

Abstract
Lysophospholipids are important signaling molecules in animals and metazoan cells. They are widely distributed among marine invertebrates, where their physiological roles are unknown. Sea cucumbers produce unique lysophospholipids. In this study, two lysophospholipids were detected in Holothuria atra for the first time, lyso-platelet activating factor and lysophosphatidylcholine, with nuclear magnetic resonance and liquid chromatography–time-of-flight mass spectrometric analyses. The lipid fraction of H. atra contained lyso-platelet activating factor and lysophosphatidylcholine, and inhibited H2O2-induced apoptosis in the macrophage cell line J774A.1. The antioxidant activity of the lysophospholipid-containing lipid fraction of H. atra was confirmed with the oxygen radical absorbance capacity method. Our results suggest that the lysophospholipids from H. atra are potential therapeutic agents for the inflammation induced by oxidative stress.
Introduction
Biologically active products from marine organisms have recently become the focus of pharmaceutical research and health food development. Among the aquatic invertebrates, sea cucumbers produce a diversity of secondary metabolites with valuable biological activities. The sea cucumber, Holothuria, which belongs to the phylum Echinodermata and the class Holothuroidea, is found on seafloors throughout the world [1]. It is considered a healthy food because it contains various physiologically active substances, including vitamins (A, C, B1, B2, and B3), trace elements (calcium, iron, magnesium, and zinc), polysaccharides (chondroitin sulfate), and saponin glycosides [2]. Sea cucumbers are also commonly used to treat wounds, eczema, arthritis, hypertension, and impotence [3].
Some bioactive compounds extracted from sea cucumbers are reported to have anti-inflammatory [4], antitumor [5], and fungicidal activities [6]. For instance, the sphingoid base composition of cerebrosides prepared from the sea cucumber is cytotoxic to human colon cancer cell lines [7]. Holotoxins from sea cucumbers are also well-known antifungal glycosides [8,9]. The characteristics of glycosphingolipids and their structure–activity relationships conferred neuritogenic activity against the rat pheochromocytoma cell line PC12 [10]. Further novel bioactive products may be discovered in the sea cucumber. In this study, we investigated the lipids of Holothuria atra, which is reported to have medicinal value [11]. A mixed extract of H. atra contained physiologically active phenolic compounds with antioxidant activity, which exerted potent hepatoprotective effects against thioacetamide-induced liver injury in a rat model [3]. Therefore, we undertook this study to identify the bioactive lipids in a mixed extract of the H. atra body wall and to evaluate their cytoprotective potential against oxidative stress. We found that lysophospholipids from H. atra inhibit H2O2-induced apoptosis in macrophages.
Materials and Methods
Ethics statement
Sea cucumbers were sampled after the appropriate permission was obtained from the Fishermen’s Cooperative Association in Okinawa Prefecture.
Extraction and purification of lipids from H. atra
Sea cucumbers (H. atra) were collected from the ocean at Okinawa, Japan, and transported frozen on dry ice to our laboratory. The whole tissues (792.3 g) were homogenized in a blender with 1.5 L of chloroform–methanol (1:2, v/v) and mixed for 1 h at room temperature. The extract was filtered under reduced pressure to produce an aqueous suspension. The residue remaining after filtering was treated with 1.5 L of chloroform–methanol mixtures (1:4 and then 2:1, v/v) for 1 h each at room temperature, and the aqueous suspension was collected as described above. The aqueous suspension of the whole tissues was treated overnight with chloroform–methanol–water (8:4:3, v/v) using the Folch method. The lipid layer from the whole tissues was mixed to form the total lipid (TL).
The TL (8.965 g) was applied to a silica-gel column (Wakogel C-200, diameter 4 cm, height 40 cm, volume 502.4 cm3; Wako Pure Chemical, Osaka, Japan) in chloroform and eluted sequentially with chloroform–methanol (4:1, 2:1, 1:1, and 1:4, v/v), methanol, chloroform–methanol–water (3:6:1, v/v), and chloroform–water (4:1, v/v).
To detect the lipids, the TL was analyzed with silica gel thin-layer chromatography (TLC; silica gel 60, 0.25 mm; Merck, Darmstadt, Germany) with a chloroform–methanol–water mixture (65:25:4 or 65:35:8, v/v/v). The lipid species were visualized by spraying the plate with 50% sulfuric acid and heating it briefly. The carbohydrates and phospholipids were detected on the TLC plate with anthrone–sulfuric acid reagent and Dittmer reagent, respectively.
The fractionated lipids were evaporated with N2 and suspended in Tocrisolve 100 (Tocris BioScience, Bristol, UK) at 200 mg/mL.
Reagents
Lyso-platelet activating factor (Lyso-PAF C18:0 and (9Z)-C18:1) were purchased from Avanti Polar Lipids (Alabaster, AL). Lysophosphatidylcholine (LPC, C18:0) and sphingomyelin were from Sigma (St Louis, MO).
Measurement of cell viability
The mouse macrophage cell line J774A.1 (ATCC TIB-67) was cultured in Dulbecco’s modified Eagle’s medium (Sigma) supplemented with 10% heat-inactivated fetal bovine serum. The J774A.1 cells were seeded in 96-well flat-bottom plates (3.8 × 105 cells/mL), incubated overnight at 37°C., and then preincubated with the lipids for 12 h. To induce apoptosis, the cells were treated with 112.5 μM or 25 μM H2O2. After 12 h, 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt and 1-methoxy-5-methylphenazinium methyl sulfate (Cell Counting Kit-8; Dojin Laboratories, Kumamoto, Japan) were added to final concentrations of 5 mM and 0.2 mM, respectively, and the cells were incubated for another 4 h. Proliferation was measured as the mean absorbance at 450 nm. Cell viability was expressed as the relative absorbance of the lipid-treated cells normalized to the absorbance of the untreated control cells (×100).
Detection of apoptotic cells using terminal deoxynucleotidyl transferase (TdT)-mediated dUTP–biotin nick-end labeling (TUNEL)
To detect apoptosis directly, the cells were analyzed with TUNEL (In Situ Cell Death Detection Kit, TMR red; Roche Diagnostics GmbH, Mannheim, Germany). Briefly, the cells were placed on glass slides and dried. They were then fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 30 min at room temperature, washed in PBS, and permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate for 2 min at 4°C. TdT labeling was performed with the commercially available kit, according to the manufacturer’s protocol. The apoptotic cells were directly detectable by their red color with fluorescence microscopy (Eclipse E400, Nikon Inc., Melville, NY). The images were captured with a CCD camera and processed with a digital fluorescence microscope (VB-6000, Keyence, Osaka, Japan).
Nuclear magnetic resonance (NMR)
All NMR experiments were performed with a JEOL ECA-600 NMR spectrometer with a 5 mm inverse triple-resonance 1H/13C/X probe, with a z-axis pulsed-field gradient for two-dimensional (2D) experiments or with a 5 mm tunable double-resonance probe for 1D experiments, operating at 600.17 MHz for 1H, 150.91 MHz for 13C, and 242.95 MHz for 31P at 298 K. The 1H chemical shift was referenced to the residual CD2HOD signal at 3.30 ppm. The 13C chemical shift was referenced to the solvent CD3OD signal at 49.0 ppm. The 31P chemical shift was referenced to an external reference of 85% H3PO4 at 0.0 ppm. The NMR spectra were measured and processed using a standard pulse sequence and the Delta version 5.0 software (JEOL, USA).
Liquid chromatography and time-of-flight mass spectrometry (LC/TOF-MS)
The seventh lipid fraction (F7) was injected into the mass spectrometer with a high-pressure LC system (Acquity UPLC System; Waters, MA), with a BEH C18 column (2.1 × 50 mm, 1.7 mm) at a flow rate of 0.25 mL/min. Gradient elution was conducted with mobile phases A (0.1% formic acid in water) and B (acetonitrile). The gradient used commenced with 40% mobile phase B, isocratic for 0.7 min; increased to 90% B over 5.3 min; isocratic for 2.5 min; and then back to 40% B over 0.5 min. All mass spectrometric analyses were performed with the SYNAPT G2 HDMS platform (Waters, Manchester, UK). A voltage (3.0 kV) was applied to the stainless steel electrospray ionization (ESI) capillary under positive ion conditions. The TOF analyzer was set to resolution mode with a resolving power of 20,000 at m/z 556 (leucine enkephalin) and the m/z range of 50–1500 was calibrated with sodium formate. The capillary, extraction cone, and cone voltages were set to 3 kV, 104 kV, and 4 kV, respectively. The desolvation gas (nitrogen) was used at a flow rate of 800 L/h, and the source and desolvation temperatures were set to 100°C and 250°C, respectively.
Ozonolysis
Excess ozone was passed through a solution of the F7 lipid fraction (1.6 mg) in CD3OD (1.1 mL) at –78°C for 1.1 h. After nitrogen gas was bubbled through the solution for 10 min to remove the excess ozone, dimethyl sulfide (0.5 mL) was added slowly with stirring at –78°C. The mixture was allowed to warm gradually to room temperature and stirring was continued overnight. An aliquot (0.2 mL) was analyzed with LC/TOF-MS, and the remaining solution was concentrated to a syrupy gum (1.0 mg) in vacuo.
Oxygen radical absorbance capacity (ORAC) assay
The ORAC assay was performed in triplicate on standard lipids and the extracts, according to the manufacturer’s recommendations (OxiSelect ORAC Activity Assay Kit; Cell Biolabs, San Diego, CA). The diluted samples (0, 2.5, 5, 10, 20, 30, 40, and 50 μM) were analyzed in triplicate in a 96-well plate. To each well were added 150 μL of fluorescein solution (0.036 mg/L; Cell Biolabs) and 25 μL of the diluted sample or 25 μL of the Trolox standard (6-hydroxy-2,5,7,8-tetramethylcroman-2-carboxylic acid; Cell Biolabs), and the plate was incubated for 30 min at 37°C, after which 25 μL of 2,2′-azobis-2-aminopropane dihydrochloride (AAPH) solution (80 mg/mL; Cell Biolabs) was added. The ORAC assay quantifies the inhibition (expressed as a percentage over time) of the fluorescence produced by peroxyl radicals generated at a constant rate by the thermal decomposition of AAPH. The antioxidant capacity is expressed in μmol Trolox equivalents (TE)/g, and is calculated from a Trolox standard curve.
Statistical analysis
All statistical analyses were performed with the GraphPad Prism 5 software (GraphPad Software Inc., La Jolla, CA). The results are presented as means ± standard deviations. The significance of differences was evaluated with one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons procedure or Student’s t test.
Results
Lipid fraction extracted with chloroform–water (4:1, v/v) prevented H2O2-induced cell death
Fig 1A shows the seven lipid fractions (F1–F7) eluted sequentially from a silica gel column with the following eluents: chloroform–methanol (4:1, 2:1, 1:1, 1:4, v/v), methanol, chloroform–methanol–water (3:6:1, v/v), and chloroform–water (4:1, v/v), respectively. We tested the protective activities of these lipid fractions in J774A.1 cells treated with H2O2 (Fig 1B). Pretreatment of the cells with TL from H. atra inhibited H2O2-induced cell death compared with that in the control cells treated with H2O2 only. The F1, F3, F4, F6, and F7 lipid fractions significantly protected the J774A.1 cells from H2O2 stress. Because F7 appeared as a single spot on TLC, we characterized this fraction in a further experiment. As shown in Fig 2, H2O2 treatment induced apoptosis in the J774A.1 cells of the control group. However, treatment with the F7 lipid fraction inhibited the apoptosis induced by H2O2. These results suggest that the F7 lipid fraction had antiapoptotic activity.
Fig 1. Thin-layer chromatogram (TLC) and cytoprotective effects of the lipids extracted from Holothuria atra.

(A) TLC analysis of the lipids extracted from H. atra using a chloroform–methanol–water mixture (65:25:4, v/v/v). The total lipid (TL) was applied to a silica gel column in chloroform and eluted with: (F1) chloroform–methanol (4:1 v/v); (F2) chloroform–methanol (2:1 v/v); (F3) chloroform–methanol (1:1 v/v); (F4) chloroform–methanol (1:4 v/v); (F5) methanol; (F6) chloroform–methanol–water (3:6:1 v/v); or (F7) chloroform–water (4:1 v/v). (B) Viability of J774A.1 cells treated with the extracted lipids. J774A.1 cells were preincubated with each lipid fraction (10 μg/mL) for 12 h. H2O2 was then added to the cells (112.5 μM). After 12 h, cell viability was measured and expressed as the mean viability ± standard deviation of triplicate experiments. (*) According to Student’s t test, the difference between the lipid-treated cells and the untreated cells (control) was significant (P < 0.05).
Fig 2. Antiapoptotic activity of the F7 fraction in H2O2-treated cells.

J774A.1 cells were preincubated with the F7 lipid fraction for 12 h. H2O2 was then added to the cells (112.5 μM). After 12 h, the TUNEL-positive cells were quantified and the results expressed as the mean ± standard deviation of triplicate experiments. (A) TUNEL staining of J774A.1 cells treated with the F7 lipid fraction. (B) Frequency of apoptosis. (*) According to Student’s t test, the difference between the lipid-treated cells and untreated cells (control) was significant (P < 0.05).
Characterization of the lipid fraction extracted with chloroform–water (4:1, v/v)
To examine the contents of the F7 fraction, TLC using anthrone–sulfuric acid reagent and Dittmer reagent was performed to detect the carbohydrates and phospholipids, respectively, and 50% sulfuric acid was used to detect organic compounds. The anthrone–sulfuric acid reagent did not detect the spot of the F7 fraction or that of sphingomyelin, Lyso-PAF, or LPC. However, the spot of the F7 fraction was detected with Dittmer reagent, with Rf values similar to those for LPC and Lyso-PAF, but not with an Rf value similar to that of sphingomyelin (Fig 3), indicating the presence of phospholipids in the F7 fraction.
Fig 3. Thin-layer chromatogram of the F7 lipid fraction.

The F7 lipid fraction was analyzed with Dittmer reagent (A) or 50% sulfuric acid (B) with a chloroform–methanol–water mixture (65:35:8, v/v/v). 1, Sphingomyelin; 2, Lyso-PAF; 3, F7 lipid fraction; 4, LPC.
To analyze the phospholipids in the F7 fraction, we performed detailed NMR experiments, including 2D NMR with DQF-COSY, TOCSY, NOESY, HSQC, HSQC-TOCSY, 1H-13C HMBC, and 1H-31P HMBC (S1 File) [12]. The 1H NMR spectrum of the F7 fraction was quite similar to that of authentic Lyso-PAF (9Z)-C18:1. However, the integration values for the olefinic methine and allylic methylene signals were only half those expected for Lyso-PAF C18:1, and there were small minor signals. This indicates that the F7 fraction was a mixture of saturated and unsaturated Lyso-PAF, with minor amounts of other lipids. The connectivity between the glycerol unit and the alkyl chain portion through the ether bond in the Lyso-PAF structure was determined from long-range correlations in the 1H-13C HMBC spectra. The presence of a choline unit and the connectivity between the choline and glycerol units through phosphate were confirmed with NOESY, 1H-13C HMBC, and 1H-31P HMBC spectral data. However, it was difficult to clarify the double bond position with TOCSY, HSQC-TOCSY, and HMBC. A direct comparison showed that the 13C NMR spectrum of the F7 fraction was not identical to that of Lyso-PAF (9Z)-C18:1, whereas it closely matched the 13C signals from the C7–C12 portion of cis-5-dodecenoic acid, used as a model of the (11Z)-C18:1 alkyl chain. These observations suggested that the F7 fraction contained an isomer of Lyso-PAF C18:1, and its double bond position was tentatively assigned to C11. The 13C chemical shift values for the allylic methylene carbon signals at 28.1 ppm indicated that the stereochemistry of the double bond was Z. An LC/TOF-MS analysis of the F7 fraction (Table 1 and Supporting Information) showed two major peaks at retention times (RTs) of 4.95 and 5.83 min in the base peak ion chromatogram. The RT and m/z were identical to those of a standard sample, indicating that the peak at RT = 5.83 corresponded to Lyso-PAF C18:0 (1). The other main peak at RT = 4.95 was probably an isomer of Lyso-PAF (9Z)-C18:1 because it had the same m/z 530 for [M+Na]+, with a slightly shorter RT. Ozonolysis was performed to determine the position of the double bond in Lyso-PAF C18:1 (RT = 4.95). To clearly identify the expected dimethyl acetal derivative, the reaction was performed in CD3OD solution. The three products were identified with LC/TOF-MS as C11-carboxylic acid (7), C11-aldehyde (8), and its d6-dimethyl acetal derivative (9). These data are consistent with the NMR data described above. Therefore, the peak at RT = 4.95 was identified as Lyso-PAF (11Z)-C18:1 [13,14].
Table 1. LC/TOF-MS data for the F7 fraction and its ozonolysis products.
| F7 fraction | RT (min) | Formula | Observed m/z | Theoretical [M+H]+ m/z | Observed m/z | Theoretical [M+Na]+ m/z |
| LPC C16:0 (4) | 4.49 | C24H50NO7P | 496.3417 | 496.3398 | 518.3230 | 518.3217 |
| Lyso-PAF C16:0 (5) | 4.76 | C24H52NO6P | 482.3615 | 482.2605 | 504.3437 | 504.3424 |
| Lyso-PAF (11Z)-C18:1 (2) | 4.95 | C26H54NO6P | 508.3752 | 508.3762 | 530.3581 | 530.3581 |
| LPC C18:0 (3) | 5.57 | C26H54NO7P | 524.3742 | 524.3711 | 546.3538 | 546.3530 |
| Lyso-PAF C18:0 (1) | 5.83 | C26H56NO6P | 510.3920 | 510.3918 | 532.3733 | 532.3737 |
| Lyso-PAF 17'-Me-C18:0 (6) | 6.23 | C27H58NO6P | 524.4068 | 524.4075 | 546.3897 | 546.3894 |
| standard samples | RT (min) | formula | Observe m/z | Theoretical [M+H]+ m/z | Observed m/z | Theoretical [M+Na]+ m/z |
| LPC C16:0 (4) | 4.49 | C24H50NO7P | 496.3395 | 496.3398 | 518.3223 | 518.3217 |
| LPC (9Z)-C18:1 | 4.76 | C26H52NO7P | 522.3547 | 522.3554 | 544.3392 | 544.3374 |
| Lyso-PAF (9Z)-C18:1 | 5.00 | C26H54NO6P | 508.3744 | 508.3762 | 530.3583 | 530.3581 |
| LPC C18:0 (3) | 5.55 | C26H54NO7P | 524.3706 | 524.3711 | 546.3537 | 546.3530 |
| Lyso-PAF C18:0 (1) | 5.85 | C26H56NO6P | 510.3920 | 510.3918 | 532.3735 | 532.3737 |
| ozonolysis products of F7 fraction | RT (min) | formula | Observe m/z | Theoretical [M+H]+ m/z | Observed m/z | Theoretical [M+Na]+ m/z |
| C11-carboxylic acid (7) | 0.70 | C19H40NO8P | 442.2568 | 442.2564 | ND | |
| C11-aldehyde (8) | 0.78 | C19H40NO7P | 426.2614 | 462.2615 | ND | |
| C11-d6-dimethyl acetal (9) | 1.01 | C21H40D6NO8P | 478.3390 | 478.3410 | ND |
ND; not done.
Some of the minor components were inferred to be LPC based on 1H-1H correlations in the DQF-COSY and TOCSY analyses of the glycerol and fatty acid units and the 13C NMR data assigned from the 1H-13C HSQC and HMBC spectra. LC/TOF-MS also detected LPC C18:0 (3), LPC C16:0 (4), Lyso-PAF C16:0 (5), and Lyso-PAF C19:0, and accurately determined their masses (Table 1 and Supporting Information). An odd number of alkyl chains in the linear form is relatively rare, so the minor signals in the NMR spectra were analyzed carefully. In the 1H-13C HMBC spectra, a minor doublet methyl signal at 0.87 ppm showed 1H-13C long-range correlations with carbon signals at 40.24 (CH2), 29.15 (CH), and 23.04 (CH3), and the last carbon was identical to the methyl carbon itself. These data, and especially the 13C NMR chemical shift values, suggested the presence of an isopropyl group at the end of the alkyl chain. Based on this evidence, the peak at RT = 6.23, Lyso-PAF C19:0, was tentatively assigned as the 17′-methyl derivative of Lyso-PAF C18:0 (6). In summary, NMR and LC/TOF-MS analyses identified Lyso-PAF C18:0 (1), Lyso-PAF (11Z)-C18:1 (2), LPC C18:0 (3), LPC C16:0 (4), Lyso-PAF C16:0 (5), and 17′-methyl Lyso-PAF C18:0 (6) in the F7 fraction (Fig 4). Based on the integration values of the 1H-NMR spectrum of the F7 fraction, the ratio of Lyso-PAF (compounds 1, 2, 5, and 6) to LPC (compounds 3 and 4) was 88:12. The content of the minor compound 6 in the F7 fraction was estimated to be ca. 5% from the integration value for the doublet methyl signal. Determining the ratio of C18:0 to C16:0, such as (1):(5) and (3):(4), was impossible with NMR. However, the ratio of (3) or (4) was roughly estimated to be ca. 1:1 with a gas chromatographic analysis of the alkaline hydrolysate.
Fig 4. Structures of lysophospholipids.

Compounds 1–6 were identified as Lyso-PAF and LPC in the F7 lipid fraction. Compounds 7–9 were the products of ozonolysis.
Roles of Lyso-PAF and LPC in the cytoprotective and antioxidative activities of F7
Commercially available C18 lipids were tested to confirm the roles of Lyso-PAF and LPC in the cytoprotection of J774A.1 cells (Fig 5). Treatment of the J774A.1 cells with the F7 fraction or with a 10 μg/mL lipid mixture containing Lyso-PAF C18:0 (44%), Lyso-PAF (9Z)-C18:1 (44%), and LPC C18:0 (12%) significantly increased cell viability in the presence of H2O2 compared with that of cells not treated with the lipids. However, treatment with Lyso-PAF C18:0, Lyso-PAF (9Z)-C18:1, or LPC C18:0 alone exerted no cytoprotective effect against H2O2 stress. In contrast, the ORAC assay revealed that Lyso-PAF (9Z)-C18:1 and LPC C18:0 each had antioxidative activity (Fig 6). These data indicate that the additive effects of Lyso-PAF and LPC may be required for their cytoprotective effect against oxidative stress.
Fig 5. Viability of J774A.1 cells treated with LPC or Lyso-PAF.

J774A.1 cells were preincubated with LPC or Lyso-PAF for 12 h, and H2O2 was then added to the cells (25 μM). After 12 h, cell viability was measured and is expressed as the mean viability ± standard deviation of triplicate experiments. (A) F7 lipid fraction. (B) Lipid mixture containing Lyso-PAF C18:0 (44%), Lyso-PAF (9Z)-C18:1 (44%), and LPC C18:0 (12%). (C) Lyso-PAF C18:0. (D) Lyso-PAF (9Z)-C18:1. (E) LPC. (*) Significant (P < 0.05) according to ANOVA followed by Tukey’s multiple comparisons.
Fig 6. ORAC assay.
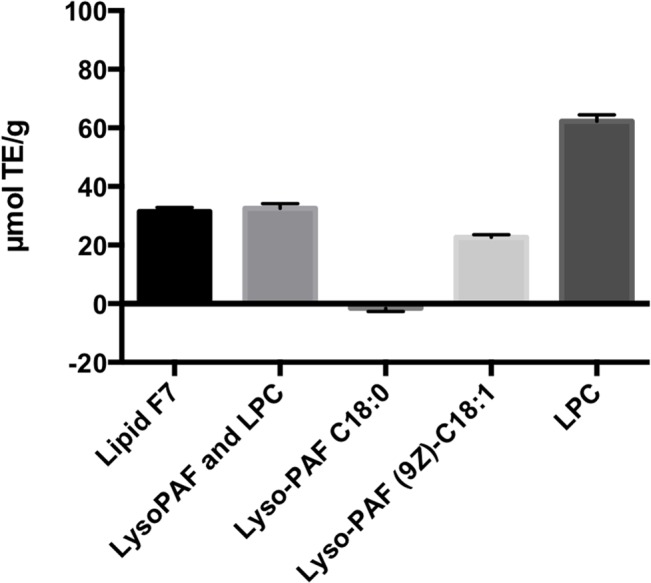
Antioxidant capacities of the F7 lipid fraction and a lipid mixture containing Lyso-PAF C18:0 (44%), Lyso-PAF (9Z)-C18:1 (44%), and LPC C18:0 (12%). Lyso-PAF C18:0, Lyso-PAF (9Z)-C18:1, and LPC are expressed in μmol Trolox equivalents (TE)/g, calculated from a Trolox standard curve.
Discussion
Various species of invertebrates contain large amounts of both phosphocholine and phosphoethanolamine. Platelet-activating factor (PAF)-like lipids are also widely distributed among various invertebrates. Therefore, phospholipids, including PAF, may be physiologically important molecules, particularly for invertebrates [15]. In this study, we detected Lyso-PAF and LPC in the lipid fraction with antioxidative activity extracted from the sea cucumber H. atra for the first time. NMR and LC/TOF-MS analyses identified Lyso-PAF C18:0 (1), Lyso-PAF (11Z)-C18:1 (2), LPC C18:0 (3), LPC C16:0 (4), Lyso-PAF C16:0 (5), and 17′-methyl Lyso-PAF C18:0 (6) in the bioactive fraction. A Lyso-PAF-related molecule has also been detected in the sea cucumber Cucumaria frondosa [16]. Interestingly, a series of Lyso-PAF analogues and LPCs were isolated from the marine sponge Spirastrella abata [14]. LPC C16:0 and Lyso-PAF (11Z)-C18:1 have also been identified in S. abata [14] and an Australian sponge, Crella incrustans [17]. These results suggest that Lyso-PAF analogues and LPCs are widely distributed among marine invertebrates, and some compounds may be species-specific.
Lyso-PAF is produced when PAF is cleaved at the sn-2 position by phospholipase A2. A highly specific PAF-acetyltransferase then adds an acetyl group to the sn-2 position to yield PAF [18]. The presence of acetyltransferase activity has been confirmed in several marine invertebrates [15], suggesting the biosynthesis of Lyso-PAF in the lower animals. Importantly, lyso-PAF can act as a bioactive lipid, with functions opposite those of PAF [19]. The PAF generated by basophils induces platelet aggregation and anaphylaxis [20,21], and the PAF produced by neutrophils plays important roles in inflammation, including in the activation of NADPH oxidase [19]. The accumulation of PAF is also thought to be a principal initiator of neuronal dysfunction and death in human immunodeficiency virus (HIV)-associated dementia and a secondary mediator of neural loss in ischemia and epilepsy [22,23]. Therefore, the intrinsic regulatory mechanisms that balance PAF activities are potential targets for therapeutic interventions. Lyso-PAF also seems to have antioxidative activity. In this study, treatment with Lyso-PAF C18:0 or Lyso-PAF (9Z)-C18:1 alone exerted no cytoprotective effect against H2O2 stress on J774A.1 cells. In contrast, according to an ORAC assay, Lyso-PAF (9Z)-C18:1 has antioxidative activity. Further research is required to evaluate the different activities of saturated and unsaturated Lyso-PAF.
In mammals, LPC is a highly abundant bioactive lysoglycerophospholipid in the circulation, predominantly associated with albumin [24] and lipoproteins, such as oxidized low-density lipoprotein (oxLDL) [25]. The hydrolysis of the fatty acid of membrane phosphatidylcholine at the sn-2 position by the superfamily of phospholipase A2 enzymes generates LPC and fatty acids [26]. Although the activity of phospholipase A2 has not been demonstrated in the sea cucumber, the expression of phospholipase has been confirmed with a proteomic analysis in the sea cucumber Apostichopus japonicus coelomocytes after Vibrio splendidus infection [27]. This suggests the presence of the biosynthetic pathway for LPC in the sea cucumber H. atra. Generally, LPC is an important signaling lipid with diverse functions in inflammation. LPC is considered a causative factor in atherosclerosis, the promotion of demyelination in the nervous system, the induction of hepatitis, and protection from sepsis. LPC also acts as a chemoattractant signal for phagocytic cells, which function as immunoregulatory cells [28]. Our results indicate that the treatment of J774A.1 cells with LPC C18:0 conferred no cytoprotective effect against H2O2 stress, whereas the ORAC assay demonstrated the antioxidative activity of LPC C18:0. Although stearoyl LPC significantly attenuates circulating high-mobility group box 1 (HMGB1) levels in endotoxemia and sepsis by suppressing the release of endotoxin-induced HMGB1 from macrophages/monocytes [29], there has been no report of the antioxidative activity of LPC. Therefore, the mechanism underlying the antioxidative activity of LPC must be addressed in a future study.
Although the effects of the lysophospholipids of sea cucumbers are largely unknown, this study has shown that Lyso-PAF and LPC in the sea cucumber H. atra have cytoprotective activity in vitro. Importantly, the additive effects of Lyso-PAF and LPC are required for their cytoprotective activity against oxidative stress. Among the analogues of lysophospholipids, Lyso-PAF exhibits cancerostatic properties [30,31] and potent antimicrobial activity [32]. Our results suggest that the combination of Lyso-PAF and LPC from the sea cucumber has activities that can control inflammation in animals, including humans. Lyso-PAF and LPC analogues are abundant in invertebrates and are widespread among marine invertebrates [15], so our novel findings should prompt the investigation of the therapeutic uses of lysophospholipids from these organisms.
Supporting Information
Fig A 1H NMR spectra of F7 fraction, and authentic Lyso-PAF (9Z)-C18:1. Fig B 13C NMR spectra of F7 fraction, and authentic Lyso-PAF (9Z)-C18:1 and cis-5-dodecenoic acid. Fig C DQF-COSY spectrum of F7 fraction. Fig D TOCSY spectrum of F7 fraction. Fig E NOESY spectrum of F7 fraction. Fig F HSQC spectrum of F7 fraction. Fig G HSQC-TOCSY spectrum of F7 fraction. Fig H 1H-13C HMBC spectrum of F7 fraction. Fig I 1H-31P HMBC spectrum of F7 fraction. Fig J Base peak ion chromatograms and mass chromatograms on LC/TOF-MS. Fig K Mass spectrum for RT 5.83 of F7 fraction, Lyso-PAF C18:0 (1). Fig L Mass spectrum for RT 4.95 of F7 fraction, Lyso-PAF (11Z)-C18:1 (2). Fig M Mass spectrum for RT 5.57 of F7 fraction, LPC C18:0 (3) Fig N Mass spectrum for RT 4.49 of F7 fraction, LPC C16:0 (4) Fig O Mass spectrum for RT 4.76 of F7 fraction, Lyso-PAF C16:0 (5). Fig P Mass spectrum for RT 6.23 of F7 fraction, Lyso-PAF 17’- Methyl-C18:0 (6). Fig Q Mass spectrum for Lyso-PAF C18:0 (1) of a standard mixture. Fig R Mass spectrum for Lyso-PAF (9Z)-C18:1 of a standard mixture. Fig S Mass spectrum for LPC C18:0 (3) of a standard mixture. Fig T Mass spectrum for LPC C16:0 (4) of a standard mixture. Fig U Mass spectrum for LPC (9Z)-C18:1 of a standard mixture. Fig V Mass spectrum for RT 0.70 of ozonolysis products, C11-carboxylic acid (7). Fig W Mass spectrum for RT 0.78 of ozonolysis products, C11-aldehyde (8). Fig X Mass spectrum for RT 1.01 of ozonolysis products, C11-d6-dimethyl acetal (9).
(PDF)
Acknowledgments
We thank Youko Matsushita (Obihiro University of Agriculture and Veterinary Medicine) and Kouji Yamada (Nagasaki University) for their technical support with lipid extraction.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported in part by Global COE Program “Frontier Program for Animal Global Health and Hygiene”, MEXT, Japan. This work was also supported in part by the Lipid Dynamics Program of RIKEN. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Althunibat OS, Hashim RB, Taher M, Daud JM, Ikeda M, Zali I. In vitro antioxidant and antiproliferative activities of three Malaysian sea cucumber species. Eur J Sci Res. 2009;37: 376–387. [Google Scholar]
- 2. Hamel JF, Mercier A. Synchronous gamete maturation and reliable spawning induction method in holothurians In: Lovatelli A, Conand C, Purcell S, Uthicke S, Hamel JF, Mercier A, editors. Advances in sea cucumber aquaculture and management. FAO fisheries technical reports no. 463 Rome: FAO; 2004. pp. 359–372. [Google Scholar]
- 3. Esmat AY, Said MM, Soliman AA, El-Masry KS, Badiea EA. Bioactive compounds, antioxidant potential, and hepatoprotective activity of sea cucumber (Holothuria atra) against thioacetamide intoxication in rats. Nutrition. 2013;29: 258–267. 10.1016/j.nut.2012.06.004 [DOI] [PubMed] [Google Scholar]
- 4. Herencia F, Ubeda A, Ferrándiz ML, Terencio MC, Alcaraz MJ, García-Carrascosa M, et al. Anti-inflammatory activity in mice of extracts from Mediterranean marine invertebrates. Life Sci. 1998;62: PL115–120. [DOI] [PubMed] [Google Scholar]
- 5. Ogushi M, Yoshie-Stark M, Suzuki T. Cytostatic activity of hot water extracts from the sea cucumber in Caco-2. Food Sci Technol Res. 2005;11: 202–206. [Google Scholar]
- 6. Darah I, Stheesh N, Ibrahim CO. Growth inhibition of dermathophites by atratoxins of Holothuria atra . Biosci Penang. 1995;6: 40–48. [Google Scholar]
- 7. Sugawara T, Zaima N, Yamamoto A, Sakai S, Noguchi R, Hirata T. Isolation of sphingoid bases of sea cucumber cerebrosides and their cytotoxicity against human colon cancer cells. Biosci Biotechnol Biochem. 2006;70: 2906–2912. [DOI] [PubMed] [Google Scholar]
- 8. Kitagawa I, Sugawara T, Yosioka I, Kuriyama K. Saponin and sapogenol. XIV. Antifungal glycosides from the sea cucumber Stichopus japonicus selenka. (1). Structure of stichopogenin A4, the genuine aglycone of holotoxin A. Chem Pharm Bull (Tokyo). 1976;24: 266–274. [DOI] [PubMed] [Google Scholar]
- 9. Kitagawa I, Sugawara T, Yosioka I. Saponin and sapogenol. XV. Antifungal glycosides from the sea cucumber Stichopus japonicus selenka. (2). Structures of holotoxin A and holotoxin B. Chem Pharm Bull (Tokyo). 1976;24: 275–284. [DOI] [PubMed] [Google Scholar]
- 10. Yamada K. Chemo-pharmaceutical studies on the glycosphingolipid constituents from echinoderm, sea cucumbers, as the medicinal materials. Yakugaku Zasshi. 2002;122: 1133–1143. [DOI] [PubMed] [Google Scholar]
- 11. Hashim RB, Azizan NA, Zamli Z, Zulkipli FH, Mazlan N, Althunibat OY. Toxicity effects of water extracts of Holothuria atra Jaeger in mice. Asian Pac J Trop Biomed. 2014;4: 614–617. 10.12980/APJTB.4.201414B154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Convert C, Michel E, Heymans F, Godfroid JJ. 1H- and 13C-NMR studies of platelet-activating factor (PAF-acether) and analogs. Influence of solvent. Biochim Biophys Acta. 1984;794: 320–325. [Google Scholar]
- 13. Yayli N, Findlay JA. Polar metabolites from the Sea Cucumber Cucumaria Frondosa . J Nat Prod. 1994;57: 84–89. [DOI] [PubMed] [Google Scholar]
- 14. Shin BA, Kim YR, Lee IS, Sung CK, Hong J, Sim CJ, et al. Lyso-PAF analogues and lysophosphatidylcholines from the marine sponge Spirastrella abata as inhibitors of cholesterol biosynthesis. J Nat Prod. 1999;62: 1554–1557. [DOI] [PubMed] [Google Scholar]
- 15. Sugiura T, Fukuda T, Miyamoto T, Waku K. Distribution of alkyl and alkenyl ether-linked phospholipids and platelet-activating factor-like lipid in various species of invertebrates. Biochim Biophys Acta. 1992;1126: 298–308. [DOI] [PubMed] [Google Scholar]
- 16. Yayli N, Findlay JA. Polar metabolites from the Sea Cucumber Cucumaria Frondosa . J Nat Prod. 1994;57: 84–89. [DOI] [PubMed] [Google Scholar]
- 17. Butler AJ, van Altena IA, Dunne SJ. Antifouling activity oflyso-platelet-activating factor extracted from Australian sponge Crella incrustans . J Chem Ecol. 1996;22: 2041–2061. 10.1007/BF02040094 [DOI] [PubMed] [Google Scholar]
- 18. Shindou H, Hishikawa D, Nakanishi H, Harayama T, Ishii S, Taguchi R, et al. A single enzyme catalyzes both platelet-activating factor production and membrane biogenesis of inflammatory cells. Cloning and characterization of acetyl-CoA:LYSO-PAF acetyltransferase. J Biol Chem. 2007;282: 6532–6539. [DOI] [PubMed] [Google Scholar]
- 19. Welch EJ, Naikawadi RP, Li Z, Lin P, Ishii S, Shimizu T, et al. Opposing effects of platelet-activating factor and lyso-platelet-activating factor on neutrophil and platelet activation. Mol Pharmacol. 2009;75: 227–234. 10.1124/mol.108.051003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Benveniste J, Henson PM, Cochrane CG. Leukocyte-dependent histamine release from rabbit platelets. The role of IgE, basophils, and a platelet-activating factor. J Exp Med. 1972;136: 1356–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vadas P, Gold M, Perelman B, Liss GM, Lack G, Blyth T, et al. Platelet-activating factor, PAF acetylhydrolase, and severe anaphylaxis. N Engl J Med. 2008;358: 28–35. 10.1056/NEJMoa070030 [DOI] [PubMed] [Google Scholar]
- 22. Bazan NG, Rodriguez de Turco EB, Allan G. Mediators of injury in neurotrauma: intracellular signal transduction and gene expression. J Neurotrauma. 1995;12: 791–814. [DOI] [PubMed] [Google Scholar]
- 23. Perry SW, Hamilton JA, Tjoelker LW, Dbaibo G, Dzenko KA, Epstein LG, et al. Platelet-activating factor receptor activation. An initiator step in HIV-1 neuropathogenesis. J Biol Chem. 1998;273: 17660–17664. [DOI] [PubMed] [Google Scholar]
- 24. Croset M, Brossard N, Polette A, Lagarde M. Characterization of plasma unsaturated lysophosphatidylcholines in human and rat. Biochem J. 2000;345: 61–67. [PMC free article] [PubMed] [Google Scholar]
- 25. Wiesner P, Leidl K, Boettcher A, Schmitz G, Liebisch G. Lipid profiling of FPLC-separated lipoprotein fractions by electrospray ionization tandem mass spectrometry. J Lipid Res. 2009;50: 574–585. 10.1194/jlr.D800028-JLR200 [DOI] [PubMed] [Google Scholar]
- 26. Burke JE, Dennis EA. Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res. 2009;50: S237–S242 (Suppl.). 10.1194/jlr.R800033-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang P, Li C, Li Y, Zhang P, Shao Y, Jin C, et al. Proteomic identification of differentially expressed proteins in sea cucumber Apostichopus japonicus coelomocytes after Vibrio splendidus infection. Dev Comp Immunol. 2014;44: 370–377. 10.1016/j.dci.2014.01.013 [DOI] [PubMed] [Google Scholar]
- 28. Sevastou I, Kaffe E, Mouratis MA, Aidinis V. Lysoglycerophospholipids in chronic inflammatory disorders: the PLA(2)/LPC and ATX/LPA axes. Biochim Biophys Acta. 2013;1831: 42–60. 10.1016/j.bbalip.2012.07.019 [DOI] [PubMed] [Google Scholar]
- 29. Chen G, Li J, Qiang X, Czura CJ, Ochani M, Ochani K, et al. Suppression of HMGB1 release by stearoyl lysophosphatidylcholine:an additional mechanism for its therapeutic effects in experimental sepsis. J Lipid Res. 2005;46: 623–627. [DOI] [PubMed] [Google Scholar]
- 30. Mangold HK, Weber N. Biosynthesis and biotransformation of ether lipids. Lipids. 1987;22: 789–799. [DOI] [PubMed] [Google Scholar]
- 31. Brachwitz H, Vollgraf C. Analogs of alkyllysophospholipids: chemistry, effects on the molecular level and their consequences for normal and malignant cells. Pharmacol Ther. 1995;66: 39–82. [DOI] [PubMed] [Google Scholar]
- 32. Steel HC, Cockeran R, Anderson R. Platelet-activating factor and lyso-PAF possess direct antimicrobial properties in vitro. APMIS. 2002; 110: 158–164. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig A 1H NMR spectra of F7 fraction, and authentic Lyso-PAF (9Z)-C18:1. Fig B 13C NMR spectra of F7 fraction, and authentic Lyso-PAF (9Z)-C18:1 and cis-5-dodecenoic acid. Fig C DQF-COSY spectrum of F7 fraction. Fig D TOCSY spectrum of F7 fraction. Fig E NOESY spectrum of F7 fraction. Fig F HSQC spectrum of F7 fraction. Fig G HSQC-TOCSY spectrum of F7 fraction. Fig H 1H-13C HMBC spectrum of F7 fraction. Fig I 1H-31P HMBC spectrum of F7 fraction. Fig J Base peak ion chromatograms and mass chromatograms on LC/TOF-MS. Fig K Mass spectrum for RT 5.83 of F7 fraction, Lyso-PAF C18:0 (1). Fig L Mass spectrum for RT 4.95 of F7 fraction, Lyso-PAF (11Z)-C18:1 (2). Fig M Mass spectrum for RT 5.57 of F7 fraction, LPC C18:0 (3) Fig N Mass spectrum for RT 4.49 of F7 fraction, LPC C16:0 (4) Fig O Mass spectrum for RT 4.76 of F7 fraction, Lyso-PAF C16:0 (5). Fig P Mass spectrum for RT 6.23 of F7 fraction, Lyso-PAF 17’- Methyl-C18:0 (6). Fig Q Mass spectrum for Lyso-PAF C18:0 (1) of a standard mixture. Fig R Mass spectrum for Lyso-PAF (9Z)-C18:1 of a standard mixture. Fig S Mass spectrum for LPC C18:0 (3) of a standard mixture. Fig T Mass spectrum for LPC C16:0 (4) of a standard mixture. Fig U Mass spectrum for LPC (9Z)-C18:1 of a standard mixture. Fig V Mass spectrum for RT 0.70 of ozonolysis products, C11-carboxylic acid (7). Fig W Mass spectrum for RT 0.78 of ozonolysis products, C11-aldehyde (8). Fig X Mass spectrum for RT 1.01 of ozonolysis products, C11-d6-dimethyl acetal (9).
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.