

SUMMARY
STING is an ER-associated membrane protein that is critical for innate-immune sensing of pathogens. STING-mediated activation of the IFN-I pathway through the TBK1/IRF3 signaling axis involves both cyclic-dinucleotide binding, and its translocation from the ER to vesicles. However, how these events are coordinated, and the exact mechanism of STING activation, remain poorly understood. Here, we found that the Shigella effector protein IpaJ potently inhibits STING signaling by blocking its translocation from the ER to ERGIC, even in the context of dinucleotide binding. Reconstitution using purified components revealed STING translocation as the rate-limiting event in maximal signal transduction. Furthermore, STING mutations associated with autoimmunity in humans were found to cause constitutive ER exit, and to activate STING independent of cGAMP binding. Together, these data provide compelling evidence for an ER-retention and ERGIC/Golgi-trafficking mechanism of STING regulation that is subverted by bacterial pathogens and is deregulated in human genetic disease.
Graphical Abstract
INTRODUCTION
Vertebrates are constantly facing challenges from pathogenic microbes. To counter this, eukaryotic cells express pattern-recognition receptors that detect microbes through pathogen-associated molecular patterns (PAMPs), which then activate interferon (IFN) and proinflammatory responses. One important protein that plays a central role in sensing a wide variety of cytosolic pathogens is STING. STING plays a key role in innate immune sensing of cytosolic DNA and cyclic dinucleotides (Burdette et al., 2011; Cai et al., 2014). DNA produced by retrovirus (e.g. HIV-1, (Gao et al., 2013; Yan et al., 2010)) and DNA viruses (e.g. Herpes simplex virus, (Ishikawa et al., 2009)) are recognized by cGAS, which converts ATP and GTP to the second messenger 2′3′ cyclic GMP-AMP (cGAMP). cGAMP directly activates STING through a mechanism that is still poorly understood. In addition, cyclic dinucleotides associated with bacterial infection (e.g. Listeria monocytogenes, (Burdette et al., 2011)) can also activate STING, providing a broad immunological sensor for host defense against remarkably diverse pathogen types.
In context of normal physiology, STING activation needs to be carefully regulated as constitutive or chronic activation due to genetic mutations are associated with autoimmune and autoinflammatory disorders (Hasan et al., 2013; Liu et al., 2014). For example, several gain-of-function mutations in TMEM173 (encodes STING) were found in patients with an autoinflammatory disease called STING-associated vasculopathy with onset in infancy (SAVI, (Liu et al., 2014)), and more recently in patients with lupus-like syndromes (Jeremiah et al., 2014). The molecular mechanisms of these diseases are unclear, although constitutive activation of STING signaling is likely involved. Thus, elucidating STING activation mechanism is of great importance in understanding both microbial pathogenesis and autoimmune disorders.
Previous studies indicated that STING activation involves trafficking from the ER to autophagic-like vesicles (Ishikawa et al., 2009), and the phosphorylation of TBK1 and IRF3 leading to NFκB and IFN gene expression (Burdette and Vance, 2013). Despite the importance of these atomic models for revealing conformational changes induced by cyclic dinucleotide ligand binding, the structure have offered very few clues on the mechanism of STING activation (Burdette and Vance, 2013; Zhang et al., 2013). Here, we took advantage of two recently characterized Shigella Type 3 Secretion System (T3SS) effector proteins, IpaJ and VirA, that uniquely target different steps of the host secretory pathway (Burnaevskiy et al., 2013), and engineered several deletion and chimeric bacterial strains that are capable of ‘pausing’ STING trafficking and signaling at intermediate stages during an ongoing infection. We found that Shigella effector IpaJ inhibits STING activation by blocking its translocation from ER to ER-Golgi intermediate compartments (ERGIC), a complex system of tubulovesicular membrane clusters found near ER exit sites. We also found that STING-mediated TBK1/IRF3 activation reaches the maximum at the ERGIC. Importantly, we found that STING disease-associated mutants cause constitutively ER exit, stably associate with ERGIC and Golgi, and activate STING signaling independent of cGAMP binding. Thus our findings provide a spatial model for STING activation and critical insights on the pathogenesis of STING diseases.
Results
Shigella flexneri antagonizes STING-mediated IFN activation
Shigella flexneri is a Gram-negative bacterial pathogen that utilizes a Type 3 Secretion System (T3SS) to inject effector proteins that regulate innate immune signaling pathways (Raymond et al., 2013; Reddick and Alto, 2014). Because Shigella invades non-phagocytic cells, escapes the vacuole, and replicates in the host cytoplasm it was reasonable to think that it would be recognized by cytosolic immune receptors such as cGAS and activates STING signaling. Shigella flexneri M90T (here foreword Shigella) infection in mouse embryonic fibroblasts (MEFs) induced a modest increase in IFNβ expression that was not observed in the non-invasive Shigella ΔmxiD strain (an essential component of the Mxi/Spa T3SS). We next tested if immune regulatory T3SS effectors may dampen the Type I IFN response. Because STING trafficking from the Endoplasmic Reticulum (ER) is essential to its activation mechanism, we performed loss-of-function genetic studies focusing on Shigella effectors IpaJ and VirA that inhibit Golgi apparatus structure and function (Burnaevskiy et al., 2013; Dong et al., 2012). As shown in Figure 1A, ShigellaΔipaJ strain induced a 10-fold increase in IFNβ expression compared to WT Shigella. This finding was consistent over 2-log range of bacterial challenge, indicating that host cells respond to cytosolic Shigella in a dose dependent manner and that IpaJ dampens the immune response even at high burdens of infection (Figure 1B). IFN-stimulated genes (ISGs) were also highly induced in ShigellaΔipaJ infected cells, whereas there was little change in inflammatory transcripts likely induced by LPS (Figure 1C, 1D).
Figure 1. Shigella effector IpaJ inhibits STING-mediated IFN activation.

(A) Quantitative RT-PCR analysis of Ifnb mRNA in MEFs infected with indicated Shigella strains for 8 hours. Individual gene expression value was normalized to housekeeping gene Gapdh (same throughout). (B) Quantitative RT-PCR analysis of Ifnb mRNA in MEFs infected with Shigella WT or ΔipaJ at indicated m.o.i. for 8 hours. (C) Quantitative RT-PCR array analysis of immune gene expression (indicated on the right) in MEFs infected with various Shigella strains (indicated on the top). (D) Quantitative RT-PCR analysis of Tnf mRNA in MEFs infected with indicated Shigella strains for 8 hours as in A. (E) Quantitative RT-PCR analysis of Ifnb mRNA in siRNA knockdown MEFs infected with Shigella WT or ΔipaJ. MEFs were transfected with indicated siRNA, and two days later infected with Shigella WT or ΔipaJ. Ifnb mRNA was measured 8 h after infection. (F) Quantitative RT-PCR analysis of Ifnb mRNA in WT or knockout MEFs (indicated on the bottom) infected with Shigella WT or ΔipaJ. *P < 0.05, **P < 0.01, ***P < 0.001 (same throughout). Data are representative of at least three independent experiments. Error bars, SEM. Unpaired t-test.
To then determine if the Type I IFN response to Shigella infection depended on the STING-TBK1-IRF3 signaling axis, the expression of each signaling component was depleted in cells. ShigellaΔipaJ-induced IFNβ expression was significantly reduced in cells treated with RNAi targeting STING, TBK1, and IRF3 (Figure 1E) and was almost completely eliminated in cGAS-/-, Sting-gt/gt or Irf3-/- MEFs (Figure 1F). In contrast, Mavs-/-, Myd88/Trif-/-, or Irf7-/- had no effect (Figure 1F), highlighting the specificity of IpaJ at inhibiting cGAS/STING cytosolic DNA sensing pathway in this cellular model of infection. These data also demonstrate that ShigellaΔipaJ-induced IFN response in MEFs is stimulated by bacteria DNA. Recently, we demonstrated that IpaJ inhibits ER to Golgi transport by selectively cleaving N-myristoylated glycine of human ARF-family GTPases (Burnaevskiy et al., 2013; Burnaevskiy et al., 2015). This proteolytic function of IpaJ was conserved in a murine model of cellular infection (Figure S1). Based on these findings it was attractive to hypothesize that Shigella IpaJ inhibits STING signaling at the level of ER to Golgi transport. Interestingly however, the Shigella T3SS effector VirA also inhibits cargo transport through the Golgi apparatus, a phenotype dependent on its ability to stimulate GTP hydrolysis on Rab1 GTPase (Dong et al., 2012). Unexpectedly, cells infected with ShigellaΔvirA strain exhibited low levels of IFNβ and ISG expression comparable to cells infected with WT Shigella strain, whereas ShigellaΔipaJ and ShigellaΔipaJ/virA induced a robust IFN response (Figure 1A, 1C). These data provide compelling evidence that STING signal transduction can be discriminated at the level of ARF and Rab GTPase regulation.
Reconstitution of IFN regulation in a Listeria model of STING activation
Since Shigella secretes a large number effector proteins into host cells, it is difficult to draw unequivocal conclusions about the role of IpaJ using this infection models (Figure 2A). Listeria monocytogenes (here forward referred to as Listeria) is a Gram-positive bacterial pathogen that directly activates STING by secreting c-di-AMP into the host cytosol (Burdette et al., 2011; Witte et al., 2012; Woodward et al., 2010) (Figure 2A). While these two evolutionarily unrelated bacterial pathogens exhibit intracellular life cycles, Listeria does not encode homologs of ipaJ or virA, nor does it disrupt ER to Golgi trafficking (Burnaevskiy et al., 2013). To then verify that IpaJ truly inhibits STING signaling in context of a well-established activation mechanism, we engineered the human attenuated Listeria vaccine strain ΔactA to secrete exogenous Shigella effector proteins into the cytosol of infected target cells. This was accomplished by fusing the promoter and the Type II secretion sequence (residues 1–100) of actA to the protease domain of ipaJ and ipajC64A (Figure 2A) or the GAP domain of virA (see materials and methods). These chimeras were recombined into the genome of ListeriaΔactA (Lauer et al., 2002b). In principle, the Shigella T3SS effector proteins should be expressed as Listeria escapes the vacuole and enters the host cell cytoplasm due to cytosolic stimulation of the actA promoter and subsequently secreted by the Type II Secretion System (due to the ActA signal sequence). As shown in Figure 2B, ListeriaΔactA::ipaJ and ListeriaΔactA::virA disrupted Golgi architecture at early stages of infection, indicating these Shigella effectors are functional when produced by Listeria. The engineered Listeria strains also invaded host cells and replicated to similar levels as the ListeriaΔactA parent strain (Figure 2C). To then assess the role of ER to Golgi transition on a natural route of STING activation, we infected WT MEFs with Listeria strains and assessed STING activation by measuring IFNβ expression (Figure 2D). Compared to ListeriaΔactA, cells infected with ListeriaΔactA::ipaJ exhibited significantly lower IFNβ gene expression whereas ListeriaΔactA::virA nor ListeriaΔactA::ipaJC64A chimeras inhibited IFNβ expression. Consistent with the notion that STING inhibition occurred by proteolytic demyrstoylation of ARF1, Listeria ΔactA::ipaJ induced the removal of GFP-ARF1 from Golgi membranes, whereas neither Listeria ΔactA::ipaJC64A nor Listeria ΔactA::virA disrupted ARF1 localization (Figure S2). Together, these findings confirm the STING inhibitory function of IpaJ in a well-defined model of microbial immune regulation.
Figure 2. Listeria/ipaJ chimeric bacteria infection blocks STING signaling.

(A) A schematic diagram of Shigella subversion and Listeria activation of the STING pathway (left) and Listeria chimera design for expressing Shigella effectors (right). (B) Fluorescent microscope of HeLa cells infected with indicated Listeria strains. The cis-Golgi (red) was detected by α-GM130 antibody. Arrows indicate disrupted Golgi apparatus. (C) Bacteria load in MEFs infected with indicated Listeria strains in a time course. (D) Quantitative RT-PCR analysis of Ifnb and Tnf mRNA in MEFs infected with indicated Shigella strains for 8 hours. Data are representative of at least three independent experiments. Error bars, SEM. Unpaired t-test (D). See also Figure S2.
Maximum STING activation on the ERGIC
The differential regulation of STING activation by bacteria secreting IpaJ or VirA suggest that subtle differences in ARF1 and Rab1 GTPase regulation at the ER to Golgi transition have profound consequences on immune signal transduction. These differences may shed light on the enigmatic activation mechanism of STING. To examine cytosolic dsDNA-stimulated STING trafficking in precise spatial and temporal resolution, we reconstituted the fundamental aspects of bacterial infection using purified components and a surrogate microinjection system (Figure 3A). To visualize STING activation at the level of ER transport in single cells, mouse STING-GFP was stably integrated into the genome of Sting-/- MEFs. As expected, STING-GFP localized to the ER in quiescent cells and rapidly transported to vesicles upon activation of cGAS by microinjected purified Shigella genomic DNA (herein dsDNA) (Figure 3A). Next, dsDNA was mixed with purified recombinant IpaJ or VirA, loaded into the micropipette, and microinjected into the cytoplasm of STING-GFP cells (Figure 3A). IpaJ effectively arrested STING trafficking out of the ER even in presence of high concentrations of dsDNA. This trafficking defect was due to proteolytic demyristoylation of ARF GTPases since recombinant IpaJ C64A failed to block STING trafficking. Interestingly, STING-GFP exited from the ER, but its transport was arrested at the ERGIC in cells microinjected with VirA and dsDNA. Because VirA does not inhibit the IFN-response (Figure 1A), it now appears the STING activation occurs immediately after ER-exit and its transition into the ERGIC. Consistent with these reconstitution studies, ShigellaΔipaJ infection activated substantial STING exit from the ER, whereas WT Shigella infection arrested STING-GFP on the ER (Figure 3B, S3). Activated STING in ShigellaΔipaJ infected cells localize to intermediate membrane compartments as shown by colocalization with both ERGIC and cis-Golgi markers (P58 and GM130, respectively), Thus, STING activation directly correlates with its ER to ERGIC/Golgi transition (Figure 3B, S3).
Figure 3. Reconstituting cytosolic DNA sensing through microinjection.

(A) Fluorescent micrographs show STING-GFP localization in MEFs after microinjection. STING-GFP MEFs were microinjected with buffer alone, DNA alone or DNA plus recombinant effector proteins (indicated on the left). Cells were fixed 6 h later and co-strained with an ERGIC marker, ERGIC/p58 (in red). * marks injected cells. Arrows indicate activated STING in vesicles, Arrowheads indicate activated STING on the ERGIC. Schematic models of STING localization under various conditions are shown on the right. Quantitation is shown below (n=30). (B) Fluorescent micrographs show STING-GFP localization in MEFs after DNA transfection or Shigella infection. STING-GFP MEFs were left untreated, transfected with HT-DNA, or infected with Shigella WT or ΔipaJ. Cells were fixed 8 h later and co-stained with ER (Calnexin), ERGIC (P58) or Golgi (GM130). Schematic models of STING localization under various conditions are shown on the right. Quantitation of colocalization was calculated as Pearson’s correlation coefficient (r) shown below (n=15). Images are representative of at least three independent experiments. See also Figure S3.
To then determine if STING transduces signal from the intermediate membrane compartments, we measured TBK1/IRF3 phosphorylation and TBK1 recruitment to STING. In control experiments, transfection of dsDNA induced IRF3 and TBK1 phosphorylation that peaked at 2-4 hours and dissipated over the next 24 hours (Figure 4A), similar to those previously described (Burdette and Vance, 2013). Endogenous TBK1 normally distributed throughout the cytoplasm was recruited to STING-GFP positive vesicles upon dsDNA transfection as expected (Figure 4C). Interestingly, we found that TBK1 was recruited to STING at intermediate membrane compartments in ShigellaΔipaJ infected cells (Figure 4D), further supporting that STING is active on these intermediate membrane compartments. We also observed stronger phosphorylation of both TBK1 and IRF3 in ShigellaΔipaJ infected cells compared to WT Shigella infected cells (Figure 4B). Conversely, ListeriaΔactA::ipaJ infection induced less TBK1 and IRF3 phosphorylation compared to ListeriaΔactA infection (Figure 4E). These data are consistent with stronger IFN expression induced by ShigellaΔipaJ infection (compared to WT) and by ListeriaΔactA infection (compared to ListeriaΔactA::ipaJ) (Figure 1A, 2D), and that IpaJ blocks STING-mediated IFN activation. Of note, we consistently detected low level of IFN activation and TBK1 and IRF3 phosphorylation in WT infected cells compared to uninfected or to ΔmixD that does not infect host cells (data not shown), despite the presence of both IpaJ and VirA in the WT strain. Nevertheless, these spatial and biochemical analysis of STING demarcates ERGIC as a primary cellular location for peak STING signal transduction.
Figure 4. STING signaling kinetics and activation on ERGIC.

(A, B) Immunoblots show kinetics of IRF3 and TBK1 phosphorylation in MEFs after HT-DNA transfection (A) or Shigella infection (B). (C, D) Fluorescent micrographs show STING-GFP and endogenous TBK1 localization in MEFs after DNA transfection (C) or Shigella infection (D). MEFs were transfected or infected as in Figure 3B. Cells were fixed 8 h later and co-stained with an TBK1 antibody. Quantitation of colocalization was calculated as Pearson’s correlation coefficient (r) shown below. (E) Immunoblots show kinetics of IRF3 and TBK1 phosphorylation in MEFs after infection of indicated Listeria strains. Data are representative of at least three independent experiments. See also Figure S4.
To examine the kinetics of STING trafficking after DNA transfection or bacterial infection, we also performed live-cell time lapse microscopy (DNA transfection) using STING-GFP cells as well as fixed-cell time course (Shigella infection) (Movie 1. Figure S4A). We found that DNA transfection caused strong activation of STING trafficking, and STING moved from the ER to vesicles in 90 minutes (Figure S4A). In contrast, ShigellaΔIpaJ infection promoted STING translocation from the ER to ERGIC/Golgi by 6 hours post infection (Figure S4B). The slower kinetics of STING trafficking after Shigella infection is likely due to the very small amount of DNA introduced by bacterial infection comparing to DNA transfection (1 microgram).
We showed previously that Nocodazole prevents membrane trafficking beyond ERGIC in a manner analogous to Rab1 inhibition by the VirA homolog EspG (Selyunin et al., 2014). Brefeldin A (BFA) potently inhibits ARF1 GTPase and can therefore be considered ‘IpaJ-like’. To then confirm previous results (Ishikawa et al., 2009) that BFA inhibits STING signaling in our model system, we treated MEFs with increasing doses of BFA or Nocodazole for 1 hour and transfected DNA to activate STING signaling and IFN expression. BFA inhibited DNA-induced IFN activation in a dose-dependent manner (Figure S4C–D). Interestingly, Nocodazole only marginally affected DNA-induced IFN activation (Figure S4C–D). These data further suggest that STING activation does not require the full extent of membrane trafficking pathway, and is consistent with maximum STING activation on intermediate membrane compartments.
Disease-associated STING mutations cause constitutive ER exit leading to ligand-independent STING activation
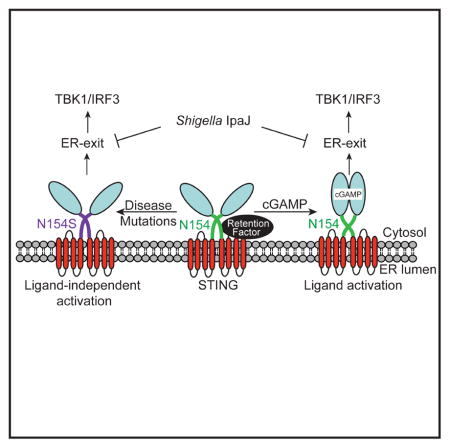
Our findings suggest a two-step activation mechanism that first involves cGAMP binding to ER localized STING and that second involves STING transport from the ER to ERGIC. The observation that IpaJ inhibited STING signal transduction by blocking its transport out of the ER in the presence of high levels of cGAMP (produced by dsDNA) or cyclic di-AMP (secreted by Listeria) suggests that nucleotide binding triggers ER-exit of STING and that STING activation of TBK1 results from its change in subcellular location (Figure S5A). Recently, the genetic lesions of patients exhibiting autoinflammatory vasculopathy and autoimmunity were mapped to single amino acids substitutions in STING. Interestingly, these residues make up a small linker region connecting the N-terminal transmembrane domain of STING to the C-terminal cyclic dinucleotide-binding domain, away from the cyclic dinucleotide binding pocket (Zhang et al., 2013). In addition, these substitutions lead to constitutive TBK1/IRF3 activation and uncontrolled interferon response (Jeremiah et al., 2014; Liu et al., 2014). We hypothesized that these residues may play an important role in retaining STING on the ER in unstimulated cells, and disease mutations disrupt ER retention resulting STING translocation to the ERGIC, bypassing the need for cGAMP binding for TBK1/IRF3 activation (Figure S5B).
To test this two-step model of STING activation in context of human disease progression, retroviral expression vectors were used to establish murine STING-GFP or the STING mutants N153 and V154 (that are highly conserved with humans) in Sting-/- MEFs (Figure 5A). Remarkably, WT STING localized to the ER as expected whereas disease-associated STING mutants clearly localized to the ERGIC and Golgi in unstimulated cells (Figure 5A). The localization of STING disease mutants on the ERGIC/Golgi did not change after dsDNA stimulation, whereas WT STING quickly translocated to post-Golgi vesicles (Figure 5A). To confirm this localization, we performed membrane fractionation by differential centrifugation (see Method, modified from (Ge et al., 2013)). We were able to clearly separate ER from the Golgi proper, with the ERGIC migrating in-between these fractions (Figure 5B). Consistent with the fluorescence microscopy images, WT STING co-migrated with the ER whereas the disease mutant N153S induced STING distribution toward ERGIC and Golgi fractions. TBK1 also shifted towards ERGIC and Golgi fractions in N153S reconstituted cells, consistent with its constitutive active state. We also tested the corresponding human mutations in an IFNβ promoter-driven luciferase reporter (IFN-Luc) assay. All three disease-associated STING mutants, V147L, N154S and V155M, but not the wild type, constitutively activated the IFN-Luc reporter (Figure 6A), consistent with published findings (Jeremiah et al., 2014; Liu et al., 2014) and further supports constitutive ER exit as a mechanism for STING activation.
Figure 5. Disease-associated STING mutants constitutively localize to the ERGIC.

(A) Fluorescent micrographs show WT mSting-GFP or mSting-N153S-GFP localization in MEFs. N153S in mouse Sting corresponds to N154S disease mutation in human STING. WT or mutant mSTING-GFP is stably expressed in Sting-/- MEFs using a retroviral vector following by selection. Each reconstituted MEFs were mock treated or transfected with HT-DNA. Cells were fixed 8 hours later and co-stained with ER (Calnexin), ERGIC (P58) and Golgi (GM130) markers. Quantitation of colocalization was calculated as Pearson’s correlation coefficient (r) shown below (n=15). (C) Membrane fractionation of WT or N153S STING-GFP reconstituted cells by differential centrifugation (see Method for details). Cell organelle pellets at indicated centrifugation speed (top) were analyzed by immunobloting. Distribution of WT and N153S STING in relationship to ER, ERGIC and Golgi markers is shown on the right based on quantitation of immunoblot in ImageJ. Data are representative of at least two independent experiments.
Figure 6. STING disease mutants activate IFN signaling independent of ligand binding.

(A) IFNβ-Luc reporter assay. HEK293T cells were transfected with IFNβ-Firefly Luc, CMV-Renilla Luciferase and increasing amount of indicated human STING plasmid (50–200 ng). Luciferase activity was determined by Dual-luc assay 24 h post transfection. (B) IFNβ-Luc reporter assay. Similar to A, except that various WT and mutant STING plasmids are fixed at 100 ng, and 24 h after STING expression, cells were stimulated with increasing amount of 2′3′-cGAMP for 20 h. (C) IFNβ-Luc reporter assay. Similar to A, except that IpaJ or IpaJ(C46A) plasmid (as indicated) were also co-transfected with STING plasmids. (D) FACS analysis of WT or N153S STING-GFP degradation after HT-DNA transfection. WT or N153S STING-GFP cells were mock treated (Mock), transfected with HT-DNA (DNA), or treated with BFA then transfected with HT-DNA (DNA+BFA). GFP fluorescent intensity was measured 24 hours later by FACS. (E) A model that illustrates two modes of STING activation: by cGAMP binding or by disease mutations. Data are representative of at least three independent experiments. Error bars, SEM. Paired t-test (A–D). See also Figure S6.
To then test whether cGAMP binding is required for activating disease-associated STING mutants, we introduced a second mutation R232A that disrupts cGAMP binding and prevents STING activation by DNA or cGAMP (Zhang et al., 2013). In transient transfection studies, all three disease mutants potently induce IFN-Luc reporter even with a secondary R232A mutation, providing strong evidence that these disease mutants activate STING independent of cGAMP binding. We also validated that human STING-R232A no longer respond to cGAMP stimulation (Figure 6B). We used 2′3′-cGAMP to stimulate 293T cells expressing wild type STING, R232A alone, individual disease mutants alone or disease mutants with R232A. Wild type STING, but not R232A, responded robustly to cGAMP stimulation as shown previously (Zhang et al., 2013). Disease mutants alone only marginally responsive to cGAMP stimulation, and disease mutants with R232A become completely irresponsive to cGAMP stimulation (Figure 6B).
Disease causing mutations in STING cause constitutive IFN activation by ERGIC/Golgi sequestration
A major prediction of these findings is that human disease mutations result in constitutive STING signaling by driving its trafficking from ER to ERCIC (bypassing the need for cGAMP), whereupon it is activated. To directly test this hypothesis, we co-expressed IpaJ or IpaJ(C64A) with each STING mutant. Remarkably, IpaJ completely inhibited STING activation by disease mutations, whereas enzymatically-dead IpaJ(C64A) had no effect (Figure 6C). Consistent with ARF1-mediating trafficking as the site of STING regulation, BFA treatment also reversed disease mutant localization back to the ER (Figure S6B). In control experiments, IpaJ did not inhibit RNA-stimulated IFN production under these conditions (Figure S6A), thereby confirming ER-exit of STING as the primary mechanism of both ligand-dependent (cGAMP) and ligand-independent (disease mutations) IFN stimulation.
Previous studies have shown that DNA-activated STING traffics to a post-Golgi vesicle compartment where it is degraded by an autophagic-like mechanism (Konno et al., 2013). Thus, another predication from our microscopy and IFN reporter studies is that STING disease mutants stably associated with the ERGIC and Golgi would not respond to DNA-mediated STING degradation. We examined Sting-KO/STING-GFP cells by FACS, and indeed we found that DNA transfection reduced wild type STING-GFP level 5-fold by 24 hours. Pretreatment with BFA prevented STING-GFP degradation, consistent with the notion that STING trafficking is required for degradation. In contrast, DNA transfection of Sting-KO/STING-N153S-GFP cells did not cause degradation of the STING disease mutant (Figure 6D). These data reveals an additional feature of the STING disease mutants – that they not only activates STING ER exit, they also stably associates with intermediate membrane compartments to prevent degradation. Collectively, our data provide a molecular explanation for gain-of-function STING diseases (Figure 6E) and suggest that blocking ER-exit (by IpaJ or BFA) could be potentially useful therapeutic avenues.
Discussion
STING activation involves translocation from the ER to vesicles, although how immune signaling and trafficking are coordinated remains unclear. The combination of experiments examining the IFN response to bacterial infection and human disease mutations provides a spatiotemporal model of STING signaling activation. Our results suggest that Shigella IpaJ antagonize STING signaling by blocking ER to ERGIC translocation through ARF GTPase demyristoylation. Studies inhibiting Rab1 GTPase by VirA further indicates that maximum STING activation occurs immediately after its translocation from ER to ERGIC. Consistent with this idea, constitutively active mutants of STING almost exclusively localize to the ERGIC and Golgi, where from they activate TBK1/IRF3 and downstream immune signaling. ERGIC was recently demonstrated as one of the main membrane sources of autophagosomes (Ge et al., 2013; Ge and Schekman, 2013). STING positive vesicles formed after DNA stimulation also depend on an autophagy-related process (Konno et al., 2013) and proteins such as Atg9a, Beclin1, ULK1 also regulate cytosolic DNA sensing and STING signaling (Konno et al., 2013; Liang et al., 2014; Saitoh et al., 2009). Our finding reveals intermediate membrane compartments such as the ERGIC as a primary organelle site for STING maximal signal transduction. The traditionally defined STING activation vesicles are likely the final destination of STING trafficking after traversing through ERGIC/Golgi as well as the membrane location for initiating STING degradation (to turn signaling off). Consistent with this notion, STING activation by DNA transfection or ShigellaΔipaJ/virA infection allows STING trafficking all the way to vesicles (Movie 1), thus turns on and then off downstream signaling. In contrast, STING activation by ShigellaΔipaJ infection or disease mutants arrest STING trafficking on the intermediate membrane compartments, allowing it to turn on downstream signaling and remains on.
We now propose an ER retention-and-release mechanism of STING activation (Figure 6E). In support of this mechanism, all three diseases-associated residues, V147, N154 and V155, appear to play non-redundant roles in retaining STING on the ER, and mutation of either one of these residues causes STING to constitutively localize to the ERGIC and Golgi and activate downstream signaling. Published STING structures suggested an ‘up-lifting’ motion of the overall cytosolic domain due to the ‘clamping’ of cGAMP (Figure S5B). These conformational changes may disrupt interactions between critical ‘ER retention residues’ on STING (i.e. diseases-associated residues) and unknown protein partners on the ER membrane, which would allow STING to exit the ER and translocate to the ERGIC. Therefore, we propose that STING normally resides in the ‘ER retention’ mode and possibly interacting with critical partners on the ER. Our findings suggest that primary function of cyclic dinucleotide binding to the STING dimer on the ER is to reorient ER retention residues (potentially disrupting critical interactions) that would allow translocation from the ER to ERGIC and Golgi. In the absence of cyclic dinucleotide binding, STING can be strongly activated by disease mutations that disrupts ER retention, or by other membrane perturbations that had been previously reported to trigger the STING signaling pathway without introducing DNA or cyclic dinucleotides (Holm et al., 2012; Noyce et al., 2006).
Our model provides an exciting molecular explanation for STING diseases, although further questions do remain, including what proteins on the ER are involved in STING retention, what features of the intermediate membrane compartments such as ERGIC make it the most fertile ground for STING signal transduction, and why do disease mutations appear to be stably associated with the ERGIC and Golgi and are constantly ‘on’ whereas dsDNA stimulation activates STING and quickly turns it ‘off’. Addressing these questions will provide further insights into the STING activation mechanism and better understanding of pathogenesis of microbial infection and STING-associated immune disorders. The two Shigella effectors, IpaJ and VirA, are useful tools for dissecting STING signaling through membrane trafficking systems, as we have clearly demonstrated using a variety of experimental settings including microinjection of recombinant proteins or infection with engineered loss-of-function Shigella strains or gain-of-function Listeria strains. Our study with these engineered bacteria strains also established a unique example of how the secretory pathway supports dynamic signaling events beyond its classical role in protein sorting and distribution. The molecular action of IpaJ can be mimicked in the future as a novel therapeutic avenue for STING-mediated autoimmune and autoinflammatory diseases.
EXPERIMENTAL PROCEDURES
Bacterial Strains
Wild type parental strain for Shigella flexneri strain M90T and all mutant strains (ΔIpaJ, ΔVirA, ΔIpaJΔVirA and ΔMxiD) were described in (Burnaevskiy et al., 2013). Listeria monocytogenes ΔactA mutant parental strain was a kind gift from Dan Portnoy. For construction Listeria monocytogenes expressing IpaJ or VirA we used previously described site-specific phage integration vector pPL1, which inserts at ComK attachment site (Lauer et al., 2002a). Promoter region and 100 N-terminal amino acids of ActA were PCR-amplified from Listeria genome using primers AAAAGAGCTCTGAAGCTTGGGAAGCAG and AAAAGCGGCCGCACCTTTCTCTGCTTTTGC (SacI and NotI restriction sites are underlined) and were cloned into pPL1, resulting in pActA N100-pPL1 cloning vector. IpaJΔ50 and VirAΔ26 (50 and 26 N-terminal amino acids are truncated respectively) were PCR amplified (AAAAGCGGCCGCGGGTTGTTTTTTGGCGAGGAAGCAGATG and ATATGTCGACTTACAAAGCCTCATTAGTTATAACTATGGAAAT primers for IpaJ; AAAAGCGGCCGCGAGTTGGAATAAATTGAGTTTTTGTGACAT and ATATGTCGACTTAAACATCAGGAGATATGATGGCAAAT primers for VirA) and cloned into pActA N100-pPL1 in frame with N-terminal region of ActA using NotI and SalI restriction sites. The resulting constructs were called IpaJΔ50 - pActA N100 - pPL1 and VirAΔ26 - pActA N100 - pPL1. For transformation of Listeria, we prepared electrocompetent cells using the protocol described previously (Monk et al., 2008). Listeria cells were incubated overnight in brain-heart infusion (BHI) media at 30°C without shaking and were diluted 1:100 in 500 ml of BHI containing 500 mM sucrose (BHIS) in the morning. Cells were grown to an OD600 of 0.1 and ampicillin was added to a concentration of 10 μg/ml. The culture was further incubated with shaking for 2 h. Cells were then cooled on ice for 10 min and centrifuged (5,000 ×g for 10 min at 4°C). Cell pellet was resuspended in 200 ml of ice-cold sucrose-glycerol wash buffer (SGWB) (10% glycerol, 500 mM sucrose; pH 7, filtered). Cells were centrifuged two more times and were resuspended in 70 ml of SGWB and in 20 ml of SGWB after the first and the second centrifugations respectively. Ten μg/ml of filter-sterilized lysozyme was added and cells were incubated at 37°C for 20 min. Cells were centrifuged (3,000 × g for 10 min at 4°C) and resuspended in 10 ml SGWB. Cells were centrifuged and resuspended in SGWB with the final volume of 2.5 ml and the 50-μl aliquots were frozen at −80°C. For transformation a 50-μl aliquot of electrocompetent cells was mixed with 1 μg of plasmid DNA and the mixture was transferred to a chilled 1-mm electroporation cuvette and pulsed at 10 kV/cm. For recovery, the cells were supplemented with 1 ml of BHIS, transferred into eppendorf tube and incubated statically at 30°C for 1.5 h. The cells were then concentrated by centriguation (3,000 × g for 2 min) into 100 μl and plated on BHI agar containing 7.5ug/ml of chloramphenicol and incubated for 3 days at 30°C. Individual Listeria colonies were screened by PCR using either CTCATGAACTAGAAAAATGTGG and TGTAACATGGAGGTTCTGGCAATC primers to confirm specific integration (Lauer et al., 2002a) or IpaJ/VirA cloning primers to confirm the presence of the transgenes. Positive clones were selected for frozen stocks and infection studies.
Cell Culture
Primary wild type, STING golden-ticket (gt/gt) (kind gift from Russell Vance), Sting-/- (kind gift from Glen Barber), Mavs-/-, Myd88/Trif-/- (kind gift from Zhijian ‘James’ Chen), Irf3-/- and Irf7-/- mouse embryonic fibroblasts (MEF) were generated from embryos on embryonic day E15.5. All MEF were grown in complete Dulbecco’s Modified Eagle’s Medium (DMEM, Sigma-Aldrich) supplemented with 10% (v/v) Fetal Bovine Serum (FBS, Sigma-Aldrich), 2 mM L-glutamine (Sigma-Aldrich), 10 mM HEPES (Sigma-Aldrich), 1 mM sodium pyruvate (Sigma-Aldrich) 100 U/mL penicillin (Sigma-Aldrich) and 100 mg/mL streptomycin (Sigma-Aldrich) at 37°C and 5% CO2. Prior to infection, MEF would be plated in a 12-well at 50,000 MEF/well in complete DMEM without antibiotics for bacterial infection.
Shigella infection
Shigella strains were streaked on a BHI agar plate containing 0.3% congo red (Sigma-Aldrich) from a frozen stock. A single red colony was picked and used to start a culture in BHI broth. The culture was grown overnight at 30°C and shaking at 200 rpm. The next day the overnight culture was restarted by diluting the overnight culture 1:25 into fresh BHI broth and grown at 37°C, shaking at 200 rpm for approximately 2 hours until the optical density (OD) reached 0.5 – 0.6. Once the OD is reached, the Shigella was centrifuged at 10,000 x g to pellet bacterial cells. The BHI supernatant was removed and the cell pellet was washed twice with Dulbecco’s Phosphate Buffered Saline (PBS) (Sigma-Aldrich) to remove any media contaminants. The Shigella cell pellet was resuspended in PBS with a 0.03% congo red, and this suspension was incubated at 37°C for 15 minutes to activate the Shigella for infection. Each well was infected with 0.1 mL of Shigella suspension for infection, and then the plates were centrifuged at 1000 x g for 10 minutes to facilitate contact between bacteria and host cells. The plates were then incubated for 90 minutes at 37°C and 5% CO2. Then, the cells were washed three times (1 mL per 12 well) with PBS and gentamicin (150 mg/mL) to remove any unbound bacteria. After the third wash, complete DMEM without antibiotics was replaced and infection continued until time points were reached. Most interferon mRNA expression was measured 6 – 8 hours post infection. For analysis of proteins in the STING-TBK1-IRF3 pathway, 10 cm dishes were used. MEF were plated at 1.5 × 106 per 10 cm plate, and initial dose of Shigella was 1 mL of activated culture.
Microinjection
Microinjection experiments was done as described in (Selyunin et al., 2014). Briefly, microinjections of IpaJΔ50 and VirAΔ26 were performed using a semiautomatic InjectMan NI2 micromanipulator (Eppendorf). Recombinant proteins were diluted in 1X Tris buffer saline (TBS) with Cascade Blue (Invitrogen) fluorescent dye (2 mg/ml). The final concentration of the proteins was 1 mg/ml. Concentration of DNA was 4 ng/ul.
RNA isolation and Quantitative RNA analysis
Total RNA from cells was isolated with TRI reagent according to the manufacturer’s instructions (Sigma-Aldrich). RNA quantity and quality were confirmed with a NanoDrop ND-1000 Spectrophotometer. cDNA was synthesized from 1 μg of RNA using the iScriptTM Reverse Transcription Supermix for RT-qPCR (Bio-Rad) per manufacturer’s instructions. iTaqTM Universal SYBR Green Supermix, and ABI-7500 Fast Real-Time PCR system (Applied Bioscience) and specific gene primers were used for real-time PCR analysis as in(Hasan et al., 2013). The results are presented relative to those of Gapdh. Qiagen qPCR Array was used for the generation of the heat map. The array plate contains validated primer sets for interferon beta gene (IFN-beta), Interferon stimulated genes (ISG: CxCL10, IFIT1, IRF7, OasI2, ISG15), pro-inflammatory genes (IL1-beta, IL1-alpha, IL-6, CCL2) and house keeping genes (Gapdh, Actin and HPRT). Heat map was generated using GENE-E program (Broad Institute).
Microscopy
For Sting-/- MEFs reconstituted with various Sting rescuing constructs, WT or mutant Sting-GFP was cloned into retroviral MRX-ibsr vector (Saitoh et al., 2009) (a kind gift from S. Akira) using Eco RI and Not I sites. Retroviruses were packaged in 293T cells and used for infection of Sting-/- MEFs followed by selection with blasticidine (Sigma). For microscopy, Sting-GFP MEFs were grown on cover slides in 12 well plates, and transfected with DNA or infected with bacteria. Cells grown on coverslips were fixed in 4% (wt/vol) paraformaldehyde and were permeabilized and stained by standard protocols. Samples mounted in Vectashield mounting medium containing DAPI (4,6-diamidino-2-phenylindole; Vector Laboratories) were imaged with a Zeiss Imager.M2 fluorescence microscope equipped with AxioVision software. Colocalization quantification by Pearson’s correlation coefficiency was done in SlideBook 5.0 software.
Reporter assays
IFN-Luciferase reporter assays were performed as described in (Jeremiah et al., 2014). pGL3-IFN-Luc and MCSV-hSTING plasmids were kind gifts from Nicolas Manel. STING disease mutations were generated by site-directed mutagenesis. For the reporter assay, pGL3-IFN-Luc (firefly), CMV-Luc (renilla), and indicated STING plasmids were transfected to 293T cells in 24 well plates. Twenty four hours later, IFN activation was measured by Dual-Luc assay (Promega). To test how various STING mutants respond to cGAMP, cells were transfected again with indicated amount of 2′3′-cGAMP (Invivogen) and IFN activation was measured 24 h after transfection.
SDS-PAGE and Western Blot Analysis
Cells were removed from the plate with Trypsin (Sigma-Aldrich), and collected by centrifugation at 500 x g for 5 minutes. Cell pellets were washed twice with equal volumes of PBS to remove any media contaminants. Cell pellet was lysed with 0.1 mL of 1X RIPA lysis buffer (50mM Tris, pH 7.5, 150mM sodium chloride, 0.5% sodium deoxycholate, 1% NP-40) containing 1X cOmplete EDTA-free Protease inhibitor cocktail tablets (Roche Diagnostics), 1X Phosphatase Inhibitor Cocktail 2 (Sigma-Aldrich), 1 mM sodium orthovanadate (Sigma-Aldrich), 25 mM sodium fluoride (Sigma-Aldich), 1 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich). The lysates were centrifuged at 20,000 x g for 20 minutes at 4°C to remove insoluble proteins. Protein concentration was quantified by using the Pierce™ BCA Protein Assay Kit per manufacturer’s instructions. 100 μg of total protein was loaded per sample on a 10% polyacrylamide gel and run at 150V for 1 hour using the Bio-Rad Mini-PROTEAN® Tetra Cell. Proteins from the gel were transferred to nitrocellulose using a Trans-Blot® SD Semi-Dry Transfer Cell (Bio-Rad) at 15V for 1 hour. Successful transfer of proteins to the nitrocellulose membrane was confirmed by using the 1X Red AlertTM Western Blot Stain (Novagen, EMD Millipore). Membranes blocked with 5% non-fat milk in 1X TBS-T (20 mM Tris Base, 150 mM sodium chloride, 0.05% Tween-20, pH 7.6). Then the membrane was cut into sections to allow for primary antibody detection. Primary antibodies used were rabbit anti-STING (Cell Signaling), rabbit anti-Phospho-TBK1/NAK (Ser172) (D52C2) XP® (Cell Signaling), rabbit anti-Phospho-IRF-3 (Ser396) (4D4G) (Cell Signaling), rabbit anti-HMGB2 (Abcam). Antibodies were used per the manufacturer’s instructions. Portions of the membrane were incubated overnight at 4°C in either TBS-T or 5% non-fat milk in TBS-T at the appropriate dilutions. Membranes were washed three times for 10 minutes each at room temperature. Membranes were incubated with goat anti-rabbit-IgG-HRP conjugated secondary antibody (Bio-Rad) diluted in 5% non-fat milk in TBS-T for 2 hours at room temperature. After which, the membranes were washed three times for 10 minutes each in TBS-T, and SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) was used to develop the blots on film. The pTBK1 and pIRF-3 membranes were stripped using Restore Western Blot Stripping Buffer (Thermo Scientific) per manufacturer’s instructions, and total TBK1 and total IRF-3 protein was determined by using rabbit anti-TBK1/NAK (D1B4)(Cell Signaling) and rabbit anti-IRF-3 antibody (a kind gift from Takashi Fujita) for detection.
Membrane Fractionation
Membrane fractionation by differencial centrifugation were performed as in (Ge et al., 2013) with the following modification. Five 15 cm plates were combined and cell pellets were washed one time with PBS to remove any media contaminants. The cell pellet was resuspended in 200 μL of membrane fractionation buffer (20 mM HEPES-NaOH pH 7.2, 400 mM sucrose, 1 mM EDTA) per 5 × 106 cells. Cells were lysed by passing the homogenate through a 25 gauge needle thirty times. Ten percent of the volume was then retained as a representation of the total fraction. Homogenates were subjected to sequential centrifugations 1000xg (15 minutes), 3000xg for (15 minutes), 10,000xg (15 minutes), 25,000xg (20 minutes), 50,000xg (20 min), and 100,000xg (30 minutes) using a Beckman TLA100.3 rotor and ultracentrifuge. Each pellet was washed with 500 μL of membrane fractionation buffer and centrifuged for the same speed to remove any contaminants.
Statistical methods
Data are presented as the mean ± SEM. Graphpad Prism 6 was used for statistical analysis. Statistical tests performed were indicated in figure legend. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Supplementary Material
Acknowledgments
We thank Dan Portnoy (UC Berkerly) for ListeriaΔactA, reagents and technical advice, Zhijian ‘James’ Chen (UTSW) for Mavs-/- and Myd88/Trif-/- mice, Mike Diamond (Wash U) for cGAS-/- MEFs, Russell Vance (UC Berkerly) for Sting-gt/gt mice, Glen Barber (U Miami) for Sting-/- mice, Nicolas Manel (Institut Curie, France) for IFN-Luc and STING plasmids, Xuewu Zhang (UTSW) for STING structure modeling. We thank members of the Yan laboratory and Alto laboratory for helpful discussions. This work was supported by grants from the National Institute of Health (AI098569, AR067135 to N.Y., AI083359 and GM100486 to N.M.A.), the Welch Foundation (#I-1831 to N.Y., #I-1704 to N.M.A.), Alliance for Lupus Foundation (N.Y.), and the Burroughs Wellcome Fund (N.Y., N.M.A).
Footnotes
AUTHOR CONTRIBUTIONS
N.Y and N.M.A designed the study. N.D., N.B., and N.Y. conducted the majority of experiments. D.C. and V.K.G. performed microinjection experiments and characterization of STING mutants. All authors discussed the results and commented on the manuscript.
The authors have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011 doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol. 2013;14:19–26. doi: 10.1038/ni.2491. [DOI] [PubMed] [Google Scholar]
- Burnaevskiy N, Fox TG, Plymire DA, Ertelt JM, Weigele BA, Selyunin AS, Way SS, Patrie SM, Alto NM. Proteolytic elimination of N-myristoyl modifications by the Shigella virulence factor IpaJ. Nature. 2013;496:106–109. doi: 10.1038/nature12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnaevskiy N, Peng T, Reddick LE, Hang HC, Alto NM. Myristoylome profiling reveals a concerted mechanism of ARF GTPase deacylation by the bacterial protease IpaJ. Molecular Cell. 2015 doi: 10.1016/j.molcel.2015.01.040. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell. 2014;54:289–296. doi: 10.1016/j.molcel.2014.03.040. [DOI] [PubMed] [Google Scholar]
- Dong N, Zhu Y, Lu Q, Hu L, Zheng Y, Shao F. Structurally distinct bacterial TBC-like GAPs link Arf GTPase to Rab1 inactivation to counteract host defenses. Cell. 2012;150:1029–1041. doi: 10.1016/j.cell.2012.06.050. [DOI] [PubMed] [Google Scholar]
- Gao D, Wu J, Wu Y-T, Du F, Aroh C, Yan N, Sun L, Chen ZJ. Cyclic GMP-AMP Synthase Is an Innate Immune Sensor of HIV and Other Retroviruses. Science (New York, NY) 2013 doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge L, Melville D, Zhang M, Schekman R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife. 2013;2:e00947. doi: 10.7554/eLife.00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge L, Schekman R. The ER-Golgi intermediate compartment feeds the phagophore membrane. Autophagy. 2013:10. doi: 10.4161/auto.26787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan M, Koch J, Rakheja D, Pattnaik AK, Brugarolas J, Dozmorov I, Levine B, Wakeland EK, Lee-Kirsch MA, Yan N. Trex1 regulates lysosomal biogenesis and interferon-independent activation of antiviral genes. Nat Immunol. 2013;14:61–71. doi: 10.1038/ni.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm CK, Jensen SB, Jakobsen MR, Cheshenko N, Horan KA, Moeller HB, Gonzalez-Dosal R, Rasmussen SB, Christensen MH, Yarovinsky TO, et al. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat Immunol. 2012;13:737–743. doi: 10.1038/ni.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg M-C, Goudin N, Frémond M-L, Nitschke P, Molina TJ, et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. The Journal of clinical investigation. 2014 doi: 10.1172/JCI79100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno H, Konno K, Barber GN. Cyclic Dinucleotides Trigger ULK1 (ATG1) Phosphorylation of STING to Prevent Sustained Innate Immune Signaling. Cell. 2013;155:688–698. doi: 10.1016/j.cell.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer P, Chow MY, Loessner MJ, Portnoy DA, Calendar R. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. Journal of bacteriology. 2002a;184:4177–4186. doi: 10.1128/JB.184.15.4177-4186.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer P, Chow MYN, Loessner MJ, Portnoy DA, Calendar R. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. Journal of bacteriology. 2002b;184:4177–4186. doi: 10.1128/JB.184.15.4177-4186.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Seo GJ, Choi YJ, Kwak MJ, Ge J, Rodgers MA, Shi M, Leslie BJ, Hopfner KP, Ha T, et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe. 2014;15:228–238. doi: 10.1016/j.chom.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371:507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk IR, Gahan CG, Hill C. Tools for functional postgenomic analysis of listeria monocytogenes. Applied and environmental microbiology. 2008;74:3921–3934. doi: 10.1128/AEM.00314-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyce RS, Collins SE, Mossman KL. Identification of a novel pathway essential for the immediate-early, interferon-independent antiviral response to enveloped virions. Journal of virology. 2006;80:226–235. doi: 10.1128/JVI.80.1.226-235.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond B, Young JC, Pallett M, Endres RG, Clements A, Frankel G. Subversion of trafficking, apoptosis, and innate immunity by type III secretion system effectors. Trends in microbiology. 2013;21:430–441. doi: 10.1016/j.tim.2013.06.008. [DOI] [PubMed] [Google Scholar]
- Reddick LE, Alto NM. Bacteria fighting back: how pathogens target and subvert the host innate immune system. Molecular cell. 2014;54:321–328. doi: 10.1016/j.molcel.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA. 2009;106:20842–20846. doi: 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyunin AS, Reddick LE, Weigele BA, Alto NM. Selective Protection of an ARF1-GTP Signaling Axis by a Bacterial Scaffold Induces Bidirectional Trafficking Arrest. Cell reports. 2014;6:878–891. doi: 10.1016/j.celrep.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte CE, Archer KA, Rae CS, Sauer JD, Woodward JJ, Portnoy DA. Innate immune pathways triggered by Listeria monocytogenes and their role in the induction of cell-mediated immunity. Advances in immunology. 2012;113:135–156. doi: 10.1016/B978-0-12-394590-7.00002-6. [DOI] [PubMed] [Google Scholar]
- Woodward JJ, Iavarone AT, Portnoy DA. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science (New York, NY) 2010;328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol. 2010;11:1005–1013. doi: 10.1038/ni.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen ZJ. Cyclic GMP-AMP Containing Mixed Phosphodiester Linkages Is An Endogenous High-Affinity Ligand for STING. Molecular cell. 2013 doi: 10.1016/j.molcel.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.