

Abstract
Hypertension is recognized as an immune disorder whereby immune cells play a defining role in the genesis and progression of the disease. The innate immune system and its component toll-like receptors (TLRs) are key determinants of the immunological outcome through their pro-inflammatory response. TLR activated signaling pathways utilize several adaptor proteins of which adaptor proteins MyD88 and TRIF define two major inflammatory pathways. In this study, we compared the contributions of MyD88 and TRIF adaptor proteins to angiotensin II (Ang II)-induced hypertension and cardiac hypertrophy in mice. Deletion of MyD88 did not prevent cardiac hypertrophy and the pressor response to Ang II tended to increase. Moreover, the increase in inflammatory gene expression (Tnfa, Nox4 and Agtr1a) was significantly greater in the heart and kidney of MyD88-deficient mice compared with wild type mice. Thus, pathways involving MyD88 may actually restrain the inflammatory responses. On the other hand, in mice with non-functional TRIF (Trifmut mice), Ang II induced hypertension and cardiac hypertrophy were abrogated, and pro-inflammatory gene expression in heart and kidneys was unchanged or decreased. Our results indicate that Ang II induces activation of a pro-inflammatory innate immune response, causing hypertension, and cardiac hypertrophy. These effects require functional adaptor protein TRIF-mediated pathways. However, the common MyD88 dependent signaling pathway, which is also activated simultaneously by Ang II, paradoxically exerts a negative regulatory influence on these responses.
Keywords: Hypertension, innate immune system, toll-like receptors, MyD88, TRIF, TICAM1, Angiotensin, gene expression
Introduction
In recent years, hypertension has increasingly been recognized as an immunological disorder whereby the components of the immune system play an important role in determining two key aspects of the disease, blood pressure and end organ damage 1–8. Greater understanding of the immune system itself has accelerated the progress in defining the cell populations of the adaptive immune system that play a role in hypertension 9–15 and the neuro-immune axis that may activate these cells 1, 16–20. More recently, the innate immune system, specifically the toll-like receptors (TLRs), has been shown to be the mediators of neuro-inflammatory response that can be modulated by angiotensin II (Ang II) 16, 17, 19. The goal of this study was to determine the innate immune mechanisms involved in Ang II induced hypertension and cardiac hypertrophy.
Ang II is a major product of the renin-angiotensin system (RAS) that plays a crucial role in the pathology and treatment of hypertension. It causes vasoconstriction, promotes sodium retention in the kidneys, acts as a trophic factor in the myocardium, activates the sympathetic nervous system, and induces a pro-inflammatory state that results in organ damage 21–24. Ang II promotes inflammation by inducing cytokine release, immune cell infiltration in the kidney, expression of genes in kidneys and the heart that are pro-inflammatory, pro-hypertrophic and pro-fibrotic, such as Tumor Necrosis Factor-α (TNF-α), Interleukin-6 (IL-6), Interleukin-1β (IL-1β) and Transforming Growth Factor-β (TGF-β) 1, 25.
TLRs are a major class of innate immune receptors that constitute a diverse family of pattern recognizing receptor (PRR). They bind to exogenous pathogen associated molecular patterns (PAMP) or endogenous damage associated molecular patterns (DAMP) to initiate intracellular signaling cascades culminating in expression of pro-inflammatory genes as well as regulate the activity of adaptive immune cells 26. TLR signaling is dependent on the association of TLRs with one of several adaptor proteins that determine the specificity of signaling 27. Activation of TLRs often initiates two major adaptor-dependent signaling pathways: one is myeloid differentiation protein 88 (MyD88)-dependent, and the other is TICAM1 (Toll Interleukin Receptor-domain-containing adaptor molecule 1), also known as TRIF (TIR-domain containing adaptor-inducing interferon-β)-dependent 28. Besides their role in induction of pro-inflammatory genes, signaling by different TLRs may be complementary, synergistic or even antagonistic 29–31 resulting in complex phenotypes and gene expression patterns.
We have previously shown a link between TLR/MyD88 signaling and myocardial infarction induced cardiac hypertrophy 32. Mice lacking MyD88 adaptor protein had significantly improved survival and reduced inflammation, cardiac fibrosis and cardiac hypertrophy after myocardial infarction. Prompted by these results we tested the hypothesis that TLR/MyD88 signaling is essential in Ang II-induced hypertension, cardiac hypertrophy and end organ inflammation.
Our results revealed a selective and paradoxical dependence of Ang II hypertension responses on the two dominant adaptor proteins of the innate immune pathways TRIF and MyD88. Whereas TRIF-dependent pathways are essential for Ang II-mediated hypertension, cardiac hypertrophy and the inflammatory response, MyD88 pathways, in contrast, appear to be anti-inflammatory and restrain those responses.
Materials and methods
Animals
All experiments with animals were done in accordance with the regulations put forth by the Institutional Animal Care and Use Committee (IACUC) of University of Iowa. The wild type C57BL/6J (WT), and Trifmut (C57BL/6J-Ticam1Lps2/J) 33 mice were obtained from Jackson Labs. MyD88−/− mice were obtained from Dr. Shizuo Akira’s group 34 and were bred in the University of Iowa animal facility in specific pathogen free environment. All mice were in C57BL/6 background.
Ang II infusion and tail-cuff recordings of blood pressure
Mini-osmotic pumps (Alzet model 1004, 0.11 μl/h, 28 days), containing saline or Ang II (730, 1000 or 3000 ng/kg/min), were inserted subcutaneously in mice under anesthesia (2.0–2.5% isoflurane). Male mice of 8–12 weeks age were used for the experiments.
Systolic arterial blood pressure was measured by tail cuff using the Visitech-2000 system. A rigorous system of recording tail cuff pressures has been in effect in our laboratories over decades and found to be reliable for steady state blood pressure measurements over long periods of time.
Baseline blood pressure was measured prior to pump insertion. Tail-cuff recordings were performed at the same time of the day (before noon) to avoid diurnal changes in the pressure. To acclimatize the mice to the procedure, one week prior to the start of the experiments, tail-cuff pressures were measured on two days. During the experiments, twenty tail cuff measurements for each mouse were recorded for each session (3 times a week). Average measurements for each mouse were then used for the groups (n ≥ 3 mice per group). At the end of the experiments (3 weeks), mice were euthanized and organs were obtained for further analyses.
Cardiac hypertrophy
Cardiac hypertrophy was assessed by measurement of heart weight to body weight ratio (HW/BW) in hearts from mice after three weeks of saline or Ang II infusion. For measurement of cardiomyocyte diameter and cardiac fibrosis, heart tissues were examined from 4 separate cross-sections collected at different levels (each 250 μm apart) from the center of the heart. In each tissue sample, a pathologist using post examination masking techniques 35 screened each heart on low magnification to identify areas with the largest cardiomyocytes diameter. At these sites, the 5 largest cardiomyocyte diameters were recorded. Thus for each heart 20 diameters were collected and the mean of these data calculated for each animal.
RNA isolation and RT-QPCR
Total RNA from mouse heart and kidneys were isolated using mirVana RNA isolation kit (Ambion) or RNeasy RNA Isolation Kit (Qiagen). A 2 μg aliquot of RNA sample was used to synthesize cDNA in 50 μl reactions using Oligo (dT) as primers and SuperScript III Reverse Transcriptase (Invitrogen). Quantitative real-time PCR was performed on QPCR cycler (ABI) using SYBR green based PCR reactions, as described 32. Quantifications were done using ΔΔCT method where Gapdh was used as a reference gene for normalization of RNA expression 36. The primers used in this study have been previously described 32, 37 and are listed in Supplemental Table-S1.
Statistical analyses
Statistical analyses were performed using an unpaired t-test or ANOVA as shown in respective figures. Post-hoc analyses on multiple group data were done using Tukey’s test. A p-value of 0.05 or less was considered to be statistically significant. All results are presented as mean ± SEM.
Results
Ang II infusion increases blood pressure in MyD88−/− but not in Trifmut mice
Compared to saline infusions, which did not alter systolic blood pressure (SBP) over three weeks, Ang II infusion (3000 ng/kg/min) during the same period increased SBP in both WT and in MyD88−/− mice (Figure 1A and 1B). In WT, blood pressure peaked at 147 ± 4 mm Hg (Figure 1A) whereas in Ang II-infused MyD88−/− mice SBP reached a peak value of 163 ± 6 mm Hg (Figure 1B). The increase in SBP during the third week of infusion was significantly greater in MyD88−/− mice than in WT mice (p < 0.05). In contrast to the WT and MyD88−/− mice, Ang II did not increase blood pressure in Trifmut and the response to Ang II was identical to the response to saline during the last two weeks of infusion averaging 116 ± 3 mm Hg with saline and 118 ± 4 mm Hg with Ang II (Figure 1C).
Figure 1.
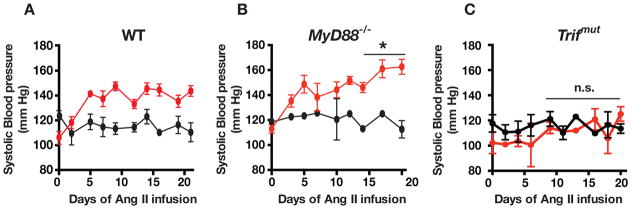
Systolic blood pressure (SBP) measured during infusion of saline (black lines) or Ang II (3000 ng/kg/min, red lines) for 3 weeks in (A) WT, (B) MyD88−/− and (C) Trifmut mice. Values represent means ± SEM (n ≥ 3 mice in each group). The pressor response was significantly greater in the last week of Ang II infusion in MyD88−/− than in WT (*p <0.05). Conversely, the pressor response during Ang II infusion was abrogated in the Trifmut mice; the SBP was unchanged during infusion of saline and Ang II for the last two weeks (n.s.= p > 0.05).
In addition, at lower Ang II dosage (730–1000 ng/kg/min) in a separate group of studies the average SBP over the last two weeks of infusion of Ang II was significantly lower in Trifmut (129 ± 3) vs. WT 135 ± 5) and highest in MyD88 −/− (142 ± 5 mm Hg; Suppl. Figure 1).
These results indicate that Ang II-induced hypertension is MyD88-independent and that the presence of MyD88 restrains the hypertensive response to Ang II, which is TRIF-dependent.
Ang II-induced cardiac hypertrophy is abrogated in Trifmut mice
The heart weight to body weight ratio (HW/BW) in WT mice infused with saline, low dose (730 ng/kg/min), and high dose (3000 ng/kg/min) of Ang II were 4.88 ± 0.16, 5.23 ± 0.15 and 6.83 ± 0.21 mg/g respectively (Figure 2A). Corresponding values in MyD88−/− mice were 3.93 ± 0.15, 4.70 ± 0.06 and 6.30 ± 0.86 (Figure 2B) and in Trifmut mice they were 4.39 ± 0.19, 4.72 ± 0.11, and 5.10 ± 0.29 (Figure 2c). The increase in HW/BW ratio with the high dose Ang II vs. saline was greater in MyD88−/− than WT but lowest in Trifmut. The high dose of Ang II infusion induced a 60% increase in HW/BW over saline infusions in MyD88−/− mice, a 40% increase in WT, and a 22% increase in Trifmut (Figure 2). Analyses of variance and linear regressions confirming the differences in the three genotypes are in Supplemental Table-S2). Cardiomyocyte diameter and fibrosis confirmed Ang II-induced cardiac structural changes (Supplemental Table-S3).
Figure 2.
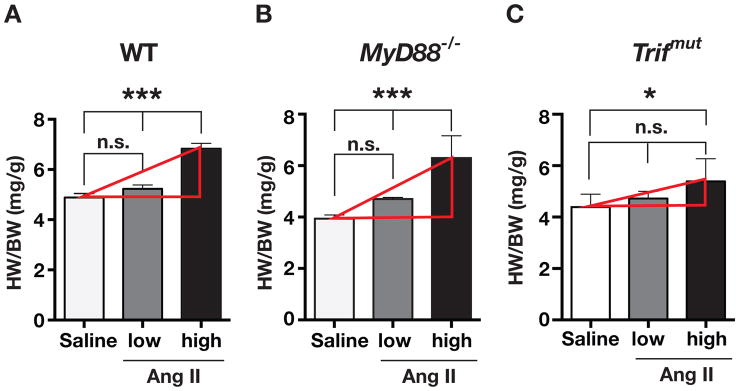
Measurement of heart weight to body weight ratios (HW/BW ratio) after 3 weeks of saline or Ang II infusions were determined in (A) WT, (B) MyD88−/−, and (C)Trifmut hearts. Low (730 or 1000 ng/kg/min) or high (3000 ng/kg/min) doses of Ang II were used (ANOVA with post-hoc Tukey’s test; n ≥3; n.s. = not significant, asterisks *= P< 0.05, ***= P< 0.001). Increase in heart weight to body weight ratios with Ang II vs. saline were greater in MyD88−/− and lesser in Trifmut compared to WT. Linear regression analyses and slopes of dose response curves are in supplemental Table S2.
Cardiac gene expression in Ang II-infused mice
We compared the expression of inflammation related genes in the hearts from saline and Ang II-infused WT, MyD88−/−, and Trifmut mice. Tumor necrosis factor-α (Tnfa) expression was significantly increased by Ang II in WT and MyD88−/− mice, but the increase was much greater in the MyD88−/− hearts than in WT hearts (Figure 3A and B). However, Ang II-infusion had no effect on Tnfa expression in Trifmut hearts (Figure 3C).
Figure 3.

Comparison of pro-inflammatory gene expression in hearts from WT, MyD88−/− and Trifmut mice after chronic infusion (3 weeks) of saline or low dose Ang II. Fold change in RNA expression was measured by ΔΔCT method using Gapdh RNA as a reference. Values in graphs are normalized to measurements in saline-infused mice. Tnfa= tumor necrosis factor-alpha, Nox4= NADPH oxidase 4, and Agtr1a= angiotensin receptor type 1a. (n≥3 samples each group; Asterisks show P≤0.05, n.s. = not significant). In general Ang II induced increase in gene expressions were enhanced in MyD88−/− and reduced in Trifmut compared to WT.
Expression of NADPH oxidase-4 (Nox4) was significantly increased to a similar extent in hearts from WT and MyD88−/− mice with Ang II-infusions (Figure 3D and E). However, Nox4 expression was unaffected in Trifmut hearts upon Ang II-infusion (Figure 3F).
The angiotensin type 1-receptor (AT1 receptor, Agtr1a) expression in heart was unaffected by Ang II infusion in WT mice (Figure 3G), was significantly increased by Ang II in MyD88−/− mice (Figure 3H) and was slightly decreased by Ang II in Trifmut mice (Figure 3I). Similarly the expression of Cxcl10 a gene that is selectively induced by the TRIF/IFN pathway was enhanced in MyD88−/− supporting a negative regulatory effect of MyD88 on the TRIF pathway. Expression of Cxcl10 was actually decreased in Trifmut mice (Suppl. Fig. 2A).
The muted effect of Ang II infusion on cardiac gene expressions was not due to a general unresponsiveness of Trifmut mice because Ang II induced significant over expression of skeletal muscle actin-α1 (Acta1) in all three strains of mice and particularly in Trifmut (Suppl. Figure 2B). Thus, the intact MyD88 pathway in WT mice has a restraining inhibitory effect on Ang II-induced inflammatory gene expression, whereas the intact TRIF pathway is required for the inflammatory effect of Ang II.
Renal gene expression in Ang II-infused mice
We compared gene expression in the kidneys of saline and Ang II-infused mice. Ang II infusion resulted in an insignificant increase in expression of Tnfa in the kidneys of WT mice (Figure 4A), however, there was a clear and significant increase in Tnfa in kidneys of MyD88−/− mice (Figure 4B), and a much smaller negligible increase in kidneys of Trifmut mice (Figure 4C).
Figure 4.

Comparison of pro-inflammatory gene expression in kidneys of WT, MyD88−/− and Trifmut after chronic infusion (3 weeks) of saline or low dose Ang II. Fold change in RNA expression was measured by ΔΔCT method using Gapdh RNA as a reference. Values in graphs were normalized to measurements in saline-infused mice (n ≥ 3 each group; Asterisks show P ≤ 0.05, n.s. = not significant). Ang II induced increase in gene expressions were enhanced in MyD88−/− and reduced in Trifmut compared to WT.
Ang II-infusion did not increase expression of Nox4 in the kidneys of WT mice but it significantly increased the expression in kidney of MyD88−/− mice (Figure 4D and 4E) and, conversely, significantly decreased Nox4 in kidney of Trifmut mice (Figure 4F).
The renal expression of Agtr1a was unaltered in kidney of Ang II-infused WT (Figure 4G), significantly increased in kidney of MyD88−/− mice (Figure 4H), but significantly decreased in kidney of Trifmut mice (Figure 4I).
Thus, the absence of MyD88 caused a greater magnitude of pro-inflammatory gene expression with Ang II in both heart and kidney. In contrast, in the absence of TRIF, the inflammatory gene overexpression was eliminated or even reduced when compared to WT, confirming the inhibitory effect of the MyD88 pathway and the TRIF dependence of the inflammatory response.
Changes in Cardiac TLR and adaptor protein expressions in MyD88−/− and Trifmut mice
Since TLR3 and TLR4 are likely to be involved in signaling pathways that use the two adaptor proteins we measured their cardiac expressions in MyD88−/− and Trifmut. The results in Figure 5 indicate that deletion of MyD88 was associated with an increase in Trif expression, which along with increases in both Tlr3 and Tlr4 expression, must have contributed to the enhanced inflammatory gene expressions observed in MyD88−/− mice. In Trifmut mice, expression of MyD88 was increased, whereas Tlr4 expression was decreased and Tlr3 expression was unchanged. Together, the negative regulatory effect of MyD88 and decreased Tlr4 expression must have contributed to the suppressed inflammatory gene expression in Trifmut.
Figure 5.
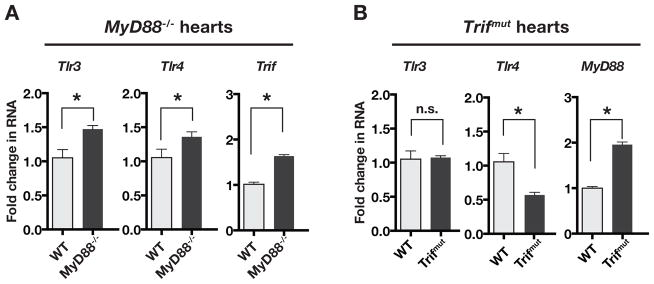
Baseline RNA expression of TLR3, TLR4 and their adaptor proteins MyD88 and TRIF in the hearts of WT vs. MyD88−/− and WT vs. Trifmut mice. Comparisons of RNA expressions between WT and MyD88−/− or WT and Trifmut hearts were done by QPCR and ΔΔCT method. Statistical significance was tested by unpaired t-test. Asterisks show significant differences from WT (P < 0.05) whereas n.s. denotes no significant change. In MyD88−/− the expressions of Trif, Tlr3, and Tlr4 were increased. In Trifmut, the expression of MyD88 was increased, Tlr4 was decreased and Tlr3 unchanged.
Discussion
We tested the contribution of the innate immune signaling by the two major adaptor proteins of TLRs, MyD88 and TRIF, in Ang II-induced hypertension. Consistent with our earlier demonstration that the innate immune system plays a major role in a genetic model of hypertension, our results show that pathways involving both adaptor proteins contributed to Ang II pressor responses in WT mice albeit in a mutually opposite manner.
Our results show that: (a) MyD88-dependent innate immune signaling pathways are not responsible for Ang II-induced hypertension, cardiac hypertrophy, and pro-inflammatory gene expression, (b) TRIF-mediated pathways, on the other hand, are the essential determinants of Ang II-induced responses, and (c) the absence of MyD88 unmasks exaggerated Ang II responses indicating that MyD88 signaling is simultaneously activated by Ang II and functions as a negative regulator of pro-inflammatory pathological responses mediated by TRIF. These results are summarized in the schematic in Figure 6. We arrived at an original and provocative finding that TRIF-dependent pathways are essential and pro-inflammatory in evoking Ang II hypertension, whereas MyD88-dependent pathways are negative regulators of TRIF pathways, and therefore may be considered anti-inflammatory. These results provide novel mechanistic insight in one of the most commonly used models of hypertension and identify putative targets of pathologic and therapeutic significance.
Figure 6.

Schematic portrays Ang II induced hypertension, cardiac hypertrophy and pro-inflammatory gene expression in innate immune cells of WT, MyD88−/− and Trifmut mice. The dual activation of MyD88 and TRIF pathways through TLR and AT1 receptors is shown in WT cells to induce NF-κB and interferon (IFN), respectively. The TRIF-dependent and negative regulatory influence of MyD88 are highlighted.
Dominance of the Immune System in Ang II Responses
Ang II is the major product of the renin-angiotensin system (RAS). It plays a major role in cardiovascular regulation of arterial pressure and blood volume, salt retention, and sympathetic activation. It adversely alters the course of several pathological states including hypertension, heart failure, atherosclerosis, and the metabolic syndrome.
The contribution of Ang II to hypertension has long been ascribed to its powerful vasoconstrictor action, its salt retaining properties and its central and peripheral sympatho-excitatory effects. A seminal notion was advanced by the work of Harrison and colleagues that Ang II induced hypertension is dependent on the immune system 13. A complementary finding has been our previous results that in a genetic hypertension model, the spontaneously hypertensive rat (SHR), Ang II dramatically enhances cytokine release (IL-6 and IL-1β) by splenocytes upon their activation by ligands of TLR 7, 8 and 9 17. That the pro-inflammatory effect of Ang II was evident in SHR splenocytes prior to the onset of hypertension suggests a genetic abnormality of the innate immune cells that links their AT1 receptors to the TLR signaling pathways and potentiates pro-inflammatory responses. More recently, Harrison’s group has also identified an important role of the innate immune system, specifically the dendritic cells, in Ang II hypertension 25.
The processes that mediate cardiac hypertrophy in response to Ang II have been ascribed to the mechanical afterload of a raised arterial pressure and also to trophic effects on cardiac myocytes with overexpression of contractile proteins by direct activation of AT1 receptors or indirectly by the adrenergic stimulus of excessive sympathetic activation. However, in a manner similar to the Ang II pressor response the trophic cardiac effect seems to be also mediated by immunologic pathways 24, 38, 39
The results of this study provide strong confirmation that Ang II hypertension, cardiac hypertrophy, and the pro-inflammatory gene expressions in both the heart and kidney, depend on innate immune signaling pathways that require the presence of the specific adaptor protein TRIF.
Ang II hypertension and cardiac hypertrophy are MyD88-independent but TRIF-dependent
An unexpected finding of this study was that MyD88, a widely recognized essential adaptor protein for the activation of pro-inflammatory TLR pathways 32, 34, 40, negatively regulated the pro-inflammatory TRIF-mediated response to Ang II. In fact, we had previously reported a beneficial reduction of cardiac hypertrophy in the MyD88−/− mice after myocardial infarction 32. Similarly Wang, et al. 41 reported a reduction in hypertrophy in MyD88−/− mice upon Ang II-infusion. However, they used a subpressor dose of Ang II (~400ng/kg/min). In our present study our model was primarily a model of hypertension using Ang II infusions at high pressor rates (3000ng/kg/min) and the resulting hypertension, cardiac hypertrophy and particularly the pro-inflammatory gene expressions were exaggerated.
These doses of Ang II cause significant vasoconstriction, neuro-humoral and central nervous system (CNS) activation with increased sympathetic activity in addition to the renal and cardiac effects. Thus, the differences in results may represent the release of different endogenous ligands that may engage different adaptor mediated pathways. Furthermore, a recent abstract 42, indicates that the hypertension and hypertrophy caused by Ang II infusions (1000ng/kg/min) were preserved in MyD88−/− mice. This study’s conclusion was in accordance with ours that MyD88 does not contribute to hypertension.
Contrasting Effects of Signaling through TRIF vs. MyD88 Adaptors on Gene Expression in Heart and Kidney
We observed contrasting changes in pro-inflammatory gene expression in MyD88−/− and Trifmut mice compared to WT mice. Tnfa is a key cytokine in Ang II induced increases in blood pressure and cardiac hypertrophy 38, 39, 43, 44. We found that in both heart and kidney the magnitude of induction of Tnfa expression was much higher in MyD88−/− than in WT and Trifmut.
Similarly reactive oxygen species (ROS) production resulting from activation of RAS system in renal tissue leads to hypertension 16 and the expression and activation of Nox4 RNA 45, 46 promotes cardiac hypertrophy 47–49. We found that Nox4 expression was slightly increased in heart of WT mice after three weeks of Ang II but significantly enhanced in both kidney and heart of MyD88−/− and, in contrast, markedly downregulated in kidney and unchanged in heart of Trifmut mice.
Increased expression of Ang II type 1 receptors (Agtr1a) was also significantly increased during the 3 week infusion of Ang II in both heart and kidney of MyD88−/− and conversely reduced significantly in Trifmut mice whereas no significant changes were observed in WT mice.
Taken together these expressions of pro-inflammatory genes correlate with the development of hypertension and cardiac hypertrophy, and demonstrate that the TRIF adaptor protein is the essential mediator of those responses whereas MyD88 is an effective negative regulator.
TLR Mediated Dual Activation of MyD88 Dependent and TRIF-dependent Signaling Pathways
Multiple TLRs interact selectively and specifically with ligands to initiate signaling cascades through adaptor protein molecules resulting in pro- or anti-inflammatory responses 50, 51. The most common adaptor protein is the MyD88 that was initially recognized as a component of the IL-1 receptor signaling complex 27. All TLRs except TLR3 and a subset of TLR4 signaling events depend on MyD88 27, 50, 51. MyD88-independent TLR4 pathways are activated by LPS in macrophages causing induction of several genes, including CXCL10, RANTES and macrophage colony stimulating factor 52. The adaptor molecule of the MyD88 independent signaling events is TRIF 27, 51. TRIF induction of IRF-3 activation and IFN-β and IFN-inducible genes are MyD88 independent 53–55.
Our results indicate that Ang II hypertension induces simultaneous activation of TLR pathways that are dependent on both adaptor proteins and have opposing effects in the WT mice with a net increase in arterial pressure, cardiac hypertrophy, and modest increases in cardiac and renal pro-inflammatory gene expression. The hypertension, cardiac hypertrophy, and the pro-inflammatory gene expression caused by Ang II were predominantly TRIF mediated. The expression of a TRIF-dependent chemokine Cxcl10, which was enhanced in the hearts of MyD88−/− mice compared to WT but decreased in Trifmut mice, further supports the induction of TRIF dependendent inflammatory signaling, which is negatively regulated by MyD88-dependent signaling.
Since TLRs are selectively activated by a large number of specific ligands of either exogenous or endogenous origin, the determination of which ligands are generated during the prolonged infusion of Ang II is challenging. One might suggest that the early rise in pressure during the first days of Ang II infusion may provoke cellular damage which releases DAMPs into the circulation which then activate TLRs on innate immune cells initiating the inflammatory cascade. An important question is how MyD88 exerts its anti-inflammatory effects.
Negative regulation of TRIF responses by MyD88 is anti-inflammatory
TLR signaling via different adaptor proteins may function in context-specific 56, 57 as well as tissue-specific manner 58. Moreover, multiple mechanisms of negative regulation of these adaptor pathways have been described 59. It is not clear whether the MyD88 anti-inflammatory effect results from direct stimulation of specific inhibitors or from gene repression effects of transcription factor NF-κB 60–62. Our results, however, suggest that deletion of MyD88 results in increased cardiac expression of TLR3 and TLR4 as well as TRIF, all of which would contribute to exaggerated responses and support a negative regulatory influence of MyD88. In contrast, the TRIF mutation decreased TLR3 without altering TLR4 expression but increased MyD88 expression. The latter would contribute to the observed suppression of responses to Ang II.
Another conceptually attractive mechanism that would explain the enhanced TRIF-dependent inflammatory responses in MyD88−/− mice is “Signaling Flux Redistribution” (SFR) 63 whereby removal of MyD88 enhances the availability and flux of signaling substrates through the alternative TRAM/TRIF dependent pathway. Enhanced inflammatory response in MyD88-deficiency has also been reported in other models as well 57, 64. Indeed, MyD88 functions as a negative regulator of the TRIF-dependent pathway in a corneal inflammation model 65. In this study, TLR3 ligand-induced inflammatory cellular infiltration in cornea was exacerbated in MyD88−/− mice. It is also noteworthy that MyD88 signaling also produces anti-inflammatory molecules, such as IL-10 66, which requires intact MyD88 for its anti-inflammatory action 67. The constitutive expression by MyD88 of A20, IRAK-M and PPAR gamma, which result in downstream repression of NF-κB transcription factor but not TRIF-dependent interferon regulatory factor 3 (IRF3), would also result in enhancement of pro-inflammatory responses in MyD88 deficiency 68, 69.
Our results showing exacerbation of Ang II hypertension and inflammatory gene expression in kidney and heart including Cxcl10 in the MyD88−/− heart supports a similar anti-inflammatory effect of MyD88. Thus, in the context of Ang II-induced hypertension and cardiac hypertrophy it is clear that MyD88 exerts a novel negative regulatory role.
Interaction of Ang II with TLRs
Exactly how the Ang II pathway interacts with the TLR pathways is not understood. Ang II-induced hypertension involves the adaptive immune system 13, which in turn is regulated by the activation of the innate immune system by TLRs that detect specific ligands (DAMPs) 70. Cytokines released by the innate immune system, such as pro-inflammatory cytokine IL-6 are essential for Ang II-induced hypertension in mice 71. Moreover, as mentioned above, we have recently shown that Ang II is capable of markedly enhancing specific pro-inflammatory TLR responses in the immune cells of SHRs but not in the immune cells of normotensive Wistar-Kyoto rats 17. The components of TLR signaling are widely expressed in multiple tissues and cell types. Although systemic Ang II infusion has wide-ranging effects on various tissues, Ang II is not known to be an agonist of the TLRs. It is possible that a downstream product of Ang II-AT1 receptor binding may generate a TLR agonist or the AT1R signaling pathway might interact with the components of the TLR pathway to activate it.
Perspective
Inflammation is a hallmark of the etiology of hypertension and end organ damage including cardiac hypertrophy. TLRs and their adaptor proteins are integral components of innate immunity whose role in hypertension and end organ damage is now fully recognized. Our finding that Ang II-responses result from activation of different pathways with pro- and anti-inflammatory adaptor proteins opens up opportunities for new molecular targets to treat recalcitrant hypertension.
Hypertension represents an abnormal immunological state that involves the adaptive as well as the innate immune system. Our finding indicates that a hypertensive state may be exacerbated as a result of suppression or impairment of MyD88-dependent pathways. Conversely, targeting the TRIF pathway may be therapeutic by blocking the inflammatory response and enhancing the negative regulatory effects of MyD88.
Supplementary Material
Novelty and Significance.
What is new?
We discovered a dual simultaneous activation by Ang II of two innate immune signaling pathways. One that includes TRIF, as its adaptor protein, is essential for Ang II-induced hypertension, cardiac hypertrophy, and pro-inflammatory gene expression, whereas the other that is dependent on adaptor protein MyD88 mediates an anti-inflammatory response that restrains Ang II hypertension, hypertrophy and inflammatory gene expression.
What is relevant?
Ang II-induced hypertension and end organ damage depend on inflammatory immune responses. However, the mechanisms of Ang II-induced inflammatory responses are not fully understood. Now, the knowledge of the specific adaptor proteins that regulate innate immune inflammatory pathways provides opportunities for novel interventions to treat hypertension and end organ damage.
Summary
We have discovered that a TRIF adaptor-dependent inflammatory pathway of the innate immune system rather than the common MyD88 adaptor-dependent pathway mediates Ang II-induced hypertension, cardiac hypertrophy, and pro-inflammatory gene expression. Paradoxically the MyD88 adaptor signaling activated by Ang II is anti-inflammatory. This study provides valuable insight into the mechanisms of hypertension and the potential for developing new therapies.
Acknowledgments
We thank Carol Whiteis, for technical assistance during the initial phase of this study. We also acknowledge discussions with and suggestions from Drs. Zuhair Ballas and John Colgan.
Source(s) of Funding
This research study was funded by the National Institutes of Health Program Project Grant to FMA (HL 14388) and VA Merit Review Award to MWC (I01 BX001414).
Abbreviations
- Ang II
Angiotensin II
- AT1R
Angiotensin II type 1 receptor
- DAMP
Damage associated molecular patterns
- PAMP
Pathogen associated molecular patterns
- PRR
Pattern recognizing receptors
- MyD88
Myeloid differentiation protein 88
- SBP
Systolic blood pressure
- TICAM1
TIR domain-containing adaptor molecule 1
- TLR
Toll-like receptor
- TRIF
Toll-interleukin receptor-domain containing adaptor protein inducing interferon-β
- Trifmut
C57BL/6J-Ticam1Lps2/J mice
Footnotes
Conflict(s) of Interest/Disclosure(s)
None
References
- 1.Singh MV, Abboud FM. Toll-like receptors and hypertension. Am J Physiol Regul Integr Comp Physiol. 2014;307:R501–504. doi: 10.1152/ajpregu.00194.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh MV, Chapleau MW, Harwani SC, Abboud FM. The immune system and hypertension. Immunol Res. 2014;59:243–253. doi: 10.1007/s12026-014-8548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM. Inflammation, immunity, and hypertension. Hypertension. 2011;57:132–140. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harrison DG, Vinh A, Lob H, Madhur MS. Role of the adaptive immune system in hypertension. Curr Opin Pharmacol. 2010;10:203–207. doi: 10.1016/j.coph.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodriguez-Iturbe B, Pons H, Quiroz Y, Lanaspa MA, Johnson RJ. Autoimmunity in the pathogenesis of hypertension. Nat Rev Nephrol. 2014;10:56–62. doi: 10.1038/nrneph.2013.248. [DOI] [PubMed] [Google Scholar]
- 6.Schiffrin EL. The immune system: role in hypertension. Canad J Cardiol. 2013;29:543–548. doi: 10.1016/j.cjca.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 7.Mian MO, Paradis P, Schiffrin EL. Innate immunity in hypertension. Curr Hypertens Rep. 2014;16:413. doi: 10.1007/s11906-013-0413-9. [DOI] [PubMed] [Google Scholar]
- 8.Ryan MJ. An update on immune system activation in the pathogenesis of hypertension. Hypertension. 2013;62:226–230. doi: 10.1161/HYPERTENSIONAHA.113.00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rudemiller N, Lund H, Jacob HJ, Geurts AM, Mattson DL PhysGen Knockout Program. CD247 modulates blood pressure by altering T-lymphocyte infiltration in the kidney. Hypertension. 2014;63:559–564. doi: 10.1161/HYPERTENSIONAHA.113.02191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang DS, Bian ZY, Zhang Y, Zhang SM, Liu Y, Zhang R, Chen Y, Yang Q, Zhang XD, Fan GC, Li H. Role of interferon regulatory factor 4 in the regulation of pathological cardiac hypertrophy. Hypertension. 2013;61:1193–1202. doi: 10.1161/HYPERTENSIONAHA.111.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quiroz Y, Johnson RJ, Rodriguez-Iturbe B. The role of T cells in the pathogenesis of primary hypertension. Nephrol Dial Transplant. 2012;27(Suppl 4):iv2–5. doi: 10.1093/ndt/gfs421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matrougui K, Abd Elmageed Z, Kassan M, Choi S, Nair D, Gonzalez-Villalobos RA, Chentoufi AA, Kadowitz P, Belmadani S, Partyka M. Natural regulatory T cells control coronary arteriolar endothelial dysfunction in hypertensive mice. Am J Pathol. 2011;178:434–441. doi: 10.1016/j.ajpath.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu Q, Horak K, Larson DF. Role of T lymphocytes in hypertension-induced cardiac extracellular matrix remodeling. Hypertension. 2006;48:98–104. doi: 10.1161/01.HYP.0000227247.27111.b2. [DOI] [PubMed] [Google Scholar]
- 15.Ba D, Takeichi N, Kodama T, Kobayashi H. Restoration of T cell depression and suppression of blood pressure in spontaneously hypertensive rats (SHR) by thymus grafts or thymus extracts. J Immunol. 1982;128:1211–1216. [PubMed] [Google Scholar]
- 16.Abboud FM, Harwani SC, Chapleau MW. Autonomic neural regulation of the immune system: implications for hypertension and cardiovascular disease. Hypertension. 2012;59:755–762. doi: 10.1161/HYPERTENSIONAHA.111.186833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harwani SC, Chapleau MW, Legge KL, Ballas ZK, Abboud FM. Neurohormonal modulation of the innate immune system is proinflammatory in the prehypertensive spontaneously hypertensive rat, a genetic model of essential hypertension. Circ Res. 2012;111:1190–1197. doi: 10.1161/CIRCRESAHA.112.277475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zubcevic J, Waki H, Raizada MK, Paton JF. Autonomic-immune-vascular interaction: an emerging concept for neurogenic hypertension. Hypertension. 2011;57:1026–1033. doi: 10.1161/HYPERTENSIONAHA.111.169748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bomfim GF, Dos Santos RA, Oliveira MA, Giachini FR, Akamine EH, Tostes RC, Fortes ZB, Webb RC, Carvalho MH. Toll-like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin Sci (Lond) 2012;122:535–543. doi: 10.1042/CS20110523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dange RB, Agarwal D, Masson GS, Vila J, Wilson B, Nair A, Francis J. Central blockade of TLR4 improves cardiac function and attenuates myocardial inflammation in angiotensin II-induced hypertension. Cardiovasc Res. 2014;103:17–27. doi: 10.1093/cvr/cvu067. [DOI] [PubMed] [Google Scholar]
- 21.Coble JP, Grobe JL, Johnson AK, Sigmund CD. Mechanisms of brain renin angiotensin system-induced drinking and blood pressure: importance of the subfornical organ. Am J Physiol Regul Integr Comp Physiol. 2015;308:R238–R249. doi: 10.1152/ajpregu.00486.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montezano AC, Nguyen Dinh Cat A, Rios FJ, Touyz RM. Angiotensin II and vascular injury. Curr Hypertens Rep. 2014;16:431. doi: 10.1007/s11906-014-0431-2. [DOI] [PubMed] [Google Scholar]
- 23.Navar LG, Prieto MC, Satou R, Kobori H. Intrarenal angiotensin II and its contribution to the genesis of chronic hypertension. Curr Opin Pharmacol. 2011;11:180–186. doi: 10.1016/j.coph.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sadoshima J, Izumo S. Molecular characterization of angiotensin II--induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ Res. 1993;73:413–423. doi: 10.1161/01.res.73.3.413. [DOI] [PubMed] [Google Scholar]
- 25.Trott DW, Harrison DG. The immune system in hypertension. Adv Physiol Educ. 2014;38:20–24. doi: 10.1152/advan.00063.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moresco EM, LaVine D, Beutler B. Toll-like receptors. Curr Biol. 2011;21:R488–493. doi: 10.1016/j.cub.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 27.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 28.Kenny EF, O’Neill LA. Signalling adaptors used by Toll-like receptors: an update. Cytokine. 2008;43:342–349. doi: 10.1016/j.cyto.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 29.Siednienko J, Halle A, Nagpal K, Golenbock DT, Miggin SM. TLR3-mediated IFN-beta gene induction is negatively regulated by the TLR adaptor MyD88 adaptor-like. Eur J Immunol. 2010;40:3150–3160. doi: 10.1002/eji.201040547. [DOI] [PubMed] [Google Scholar]
- 30.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 31.Re F, Strominger JL. IL-10 released by concomitant TLR2 stimulation blocks the induction of a subset of Th1 cytokines that are specifically induced by TLR4 or TLR3 in human dendritic cells. J Immunol. 2004;173:7548–7555. doi: 10.4049/jimmunol.173.12.7548. [DOI] [PubMed] [Google Scholar]
- 32.Singh MV, Swaminathan PD, Luczak ED, Kutschke W, Weiss RM, Anderson ME. MyD88 mediated inflammatory signaling leads to CaMKII oxidation, cardiac hypertrophy and death after myocardial infarction. J Mol Cell Cardiol. 2012;52:1135–1144. doi: 10.1016/j.yjmcc.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 34.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 35.Gibson-Corley KN, Olivier AK, Meyerholz DK. Principles for valid histopathologic scoring in research. Vet Pathol. 2013;50:1007–1015. doi: 10.1177/0300985813485099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Livak K, Schmittgen T. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 37.Singh MV, Kapoun A, Higgins L, Kutschke W, Thurman JM, Zhang R, Singh M, Yang J, Guan X, Lowe JS, Weiss RM, Zimmermann K, Yull FE, Blackwell TS, Mohler PJ, Anderson ME. Ca2+/calmodulin-dependent kinase II triggers cell membrane injury by inducing complement factor B gene expression in the mouse heart. J Clin Invest. 2009;119:986–996. doi: 10.1172/JCI35814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakamura K, Fushimi K, Kouchi H, Mihara K, Miyazaki M, Ohe T, Namba M. Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin II. Circulation. 1998;98:794–799. doi: 10.1161/01.cir.98.8.794. [DOI] [PubMed] [Google Scholar]
- 39.Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51:1345–1351. doi: 10.1161/HYPERTENSIONAHA.107.102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gomolak JR, Didion SP. A role for innate immunity in the development of hypertension. Medi Hypotheses. 2014;83:640–643. doi: 10.1016/j.mehy.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang K, Liu F, Zhou LY, Long B, Yuan SM, Wang Y, Liu CY, Sun T, Zhang XJ, Li PF. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res. 2014;114:1377–1388. doi: 10.1161/CIRCRESAHA.114.302476. [DOI] [PubMed] [Google Scholar]
- 42.Gomolak JR, Faulkner JL, Didion SP. Myloid Differentiation Factor 88 (MyD88) does not contribute to hypertension or endothelial dysfunction produced by angiotensin II. FASEB J. 2013;27:1131.15. [Google Scholar]
- 43.Torre-Amione G, Kapadia S, Lee J, Durand JB, Bies RD, Young JB, Mann DL. Tumor necrosis factor-alpha and tumor necrosis factor receptors in the failing human heart. Circulation. 1996;93:704–711. doi: 10.1161/01.cir.93.4.704. [DOI] [PubMed] [Google Scholar]
- 44.Zhang J, Patel MB, Griffiths R, Mao A, Song YS, Karlovich NS, Sparks MA, Jin H, Wu M, Lin EE, Crowley SD. Tumor Necrosis Factor-alpha Produced in the Kidney Contributes to Angiotensin II-dependent Hypertension. Hypertension. 2014;64:1275–1281. doi: 10.1161/HYPERTENSIONAHA.114.03863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim S-M, Kim Y-G, Jeong K-H, Lee S-H, Lee T-W, Ihm C-G, Moon J-Y. Angiotensin II-Induced Mitochondrial Nox4 Is a Major Endogenous Source of Oxidative Stress in Kidney Tubular Cells. PLoS ONE. 2012;7:e39739. doi: 10.1371/journal.pone.0039739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, Li H, Bodenschatz M, August M, Kleschyov AL, Tsilimingas N, Walter U, Forstermann U, Meinertz T, Griendling K, Munzel T. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res. 2002;90:E58–65. doi: 10.1161/01.res.0000012569.55432.02. [DOI] [PubMed] [Google Scholar]
- 47.Matsushima S, Kuroda J, Ago T, Zhai P, Park JY, Xie LH, Tian B, Sadoshima J. Increased oxidative stress in the nucleus caused by Nox4 mediates oxidation of HDAC4 and cardiac hypertrophy. Circ Res. 2013;112:651–663. doi: 10.1161/CIRCRESAHA.112.279760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zeng SY, Chen X, Chen SR, Li Q, Wang YH, Zou J, Cao WW, Luo JN, Gao H, Liu PQ. Upregulation of Nox4 promotes angiotensin II-induced epidermal growth factor receptor activation and subsequent cardiac hypertrophy by increasing ADAM17 expression. Can J Cardiol. 2013;29:1310–1319. doi: 10.1016/j.cjca.2013.04.026. [DOI] [PubMed] [Google Scholar]
- 49.Zhao QD, Viswanadhapalli S, Williams P, Shi Q, Tan C, Yi X, Bhandari B, Abboud HE. NADPH Oxidase 4 Induces Cardiac Fibrosis and Hypertrophy Through Activating Akt/mTOR and NFκB Signaling Pathways. Circulation. 2015;131:643–655. doi: 10.1161/CIRCULATIONAHA.114.011079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 51.West AP, Koblansky AA, Ghosh S. Recognition and signaling by toll-like receptors. Annu Rev Cell Dev Biol. 2006;22:409–437. doi: 10.1146/annurev.cellbio.21.122303.115827. [DOI] [PubMed] [Google Scholar]
- 52.Bjorkbacka H, Fitzgerald KA, Huet F, Li X, Gregory JA, Lee MA, Ordija CM, Dowley NE, Golenbock DT, Freeman MW. The induction of macrophage gene expression by LPS predominantly utilizes Myd88-independent signaling cascades. Physiol Genomics. 2005;19:319–330. doi: 10.1152/physiolgenomics.00128.2004. [DOI] [PubMed] [Google Scholar]
- 53.Doyle S, Vaidya S, O’Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, Cheng G. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 54.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 55.Kawai T, Takeuchi O, Fujita T, Inoue J, Muhlradt PF, Sato S, Hoshino K, Akira S. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol. 2001;167:5887–5894. doi: 10.4049/jimmunol.167.10.5887. [DOI] [PubMed] [Google Scholar]
- 56.Wang E, Feng Y, Zhang M, Zou L, Li Y, Buys ES, Huang P, Brouckaert P, Chao W. Toll-like receptor 4 signaling confers cardiac protection against ischemic injury via inducible nitric oxide synthase- and soluble guanylate cyclase-dependent mechanisms. Anesthesiology. 2011;114:603–613. doi: 10.1097/ALN.0b013e31820a4d5b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gobert AP, Bambou JC, Werts C, Balloy V, Chignard M, Moran AP, Ferrero RL. Helicobacter pylori heat shock protein 60 mediates interleukin-6 production by macrophages via a toll-like receptor (TLR)-2-, TLR-4-, and myeloid differentiation factor 88-independent mechanism. J Biol Chem. 2004;279:245–250. doi: 10.1074/jbc.M307858200. [DOI] [PubMed] [Google Scholar]
- 58.Cotroneo TM, Nemzek-Hamlin JA, Bayliss J, Su GL. Lipopolysaccharide binding protein inhibitory peptide alters hepatic inflammatory response post-hemorrhagic shock. Innate Immun. 2012;18:866–875. doi: 10.1177/1753425912444641. [DOI] [PubMed] [Google Scholar]
- 59.Lang T, Mansell A. The negative regulation of Toll-like receptor and associated pathways. Immunol Cell Biol. 2007;85:425–434. doi: 10.1038/sj.icb.7100094. [DOI] [PubMed] [Google Scholar]
- 60.Datta De D, Datta A, Bhattacharjya S, Roychoudhury S. NF-kappaB mediated transcriptional repression of acid modifying hormone gastrin. PLoS One. 2013;8:e73409. doi: 10.1371/journal.pone.0073409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ashburner BP, Westerheide SD, Baldwin AS., Jr The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol. 2001;21:7065–7077. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baetz D, Regula KM, Ens K, Shaw J, Kothari S, Yurkova N, Kirshenbaum LA. Nuclear factor-kappaB-mediated cell survival involves transcriptional silencing of the mitochondrial death gene BNIP3 in ventricular myocytes. Circulation. 2005;112:3777–3785. doi: 10.1161/CIRCULATIONAHA.105.573899. [DOI] [PubMed] [Google Scholar]
- 63.Selvarajoo K, Takada Y, Gohda J, Helmy M, Akira S, Tomita M, Tsuchiya M, Inoue J, Matsuo K. Signaling flux redistribution at toll-like receptor pathway junctions. PLoS One. 2008;3:e3430. doi: 10.1371/journal.pone.0003430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Obonyo M, Sabet M, Cole SP, Ebmeyer J, Uematsu S, Akira S, Guiney DG. Deficiencies of myeloid differentiation factor 88, Toll-like receptor 2 (TLR2), or TLR4 produce specific defects in macrophage cytokine secretion induced by Helicobacter pylori. Infect Immun. 2007;75:2408–2414. doi: 10.1128/IAI.01794-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johnson AC, Li X, Pearlman E. MyD88 functions as a negative regulator of TLR3/TRIF-induced corneal inflammation by inhibiting activation of c-Jun N-terminal kinase. J Biol Chem. 2008;283:3988–3996. doi: 10.1074/jbc.M707264200. [DOI] [PubMed] [Google Scholar]
- 66.de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;174:1209–1220. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang J, Kunkel SL, Chang CH. Negative regulation of MyD88-dependent signaling by IL-10 in dendritic cells. Proc Natl Acad Sci U S A. 2009;106:18327–18332. doi: 10.1073/pnas.0905815106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 69.Turer EE, Tavares RM, Mortier E, Hitotsumatsu O, Advincula R, Lee B, Shifrin N, Malynn BA, Ma A. Homeostatic MyD88-dependent signals cause lethal inflamMation in the absence of A20. J Exp Med. 2008;205:451–464. doi: 10.1084/jem.20071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM. Inflammation, Immunity, and Hypertension. Hypertension. 2010;57:132–140. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee DL, Sturgis LC, Labazi H, Osborne JB, Jr, Fleming C, Pollock JS, Manhiani M, Imig JD, Brands MW. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol. 2006;290:H935–940. doi: 10.1152/ajpheart.00708.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.