecipitation methods.8, 9 When carried out in the presence of a stabilizer, nanoprecipitation is a form of arrested precipitation wherein the kinetics of solute nucleation and growth and those of emulsifier adsorption onto the growing particle nuclei are balanced to produce particles in the nanometer range. Hence, amphiphilic polymer stabilizers allow not only size control, but also effective interfacing of CPNs with biological media through electrostatic and/or steric effects.
Tailoring surface properties of CPNs to display bioinertness or to enable biorecognition can be achieved through pre- or post-nanoprecipitation functionalization with, among others, peptide-polymer conjugates. While peptide-polymer based nanoparticles have been widely used for cellular targeting through ligand-receptor interactions, only a limited number of successful cases of nanoparticle-based ECM targeting strategies have been reported.10, 11 The ECM of a tissue is a valuable biomarker for imaging and targeted delivery, as its structural modifications are clear indicators of diseased states. Collagen is the most abundant protein in the ECM, playing a key role in the pathology of a variety of diseases and disorders, such as arthritis, fibrosis, and cancer.12
Unfolded collagen chains present in tissues undergoing normal or pathological remodeling can be targeted by single-strand collagen mimetic peptides (CMPs) consisting of (GPO)x (x=6-10, O: hydroxyproline) sequence. The targeting mechanism is analogous to DNA fragments binding to complementary DNA strands.12-16 As only single-strand CMPs are able to hybridize with collagen chains but CMPs self-assemble into homotrimers during storage at low temperatures, monomeric CMPs have to be generated by heating the trimeric peptide above its melting temperature just prior to application to collagen substrates.17-19 Strategies to circumvent self-trimerization have been examined, including installation of a light-cleavable protective group on the CMP.14 While encouraging results were obtained by this method, realizing the full potential of CMP-collagen binding is nonetheless limited by additional heat- or light-activation procedures. We speculated that immobilizing monomeric CMPs on a nanoparticle surface at low density would prevent their triple helical self-assembly due to spatial distance between the CMPs and that these CMP-conjugated nanoparticles could be directly used without activation.
Herein, we report on the synthesis of a CMP-polymer amphiphile and the preparation of CMP-stabilized conjugated polymer nanoparticles (CMP-CPN) by nanoprecipitation. The ability of these nanoparticles to either probe collagen strands or enable sensitive fluorescent imaging of collagen in fixed tissue sections is also reported. PFBT (poly(9,9-dioctylfluorenyl-2,7-diyl)-co-(1,4-benzo-(2,10,3)-thiadiazole)) was used as the conjugated polymer since it has been widely cited as exhibiting excellent photostability and high brightness.4, 20-23
The stabilizing amphiphilic polymer, poly(styrene-co-NAS) 2, was synthesized by reversible addition-fragmentation chain transfer (RAFT) polymerization of N-acryloxysuccinimide (NAS) 1 and styrene (Fig S1, ESI†). The NAS group served as conjugation site for either the CMP or poly(ethylene glycol) (PEG). Aside from affecting targeting, the hydrophilic nature of the CMP was expected to impart colloidal nanoparticle stabilization. The comonomer ratio used (8% mol 1) had a high hydrophobic content so as to effectively stabilize PFBT nanoparticles through hydrophobic interaction.
Conjugation of the CMP occurred quantitatively through the active pendant ester groups. Because of the propensity of CMPs to self-trimerize at room temperature, the CMP peptide was preheated to 80 °C and conjugation was performed at 50 °C; an average of 5-6 CMPs per polymer chain was found by 1H NMR (Fig S4, ESI†). The resultant polymer-peptide, PS-g-CMP (4) had an apparent Mn of 21,078 g/mol and a hydrophilic weight ratio of ca. 60%. As a negative control for the CMP conjugate, we used the same backbone and substituted the CMP for PEG of similar molecular weight (5, Fig S5 and S6, ESI†; MnPEG∼1980 g/mol vs. MnCMP∼2558 g/mol, total hydrophilic weight ratio of the copolymer ∼ 50%); we refer to this stabilizer as PS-g-PEG (6).
PS-g-CMP or PS-g-PEG-stabilized PFBT nanoparticles (CMP-CPNs or PEG-CPNs, respectively) were produced by flash nanoprecipitation in a multi-inlet vortex mixer (MIVM).24 A key factor in nanoprecipitation is mixing intensity, as mass transfer to achieve high supersaturation rates with uniform spatial distribution is required to ensure the formation of small particles with narrow polydispersity.25, 26 High energy mixing techniques can achieve mixing times on the order of milliseconds with controllable particle size distributions.27 In the MIVM used, spatially homogeneous supersaturation is generally achieved at Reynolds numbers >2000 (see ESI). In this study, we employed high inlet velocities (Re∼8640) so as to work in the flow field-independent regime. The stabilizing polymer (4 or 6) was dissolved in DMSO and mixed with a solution of PFBT in THF to generate the organic solution (Table S1, ESI†).
As shown in Fig 1 and Table S1, particles had a relatively narrow polydispersity and average particle size was readily controlled between ∼15 and ∼40 nm according to solute and stabilizer concentration and type. In precipitation by solvent shifting, particle size and size distribution are determined by the kinetics of nucleation and growth of the solute, the rate and magnitude of supersaturation, and mixing intensity, as well as the occurrence of secondary processes. In addition to the solute and the mutually miscible solvent/antisolvent pair, additives such as stabilizers or emulsifiers can also be present during the solvent shifting process and the exact mechanism by which they influence particle formation is complex.27 The function of each additive is complicated by the fact that they can also act as nuclei for particle growth. In this sense, we attribute the observed decrease in particle size with increasing stabilizer concentration to more nucleation sites provided by the amphiphile. This argument also explains the size of PEG-CPNs. PS-g-PEG has a larger hydrophobic content than PS-g-CMP Therefore, for a given concentration it is expected to generate more nuclei, resulting in smaller particles. Other factors contributing to the observed size difference among CMP- and PEG-based amphiphiles are molecular weight (Δ∼580 g/mol) and chain rigidity, both of which are higher for the peptide. Notably, in the absence of the amphiphilic stabilizer, macroscopic precipitates of PFBT were observed in the MIVM, particularly for solute concentrations above 100 μg/mL.
Figure 1.
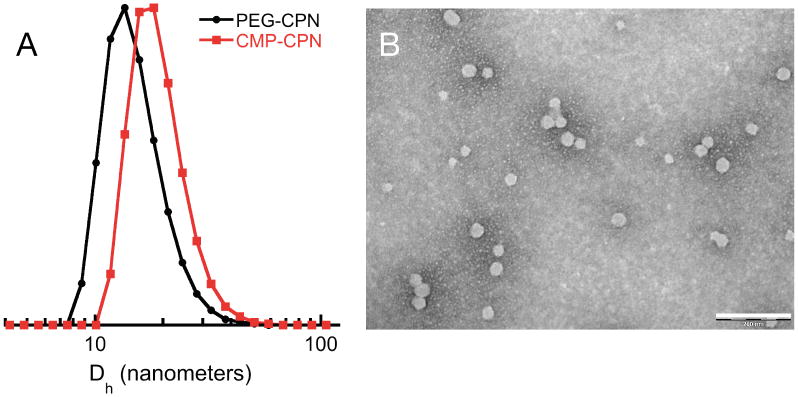
Particle size distributions by DLS of PEG- and CMP-CPNs (A) and representative TEM (B) of CMP-CPNs prepared from a PFBT solution of 200 μg/mL (scale bar: 200 nm).
Long-term stability studies revealed that both types of nanoparticles form stable dispersions in water (Fig S8, ESI†) with imperceptible formation of large aggregates for at least 90 days, suggesting that interparticle CMP trimerization did not take place, despite the low storage temperature (4 °C). This is because the CMP triple helices fold only when the peptide chains are parallel to one another; when CMP-CPN particles come together, the CMPs from each particle are in anti-parallel orientation, unsuitable for trimerization and particle aggregation.
Furthermore, zeta-potential measurements of CPNs revealed a slight negative surface charge (Table S1). The low surface charge and absence of agglomerates suggest that particle stabilization occurs by steric rather than electrostatic effects. Lastly, since PEG-CPNs are to be used as negative controls of CMP-CPNs, we measured their fluorescence properties (Fig S9, ESI†). For a given concentration of PFBT, both types of particles exhibited similar emission intensities, indicating that the stabilizing moiety does not significantly impact their optical properties. Incubation at low temperature also did not affect nanoparticle fluorescent properties (Fig S10, ESI†).
Binding of CMP-CPNs to collagen was examined on coatings of BSA and gelatin (denatured type I collagen), using PEG-CPNs as control. Nanoparticle binding levels were measured by PFBT fluorescence on the coatings after washing (Fig 2A). Both types of nanoparticles exhibited negligible binding to the BSA coating, demonstrating the extremely low non-specific binding of CMP-CPNs, comparable to that of PEG-CPN. This is attributed to the hydrophilic and neutral CMP coating on the nanoparticle. Moreover, CMP-CPNs showed a binding level an order of magnitude higher than PEG-CPN on gelatin coating, indicating that CMP-CPNs can specifically bind to collagen chains with high specificity. To rule out intraparticle CMP trimerization, we compared binding affinities of CMP-CPN on gelatin coatings with and without heat activation. A group of CMP-CPN solutions were heated to 75 °C immediately prior to the assay to ensure dissociation of any possible pre-folded CMP trimers and enhance their availability toward collagen binding. Another group of CMP-CPN samples, not subject to heat treatment, were used in parallel. The results indicated that the two groups of CMP-CPNs showed comparable levels of binding to the gelatin coating (p=0.133, student test), suggesting that nanoparticle-immobilized CMPs remain mostly monomeric and active, even after months of refrigeration. This is the result of the low density of CMPs displayed on the surface of the nanoparticles: 5-6 out of all 87 repeat units of 2 were conjugated to CMPs (Figure S3). The intra-particle self-assembly of CMPs is not possible because the CMP chains are far away from each other. Gelatin, however, with its long and flexible chain, is free to interact with the CMPs on the particle surface.
Figure 2.
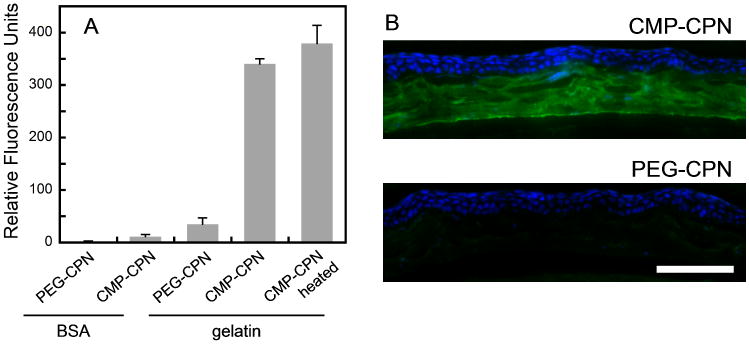
Specific binding of CMP-CPN to collagen chains. (A) Comparative fluorescence levels (ex: 460 nm, em: 535 nm) of BSA and gelatin (denatured collagen chain) coatings treated with PEG-CPN or CMP-CPN. The binding levels of CMP-CPN on gelatin under room temperature and after heating were compared. (B) Fluorescence micrographs of fixed mouse cornea sections probed by CMP-CPN or PEG-CPN (green) and co-stained with DAPI (blue) (scale bar: 100 μm).
Finally, we evaluated the ability of CMP-CPNs to visualize collagen in histology sections (Fig 2B). We chose mouse cornea tissue because it not only consists of mostly of collagen fibers in the stroma, but also because it is an important tissue target that has been heavily explored for nanoparticle-based diagnostics and therapeutics for ophthalmology healthcare.28-30 Tested cornea sections contained denatured collagen chains available for CMP-hybridization as the tissue had been preserved by chemical fixation.15 Having established the binding ability of surface-grafted CMP on CMP-CPNs, the solution of nanoparticles was used without heat treatment. As seen in Fig 2B, CMP-CPNs selectively stained the collagen-rich stroma of the cornea section (in green) with respect to the cellular epithelium (in blue). The intense green fluorescence from the semiconducting PFBT revealed the fine details of collagen fibril organization in the corneal stroma, as well as a bright green line at its interior side corresponding to the Descement's membrane that is rich in type VIII collagen. In contrast, PEG-CPNs failed to stain the tissue, showing only the DAPI staining of the epithelium.
Conclusions
Nanoparticles of a conducting polymer (PFBT), with the ability for selective collagen binding, were produced by a nanoprecipitation method using a collagen mimetic peptide (CMP)-polymer hybrid as the stabilizing amphiphile. The surface presentation of CMPs precluded the characteristic triple helical self-assembly of their monomeric form into homotrimers, attributed to the spatial distance between peptide chains. The ability of surface-bound CMPs to hybridize with denatured collagen, without any pre-activation step, was demonstrated by histological staining of mouse corneal tissue sections. The absence of intra- and inter-particle homotrimerization, along with the ability of CMPs to directly target denatured collagen molecules showcase the advantages of surface presentation of single strand CMPs.
Supplementary Material
Scheme 1.

Stabilizing copolymers PS-g-CMP (4) and PS-g-PEG (6) (A); x=0.92, y=0.08 and m=87. Structure of PFBT (B).
Acknowledgments
Financial support was provide by The Johns Hopkins University as start-up funds, and through an NSF CAREER Award to M.H.-A. (DMR 1151535).
Footnotes
Electronic Supplementary Information (ESI) available: experimental procedures and additional figures. See DOI: 10.1039/c000000x/
References
- 1.Fernando LP, Kandel PK, Yu JB, McNeill J, Ackroyd PC, Christensen KA. Biomacromolecules. 2010;11:2675–2682. doi: 10.1021/bm1007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pecher J, Huber J, Winterhalder M, Zumbusch A, Mecking S. Biomacromolecules. 2010;11:2776–2780. doi: 10.1021/bm100854a. [DOI] [PubMed] [Google Scholar]
- 3.Wu CF, Hansen SJ, Hou QO, Yu JB, Zeigler M, Jin YH, Burnham DR, McNeill JD, Olson JM, Chiu DT. Angew Chem Int Edit. 2011;50:3430–3434. doi: 10.1002/anie.201007461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng LH, Liu LB, Lv FT, Bazan GC, Wang S. Adv Mater. 2014;26:3926–3930. doi: 10.1002/adma.201305206. [DOI] [PubMed] [Google Scholar]
- 5.Feng LH, Zhu CL, Yuan HX, Liu LB, Lv FT, Wang S. Chem Soc Rev. 2013;42:6620–6633. doi: 10.1039/c3cs60036j. [DOI] [PubMed] [Google Scholar]
- 6.Wu CF, Schneider T, Zeigler M, Yu JB, Schiro PG, Burnham DR, McNeill JD, Chiu DT. J Am Chem Soc. 2010;132:15410–15417. doi: 10.1021/ja107196s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baier MC, Huber J, Mecking S. J Am Chem Soc. 2009;131:14267–14273. doi: 10.1021/ja905077c. [DOI] [PubMed] [Google Scholar]
- 8.Landfester K, Montenegro R, Scherf U, Guntner R, Asawapirom U, Patil S, Neher D, Kietzke T. Adv Mater. 2002;14:651–655. [Google Scholar]
- 9.Kietzke T, Neher D, Landfester K, Montenegro R, Guntner R, Scherf U. Nat Mater. 2003;2:408–U407. doi: 10.1038/nmat889. [DOI] [PubMed] [Google Scholar]
- 10.Rothenfluh DA, Bermudez H, O'Neil CP, Hubbell JA. Nat Mater. 2008;7:248–254. doi: 10.1038/nmat2116. [DOI] [PubMed] [Google Scholar]
- 11.Zhang B, Shen S, Liao ZW, Shi W, Wang Y, Zhao JJ, Hu Y, Yang JR, Chen J, Mei H, Hu Y, Pang ZQ, Jiang XG. Biomaterials. 2014;35:4088–4098. doi: 10.1016/j.biomaterials.2014.01.046. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Yu SM. Curr Opin Chem Biol. 2013;17:968–975. doi: 10.1016/j.cbpa.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu SM, Li Y, Kim D. Soft Matter. 2011;7:7927–7938. doi: 10.1039/C1SM05329A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Foss CA, Summerfield DD, Doyle JJ, Torok CM, Dietz HC, Pomper MG, Yu SM. P Natl Acad Sci USA. 2012;109:14767–14772. doi: 10.1073/pnas.1209721109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Ho D, Meng H, Chan TR, An B, Yu H, Brodsky B, Jun AS, Yu SM. Bioconjugate Chem. 2013;24:9–16. doi: 10.1021/bc3005842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Foss CA, Pomper MG, Yu MS. Journal of visualized experiments: JoVE. 2014:e51052. doi: 10.3791/51052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang AY, Mo X, Chen CS, Yu SM. J Am Chem Soc. 2005;127:4130–4131. doi: 10.1021/ja0431915. [DOI] [PubMed] [Google Scholar]
- 18.Wang AY, Leong S, Liang YC, Huang RCC, Chen CS, Yu SM. Biomacromolecules. 2008;9:2929–2936. doi: 10.1021/bm800727z. [DOI] [PubMed] [Google Scholar]
- 19.Wang AY, Foss CA, Leong S, Mo X, Pomper MG, Yu SM. Biomacromolecules. 2008;9:1755–1763. doi: 10.1021/bm701378k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang XL, Groff LC, McNeill JD. Langmuir. 2013;29:13925–13931. doi: 10.1021/la402767j. [DOI] [PubMed] [Google Scholar]
- 21.Wu C, Bull B, Szymanski C, Christensen K, McNeill J. Acs Nano. 2008;2:2415–2423. doi: 10.1021/nn800590n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu JB, Wu CF, Sahu SP, Fernando LP, Szymanski C, McNeill J. J Am Chem Soc. 2009;131:18410–18414. doi: 10.1021/ja907228q. [DOI] [PubMed] [Google Scholar]
- 23.Rong Y, Wu CF, Yu JB, Zhang XJ, Ye FM, Zeigler M, Gallina ME, Wu IC, Zhang Y, Chan YH, Sun W, Uvdal K, Chiu DT. Acs Nano. 2013;7:376–384. doi: 10.1021/nn304376z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Cheng CY, Liu Y, Prud'homme RK, Fox RO. Chem Eng Sci. 2008;63:2829–2842. [Google Scholar]
- 25.Texter J. J Disper Sci Technol. 2001;22:499–527. [Google Scholar]
- 26.Schwarzer HC, Peukert W. Aiche J. 2004;50:3234–3247. [Google Scholar]
- 27.Horn D, Rieger J. Angew Chem Int Edit. 2001;40:4331–4361. [Google Scholar]
- 28.Xu Q, Kambhampati SP, Kannan RM. Middle East African journal of ophthalmology. 2013;20:26–37. doi: 10.4103/0974-9233.106384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iezzi R, Guru BR, Glybina IV, Mishra MK, Kennedy A, Kannan RM. Biomaterials. 2012;33:979–988. doi: 10.1016/j.biomaterials.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 30.Liu SY, Jones L, Gu FX. Macromol Biosci. 2012;12:608–620. doi: 10.1002/mabi.201100419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.