

Abstract
Cadmium (Cd), a toxic environmental contaminant, contributes to neurodegeneration. Rapamycin, a macrocyclic lactone, has shown preventive effect on Cd-induced neuronal cell death. However, the underlying mechanism is not fully understood. Here, we show that rapamycin prevented Cd-induced apoptotic cell death in neuronal cells. Coincidently, rapamycin markedly blocked Cd-induced phosphorylation of Akt, S6K1 and 4E-BP1 in the cells. Expression of a rapamycin-resistant and kinase-active mTOR (S2035T, mTOR-T), but not a rapamycin-resistant and kinase-dead mTOR (S2035T/D2357E, mTOR-TE), conferred resistance to rapamycin inhibition of Cd-induced cell death, implying that the preventive effect of rapamycin on Cd-induced neurotoxicity is mTOR kinase activity-dependent. It appeared that both mTORC1 and mTORC2 were involved in the inhibitory activity of rapamycin, as silencing raptor, rictor or raptor/rictor enhanced rapamycin’s blockage of Cd-induced cell death. Furthermore, downregulation of S6K1, ectopic expression of constitutively hypophosphorylated 4E-BP1 or dominant negative Akt, or co-treatment with Akt inhibitor also potentiated the rapamycin’s inhibitory effect. The findings indicate that rapamycin prevents Cd-induced neuronal cell death via suppressing both mTORC1 and mTORC2 pathways. Our results highlight that rapamycin may be exploited for the prevention of Cd-induced neurodegenerative disorders.
Keywords: Rapamycin, Cadmium, Neuronal cells, Apoptosis, mTOR
Graphical Abstract
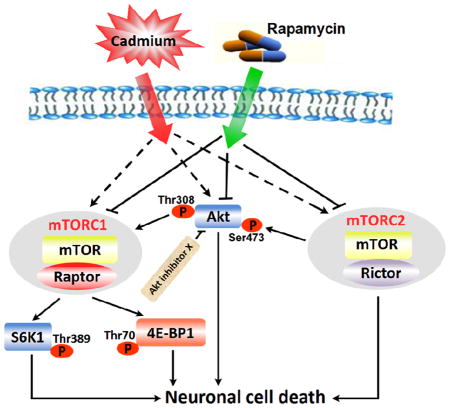
It is unclear how inhibition of mTOR by rapamycin prevents Cd-neuronal cell death. This study underscores that rapamycin prevents neuronal cells from Cd-poisoning, not only by targeting mTORC1-mediated S6K1/4E-BP1 pathways, but also via targeting mTORC2-mediated Akt pathway. The findings highlight that rapamycin may be exploited for prevention of Cd-induced neurodegenerative disorders.
1. Introduction
Cadmium (Cd), an environmental contaminant, which is mainly released from smelting and refining of metals, burning of chemical fuels and municipal wastes, and cigarette smoking, is recognized as one of the most toxic metals with very strong accumulation in the human body because its very long biological half-life (15–20 years) (Wang and Du, 2013). Cd targets several organs and tissues such as kidney (Johri et al., 2010), blood (Kocak and Akcil, 2006), bones (Akesson et al., 2006), testis (Thompson and Bannigan, 2008), and brain (Lopez et al., 2003; Mendez-Armenta and Rios, 2007; Okuda et al., 1997), resulting in nephrotoxicity, immunotoxicity, osteotoxicity, genotoxicity, neurotoxicity and tumors. As Cd has high blood-brain barrier permeability, exposure to Cd severely affects the function of the nervous system, with symptoms including headache and vertigo, olfactory dysfunction, slowing of vasomotor functioning, peripheral neuropathy, decreased equilibrium, neurobehavioral defects in attention, psychomotor speed, and learning disabilities (Jarup et al., 1993; Pihl and Parkes, 1977; Wang and Du, 2013; Wright et al., 2006). Besides, Parkinsonism after acute Cd poisoning has been documented (Okuda et al., 1997). Thus, Cd intoxication has been considered as a possible etiological factor of neurodegenerative diseases, such as Parkinson’s desease (PD), Alzheimer’s disease (AD) and Huntington’s disease (HD) (Johnson, 2001; Okuda et al., 1997; Panayi et al., 2002).
The mammalian target of rapamycin (mTOR), a serine/threonine (Ser/Thr) protein kinase, is a central controller for cell growth/proliferation and survival (Cornu et al., 2013; Laplante and Sabatini, 2012). In mammalian cells, mTOR functions at least as two complexes, mTORC1 and mTORC2, with distinct substrate specificities (Laplante and Sabatini, 2012). mTORC1 consists of mTOR, raptor (regulatory-associated protein of mTOR), mLST8 (also termed G-protein β-subunit-like protein, GβL, a yeast homolog of LST8), PRAS40 (proline-rich Akt substrate 40 kDa) and DEPTOR, whereas mTORC2 includes mTOR, rictor (rapamycin insensitive companion of mTOR), mLST8, mSin1 (mammalian stress-activated protein kinase-interacting protein 1), protor (protein observed with rictor) and DEPTOR (Cornu et al., 2013; Laplante and Sabatini, 2012; Peterson et al., 2009; Zhou et al., 2010). mTORC1 regulates phosphorylation of p70 S6 kinase 1 (S6K1) and eukaryotic initiation factor 4E (eIF4E) binding protein 1 (4E-BP1), and controls protein and lipid synthesis, cell growth, proliferation, survival and motility (Cornu et al., 2013; Jacinto et al., 2004; Kim et al., 2002; Laplante and Sabatini, 2012; Polak and Hall, 2009; Sarbassov et al., 2004), while mTORC2 regulates phosphorylation or activity of Akt, serum and glucocorticoid-induced kinase 1 (SGK1), protein kinase C α (PKCα), focal adhesion proteins and small GTPases, and controls cell survival and actin cytoskeleton (Cornu et al., 2013; Laplante and Sabatini, 2012).
Studies have demonstrated that mTOR regulates differentiation, development and survival in neurons, and exerts an important role in synaptic plasticity, learning and memory, and food uptake in adult brain (Jaworski and Sheng, 2006; Swiech et al., 2008). mTOR activity is modified in various pathologic states of the nervous system, including brain tumors, tuberous sclerosis, cortical displasia and neurodegenerative disorders such as PD, AD, and HD (Swiech et al., 2008). We have recently found that Cd induces neuronal apoptosis in part through activation of mTOR signaling pathway (Chen et al., 2008; Chen et al., 2011a; Chen et al., 2014; Chen et al., 2011b). Rapamycin, a macrocyclic lactone, is a potent and specific mTORC1 inhibitor (Zhou et al., 2010). Extensive data have indicated that mTORC1 is sensitive to short rapamycin exposure (Hara et al., 2002; Laplante and Sabatini, 2012). However, the effect of rapamycin on mTORC2-mediated Akt phosphorylation depends on the concentration and duration of rapamycin treatment (Sarbassov et al., 2006). Recent studies have shown that rapamycin is neuroprotective in various neurological diseases (Bove et al., 2011; Erlich et al., 2007; Malagelada et al., 2010; Pan et al., 2009). Thus, we hypothesized that rapamycin that can inhibit mTORC1 and/or mTORC2 may have preventive effect on Cd-induced neurotoxicity. As expected, inhibition of mTOR by pretreatment with rapamycin for 48 h indeed prevented Cd-induced death in neuronal cells (Chen et al., 2008; Chen et al., 2014). Administration of rapamycin in vivo also potently attenuated Cd-induced activation of mTOR signaling, brain damage and neuronal cell death in mice (Chen et al., 2014). However, so far, it remains unclear whether the preventive activity is attributed to rapamycin’s targeting mTORC1 and/or mTORC2.
Here, for the first time, we show that rapamycin prevented Cd-induced neuronal cell death, not only by targeting both mTORC1-mediated S6K1/4E-BP1 pathways, but also via targeting mTORC2-mediated Akt pathway. The findings indicate that rapamycin rescues neuronal cells from Cd-poisoning via inhibiting Cd-induced activation of both mTORC1 and mTORC2. Our results highlight that rapamycin may be exploited for the prevention of Cd-induced neurodegenerative disorders.
2. Materials and methods
2.1. Reagents
Cadmium chloride, poly-D-lysine (PDL), 4′,6-diamidino-2-phenylindole (DAPI), and protease inhibitor cocktail were purchased from Sigma (St Louis, MO, USA). Rapamycin was from ALEXIS (San Diego, CA, USA). Dulbecco’s modified Eagle medium (DMEM), 0.05% Trypsin-EDTA, NEUROBASAL™ Media, and B27 Supplement were purchased from Invitrogen (Grand Island, NY, USA). Horse serum and fetal bovine serum (FBS) were supplied by Hyclone (Logan, UT, USA). Enhanced chemiluminescence solution was from Millipore (Billerica, MA, USA). Akt inhibitor X was provided by Santa Cruz Biotechnology (Santa Cruz, CA, USA). The following antibodies were used: phospho-Akt (Ser473), 4E-BP1, phospho-4E-BP1 (Thr70), S6 ribosomal protein, phospho-S6 ribosomal protein (Ser235/236), phospho-S6K1 (Thr389), caspase-3, and PARP (all from Cell Signaling Technology, Beverly, MA, USA); Akt, GSK3β, and S6K1 (all from Santa Cruz Biotechnology); phospho-GSK3β (Ser9) (Epitomics, Burlingame, CA, USA); raptor and rictor (Bethyl Laboratories, Montgomery, TX, USA); HA, mTOR, phospho-Akt (Thr308), and β-tubulin (all from Sigma); goat anti-rabbit IgG-horseradish peroxidase (HRP), goat anti-mouse IgG-HRP, and rabbit anti-goat IgG-HRP (Pierce, Rockford, IL, USA). Other chemicals were purchased from local commercial sources and were of analytical grade.
2.2. Cells
Rat pheochromocytoma (PC12) and human neuroblastoma SH-SY5Y cell lines were from American Type Culture Collection (ATCC) (Manassas, VA, USA). PC12 cells were cultured in antibiotic-free DMEM supplemented with 10% horse serum and 5% FBS, whereas SH-SY5Y cells were grown in antibiotic-free DMEM supplemented with 10% FBS. Cells were maintained in a humid incubator (37°C, 5% CO2). For isolation of primary neurons, fetal mice at 16–18 days of gestation were chosen and primary cortical neurons were isolated and cultured as described (Chen et al., 2010). Fresh medium was replaced every 3 days. The primary neurons were used for experiments after 6 days of culture.
2.3. Recombinant adenoviral constructs and infection of cells
The recombinant adenoviruses expressing FLAG-tagged rapamycin-resistant and kinase-active mTOR mutant (S2035T; designated mTOR-T), FLAG-tagged rapamycin-resistant and kinase-dead mTOR-T (S2035T/D2357E, designated mTOR-TE), hemagglutinin (HA)-tagged constitutively hypophosphorylated 4E-BP1 (Ad-4EBP1-5A) and green fluorescence protein (Ad-GFP) were described previously (Chen et al., 2014; Liu et al., 2008; Liu et al., 2006; Liu et al., 2010). Recombinant adenovirus encoding HA-tagged dominant negative Akt (dn-Akt, T308A/S473A) was a generous gift from Dr. Kenneth Walsh (Boston University, Boston, MA). The viruses were amplified, titrated and used as described (Huang et al., 2003; Liu et al., 2008). For experiments, PC12 cells or SH-SY5Y cells were grown in the growth medium and infected with the individual adenovirus for 24 h at 5 of multiplicity of infection (MOI = 5). Subsequently, cells were used for experiments. Ad-GFP served as a control. Expression of HA-tagged 4E-BP1-5A and dn-Akt, as well as FLAG-tagged mTOR-T and mTOR-TE was determined by Western blot analysis with antibodies to HA and FLAG, respectively.
2.4. Lentiviral shRNA cloning, production and infection
Lentiviral shRNAs to raptor, rictor, S6K1, and GFP were described previously (Chen et al., 2010; Liu et al., 2006). The lentivirus-expressing GFP shRNA served as a control. A monolayer of PC12 and/or SH-SY5Y cells, when grown to about 70% confluence, were infected with above lentivirus-containing supernatant in the presence of 8 μg/ml polybrene for 24 h, and then exposed to 2 μg/ml puromycin. In 5 days, cells were used for experiments.
2.5. Live cell assay by trypan blue exclusion
PC12 and/or SH-SY5Y cells were seeded at a density of 5 × 105 cells/well in a 6-well plate, pre-coated with (for PC12) or without (for SH-SY5Y) PDL (0.2 μg/ml). The next day, cells were treated with/without Cd (10 and 20 μM) for 24 h following pre-incubation with/without rapamycin (0.2 μg/ml) for 48 h, with 5 replicates of each treatment. In some cases, following infection with Ad-mTOR-T, Ad-mTOR-TE, Ad-4EBP1-5A, Ad-dn-Akt or Ad-GFP, or infection with lentiviral shRNA to raptor, rictor, raptor/rictor, S6K1 or GFP, the cells were pretreated with rapamycin (0.2 μg/ml) for 48 h and then exposed to Cd (20 μM) for 24 h. Subsequently, live cells were monitored by counting viable cells using trypan blue exclusion test.
2.6. DAPI staining
PC12 cells, SH-SY5Y cells and/or primary neurons, seeded at a density of 5×105 cells/well in a 6-well plate containing a PDL-uncoated or -coated glass coverslip per well, were treated with/without Cd (10 and 20 μM) for 24 h following pre-incubation with/without rapamycin (0.2 μg/ml) for 48 h, with 5 replicates of each treatment. In some cases, cells were pretreated with/without rapamycin (0.2 μg/ml) for 48 h, and then with/without Akt inhibitor X (20 μM) for 2 h, followed by exposure to Cd (20 μM) for 24 h; in other cases, after infection with Ad-mTOR-T, Ad-mTOR-TE, Ad-4EBP1-5A, Ad-dn-Akt or Ad-GFP, or infection with lentiviral shRNA to raptor, rictor, raptor/rictor, S6K1 or GFP, the cells were pretreated with rapamycin (0.2 μg/ml) for 48 h and then exposed to Cd (20 μM) for 24 h. Subsequently, the cells with fragmented and condensed nuclei were stained by adding DAPI (4 μg/ml in deionized water) as described (Chen et al., 2008). Photographs were taken under a fluorescence microscope (Nikon 80i, Tokyo, Japan) equipped with a digital camera.
2.7. TUNEL staining
The terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP) nick-end labeling (TUNEL) staining was performed according to the manufacture’s instructions of In Situ Cell Death Detection Kit® (Roche, Mannheim, Germany). In brief, PC12 cells, seeded at a density of 5×105 cells/well in a 6-well plate containing a PDL-coated glass coverslip per well, were treated with/without Cd (10 and 20 μM) for 24 h following pre-incubation with/without rapamycin (0.2 μg/ml) for 48 h. After that, cells were fixed with 4% paraformaldehyde prepared in PBS for 2 h at 4°C. The fixed cells of each slide were washed 3 times with PBS, and then incubated in permeabilization solution (0.1% Triton 100-X, 0.1% sodium citrate) for 2 min on ice, followed by addition of TUNEL reaction mixture (TdT enzyme solution and labeling solution) to the samples and incubation for 1 h in a dark and humidified incubator at 37°C. After TUNEL labeling reaction, all stained specimens were rinsed 3 times with PBS and mounted with coverslips containing a mounting medium. Finally, the samples were analyzed under a fluorescence microscope (Nikon 80i, Tokyo, Japan) equipped with a digital camera. For quantitative analysis of the fluorescence staining, the integral optical density (IOD) was measured by Image-Pro Plus 6.0 software (Media Cybernetics Inc., Newburyport, MA, USA).
2.8. Western blot analysis
The indicated cells, after treatment, were briefly washed with cold PBS, and then on ice, lysed in RIPA buffer [50 mM Tris, pH 7.2; 150 mM NaCl; 1% sodium deoxycholate; 0.1% sodium dodecyl sulfate (SDS); 1% Triton X-100; 10 mM NaF; 1 mM Na3VO4; protease inhibitor cocktail (1:1000)]. Lysates were sonicated for 10 s and centrifuged at 14,000 rpm for 10 min at 4°C. The supernatants were collected. Protein concentration was determined by bicinchoninic acid assay with bovine serum albumin as a standard (Pierce). After that, Western blotting was performed as described previously (Chen et al., 2010).
2.9. Statistical analysis
Results were expressed as mean ± SEM. Student’s t-test for non-paired replicates was used to identify statistically significant differences between treatment means. Group variability and interaction were compared using either one-way or two-way ANOVA followed by Bonferroni’s post-tests to compare replicate means. Significance was accepted at p < 0.05.
3. Results
3.1. Rapamycin attenuates Cd-induced apoptotic cell death in neuronal cells
We have recently shown that Cd induces neuronal apoptosis in part through activation of mTOR signaling pathway (Chen et al., 2008; Chen et al., 2011a; Chen et al., 2014; Chen et al., 2011b), and inhibition of mTOR by rapamycin prevents Cd-induced neuronal cell death (Chen et al., 2008; Chen et al., 2014). In line with the above findings, here we also observed that pretreatment of neuronal cells (PC12 cells, SH-SY5Y cells and primary neurons) with rapamycin (0.2 μg/ml) for 48 h attenuated apoptotic cell death induced by 24-h exposure with Cd (10 and 20 μM). This is evidenced by the following findings. 1) The number of live cells in the Cd/rapamycin group was significantly higher than that in the Cd alone group (Fig. 1A), as determined by trypan blue exclusion. 2) Treatment with Cd for 24 h markedly increased nuclear fragmentation and condensation (arrows), a hallmark of apoptosis (Hao et al., 2013), in PC12 cells, which was potently diminished by rapamycin pretreatment (Fig. 1B and C), as detected by DAPI staining. 3) Pretreatment with rapamycin for 48 h dramatically decreased the number of TUNEL-positive cells (in green) in PC12 cells exposed to Cd (Fig. 1B and C). Similar results were also seen in SH-SY5Y cells and primary neurons (data not shown). 4) Treatment with Cd for 4 h resulted in robust cleavages of caspase-3 and PARP in PC12 cells, SH-SY5Y cells and primary neurons, which were strikingly attenuated by rapamycin (Fig. 1D). Collectively, our results strongly support the notion that rapamycin can prevent against Cd-induced neuronal cell death.
Fig. 1.

Rapamycin attenuates Cd-induced apoptotic cell death in neuronal cells. PC12 cells, SH-SY5Y cells and/or primary neurons were pretreated with rapamycin (Rap, 0.2 μg/ml) for 48 h, and then exposed to Cd (10 and 20 μM) for 4 h (for Western blotting) or 24 h (for live cell assay, DAPI and TUNEL staining). A) Live cells were detected by counting viable cells using trypan blue exclusion, showing that rapamycin reversed Cd-induced death of PC12 and SH-SY5Y cells. B) Apoptosis in PC12 cells was evaluated by nuclear fragmentation and condensation (arrows) using DAPI staining (upper panel) and concurrently by in situ detection of fragmented DNA (in green) using TUNEL staining (lower panel). Scale bar: 20 μm. C) The percentages of cells with fragmented nuclei and the number of TUNEL-positive cells were quantified, showing that rapamycin markedly attenuated Cd-induced apoptosis in PC12 cells. D) Indicated cell lysates were subjected to Western blot analysis using antibodies to cleaved-caspase-3 and cleaved-PARP. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. Results are presented as mean ± SEM (n = 5). a p < 0.05, difference with control group; b p < 0.05, difference with 10 μM Cd group; c p < 0.05, difference with 20 μM Cd group.
3.2. Rapamycin prevention against Cd-induced neuronal cell death is mTOR kinase activity dependent
Because a kinase activity-independent function for mTOR in the regulation of cell differentiation has been reported (Erbay and Chen, 2001), we next examined whether rapamycin prevention against Cd-induced neuronal cell death is mTOR kinase activity dependent. For this, PC12 cells were infected with recombinant adenoviral vectors encoding GFP (control), FLAG-tagged rapamycin-resistant and kinase-active mTOR (S2035T, mTOR-T), or kinase-dead mTOR-T (S2035T/D2357E, mTOR-TE) for 24 h, and then pretreated with/without rapamycin (0.2 μg/ml) for 48 h, followed by exposure to Cd (20 μM) for 4 h or 24 h. We observed that ectopic expression of FLAG-tagged mTOR-T, but not FLAG-tagged mTOR-TE or GFP, rendered resistance to rapamycin inhibition of phosphorylation of S6K1 and 4E-BP1, two best known substrates of mTOR, in PC12 cells (Fig. 2A). The data reveal that mTOR-T functioned as a rapamycin-resistant mutant, and mTOR-TE as a kinase-dead mutant in PC12 cells, as seen in other cell lines (Erbay and Chen, 2001; Liu et al., 2010). In addition, we noticed that expression of mTOR-T, but not mTOR-TE or GFP, also rendered potent resistance to rapamycin blockage of cleavages of caspase-3 and PARP in the cells (Fig. 2A). Furthermore, expression of mTOR-TE more substantially inhibited phosphorylation of S6K1 and 4E-BP1, including cleavages of caspase-3 and PARP, compared to GFP control in PC12 cells in response to Cd or Cd/rapamycin (Fig. 2A). Interestingly, ectopic expression of mTOR-T conferred high resistance to rapamycin’s prevention of Cd-induced cell death in PC12 cells, whereas expression of mTOR-TE remained sensitive to rapamycin and slightly enhanced the preventive effect of rapamycin against Cd-induced neurotoxicity compared to the GFP control (Fig. 2B and C), implying that rapamycin prevents Cd-induced neuronal cell death in an mTOR kinase activity-dependent manner.
Fig. 2.

Rapamycin prevents Cd-induced neuronal cell death in an mTOR kinase activity-dependent manner. PC12 cells, infected with Ad-mTOR-T, Ad-mTOR-TE, and Ad-GFP (as control), respectively, were pretreated with rapamycin (Rap, 0.2 μg/ml) for 48 h, followed by exposure to Cd (20 μM) for 4 h (for Western blotting) or 24 h (for live cell assay and DAPI staining). A) Ectopic expression of FLAG-tagged mTOR-T, but not FLAG-tagged mTOR-TE or GFP, rendered resistance to rapamycin inhibition of Cd-induced phosphorylation of S6K1 and 4E-BP1 in PC12 cells. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. B and C) Expression of mTOR-T, but not mTOR-TE or GFP, resisted to rapamycin’s prevention against Cd-induced B) cell viability reduction and C) apoptosis in PC12 cells. Results are presented as mean ± SEM (n = 5). a p < 0.05, difference with control group; b p < 0.05, Ad-mTOR-T group or Ad-mTOR-TE group versus Ad-GFP group.
3.3. Rapamycin prevents Cd-induced neuronal cell death by targeting both mTORC1 and mTORC2
mTOR functions in mammalian cells as two complexes, mTORC1 and mTORC2, which are well known to regulate the phosphorylation of S6K/4E-BP1 and Akt, respectively (Cornu et al., 2013; Laplante and Sabatini, 2012). To determine how rapamycin prevents Cd-induced neuronal cell death, first of all, PC12 cells and primary neurons were treated with Cd (10 and 20 μM) for 4 h following pretreatment with/without rapamycin (0.2 μg/ml) for 48 h. By Western blot analysis, we found that pretreatment with rapamycin not only profoundly inhibited Cd-induced phosphorylation of S6K1 (including S6, a substrate of S6K1) and 4E-BP1, but also remarkably suppressed Cd-induced phosphorylation of Akt in the cells (Fig. 3A), suggesting that rapamycin rescues neuronal cells from Cd-induced cell death probably by targeting both mTORC1 and mTORC2.
Fig. 3.

mTORC1 and mTORC2 are involved in rapamycin’s prevention from Cd-induced neuronal cell death. PC12 cells and primary neurons, or PC12 cells infected with lentiviral shRNAs to raptor, rictor, raptor/rictor or GFP (as control), were pretreated with rapamycin (Rap, 0.2 μg/ml) for 48 h, followed by exposure to Cd (10 and/or 20 μM) for 4 h (for Western blotting) or 24 h (for live cell assay and DAPI staining). A) Rapamycin strongly blocked Cd-induced phosphorylation of Akt, S6K1 and 4E-BP1 in PC12 cells and primary neurons. B) Lentiviral shRNA to raptor, rictor or raptor/rictor, but not GFP, downregulated raptor, rictor or raptor/rictor in PC12 cells, respectively. For A) and B), the blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. C and D) Silencing raptor, rictor or raptor/rictor potentiated rapamycin resistance to Cd-induced C) cell viability reduction and D) apoptosis, and this effect was more potent in the double raptor/rictor-silenced cells than in the single raptor or rictor-silenced cells. Results are presented as mean ± SEM (n = 5). a p < 0.05, difference with control group; b p < 0.05, difference with 10 μM Cd group; c p < 0.05, difference with 20 μM Cd group. d p < 0.05, Raptor shRNA group, Rictor shRNA group or Raptor/Rictor shRNA group versus GFP shRNA group; e p < 0.05, Raptor/Rictor shRNA group versus Raptor shRNA group or Rictor shRNA group.
To dissect whether and how mTORC1 and mTORC2 were involved in the preventive effect of rapamycin on Cd-induced neuronal cell death, mTORC1 and mTORC2 was disrupted by silencing raptor and rictor, respectively. Western blot analysis showed that lentiviral shRNA to raptor or rictor, but not GFP, downregulated raptor or rictor protein expression by ~90% in PC12 cells (Fig. 3B). Downregulation of raptor inhibited the basal and Cd-induced phosphorylation of S6K1 and 4E-BP1, whereas downregulation of rictor obviously inhibited the basal and Cd-induced phosphorylation of Akt (Ser 473) (Fig. 3B). Of note, pretreatment with rapamycin for 48 h not only potently inhibited Cd-induced phosphorylation of S6K1 and 4E-BP1, but also obviously inhibited Cd-induced phosphorylation of Akt in PC12 cells infected with lentiviral shRNA to raptor, rictor or GFP (Fig. 3B). Interestingly, silencing raptor or rictor enhanced the inhibitory effect of rapamycin on Cd-induced cleavage of caspase-3 compared to the control (GFP shRNA) (Fig. 3B). Consistently, silencing raptor or rictor alone did not obviously alter the cell viability, but conferred resistance to Cd-induced cell death in PC12 cells (Fig. 3C and D). Furthermore, rapamycin was able to strengthen the inhibitory effect of raptor shRNA or rictor shRNA on Cd-induced cell death (Fig. 3C and D).
It has been recently shown that mTORC2 acts in neurons at least partly via mTORC1 (Urbanska et al., 2012). Based on our above findings, we next queried whether rapamyicn’s prevention against Cd-induced neuronal cell death is indeed by inhibiting both mTORC1 and mTORC2. To this end, we extended our studies by double knockdown of raptor and rictor. The results indicate that co-infection with lentiviral shRNAs to raptor and rictor silenced raptor/rictor protein expression by ~90% in PC12 cells (Fig. 3B). Of interest, the double silencing of raptor/rictor showed a more potent inhibitory effect on Cd-induced phosphorylation of Akt, S6K1 and 4E-BP1, cleavage of caspase-3, as well as reduction of cell viability and increase of fragmented nuclei than the single silencing of raptor or rictor (Fig. 3B–D). Furthermore, the double silencing of raptor/rictor also more strongly enhanced the inhibitory effect of rapamycin on Cd-induced cleavage of caspase-3, reduction of cell viability and increase of fragmented nuclei, compared to the single silencing of raptor or rictor (Fig. 3B–D). Taken together, the results indicate that rapamycin inhibition of both mTORC1 and mTORC2 is crucial for prevention of Cd-induced death in neuronal cells.
3.4. mTORC1-mediated S6K1 and 4E-BP1 pathways are both involved in rapamycin’s prevention of Cd-induced neuronal cell death
S6K1 and 4E-BP1 are two best-characterized downstream effector molecules of mTORC1 (Cornu et al., 2013; Laplante and Sabatini, 2012). To further investigate the roles of S6K1 and 4E-BP1 in rapamycin’s prevention from Cd-induced neuronal cell death, genetic manipulation of the activity of S6K1 or 4E-BP1 was conducted. At first, lentiviral shRNA to S6K1 was used to silence expression of cellular S6K1 protein. As shown in Figure 4A, infection of SH-SY5Y cells with lentiviral shRNA to S6K1 downregulated expression of cellular S6K1 protein by ~ 90%, compared to control cells infected with lentiviral shRNA to GFP. In line with this, silencing S6K1 expression resulted in reduction of S6K1 kinase activity, as detected by Western blotting with antibodies to phospho-S6K1 (Thr389) and phospho-S6 ribosomal protein (Ser235/236), respectively (Fig. 4A). Treatment with Cd increased phosphorylation of S6K1 and S6 in the control cells infected with lentiviral shRNA to GFP (Lane 3 vs. Lane 1). Silencing S6K1 remarkably attenuated Cd-induced phosphorylation of S6K1 and S6 (Lane 7 vs. Lane 3). Rapamycin (0.2 μg/ml) was able to almost completely block Cd-induced S6K1/S6 phosphorylation in SH-SY5Y cells, regardless of infection with lentiviral shRNA to S6K1 or not (Lane 4 vs. Lane 3; Lane 8 vs. Lane 7) (Fig. 4A). However, silencing S6K1 enhanced the inhibitory effect of rapamycin on Cd-induced cleavages of caspase-3 and PARP (Lane 8 vs. Lane 4) (Fig. 4A). Additionally, we also tested Akt activity, showing that downregulation of S6K1 failed to attenuate phosphorylation of Akt (Ser473 and Thr308) in the cells in response to Cd and/or rapamycin (Fig. 4A). Similar manifestations were also observed in PC12 cells (data not shown). Consistently, by trypan blue exclusion and DAPI staining, we observed that silencing S6K1 alone partially prevented Cd-induced cell death in PC12 and/or SH-SY5Y cells (Fig. 4B and C). Addition of rapamycin more significantly rescued cells from death induced by Cd (Fig. 4B and C). The findings suggest that rapamycin prevents Cd-induced neuronal cell death in part by suppressing mTORC1-mediated S6K1 pathway.
Fig. 4.

Rapamycin prevents Cd-induced neuronal cell death by targeting mTORC1-mediated S6K1 pathway. PC12 and SH-SY5Y cells, infected with lentiviral shRNA to S6K1 or GFP (as control), were pretreated with rapamycin (Rap, 0.2 μg/ml) for 48 h, followed by exposure to Cd (20 μM) for 4 h (for Western blotting) or 24 h (for cell morphology, live cell assay and DAPI staining). A) Total cell lysates were subjected to Western blot analysis using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. B and C) Silencing S6K1 enhanced preventive effect of rapamycin on Cd-induced B) cell viability reduction and C) apoptosis in PC12 and SH-SY5Y cells. Results are presented as mean ± SEM (n = 5). a p < 0.05, difference with control group; b p < 0.05, S6K1 shRNA group versus GFP shRNA group.
Next, to evaluate whether rapamycin acts through 4E-BP1/eIF4E pathway, we employed recombinant adenovirus encoding HA-tagged 4E-BP1 mutants where Thr36, Thr45, Ser64, Thr69 and Ser82 are replaced by Ala residues (Ad-4EBP1-5A) mimicking hypophosphorylated residues, thus tightly binding to and sequester eIF4E in cells (Mothe-Satney et al., 2000). As shown in Fig. 5A, HA-tagged 4E-BP1 and higher levels of 4E-BP1 were detected in Ad-4EBP1-5A-infected PC12 cells. As expected, exposure to rapamycin increased the mobility of 4E-BP1 (indicating dephosphorylation) in the control cells infected with Ad-GFP, but not in the cells infected with Ad-4EBP1-5A. Interestingly, expression of 4E-BP1-5A partially prevented the cells from Cd-induced cell death, and especially addition of rapamycin exhibited more inhibitory effect on Cd-induced cell death, as judged by Western blotting for cleaved caspase-3 (Fig. 5A), trypan blue exclusion (Fig. 5B), and DAPI staining (Fig. 5C). The findings imply that rapamycin prevents Cd-induced neuronal cell death also in part by targeting mTORC1-mediated 4E-BP1/eIF4E pathway.
Fig. 5.

Rapamycin prevents Cd-induced neuronal cell death by targeting mTORC1-mediated 4E-BP1 pathway. PC12 cells, infected with Ad-4EBP1-5A or Ad-GFP (as control), were pretreated with rapamycin (Rap, 0.2 μg/ml) for 48 h, followed by exposure to Cd (20 μM) for 4 h (for Western blotting) or 24 h (for live cell assay and DAPI staining). A) Total cell lysates were subjected to Western blot analysis using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. B and C) Expression of 4EBP1-5A strengthened rapamycin resistance to Cd-induced B) cell viability reduction and C) apoptosis in PC12 cells. Results are presented as mean ± SEM (n = 5). a p < 0.05, difference with control group; b p < 0.05, Ad-4EBP1-5A group versus Ad-GFP group.
3.5. Ectopic expression of dominant negative Akt or pharmacological inhibition of Akt strengthens rapamycin’s protection against Cd-induced neuronal cell death
To determine whether mTORC2-mediated Akt pathway plays a role in rapamycin’s prevention against Cd-induced neuronal cell death, PC12 cells and primary neurons were pretreated with/without rapamycin (0.2 μg/ml) for 48 h, and then treated with/without Akt inhibitor X (20 μM) for 2 h, followed by exposure to Cd (20 μM) for 4 h or 24 h. As shown in Fig. 6A, Akt inhibitor X or rapamycin obviously suppressed the basal or Cd-induced phosphorylation of Akt (Ser473 and Thr308) and its substrate GSK3β (Ser9) in the cells. In addition, similar effects for the basal or Cd-induced phosphorylation of S6K1 (Thr389) and 4E-BP1 (Thr70) were also observed in the cells treated with Akt inhibitor X or rapamycin (Fig. 6A). Especially, co-treatment with rapamycin/Akt inhibitor X exhibited a stronger inhibitory effect on Cd-induced phosphorylation of Akt, S6K1, and 4E-BP1 and cleavage of caspase-3/PARP in the cells (Fig. 6A), implying that inhibition of Akt partially inhibits mTORC1-mediated S6K1 and 4E-BP1 pathways activated by Cd exposure. Consistently, co-treatment with rapamycin/Akt inhibitor X also rescued cells from Cd-induced apoptosis more potently than rapamcyin or Akt inhibitor X alone (Fig. 6B), suggesting that mTORC2-mediated Akt pathway is involved in rapamycin’s prevention against Cd-induced neuronal cell death as well.
Fig. 6.

Pharmacological inhibition of Akt strengthens rapamycin’s prevention from Cd-induced neuronal cell death. PC12 cells and primary neurons were pretreated with/without rapamycin (Rap, 0.2 μg/ml) for 48 h, and then treated with/without Akt inhibitor X (20 μM) for 2 h, followed by exposure to Cd (20 μM) for 4 h (for Western blotting) or 24 h (for DAPI staining). A) Co-treatment with rapamycin/Akt inhibitor X showed a stronger inhibitory effect on Cd activation of Akt, S6K1, 4E-BP1 and caspase-3/PARP. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. B) Co-treatment with rapamycin/Akt inhibitor X rescued cells from Cd-induced apoptosis more effectively than rapamcyin or Akt inhibitor X alone. Results are presented as mean ± SEM (n = 5). a p < 0.05, difference with control group; b p < 0.05, difference with 20 μM Cd group; c p < 0.05, difference with Cd/Akt inhibitor X group or Cd/rapamycin group.
To corroborate the above finding, PC12 cells, infected with recombinant adenovirus encoding HA-tagged dominant negative Akt (Ad-dn-Akt) or GFP (Ad-GFP) (as control), were pretreated with/without rapamycin (0.2 μg/ml) for 48 h, and then exposed to Cd (20 μM) for 4 h or 24 h. As expected, the infection with Ad-dn-Akt, but not Ad-GFP, resulted in expression of a high level of HA-tagged Akt mutant (Fig. 7A). The basal phosphorylation level of Akt was obviously inhibited by the infection with Ad-dn-Akt, compared to the control infection with Ad-GFP (Fig. 7A). Of note, ectopic expression of dn-Akt apparently attenuated Cd-induced phosphorylation of Akt and activation of caspase-3, and potentiated the inhibitory effect of rapamycin on the above events (Fig. 7A). Using live cell assay and DAPI staining, we found that overexpression of dn-Akt alone partially prevented Cd-induced viability reduction (Fig. 7B) and apoptosis (Fig. 7C and D) in PC12 cells. However, addition of rapamycin rendered more significant resistance to Cd-induced cell death in Ad-dn-Akt-infected group than in Ad-GFP-infected group (Fig. 7B-D). Taken together, our results indicate that rapamycin prevents Cd-induced neuronal cell death also in part by inhibiting mTORC2-mediated Akt pathway.
Fig. 7.

Ectopic expression of dominant negative Akt enhances rapamycin’s prevention from Cd-induced neuronal cell death. PC12 cells, infected with recombinant adenovirus expressing dominant negative (dn) Akt (Ad-dn-Akt) or GFP (Ad-GFP) (as control), were pretreated with rapamycin for 48 h, followed by exposure to Cd (20 μM) for 4 h (for Western blotting) or 24 h (for live cell assay and DAPI staining). A) Total cell lysates were subjected to Western blot analysis using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. B) Live cells were detected by counting viable cells using trypan blue exclusion. C) Apoptotic cells were evaluated by nuclear fragmentation and condensation (arrows) using DAPI staining. Scale bar: 20 μm. D) The percentages of cells with fragmented nuclei were quantified. B and D) Rapamycin exhibited more obvious prevention from Cd-induced B) cell viability reduction and D) apoptosis in Ad-dn-Akt-infected group than in Ad-GFP-infected group. Results are presented as mean ± SEM (n = 5). a p < 0.05, difference with control group; b p < 0.05, Ad-dn-Akt group versus Ad-GFP group.
4. Discussion
Cadmium is one of the most toxic environmental and industrial pollutants. Clinical and epidemiological data have shown that Cd exerts its toxic effects not only on the kidneys, liver and testis but also on the central nervous system (CNS) (Johri et al., 2010; Jomova and Valko, 2011; Mendez-Armenta and Rios, 2007; Okuda et al., 1997; Thompson and Bannigan, 2008). Cd can penetrate the blood-brain barrier and accumulate in the brain, which contributes to the brain damage, and is thought to play a crucial role in human neurodegenerative diseases (Goncalves et al., 2010; Johnson, 2001; Mendez-Armenta and Rios, 2007; Okuda et al., 1997; Panayi et al., 2002; Watjen and Beyersmann, 2004). Therefore, it is of great importance to find a novel therapeutic target and strategy to control the neurotoxicity of Cd to brain. Rapamycin, a macrocyclic lactone, influences a variety of essential cellular processes, such as cell growth and proliferation, protein synthesis and autophagy by inhibiting mTOR (Bove et al., 2011; Zhou et al., 2010). A series of studies have recently shown that rapamycin is able to extend lifespan in various species, including mice (Bove et al., 2011; Ehninger et al., 2014; Harrison et al., 2009; Selman et al., 2009), and also provide therapeutic benefit in several experimental models of neurodegenerative diseases, including PD, AD and HD (Bove et al., 2011; Erlich et al., 2007; Malagelada et al., 2010; Pan et al., 2008). Low concentrations of rapamycin can improve learning and memory (Bove et al., 2011; Ehninger et al., 2014). However, complete inhibition of mTOR by high concentrations of rapamycin has detrimental effects on long-term memory facilitation and consolidation, and causes amnesia (Bove et al., 2011; Tischmeyer et al., 2003). In addition, rapamycin treatment in mutant superoxide dismutase 1 (SOD1) mice exacerbates ALS pathology (Zhang et al., 2011). Therefore, rapamycin may act as a neuroprotective agent for fighting neurodegenerative diseases, but some attentions should be paid not only to the dosage of rapamycin but also to the mutations of some genes (e.g. SOD1).
Our recent studies have shown that Cd results in brain damage and neuronal cell apoptosis in part through activation of mTOR signaling pathway (Chen et al., 2008; Chen et al., 2011a; Chen et al., 2014; Chen et al., 2011b). By inhibiting activation of mTOR pathway, rapamycin in vitro and in vivo may effectively attenuate Cd-induced brain damage and neuronal cell death (Chen et al., 2008; Chen et al., 2014). In mammalian cells, mTOR functions as at least two complexes, mTORC1 and mTORC2 (Cornu et al., 2013; Laplante and Sabatini, 2012). However, little is known about whether rapamycin rescues cells from Cd-induced cell death by targeting mTORC1 and/or mTORC2. Here, for the first time, we provide evidence that rapamycin prevents Cd-induced apoptotic cell death in neuronal cells by inhibiting both mTORC1 and mTORC2 signaling pathways. Our data suggest that rapamycin has an ability to detoxicate Cd via inhibiting mTORC1/2 network in neuronal cells.
mTOR plays a central role in the regulation of growth/proliferation and survival (Cornu et al., 2013; Laplante and Sabatini, 2012). Two structurally and functionally distinct mTOR-containing multiprotein complexes (mTORC1 and mTORC2) have been documented (Laplante and Sabatini, 2012). The functions of mTORC1/2 are greatly affected by the complex integrity, especially their associations with raptor and rictor, respectively (Jacinto et al., 2004; Kim et al., 2002; Laplante and Sabatini, 2012; Sarbassov et al., 2004). mTORC1 and mTORC2 were originally identified on the basis of their differential sensitivity to the inhibitory effects of rapamycin: mTORC1 is rapamycin-sensitive, and mTORC2 is rapamycin-insensitive (Jacinto et al., 2004; Sarbassov et al., 2004). However, further studies have revealed that prolonged treatment or treatment with high concentrations of rapamycin can also inhibit mTORC2 activity in a cell-type-dependent manner (Sarbassov et al., 2006). In this study, we found that pretreatment with 0.2 μg/ml of rapamycin for 48 h was able to suppress the phosphorylation of S6K1, 4E-BP1 and Akt, suggesting inhibition of both mTORC1 and mTORC2. Using a rapamycin-resistant but kinase-active mTOR mutant (S2035T, mTOR-T) or kinase-dead mTOR-T mutant (S2035T/D2357E, mTOR-TE), we found that expression of mTOR-T, but not mTOR-TE, conferred resistance to rapamycin’s inhibition of Cd-induced phosphorylation of S6K1 and 4E-BP1, as well as cell death, implying the preventive effect of rapamycin on Cd-induced neurotoxicity is mTOR kinase activity-dependent. Furthermore, we observed that single or double disruption of mTORC1 and mTORC2 by using lentivial shRNAs to raptor and rictor attenuated Cd-induced cell death. Of importance, pretreatment of rapamycin for 48 h more significantly enhanced this inhibitory effect in the cells infected with lentiviral shRNA to raptor, rictor or raptor/rictor, and especially there existed a more potent rescue for the double raptor/rictor-silenced cells than for the single raptor or rictor-silenced cells. Our data imply that rapamycin prevents Cd-induced cell death, not only by inhibiting both mTORC1 and mTORC2 but also possibly via targeting unidentified mTOR complex(es) or other signaling molecules in neuronal cells.
S6K1 and 4E-BP1 are two best-characterized downstream targets of mTORC1 (Cornu et al., 2013; Laplante and Sabatini, 2012). To gain more insights into the mechanism by which rapamycin prevents Cd-induced cell death via targeting mTORC1, S6K1 and 4E-BP1/eIF4E pathways were disrupted by infecting cells with lentiviral shRNA to S6K1 and Ad-4E-BP1-5A, respectively. We found that downregulation of S6K1 or ectopic expression of 4E-BP1-5A conferred resistance to Cd-induced cell death, and addition of rapamycin strengthened the anti-apoptotic effect of S6K1 shRNA or Ad-4EBP1-5A in the cells. The findings underscore the importance of both S6K1 and 4E-BP1/eIF4E pathways in Cd-induced death in neuronal cells. Both S6K1 and 4E-BP1/eIF4E pathways are crucial for protein and lipid synthesis (Cornu et al., 2013; Laplante and Sabatini, 2012). Recently, we have found that Cd can induce oxidative stress in neuronal cells (Chen et al., 2011a). Possibly, Cd-induced persistent phosphorylation of S6K1 and 4E-BP1 results in prolonged protein/lipid synthesis in the cells, which consumes too much energy and generates more waste, thereby accelerating cell death under such an oxidative stress condition. In contrast, inhibition of mTORC1 by rapamycin may suppress Cd-activated protein/lipid synthesis, which spares a lot of energy and produces less waste, thus attenuating Cd-induced cell death. In addition to targeting mTORC1, rapamycin was found to inhibit Cd-activated mTORC2 and MAPKs in this study (see discussion below). This may explain why rapamycin was able to potentiate the preventive effect of S6K1 shRNA or Ad-4EBP1-5A on Cd-induced cell death.
In this study, we have observed that mTORC2, particularly mTORC2-mediated Akt pathway, also plays a critical role in the neuroprotection of rapamycin. This is supported by the findings that rapamycin inhibited Cd-induced phosphorylation of Akt; disruption of mTORC2 by silencing rictor rescued cells from Cd-induced cell death; pharmacological inhibition of Akt activity with Akt inhibitor X or ectopic expression of dominant negative Akt significantly attenuated Cd-induced cell death, and strengthened the inhibitory effect of rapamycin on Cd-induced cell death in neuronal cells. In addition to Akt, mTORC2 also regulates serum- and glucocorticoid-induced protein kinase 1 (SGK1), protein kinase C-α, small GTPases, and focal adhesion proteins (Cornu et al., 2013; Laplante and Sabatini, 2012). Currently, it is unknown whether any of other mTORC2-mediated signaling molecules is involved in Cd-induced cell death. More studies are required to address this issue.
Autophagy has been documented to exert neuroprotection by enhancing clearance of harmful protein aggregates (Ghavami et al., 2014; Ravikumar et al., 2006; Sarkar et al., 2009). mTOR negatively regulates autophagy (Cornu et al., 2013). Some data have pointed to rapamycin protection in the models of PD and HD because of its stimulation of autophagy (Ghavami et al., 2014; Sarkar et al., 2009). Likely, rapamycin may prevent Cd-induced apoptosis in part by inducing autophagy.
In conclusion, we have identified that both mTORC1-mediated S6K1/4E-BP1 and mTORC2-mediated Akt pathways contribute to Cd-induced cell death. Rapamycin prevents Cd-induced apoptotic cell death not only via inhibiting mTORC1, but also by suppressing mTORC2. Our findings highlight that rapamycin may be exploited for the prevention of Cd-induced neurodegenerative disorders.
Highlights.
Rapamycin inhibits Cd-induced neuronal death by mTORC1/mTORC2 signaling pathways.
Rapamycin blocks Cd-induced neuronal death by mTORC1-mediated S6K1/4E-BP1 pathways.
Rapamycin prevents Cd-induced neuronal death via mTORC2-mediated Akt pathway.
Rapamycin may be exploited for prevention of Cd-induced neurodegenerative diseases.
Acknowledgments
This work was supported in part by the grants from NIH (CA115414; S.H.), National Natural Science Fundation of China (No. 30971486, 81271416; L.C.), American Cancer Society (RSG-08-135-01-CNE; S.H.), Louisiana Board of Regents (NSF-2009-PFUND-144; S.H.), the Scientific Research Foundation of the State Education Ministry of China (SEMR20091341, L.C.), the Project for the Priority Academic Program Development and the Natural Science Foundation of Jiangsu Higher Education Institutions of China (10KJA180027; L. C.), and Innovative Research Program of Jiangsu College Graduate of China (KYLX_0713; C.X.).
Abbreviations
- 4E-BP1
eukaryotic initiation factor 4E binding protein 1
- AD
Alzheimer disease
- Akt
protein kinase B (PKB)
- Cd
cadmium
- DAPI
4′, 6-diamidino-2-phenylindole
- DMEM
Dulbecco’s Modified Eagle’s Medium
- FBS
fetal bovine serum
- HD
Huntington’s disease
- mTOR
mammalian target of rapamycin
- mTORC1/2
mTOR complex 1/2
- PBS
phosphate buffered saline
- PD
Parkinson disease
- PDL
poly-D-lysine
- Raptor
regulatory-associated protein of mTOR
- Rictor
rapamycin insensitive companion of mTOR
- S6K1
S6 kinase 1
- TUNEL
the terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP) nick-end labeling
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akesson A, Bjellerup P, Lundh T, Lidfeldt J, Nerbrand C, Samsioe G, Skerfving S, Vahter M. Cadmium-induced effects on bone in a population-based study of women. Environ Health Perspect. 2006;114(6):830–834. doi: 10.1289/ehp.8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011;12(8):437–452. doi: 10.1038/nrn3068. [DOI] [PubMed] [Google Scholar]
- Chen L, Liu L, Luo Y, Huang S. MAPK and mTOR pathways are involved in cadmium-induced neuronal apoptosis. J Neurochem. 2008;105(1):251–261. doi: 10.1111/j.1471-4159.2007.05133.x. [DOI] [PubMed] [Google Scholar]
- Chen L, Xu B, Liu L, Luo Y, Yin J, Zhou H, Chen W, Shen T, Han X, Huang S. Hydrogen peroxide inhibits mTOR signaling by activation of AMPKalpha leading to apoptosis of neuronal cells. Lab Invest. 2010;90(5):762–773. doi: 10.1038/labinvest.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Xu B, Liu L, Luo Y, Zhou H, Chen W, Shen T, Han X, Kontos CD, Huang S. Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death. Free Radic Biol Med. 2011a;50(5):624–632. doi: 10.1016/j.freeradbiomed.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Ren Q, Zhang J, Ye Y, Zhang Z, Xu Y, Guo M, Ji H, Xu C, Gu C, Gao W, Huang S, Chen L. N-acetyl-L-cysteine protects against cadmium-induced neuronal apoptosis by inhibiting ROS-dependent activation of Akt/mTOR pathway in mouse brain. Neuropathol Appl Neurobiol. 2014;40(6):759–777. doi: 10.1111/nan.12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Xu Y, Xu B, Guo M, Zhang Z, Liu L, Ma H, Chen Z, Luo Y, Huang S, Chen L. CaMKII is involved in cadmium activation of MAPK and mTOR pathways leading to neuronal cell death. J Neurochem. 2011b;119(5):1108–1118. doi: 10.1111/j.1471-4159.2011.07493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornu M, Albert V, Hall MN. mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev. 2013;23(1):53–62. doi: 10.1016/j.gde.2012.12.005. [DOI] [PubMed] [Google Scholar]
- Ehninger D, Neff F, Xie K. Longevity, aging and rapamycin. Cell Mol Life Sci. 2014;71:4325–4346. doi: 10.1007/s00018-014-1677-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbay E, Chen J. The Mammalian Target of Rapamycin Regulates C2C12 Myogenesis via a Kinase-independent Mechanism. J Biol Chem. 2001;276(39):36079–36082. doi: 10.1074/jbc.C100406200. [DOI] [PubMed] [Google Scholar]
- Erlich S, Alexandrovich A, Shohami E, Pinkas-Kramarski R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis. 2007;26(1):86–93. doi: 10.1016/j.nbd.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, Christoffersson J, Chaabane W, Moghadam AR, Kashani HH, Hashemi M, Owji AA, Los MJ. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24–49. doi: 10.1016/j.pneurobio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Goncalves JF, Fiorenza AM, Spanevello RM, Mazzanti CM, Bochi GV, Antes FG, Stefanello N, Rubin MA, Dressler VL, Morsch VM, Schetinger MR. N-acetylcysteine prevents memory deficits, the decrease in acetylcholinesterase activity and oxidative stress in rats exposed to cadmium. Chem Biol Interact. 2010;186(1):53–60. doi: 10.1016/j.cbi.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Hao B, Cheng S, Clancy CJ, Nguyen MH. Caspofungin kills Candida albicans by causing both cellular apoptosis and necrosis. Antimicrob Agents Chemother. 2013;57(1):326–332. doi: 10.1128/AAC.01366-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110(2):177–189. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460(7253):392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Shu L, Dilling MB, Easton J, Harwood FC, Ichijo H, Houghton PJ. Sustained activation of the JNK cascade and rapamycin-induced apoptosis are suppressed by p53/p21(Cip1) Mol Cell. 2003;11(6):1491–1501. doi: 10.1016/s1097-2765(03)00180-1. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6(11):1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- Jarup L, Persson B, Edling C, Elinder CG. Renal function impairment in workers previously exposed to cadmium. Nephron. 1993;64(1):75–81. doi: 10.1159/000187282. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Sheng M. The growing role of mTOR in neuronal development and plasticity. Mol Neurobiol. 2006;34(3):205–219. doi: 10.1385/MN:34:3:205. [DOI] [PubMed] [Google Scholar]
- Johnson S. Gradual micronutrient accumulation and depletion in Alzheimer’s disease. Med Hypotheses. 2001;56(6):595–597. doi: 10.1054/mehy.2000.1301. [DOI] [PubMed] [Google Scholar]
- Johri N, Jacquillet G, Unwin R. Heavy metal poisoning: the effects of cadmium on the kidney. Biometals. 2010;23(5):783–792. doi: 10.1007/s10534-010-9328-y. [DOI] [PubMed] [Google Scholar]
- Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283(2–3):65–87. doi: 10.1016/j.tox.2011.03.001. [DOI] [PubMed] [Google Scholar]
- Kim D, Sarbassov D, Ali S, King J, Latek R, Erdjument-Bromage H, Tempst P, Sabatini D. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110(2):163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Kocak M, Akcil E. The effects of chronic cadmium toxicity on the hemostatic system. Pathophysiol Haemost Thromb. 2006;35(6):411–416. doi: 10.1159/000102047. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Chen L, Chung J, Huang S. Rapamycin inhibits F-actin reorganization and phosphorylation of focal adhesion proteins. Oncogene. 2008;27(37):4998–5010. doi: 10.1038/onc.2008.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Li F, Cardelli JA, Martin KA, Blenis J, Huang S. Rapamycin inhibits cell motility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathways. Oncogene. 2006;25(53):7029–7040. doi: 10.1038/sj.onc.1209691. [DOI] [PubMed] [Google Scholar]
- Liu L, Luo Y, Chen L, Shen T, Xu B, Chen W, Zhou H, Han X, Huang S. Rapamycin inhibits cytoskeleton reorganization and cell motility by suppressing RhoA expression and activity. J Biol Chem. 2010;285(49):38362–38373. doi: 10.1074/jbc.M110.141168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez E, Figueroa S, Oset-Gasque MJ, Gonzalez MP. Apoptosis and necrosis: two distinct events induced by cadmium in cortical neurons in culture. Br J Pharmacol. 2003;138(5):901–911. doi: 10.1038/sj.bjp.0705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J Neurosci. 2010;30(3):1166–1175. doi: 10.1523/JNEUROSCI.3944-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Armenta M, Rios C. Cadmium neurotoxicity. Environ Toxicol Pharmacol. 2007;23(3):350–358. doi: 10.1016/j.etap.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Mothe-Satney I, Yang D, Fadden P, Haystead TAJ, Lawrence JC., Jr Multiple Mechanisms Control Phosphorylation of PHAS-I in Five (S/T)P Sites That Govern Translational Repression. Mol Cell Biol. 2000;20(10):3558–3567. doi: 10.1128/mcb.20.10.3558-3567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda B, Iwamoto Y, Tachibana H, Sugita M. Parkinsonism after acute cadmium poisoning. Clin Neurol Neurosurg. 1997;99(4):263–265. doi: 10.1016/s0303-8467(97)00090-5. [DOI] [PubMed] [Google Scholar]
- Pan T, Kondo S, Zhu W, Xie W, Jankovic J, Le W. Neuroprotection of rapamycin in lactacystin-induced neurodegeneration via autophagy enhancement. Neurobiol Dis. 2008;32(1):16–25. doi: 10.1016/j.nbd.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Pan T, Rawal P, Wu Y, Xie W, Jankovic J, Le W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience. 2009;164(2):541–551. doi: 10.1016/j.neuroscience.2009.08.014. [DOI] [PubMed] [Google Scholar]
- Panayi AE, Spyrou NM, Iversen BS, White MA, Part P. Determination of cadmium and zinc in Alzheimer’s brain tissue using inductively coupled plasma mass spectrometry. J Neurol Sci. 2002;195(1):1–10. doi: 10.1016/s0022-510x(01)00672-4. [DOI] [PubMed] [Google Scholar]
- Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137(5):873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihl RO, Parkes M. Hair element content in learning disabled children. Science. 1977;198(4313):204–206. doi: 10.1126/science.905825. [DOI] [PubMed] [Google Scholar]
- Polak P, Hall MN. mTOR and the control of whole body metabolism. Curr Opin Cell Biol. 2009;21(2):209–218. doi: 10.1016/j.ceb.2009.01.024. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Berger Z, Vacher C, O’Kane CJ, Rubinsztein DC. Rapamycin pre-treatment protects against apoptosis. Hum Mol Genet. 2006;15(7):1209–1216. doi: 10.1093/hmg/ddl036. [DOI] [PubMed] [Google Scholar]
- Sarbassov D, Ali S, Kim D, Guertin D, Latek R, Erdjument-Bromage H, Tempst P, Sabatini D. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14(14):1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22(2):159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009;16(1):46–56. doi: 10.1038/cdd.2008.110. [DOI] [PubMed] [Google Scholar]
- Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, Gems D, Withers DJ. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326(5949):140–144. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiech L, Perycz M, Malik A, Jaworski J. Role of mTOR in physiology and pathology of the nervous system. Biochim Biophys Acta. 2008;1784(1):116–132. doi: 10.1016/j.bbapap.2007.08.015. [DOI] [PubMed] [Google Scholar]
- Thompson J, Bannigan J. Cadmium: toxic effects on the reproductive system and the embryo. Reprod Toxicol. 2008;25(3):304–315. doi: 10.1016/j.reprotox.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Tischmeyer W, Schicknick H, Kraus M, Seidenbecher CI, Staak S, Scheich H, Gundelfinger ED. Rapamycin-sensitive signalling in long-term consolidation of auditory cortex-dependent memory. Eur J Neurosci. 2003;18(4):942–950. doi: 10.1046/j.1460-9568.2003.02820.x. [DOI] [PubMed] [Google Scholar]
- Urbanska M, Gozdz A, Swiech LJ, Jaworski J. Mammalian target of rapamycin complex 1 (mTORC1) and 2 (mTORC2) control the dendritic arbor morphology of hippocampal neurons. J Biol Chem. 2012;287(36):30240–30256. doi: 10.1074/jbc.M112.374405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Du Y. Cadmium and its neurotoxic effects. Oxid Med Cell Longev. 2013;2013:898034. doi: 10.1155/2013/898034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watjen W, Beyersmann D. Cadmium-induced apoptosis in C6 glioma cells: influence of oxidative stress. Biometals. 2004;17(1):65–78. doi: 10.1023/a:1024405119018. [DOI] [PubMed] [Google Scholar]
- Wright RO, Amarasiriwardena C, Woolf AD, Jim R, Bellinger DC. Neuropsychological correlates of hair arsenic, manganese, and cadmium levels in school-age children residing near a hazardous waste site. Neurotoxicology. 2006;27(2):210–216. doi: 10.1016/j.neuro.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Zhang X, Li L, Chen S, Yang D, Wang Y, Zhang X, Wang Z, Le W. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy. 2011;7(4):412–425. doi: 10.4161/auto.7.4.14541. [DOI] [PubMed] [Google Scholar]
- Zhou H, Luo Y, Huang S. Updates of mTOR inhibitors. Anticancer Agents Med Chem. 2010;10(7):571–581. doi: 10.2174/187152010793498663. [DOI] [PMC free article] [PubMed] [Google Scholar]