

Abstract
Establishment of the corpus callosum involves coordination between callosal projection neurons and multiple midline structures, including the glial wedge (GW) rostrally and hippocampal commissure caudally. GW defects have been associated with agenesis of the corpus callosum (ACC). Here we show that conditional Lhx2 inactivation in cortical radial glia using Emx1-Cre or Nestin-Cre drivers results in ACC. The ACC phenotype was characterized by aberrant ventrally projecting callosal axons rather than Probst bundles, and was 100% penetrant on 2 different mouse strain backgrounds. Lhx2 inactivation in postmitotic cortical neurons using Nex-Cre mice did not result in ACC, suggesting that the mutant phenotype was not autonomous to the callosal projection neurons. Instead, ACC was associated with an absent hippocampal commissure and a markedly reduced to absent GW. Expression studies demonstrated strong Lhx2 expression in the normal GW and in its radial glial progenitors, with absence of Lhx2 resulting in normal Emx1 and Sox2 expression, but premature exit from the cell cycle based on EdU-Ki67 double labeling. These studies define essential roles for Lhx2 in GW, hippocampal commissure, and corpus callosum formation, and suggest that defects in radial GW progenitors can give rise to ACC.
Keywords: agenesis of the corpus callosum, Cre-lox, glial wedge, Lhx2, mouse
Introduction
The corpus callosum is one of the largest white matter tracts in the human brain, serving to physically and functionally connect the hemispheres of the cerebral cortex. Consisting roughly of 190 million axons (Tomasch 1954), the corpus callosum plays critical roles in normal cognitive function. In humans, the corpus callosum does not form in 1:4000 individuals (Guillem et al. 2003; Wang et al. 2004), and 3–5% of all patients evaluated for neurodevelopmental disorders have partial or complete agenesis of the corpus callosum (ACC) (Jeret et al. 1985; Bodensteiner et al. 1994). Successful genesis of the corpus callosum depends on the integrity of, and cooperation between, callosal projection neurons and midline cell populations that guide callosal axons. These axons extend toward and cross the midline in response to various molecular cues produced by midline cells, including semaphorins (Niquille et al. 2009), fibroblast growth factors (Fgfs) (Huffman et al. 2004; Smith et al. 2006; Tole et al. 2006), netrins (Serafini et al. 1996; Molyneaux et al. 2009), and slits (Bagri et al. 2002; Shu, Sundaresan et al. 2003; Andrews et al. 2006).
One of the first midline structures encountered by callosal axons is the glial wedge (GW). Located between the rostromedial cortex and septum bilaterally the GW strongly expresses the astroglial marker Glial Fibrillary Acidic Protein (GFAP). Midline glial structures were first described by Bignami and Dahl (1971) following the discovery of GFAP. The radial glial scaffold of the developing telencephalon was then extensively detailed by Levitt and Rakic (1980) as part of their investigation into GFAP expression throughout the nervous system. The glia at the midline surface of the cerebral hemispheres were later named midline zipper glia by Silver, and was hypothesized to function as a barrier (Silver et al. 1982, 1993; Hankin and Silver 1988). The glial structure in the medial wall of the lateral ventricle was later renamed by Richards as the GW (Shu and Richards 2001), who showed that the GW repels ipsilateral callosal axons towards the midline (Shu and Richards 2001). Once the axons have crossed the midline, the GW functions to guide the axons towards contralateral cortex (Keeble et al. 2006; Shu, Sundaresan et al. 2003). The GW is composed of specialized astrocytes that are locally generated from cortical radial glia between embryonic day 13 (E13) to E17 (Shu, Puche et al. 2003), and represents the first site where GFAP can be immunohistochemically detected during cortical development (by ∼E14 in mice) (Shu and Richards 2001).
The hippocampal commissure is a midline structure that is thought to facilitate caudal callosal development independent of the GW. The hippocampal commissure lies directly beneath (i.e., ventral to) the caudal corpus callosum, but does not underlie its rostral portion. Because hippocampal commissural axons cross the midline before callosal axons (Wahlsten 1981), it has been hypothesized that the hippocampal commissure serves as a scaffold for the caudal corpus callosum (Ozaki and Wahlsten 1992; Livy and Wahlsten 1997; Paul et al. 2007; Donahoo and Richards 2009). The concept of independent roles for the GW and hippocampal commissure in rostral and caudal callosal development, respectively (Donahoo and Richards 2009), is supported by mouse mutants with GW deficits, but intact hippocampal commissures, which are associated with ACC rostrally, but not caudally (Shu, Butz et al. 2003; Piper, Moldrich et al. 2009).
One molecule that is well positioned to regulate GW and callosal development is the LIM homeodomain transcription factor Lhx2. In neuroepithelial cells, Lhx2 acts as a selector gene that cell-autonomously specifies cortical identity over alternative cortical hem and antihem fates (Mangale et al. 2008). Using conditional Lhx2 null mice and different Cre recombinase strategies, we defined an E8.5–10.5 critical period for this selector gene activity. Importantly, Lhx2 inactivation driven by Emx1-Cre, which begins around E10.5, did not result in the selector gene phenotype (Mangale et al. 2008). This has allowed us to study the consequences of Lhx2 loss from specified cortical radial glia, which like neuroepithelial cells, express Lhx2 at high levels (Bulchand et al. 2001; Monuki et al. 2001; Suter et al. 2007). Lhx2 is also expressed in upper layer cortical neurons (Bulchand et al. 2003; Abellan et al. 2010), which raises the possibility of essential Lhx2 functions in the callosal projection neurons themselves.
Here we explored the role of Lhx2 in corpus callosum development by inactivating Lhx2 in cortical radial glial lineages or in cortical projection neurons. Loss of Lhx2 in cortical radial glia using either Emx1-Cre (Jin et al. 2000) or Nestin-Cre drivers (Tronche et al. 1999) resulted in complete and fully penetrant ACC. In contrast, Lhx2 inactivation in postmitotic cortical neurons with Nex-Cre (Goebbels et al. 2006) did not result in an acallosal phenotype, with our studies instead implicating the hippocampal commissure and GW—and radial GW progenitors in particular—in the ACC phenotype.
Materials and Methods
Mice
All animal work was done in accordance with UCI institutional guidelines and IACUC protocol #2001-2304. Homozygous floxed Lhx2 conditional knockout (cKO) females, Lhx2tm1(cre)Monu(Mangale et al. 2008) were mated with males carrying the fixed “standard” Lhx2 null allele, Lhx2tm1Dra (“Lhx2 sKO”) (Porter et al. 1997), and a Cre recombinase allele (Emx1-Cre (Tg(Emx1-cre)5Yql), Nestin-Cre (TgN(NesCre)1Kln), or Nex-Cre (Neurod6tm1(cre)Kan)). The Lhx2 cKO mice were derived from TG2a ES cells (129 strain) and backcrossed with C57BL/6N or CD1 mice (Charles River) for at least 10 generations, or maintained on a mixed C57BL/6N;CD1 (50:50) background via sibling matings for over 10 generations. Unless otherwise specified, Lhx2 cKO females on this mixed background were used. The Emx1-Cre line (courtesy of Yuqing Li) (Jin et al. 2000) consists of a lacZ-IRES-Cre cassette inserted into the first exon of the Emx1 gene, and was maintained on a C57BL/6N background. The Emx1Cre/+;Lhx2sKO/+ X Lhx2cKO/cKO crosses resulted in control and mutant animals that were obligate heterozygotes for the Lhx2 cKO allele; Cre-positive mutants and controls therefore differed genetically by the presence or absence of the Lhx2sKO allele alone. Heterozygosity for the Emx1-Cre allele also avoided the potentially confounding consequences of Emx1 loss of function (Guo et al. 2000). Nex-Cre (courtesy of Klaus Nave) (Goebbels et al. 2006), which has Cre recombinase in place of Nex exon 2, was also maintained on C57BL/6N background. Nestin-Cre, JAX #003771, courtesy of Grant MacGregor) (Tronche et al. 1999) is a random integration Cre line driven by the rat Nestin promoter and enhancer. Because the original Nestin-Cre line (on C57BL/6JMgr background) carried a potentially deleterious Nnt null mutation (Toye et al. 2005; Huang et al. 2006; Mekada et al. 2009), Nestin-Cre animals were outcrossed, then backcrossed with C57BL/6JEi mice (Nnt homozygous wild-type). Nestin-Cre hemizygous, Nnt homozygous wild-type offspring were selected, backcrossed to C57BL/6N mice (Nnt homozygous wild-type), and maintained on this mixed C57BL/6N;C57BL/6JMgrEi background. ROSA26 reporter (R26R) mice (Gt (ROSA)26Sortm1sor, JAX #003474) (Soriano 1999) have a floxed stop sequence upstream of lacZ in the ROSA26 locus, and were maintained on a CD1 background.
Genotypes were determined by PCR as described (Mangale et al. 2008). Mouse pregnancies were timed by vaginal plug (noon of the plug date was designated E0.5). Embryonic dissections and tissue processing were conducted as previously described (Monuki et al. 2001; Currle et al. 2005; Mangale et al. 2008). Saponin-containing fixative (Hu et al. 2008; Mangale et al. 2008) was used for tissues prepared for immunohistochemistry (IHC).
Postnatal mice were perfusion-fixed after inducing anesthesia with Euthasol (Virbac). Once anesthetized, the anterior ribs were removed to reveal the heart. The right atrium was punctured, and a 21G butterfly needle was inserted into the left ventricle. Phosphate buffered saline was injected into the left ventricle until the venous blood was significantly diluted, then fixative was injected for several seconds. After perfusion, the brains were dissected and drop-fixed.
Tract Tracing
DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate) crystals were manually placed in the neocortex of dissected brains that were perfused, then drop-fixed in 4% paraformaldehyde overnight (Godement et al. 1987). Brains were incubated in fixative at 37 °C for 3–5 weeks, and then sectioned on a vibratome at 100 μm thickness. Sections were immediately counterstained with Hoechst 33342, mounted on slides, and examined under epifluorescence on an upright Nikon E600 microscope.
In utero electroporations with pCAG-GFP plasmids (Mutch et al. 2009) were carried out at E14.5 following standard protocols (Saito and Nakatsuji 2001). Pregnant mice were anesthetized with 90 mg/kg pentobarbital intraperitoneally; 0.05 mg/kg buprenorphine was used for analgesia. One microliter of DNA (5 μg/μL) diluted in 0.01% fast green dye solution was injected into a single fetal lateral ventricle. The following electroporation settings were used (BTX ECM 830, Harvard Apparatus): LV (Low Voltage mode) 30 V, 5 × 50 ms pulses, 950 ms intervals.
Histology, Histochemistry, In Situ Hybridization, and RT-qPCR
H&E, Xgal staining, colorimetric digoxigenin-based in situ hybridization, and RT-qPCR were carried out following standard protocols (Monuki et al. 2001; Currle et al. 2005; Hu et al. 2008; Mangale et al. 2008). The following in situ hybridization probe templates were used: Bmp6 cDNA (Furuta et al. 1997), Draxin (IMAGE clone 6853328), and mouse Lhx2 cDNA (Robertson et al. 1994). Some X-gal sections were counterstained with eosin to provide additional contrast.
Immunofluorescent and EdU Labeling
Antibody labelings were performed using standard procedures (Monuki et al. 2001; Currle et al. 2005; Mangale et al. 2008). For Lhx2/phalloidin double labeling, phalloidin-TRITC (Sigma; 1:1000) was incubated concurrently with secondary antibody. For EdU visualization, the Click-IT reaction (Invitrogen) was carried out according to manufacturer's protocol after secondary antibody incubation. Tbr1 and Ki67 staining included an antigen retrieval step with sodium citrate buffer. Antibodies used were rabbit polyclonal anti-Lhx2 (1:100) (Mangale et al. 2008), mouse monoclonal anti-GFAP (1:500, Sigma G3893), rabbit polyclonal anti-Tbr1 (1:1000, Chemicon AB96818), goat polyclonal anti-Calretinin (1:1000, Millipore AB1550), mouse monoclonal anti-TuJ1 (1:500, Sigma T8660), rabbit polyclonal anti-cleaved Caspase 3 (1:1000, Cell Signaling Technology 9661S), mouse monoclonal anti-Satb2 (1:100, Abcam AB51502), rabbit polyclonal anti-Ctip2 (1:1000, Abcam AB28448), rabbit polyclonal anti-Ki67 (1:1000, Abcam AB15580), and goat polyclonal anti-Sox2 (1:500; Santa Cruz sc-17320). Photomicrographs were taken on a Nikon E600 upright microscope with Spot RT3 camera. Monochrome images were later pseudocolored in Adobe Photoshop CS. For all fluorescent image comparisons shown, identical acquisition parameters and parallel brightness/contrast adjustments were used. Postacquisition adjustments were restricted to brightness and contrast adjustments only. For brightfield images, color balance and contrast were adjusted.
For EdU-Ki67 studies, confocal images and Z-stacks were acquired using a Zeiss LSM700 confocal microscope and Zen LE software. Image analyses were performed in Imaris 7.4.2. Quantifications of EdU and Ki67 were performed by manually counting immunopositive nuclei in 320 × 320 μm stacks composed of 10 confocal images totaling 5.16 μm in thickness. Each EdU-positive cell was individually analyzed for Ki67 positivity within an individual confocal slice and cross-checked for validity in 3D renderings (Imaris). Ki67 positivity was conservatively assessed using Ki67-negative areas outside of the VZ/SVZ to threshold for specific nuclear signal. Three sections from 2 biological replicates each of E14.5 control and mutant embryos were used for the EdU-Ki67 analyses.
Callosal Thickness Quantification
Rostral and caudal H&E sections of adult Nex/Lhx2 corpus callosum were photographed under low power microscopy and subsequently coded. The region of greatest vertical thickness was measured by a blinded individual after calibration using ImageJ software. The measurements were averaged after decoding, and a Student's t-test was applied to the sample set.
Results
Emx1/Lhx2 and Nestin/Lhx2 Mice Exhibit ACC
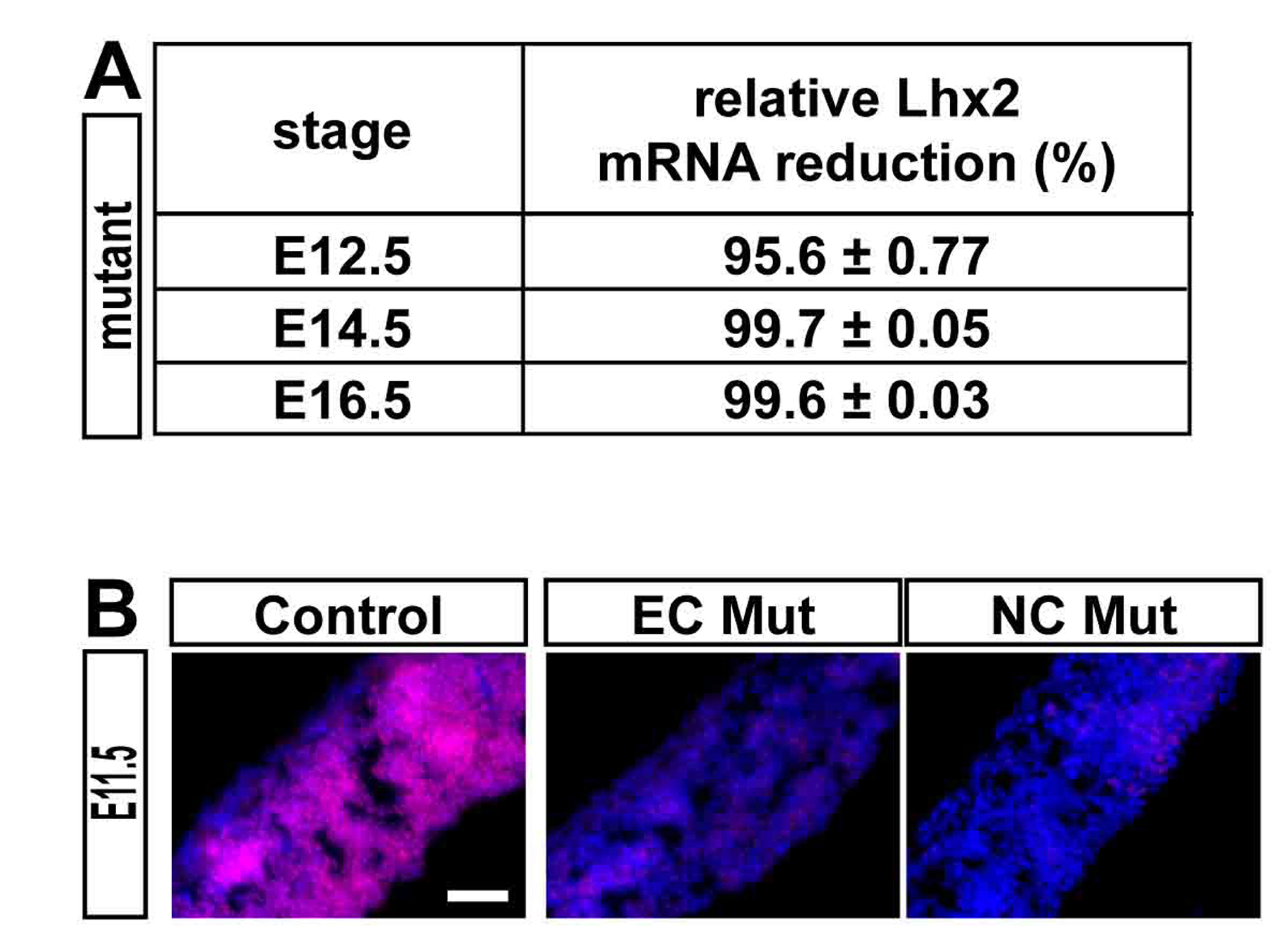
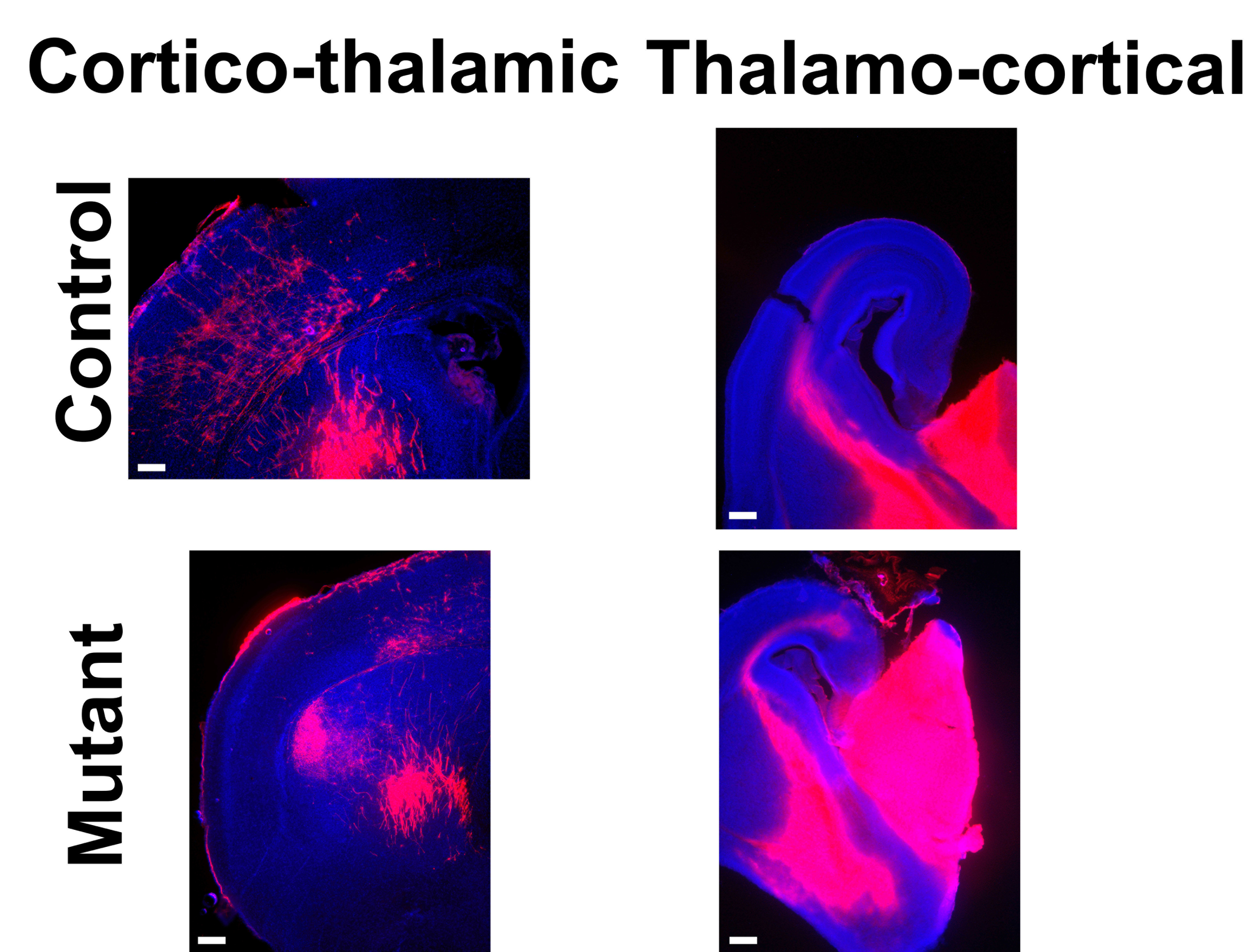
We used Emx1-Cre (Jin et al. 2000) and Nestin-Cre (Tronche et al. 1999) lines to drive Lhx2 inactivation in radial glia near the onset of cortical neurogenesis, and examined at least 2 litters at E18, P7, and adult stages (P21, P56, and P84) for both conditional null models. (These are Emx1Cre+/−;Lhx2cKO/sKO and NestinCre+/1;Lhx2cKO/sKO which we report as “Emx1/Lhx2” and “Nestin/Lhx2”, respectively.) At E18 and later stages (when the corpus callosum is normally detectable), 100% of Emx1/Lhx2 and Nestin/Lhx2 null mice had complete ACC (nlitters = 14; nmutants = 36), whereas all control littermates examined had an intact corpus callosum (ncontrol = 51) (Fig. 1A,B). In addition to ACC, all Lhx2 null mice lacked the hippocampal commissure (Fig. 1C). In contrast, both Emx1/Lhx2 and Nestin/Lhx2 mutant mice had intact anterior commissures (Fig. 1D). Corticothalamic and thalamocortical projections also appeared normal in these animals, based on DiI tract tracing (see Supplementary Fig. 2). It is worth noting that none of the Emx1/Lhx2 mutants were homozygous for the knock-in Emx1-Cre allele (i.e., all were obligate Cre heterozygotes), thus avoiding potential confounds related to Emx1 loss-of-function (Guo et al. 2000). In addition, both mutants and controls were obligate heterozygotes for the Lhx2 cKO allele; mutants and controls therefore differed solely by the presence or absence of the Lhx2 sKO allele. Mice heterozygous for Lhx2 (due to either the sKO or recombined cKO alleles) had normal callosal phenotypes that were indistinguishable from wild-type. These findings indicate that Lhx2 mutant animals have selective agenesis of the corpus callosum and hippocampal commissure.
Figure 1.

Absence of corpus callosum and hippocampal commissure, but preserved anterior commissure, in Emx1/Lhx2 and Nestin/Lhx2 mutant adults. H&E histology, coronal sections; dashed boxes in A–A″ are magnified in B–B″. (A,B) Normal corpus callosum in an Emx1-Cre-positive control littermate. (A′–B′,A″–B″) Emx1/Lhx2 and Nestin/Lhx2 mutants lack a corpus callosum, and misrouted ventrally oriented axons are present in the septum (black arrowheads). Probst bundles are not seen. (C–C″) In contrast to an Emx1-Cre-positive control, Emx1/Lhx2 and Nestin/Lhx2 mutants lack a hippocampal commissure. (D–D″) Anterior commissure is intact in Emx1-Cre control, Emx1/Lhx2 mutant, and Nestin/Lhx2 mutant mice. Abbreviations: CC, corpus callosum; HC, hippocampal commissure; AC, anterior commissure. Scale bars: 200 μm.
Mouse strain background is a well-known modifier of inbred (Wahlsten 1982; Kusek et al. 2007) and genetic forms of ACC (Magara et al. 1999; Guo et al. 2000). We therefore examined Emx1/Lhx2 animals on 2 different strain backgrounds by using Lhx2 cKO females that were backcrossed for at least 10 generations onto either CD1 or C57BL/6N background. (Note: Neither CD1 nor C57BL6 mice are associated with ACC.) These crosses resulted in complete ACC in all Lhx2 null animals, with no ACC in littermate controls (nmutants = 5, ncontrols = 4 for C57BL/6N background; nmutants = 11, ncontrols = 10 for mixed C57BL/6N;CD1 background). ACC therefore remained fully penetrant on 2 different strain backgrounds and with 2 different Cre drivers, revealing that the ACC phenotype can be fully attributed to the loss of Lhx2.
Lhx2 Mutant ACC is Characterized by Ventrally projecting Callosal Axons Rather Than Probst Bundles
In H&E sections, the Emx1/Lhx2 and Nestin/Lhx2 ACC phenotypes were similar, with callosal axons apparently entering the septum (Fig. 1B). Many ACC mouse mutants possess Probst bundles (Ozaki et al. 1987; Ren et al. 2007), which are relatively large bundles of aberrantly projecting axons near the midline that extend longitudinally along the rostrocaudal axis. However, significant Probst bundles were not detected in the mutant animals.
To further characterize the callosal axon defects, we performed tract tracing on Emx1/Lhx2 animals with DiI. These studies confirmed the absence of crossing callosal axons and delineated their ventral rather than rostrocaudal orientation (nmutants = 8, ncontrols = 14) (Fig. 2A,B). The same axonal patterns were observed when cortical radial glia were electroporated in utero with GFP at E14, then examined at E19.5 (n = 3) (Fig. 2C,D, see Supplementary Fig. 7). These findings indicate that callosal axons in Lhx2 mutant animals project ventrally toward the septum rather than forming Probst bundles.
Figure 2.
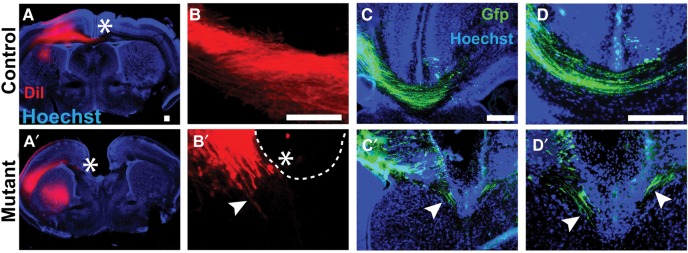
Ventrally oriented callosal axons in Emx1/Lhx2 mutant mice. DiI and in utero electroporation studies, coronal sections. (A–D) Callosal axons cross the midline in Emx1/Lhx2 control adults following neocortical placement of DiI crystals (A,B) or in control E19.5 animals following GFP electroporation at E14.5 (C,D). (A′–D′) DiI- or GFP labeled callosal axons (white arrowheads) in Emx1/Lhx2 mutant littermates do not cross the midline and are ventrally oriented. Rostrocaudally oriented axons typical of Probst bundles are not apparent. White asterisks designate the end of visible DiI labeling in callosal axons. Scale bars: 100 μm.
Loss of Lhx2 Does not Impair Callosal Projection Neuron Specification
Callosal projection neurons are normally specified by Satb2 secondary to the repression of Ctip2 (Alcamo et al. 2008; Britanova et al. 2008), which instead specifies corticospinal motor neurons (Arlotta et al. 2005; Chen et al. 2005; Molyneaux et al. 2005; Baranek et al. 2012). These 2 populations of neurons are mutually exclusive, and there is normally no co-expression of Satb2 and Ctip2. Similarly, the E18.5 Emx1/Lhx2 mutant cortex had relatively normal patterns of Ctip2 and Satb2 expression (nmutants = 2, ncontrols = 2) (Fig. 3A–D,A′–D′). Similar patterns were also seen in control and mutant sections in Nestin/Lhx2 mice at E16.5 (nmutants = 2, ncontrols = 2) and E18.5 (nmutants = 3, ncontrols = 2), and in Emx1/Lhx2 at E16.5 (nmutants = 2, ncontrols = 2) and P5 (nmutants = 1, ncontrols = 1) (data not shown). In addition, the neuronal axonal marker TuJ1, which is normally expressed in callosally projecting neurons (Chuikov et al. 2010), is also appropriately expressed in the mutant callosal tract that leads to the septum in the E16.5 Nestin/Lhx2 mouse (nmutants = 2, ncontrols = 2) (Fig. 3E–E′). These findings suggest that Lhx2 mutant callosal projection neurons are specified correctly, thus raising the possibility of non-cell autonomous factors in the acallosal phenotype.
Figure 3.
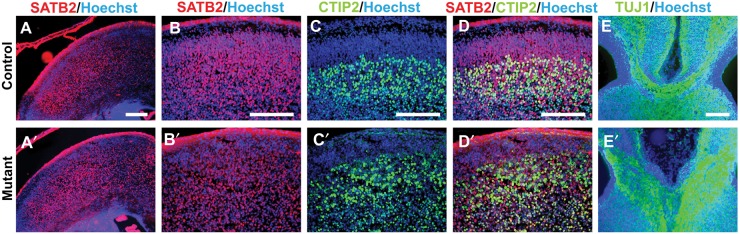
Normal specification of callosal and corticospinal projection neurons in Emx1/Lhx2 and Nestin/Lhx2 mice. (A–D,A′–D′) Satb2 expression is widespread in both Emx1/Lhx2 control and mutant cortex at E18.5. In the same sections, Ctip2 expression is confined to lower cortical layers. (D,D′) Satb2 and Ctip2 exhibit mutually exclusive staining in both control and mutant neocortical neurons, suggesting normal specification of callosal and corticospinal neurons, respectively. (E,E′) TuJ1 staining of Nestin/Lhx2 at E16.5 highlight the normal and abnormal tracts of callosal projection neurons in controls and mutants. Scale bars: 100 μm.
Corpus Callosum is Present in Nex/Lhx2 Mutants
To further address the possibility of cell autonomy, we used Nex-Cre to inactivate Lhx2 in cortical projection neurons. Nex-Cre begins to drive recombination in these neurons by E11.5, but is not expressed in ventricular zone progenitors, interneurons, astrocytes, or oligodendrocytes (Goebbels et al. 2006). We confirmed this cell type-specificity of Nex-Cre-mediated recombination using rosa26 conditional lacZ mice (n = 2 animals; Fig. 4A′) and by IHC in E14.5 Nex/Lhx2 embryos, which revealed specific Lhx2 loss from the cortical plate of neocortex, rostromedial cortex, and hippocampus, but not from the ventricular zone (nmutants = 2) (Fig. 4B′–D′).
Figure 4.
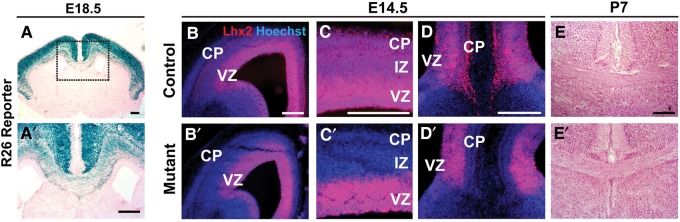
Presence of corpus callosum in Nex/Lhx2 mice. (A,A′) LacZ histochemistry, E18.5 Nex/R26R coronal sections. Widespread lacZ expression occurs in the cortical plate, but not in the VZ, glial wedge, or other forebrain regions. Xgal staining of the corpus callosum confirms Nex-Cre-mediated recombination in callosal neurons. Boxed region in A is magnified in A′. (B–D,B′–D′) Lhx2 IHC, E14.5 Nex/Lhx2 coronal sections. Lhx2 expression normally occurs in all regions of the cortical VZ and cortical plate (CP), including lateral neocortex (C) and rostromedial neocortex (D). In Nex/Lhx2 mutants, little-to-no Lhx2 expression is detected in all CP regions, while VZ expression persists (B′–D′). (E,E′) H&E stains, P7 Nex/Lhx2 coronal sections. A corpus callosum of similar caliber is present in both control and mutant littermates. Abbreviations: VZ, ventricular zone; IZ, intermediate zone; CP, cortical plate. Scale bars: 200 μm.
Despite the loss of Lhx2, the corpus callosum and hippocampal commissure were present in all Nex/Lhx2 mutants examined at E18.5 (nmutants = 3), P2 (nmutants = 2), P7 (nmutants = 4), and adult stages (P21, P56, P84) (nmutants = 5) (Fig. 4E). No obvious differences in thickness or caliber of these commissural tracts were seen between Nex/Lhx2 mutant and control littermates, which was confirmed by quantifying rostral (P = 0.36) and caudal (P = 0.16) corpus callosum thickness (nmutants = 4, ncontrols = 8) (see Supplementary Fig. 3). These observations indicate that defects in Nex-Cre-positive neurons, including callosal projection neurons, are not responsible for the ACC phenotype in Emx1/Lhx2 and Nestin/Lhx2 animals.
The GW is Deficient or Absent in Emx1/Lhx2 and Nestin/Lhx2 Mutants
The ventrally projecting axonal phenotype in Nestin/Lhx2 and Emx1/Lhx2 animals is relatively uncommon among ACC mouse mutants, but has been described in 2 related mutants (Nfia and Nfib null mice) thought to have primary defects of the GW (Shu et al. 2003; Piper et al. 2009). We therefore examined the GW in Emx1/Lhx2 animals. We found that the GW was virtually undetectable by H&E or GFAP IHC in mutant animals at stages spanning the initial crossing of callosal axons (E15-17; n = 10) (Fig. 5A–D). In these mutants, the abnormally hypocellular GW region also contained the aberrant callosal axons, consistent with a role for GW in repelling these axons. Similar GW deficiencies were also observed in Nestin/Lhx2 mutants (data not shown).
Figure 5.

GW deficits in Emx1/Lhx2 and Nestin/Lhx2 mutant mice. H&E histology and GFAP IHC, coronal sections; boxed regions are magnified in panels immediately below each low-power image. (A–C) At E16.5–18.5, the GW in control littermates (black arrowheads) is a cell-dense, wedge-shaped structure extending from the ventricle and situated between rostromedial cortex and septum. (A′–C′) Little to no GW is apparent in E16.5–18.5 Emx1/Lhx2 mutant embryos. The GW region contains eosinophilic cell-sparse zones corresponding to the misrouted callosal axons. (D,D′) GFAP-positive stellate astrocytes seen in the normal GW of E18.5 control animals (arrowheads in D) are absent from an Emx1/Lhx2 mutant (D′). (E,E′) E16.5 Nestin/Lhx2 ISH for Bmp6 in the control faintly marks the GW (black arrowheads) as well as a medial cell population dorsal to the corpus callosum (asterisks). No expression is detected in the mutant GW, while medial Bmp6 expression is maintained. Scale bars: 100 μm.
We then performed ISH for signaling molecules expressed in the GW, including Bmps, which have important roles in callosal development, including genesis of the GW (Sanchez-Camacho et al. 2011). Bmp6 was detected in the GW of Nestin/Lhx2 controls at E16.5 (Fig. 5E), but not in the mutant GW (Fig. 5E′), while its expression was maintained in other medial cell populations in both controls and mutants (asterisks in Fig. 5E,E′). The repulsive molecule Draxin (Islam et al. 2009) was detected in the ependymal lining of the GW, but not in the GW proper of mutants compared with controls (see Supplementary Fig. 4), although the normal absence of Draxin from adjacent fimbrial ependyma precluded definitive interpretation. Nonetheless, the Bmp6 and Draxin findings further support the conclusion of GW loss based on the histological and GFAP IHC studies.
Lhx2 is Expressed in GW Progenitors
Given the essential role for Lhx2 in GW formation, we studied Lhx2 expression during GW development. At E15.5, cells throughout the GW region strongly expressed Lhx2 protein, including cells in the GW ventricular zone (VZ) (Fig. 6A–C). In situ hybridizations at P4 revealed continued Lhx2 expression in the VZ as well as expression in the GW proper (Fig. 6D). Lhx2 is therefore expressed in GW cells and progenitors prior to and during corpus callosum formation.
Figure 6.

Lhx2 and Emx1-Cre expression in the developing GW. (A–C,A′–C′) Lhx2 IHC (green) and phalloidin staining (red), E15.5 coronal sections. Lhx2 expression spans the medial cortical wall, including the GW and GW VZ (white arrowheads). Lhx2 is also expressed in the septal VZ, but less so in the septal mantle zone. Phalloidin staining also demarcates the cortical-septal boundary and GW region. (D,D′) Lhx2 in situ hybridization, P4 coronal section. Lhx2 expression in the GW is maintained into the postnatal period. (E–E′,F–F′) LacZ histochemistry, E18.5 and P4 Emx1/Lhx2 coronal sections. Expression of the Emx1-Cre allele, which contains a lacZ reporter, is expressed in the GW VZ and GW proper of control littermates(E,F). In Emx1/Lhx2 mutants, far fewer Xgal-stained cells are seen in the GW region (E′,F′). (G–G′,H–H′) Sox2 IHC E12.5 and E16.5 Emx1/Lhx2 coronal sections. At both stages, Sox2 expression persists in GW progenitors of both Emx1-Cre-positive littermate controls (G,H) and Emx/Lhx2 mutants (G′–H′). Abbreviations: VZ, ventricular zone. Scale bars: 100 μm.
If GW defects account for Emx1/Lhx2 and Nestin/Lhx2 mutant ACC phenotypes, then both Emx1-Cre and Nestin-Cre drivers must also be expressed in the GW in addition to Lhx2. Nestin-Cre is well known to be expressed in all radial glia (Zimmerman et al. 1994; Jaxmice.jax.org 2012) but expression of Emx1-Cre in the GW region has not been specifically described. We therefore studied Emx1-Cre expression in the GW region using the lacZ reporter contained in the Emx1-Cre allele. Studies on Emx1-Cre-positive control littermates revealed Xgal staining in the E18.5 GW VZ and in the P4 GW (Fig. 6E,F). In Emx1/Lhx2 mutants, significantly less staining was seen in the GW region (Fig. 6E′,F′), consistent with the histologic and immunohistochemical GW defects described earlier (Fig. 5). We also studied the expression of the generic radial glial marker Sox2 (Suh et al. 2007), and found maintained Sox2 expression in the GW region at both E12.5 and E16.5 (Fig. 6G,H). These findings indicate that Lhx2 and Emx1-Cre are co-expressed in GW cells and progenitors, confirm the GW deficiency in Emx1/Lhx2 mutants, and argue against abnormal radial glial specification as the cause of this GW deficiency.
Radial GW Progenitors are Abnormal in Emx1/Lhx2 and Nestin/Lhx2 Mutants
To characterize the GW defect further, we performed birthdating studies with the thymidine analog, EdU, during the GW genesis period in mice (E13-P2) (Shu, Puche et al. 2003). We injected EdU at E14.5 or E16.5, then examined Emx1/Lhx2 or Nestin/Lhx2 animals at E18.5. Compared with controls, much less EdU labeling was detected in all mutant GW analyzed (n = 9 animals) (Fig. 7). In contrast, cell death was unchanged in the mutant GW region based on activated Caspase-3 and TUNEL stains (see Supplementary Fig. 5), suggesting a deficit in the generation of GW cells.
Figure 7.
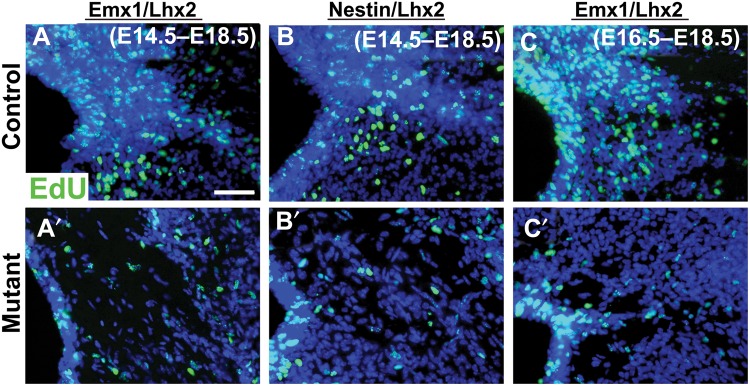
Impaired GW genesis in Emx1/Lhx2 and Nestin/Lhx2 mutant mice. EdU birthdating studies, E18 coronal sections. (A–C) EdU administration at E14 (A,B) or E16 (C) results in prominent EdU labeling (green) of the GW in control littermates. (A′–C′) Emx1/Lhx2 and Nestin/Lhx2 mutants show little-to-no EdU labeling in the GW. Scale bar: 100 μm.
We then carried out a series of 4 EdU experiments to study the radial GW progenitors in Emx1/Lhx2 mice. In the first study, EdU was administered at E11.5, then examined at E12.5. In the GW VZ of Cre-positive littermate controls, relatively little EdU labeling was seen presumably due to the fast cell cycle rate (8–10 h) (Takahashi et al. 1995) and subsequent dilution of EdU label (Fig. 8A). Also consistent with this explanation is the findings that most of the remaining EdU positive cells are located in the cortical mantle, which mainly harbors postmitotic cells. In Emx1/Lhx2 mutants, however, there were significant numbers of strongly labeled VZ cells (n = 5/5) (Fig. 8A′). This suggests a defect in the progenitors that lie in the VZ. Increased GW VZ labeling was also seen in these mutants when EdU-injected E11.5 embryos were examined at E14.5 (n = 3) (Fig. 8B,B′,C,C′); quantifications from separate E11.5–E14.5 studies revealed a >4-fold increase in EdU labeling of GW VZ cells in mutants compared with control littermates (5.13 ± 0.28% vs. 1.16 ± 0.17%, n = 3 animals each group, P = 0.0003; Fig. 8H). (Note: neocortical and hippocampal sizes are also abnormal in Emx1/Lhx2 mutants; see Discussion for further details.)
Figure 8.

Abnormal radial GW progenitors in Emx1/Lhx2 mice. EdU studies, coronal sections. Red bars approximate the GW region. (A,A′) Following E11.5 administration, EdU label is abnormally retained in the GW VZ of an E12.5 Emx1/Lhx2 mutant (A′) compared with a control littermate (A). (B–C,B′–C′) Abnormal EdU retention in the GW VZ persists at E14.5 in mutants compared with littermate controls following E11.5 administration. (D–E,D′–E′) Acute administration (9 h prior to sacrifice) reveals far fewer EdU-labeled cells in the GW VZ of mutants compared with littermate controls. (F,G) EdU/Ki67 labeling reveals Ki67-positive and -negative subpopulations of EdU-labeled cells in both control and mutant E14.5 GW VZ/SVZ. (F′,F″) Examples of Ki67-positive and -negative EdU-labeled cells, respectively, in control VZ/SVZ. (G′,G″) Examples of Ki67-positive and -negative EdU-labeled cells, respectively, in mutant VZ/SVZ. (H) Quantification of EdU-positive cells in the GW VZ/SVZ reveals a statistically significant increase in mutants compared with littermate controls indicated by asterisks (5.13 ± 0.28 vs. 1.16 ± 0.17, P = 0.0003, n = 3). (I) Quantification of the Ki67-positive fractions of EdU-labeled cells in the GW VZ/SVZ reveals a significant decrease in mutant embryos compared with control littermates (28.29 ± 0.53 vs. 82.74 ± 3.90, P = 0.002, n = 3). Scale bars: A–E,F,G, 100 μm; F′,F″,G′,G″, 10 μm.
To investigate the abnormal EdU retention further, we performed EdU/Ki67 double labeling to determine the cell cycle status of EdU-labeled cohorts. In E14.5 embryos, the number of Ki67-expressing GW cells were markedly reduced in Emx1/Lhx2 mutants compared with controls (Fig. 8F,G). EdU/Ki67 analysis revealed both Ki67-positive and -negative cohorts among EdU-labeled cells in both control and mutant embryos (Fig. 8F′,F″,G′,G″); we therefore quantified these subpopulations separately. These quantifications revealed that the majority of control EdU-retaining cells are Ki67-positive, whereas most of the mutant EdU-retaining cells are Ki67-negative (82.74 ± 3.90% vs. 28.29 ± 0.53%; n = 3 animals each group; P = 0.0002) (Fig. 8I). Thus, most of the abnormal EdU-retaining cells in the mutants were no longer in cell cycle, suggesting premature exit of radial GW progenitors in Emx1/Lhx2 mutants.
As an additional test for premature cell cycle exit, we performed a short-term EdU study (EdU injection 9 h prior to sacrifice at E14.5). In control littermates, significant EdU labeling was seen in the GW VZ (n = 3) (Fig. 8D,E), indicating recent S-phase progression of a significant GW radial glial cohort. In contrast, relatively few labeled cells were seen in the GW VZ of Emx1/Lhx2 mutant littermates (n = 3) (Fig. 8D′,E′). Thus, consistent with the Ki67 (Fig. 8F,G), long-term EdU (Fig. 8B′-C″,H), and EdU/Ki67 analyses (Fig. 8F–I), these short-term EdU studies suggest premature cell cycle exit of radial GW progenitors in Emx1/Lhx2 mutants, with progenitor defects detectable by E12.5 (Fig. 8A′,A″).
Discussion
These studies demonstrate that Lhx2 is critical for corpus callosum development. Lhx2 inactivation in cortical radial glia using 2 different Cre drivers (Emx1-Cre and Nestin-Cre) resulted in complete ACC in both rostral and caudal domains, which was fully penetrant on multiple strain backgrounds. Lhx2-dependent ACC was not due to defects in specification or in other cell-autonomous processes in callosal projection neurons, but was instead associated with a failure in GW formation, with the earliest GW defect seen in radial GW progenitors. The histologic, marker, and EdU/Ki67 studies implicate premature cell cycle exit as a principal defect of Lhx2-null GW progenitors, which is likely cell-autonomous, given the high Lhx2 expression levels in radial GW progenitors and phenotypic onset soon after conditional Lhx2 inactivation. The rostral ACC phenotype displayed all of the features expected for a primary GW defect—misrouted callosal axons projecting through the region normally occupied by the GW, and extension of these axons ventrally toward the septum rather than forming Probst bundles (Shu, Butz et al. 2003). Lhx2 is therefore essential for generating the GW, with failure in GW genesis accounting for the described ACC phenotypes.
Additional midline structures implicated in normal corpus callosum development (e.g., indusium griseum, glial sling, midline zipper glia, and guidepost neurons) (Shu, Puche et al. 2003; Smith et al. 2006; Niquille et al. 2009; Sanchez-Camacho et al. 2011) may also be abnormal in Lhx2 mutant animals. For example, midline guidepost neurons marked by Tbr1 and calretinin appear to be deficient in Lhx2 mutant mice (see Supplementary Fig. 6). However, the potential effects of these midline cell populations on callosal development may be masked by the GW defect, which causes the misrouted callosal axons in Lhx2 mutants to project ventrally rather than towards the midline. As such, the wayward axons may not have the opportunity to normally interact with other midline structures that facilitate corpus callosum formation. Similarly, hemispheric fusion often appeared defective in Lhx2 mutants, although some mutants remained fused, mutant hemispheres were prone to artifactual separation, and defects in fusion per se are generally associated with Probst bundles (Wahlsten et al. 2006). Thus, additional work will be necessary to determine the role of Lhx2 in these midline cell populations and processes.
Since the cingulate cortex is smaller than normal in both mutant models, pioneer neurons of cingulate origin that are thought to be a scaffold for later callosal projection neurons (Koester and O'Leary 1994; Rash and Richards 2001; Piper, Plachez et al. 2009) may also be reduced in number. If so, however, they must also have an abnormal ventral trajectory together with the other callosal axons. Interestingly, a population of corticoseptal projection neurons differentiates prior to callosal formation (Hankin and Silver 1988), which may serve as a substrate for the misrouted callosal axons in the Lhx2 mutants.
Our studies support current models, which suggest that the GW and hippocampal commissure have separate roles within different domains of the developing corpus callosum based on correlation of ACC defects in different mutant mice (Paul et al. 2007; Donahoo and Richards 2009). As mentioned earlier, Lhx2-dependent ACC has similarities to the acallosal phenotypes associated with Nfia and Nfib null mice (das Neves et al. 1999; Shu et al. 2003). All 3 ACC models have GW deficits and misrouted callosal axons that project ventrally towards the septum. ACC in Nfia and Nfib null mice, however, is not as severe as the ACC in Lhx2 null animals, since the caudal portion of the corpus callosum persists in the Nfia and Nfib nulls. This may be attributed to the hippocampal commissure, which is intact in the Nfia and Nfib null models, but not in Lhx2 null mice. Collectively, these models therefore further support the concept that the hippocampal commissure selectively promotes caudal callosal development, whereas the GW is essential for rostral corpus callosum formation. Anatomically, this model makes sense, since neither the hippocampal commissure nor GW cover the full rostrocaudal extent of the corpus callosum (hippocampal commissure is not present rostrally, while the GW is not present caudally).
Our studies further suggest that the Lhx2-null GW defect stems from a critical role for Lhx2 in radial GW progenitors. Specifically, in addition to histologic evidence, 4 lines of experimental evidence point to premature cell cycle exit of these progenitors in Lhx2 null embryos: (1) Ki67 (depletion of Ki67-positive cells from GW VZ by E14.5), (2) acute EdU (deficient GW VZ incorporation), (3) chronic EdU (increased GW VZ retention), and (4) EdU-Ki67 (the absence of Ki67 from most EdU-retaining null cells). Since cell cycle length of radial glia, and specifically the G1 phase, is closely linked to cell cycle exit probability (Takahashi et al. 1995; Miyama et al. 1997; Caviness et al. 2003), mutant radial GW progenitors may have prolonged cell cycles, but this remains to be directly determined. There are at least 3 nonmutually exclusive processes that could accompany the premature cell cycle exit of Lhx2-null GW progenitors: (1) misspecification of the GW progenitor domain, (2) deficient expansion of the specified GW progenitor domain, and (3) aberrant differentiation of GW radial glia. With regard to Possibility 3, elevated cell death was not detected (H&E, TUNEL, antiactivated Casp3; see Supplementary Fig. 5), and small numbers of GFAP-positive astrocytes are present in the E18.5 Lhx2 null GW (Fig. 5D′), suggesting that some Lhx2-null GW progenitors do normally differentiate into GW astrocytes; Possibilities 1 and 2 cannot be adequately addressed without marker panels that define all GW domain borders. In addition, definitive assessment of GW progenitor differentiation into nonastrocytic cell types (e.g., neurons or ependymal cells) would require GW-specific lineage tracing, ideally genetic, which is not yet available.
Interestingly, premature cell cycle exit and differentiation is associated with other Lhx2 null phenotypes in the dorsal telencephalon. In the developing hippocampus, Lhx2 inactivation at E14.5–E15.5 via electroporation results in premature astrogliogenesis, failure to maintain normal neurogenesis, and a hypoplastic hippocampus (Subramanian et al. 2011). In neocortical radial glia, genetic Lhx2 inactivation driven by Emx1-Cre causes premature cell cycle exit, early increases in neuron and neuronal progenitor numbers, and ultimately a hypoplastic neocortex (Chou and O'Leary 2013). Taken together with our findings on the GW, these studies collectively suggest that Lhx2 has a general role in maintaining radial glial progenitors of the dorsal telencephalon (neocortical, hippocampal, and GW primordia). Furthermore, the similar acallosal phenotypes of Lhx2 null and Nfia null mice discussed above, together with the previous links between Lhx2 and Notch pathway effectors (including Nfia) in the Lhx2 null neocortical and hippocampal phenotypes (Subramanian et al. 2011; Chou and O'Leary 2013), raise the possibility of Lhx2-Notch pathway interactions in the maintenance of all of these radial glial subtypes.
Additional insights can be inferred from the position-dependent differences among Lhx2 null radial glia within the dorsal telencephalon (Subramanian et al. 2011; Chou and O'Leary 2013). Lhx2 loss results in neuron-to-astrocyte fate switch in the hippocampus, but not in the neocortex or GW. In the GW, which is medial to the hippocampus, Lhx2 loss effectively results in GW aplasia rather than hypoplasia. Thus, the consequence of Lhx2 loss in the dorsal telencephalon depends on position, with the most severe defect occurring medially (GW), where Lhx2 expression levels are highest. This is reminiscent of the position-dependent effect of Lhx2 in earlier-stage cortical neuroepithelial cells, with medial and lateral cortical cells adopting either hem or antihem fate, respectively (Mangale et al. 2008). Position-dependence is therefore a consistent theme for Lhx2 loss of function in the dorsal telencephalon, which suggests Lhx2 interaction with other positional determinants in this tissue.
Interestingly, Lhx2 null zebrafish also have abnormal midline glia associated with commissural defects in the brain. In these zebrafish, known as belladonna (bel) mutants, abnormal glial bridge cells at the midline are associated with agenesis of the anterior commissure, postoptic commissure, and optic chiasm (Seth et al. 2006). (Note: zebrafish do not have structural homologs of the mouse corpus callosum or hippocampal commissure.) Zebrafish glial bridge cells, like mouse GW cells, are a GFAP-positive population that facilitate commissural axon crossing during normal development (Barresi et al. 2005). In bel mutants, however, glial bridge cells occur in excess, whereas Lhx2 null mice have reduced to absent GW cells. Thus, the consequences of Lhx2 loss to midline glia appear to be diametrically opposed in zebrafish and mice. Nonetheless, it is interesting to consider an evolutionarily conserved role for Lhx2 in midline glia that promotes commissure formation, which may have then diverged in mice and zebrafish.
Supplementary Material
Supplementary material can be found at: http://www.cercor.oxfordjournals.org/.
Authors’ contributions
G.A.C. and E.S.M. designed research; G.A.C., K.E.H., T.M.C., C.U., F.P., J.F., and N.F. performed research; G.A.C., T.M.C., and E.S.M. analyzed data and wrote the manuscript.
Funding
This work was supported by the UC Irvine MSTP program (G.A.C.); National Institutes of Health (grant R01 NS064587) to E.S.M.
Supplementary Material
Notes
We thank Yuqing Li (University of Alabama-Birmingham) for Emx1-Cre mice, Grant MacGregor (UC Irvine) for Nestin-Cre mice free of the Nnt mutation, Klaus Nave (Max Planck Institute for Experimental Medicine) for Nex-Cre mice, and Xing Dai (UC Irvine) for R26R mice. We also thank Shubha Tole (Tata Institute for Fundamental Research) for comments on the manuscript, and members of the Monuki and Flanagan labs (UC Irvine) for their input and support. Conflict of Interest: None declared.
References
- Abellan A, Menuet A, Dehay C, Medina L, Retaux S. 2010. Differential expression of LIM-homeodomain factors in Cajal–Retzius cells of primates, rodents, and birds. Cereb Cortex. 20:1788–1798. [DOI] [PubMed] [Google Scholar]
- Alcamo EA, Chirivella L, Dautzenberg M, Dobreva G, Farinas I, Grosschedl R, McConnell SK. 2008. Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron. 57:364–377. [DOI] [PubMed] [Google Scholar]
- Andrews W, Liapi A, Plachez C, Camurri L, Zhang J, Mori S, Murakami F, Parnavelas JG, Sundaresan V, Richards LJ. 2006. Robo1 regulates the development of major axon tracts and interneuron migration in the forebrain. Development. 133:2243–2252. [DOI] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. 2005. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 45:207–221. [DOI] [PubMed] [Google Scholar]
- Bagri A, Marin O, Plump AS, Mak J, Pleasure SJ, Rubenstein JL, Tessier-Lavigne M. 2002. Slit proteins prevent midline crossing and determine the dorsoventral position of major axonal pathways in the mammalian forebrain. Neuron. 33:233–248. [DOI] [PubMed] [Google Scholar]
- Baranek C, Dittrich M, Parthasarathy S, Bonnon CG, Britanova O, Lanshakov D, Boukhtouche F, Sommer JE, Colmenares C, Tarabykin V, et al. 2012. Protooncogene Ski cooperates with the chromatin-remodeling factor Satb2 in specifying callosal neurons. Proc Natl Acad Sci U S A. 109:3546–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barresi MJ, Hutson LD, Chien CB, Karlstrom RO. 2005. Hedgehog regulated Slit expression determines commissure and glial cell position in the zebrafish forebrain. Development. 132:3643–3656. [DOI] [PubMed] [Google Scholar]
- Bignami A, Dahl D. 1971. Astrocyte-specific protein and neuroglial differentiation. An immunofluorescence study with antibodies to the glial fibrillary acidic protein. J Comp Neurol. 153:27–38. [DOI] [PubMed] [Google Scholar]
- Bodensteiner J, Schaefer GB, Breeding L, Cowan L. 1994. Hypoplasia of the corpus callosum: a study of 445 consecutive MRI scans. J Child Neurol. 9:47–49. [DOI] [PubMed] [Google Scholar]
- Britanova O, de Juan Romero C, Cheung A, Kwan KY, Schwark M, Gyorgy A, Vogel T, Akopov S, Mitkovski M, Agoston D, et al. 2008. Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron. 57:378–392. [DOI] [PubMed] [Google Scholar]
- Bulchand S, Grove EA, Porter FD, Tole S. 2001. LIM-homeodomain gene Lhx2 regulates the formation of the cortical hem. Mech Dev. 100:165–175. [DOI] [PubMed] [Google Scholar]
- Bulchand S, Subramanian L, Tole S. 2003. Dynamic spatiotemporal expression of LIM genes and cofactors in the embryonic and postnatal cerebral cortex. Dev Dyn. 226:460–469. [DOI] [PubMed] [Google Scholar]
- Caviness VS, Jr, Goto T, Tarui T, Takahashi T, Bhide PG, Nowakowski RS. 2003. Cell output, cell cycle duration and neuronal specification: a model of integrated mechanisms of the neocortical proliferative process. Cereb Cortex. 13:592–598. [DOI] [PubMed] [Google Scholar]
- Chen B, Schaevitz LR, McConnell SK. 2005. Fezl regulates the differentiation and axon targeting of layer 5 subcortical projection neurons in cerebral cortex. Proc Natl Acad Sci USA. 102:17184–17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou SJ, O'Leary DD. 2013. Role for Lhx2 in corticogenesis through regulation of progenitor differentiation. Mol Cell Neurosci. 56:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuikov S, Levi BP, Smith ML, Morrison SJ. 2010. Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat Cell Biol. 12:999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currle DS, Cheng X, Hsu CM, Monuki ES. 2005. Direct and indirect roles of CNS dorsal midline cells in choroid plexus epithelia formation. Development. 132:3549–3559. [DOI] [PubMed] [Google Scholar]
- das Neves L, Duchala CS, Tolentino-Silva F, Haxhiu MA, Colmenares C, Macklin WB, Campbell CE, Butz KG, Gronostajski RM. 1999. Disruption of the murine nuclear factor I-A gene (Nfia) results in perinatal lethality, hydrocephalus, and agenesis of the corpus callosum. Proc Natl Acad Sci USA. 96(21):11946–11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahoo AL, Richards LJ. 2009. Understanding the mechanisms of callosal development through the use of transgenic mouse models. Semin Pediatr Neurol. 16:127–142. [DOI] [PubMed] [Google Scholar]
- Furuta Y, Piston DW, Hogan BL. 1997. Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development. 124:2203–2212. [DOI] [PubMed] [Google Scholar]
- Godement P, Vanselow J, Thanos S, Bonhoeffer F. 1987. A study in developing visual systems with a new method of staining neurones and their processes in fixed tissue. Development. 101:697–713. [DOI] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA. 2006. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis. 44:611–621. [DOI] [PubMed] [Google Scholar]
- Guillem F, Bicu M, Pampoulova T, Hooper R, Bloom D, Wolf MA, Messier J, Desautels R, Todorov C, Lalonde P, et al. 2003. The cognitive and anatomo-functional basis of reality distortion in schizophrenia: a view from memory event-related potentials. Psychiatry Res. 117:137–158. [DOI] [PubMed] [Google Scholar]
- Guo H, Christoff JM, Campos VE, Jin XL, Li Y. 2000. Normal corpus callosum in Emx1 mutant mice with C57BL/6 background. Biochem Biophys Res Commun. 276:649–653. [DOI] [PubMed] [Google Scholar]
- Hankin MH, Silver J. 1988. Development of intersecting CNS fiber tracts: the corpus callosum and its perforating fiber pathway. J Comp Neurol. 272:177–190. [DOI] [PubMed] [Google Scholar]
- Hu JS, Doan LT, Currle DS, Paff M, Rheem JY, Schreyer R, Robert B, Monuki ES. 2008. Border formation in a Bmp gradient reduced to single dissociated cells. Proc Natl Acad Sci U S A. 105:3398–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TT, Naeemuddin M, Elchuri S, Yamaguchi M, Kozy HM, Carlson EJ, Epstein CJ. 2006. Genetic modifiers of the phenotype of mice deficient in mitochondrial superoxide dismutase. Hum Mol Genet. 15:1187–1194. [DOI] [PubMed] [Google Scholar]
- Huffman KJ, Garel S, Rubenstein JL. 2004. Fgf8 regulates the development of intra-neocortical projections. J Neurosci. 24:8917–8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam SM, Shinmyo Y, Okafuji T, Su Y, Naser IB, Ahmed G, Zhang S, Chen S, Ohta K, Kiyonari H, et al. 2009. Draxin, a repulsive guidance protein for spinal cord and forebrain commissures. Science. 323(5912):388–393. [DOI] [PubMed] [Google Scholar]
- Jaxmice.jax.org 2012. http://jaxmice.jax.org/strain/003771.html. In. (Last accessed 31 March 2014).
- Jeret JS, Serur D, Wisniewski K, Fisch C. 1985. Frequency of agenesis of the corpus callosum in the developmentally disabled population as determined by computerized tomography. Pediatr Neurosci. 12:101–103. [DOI] [PubMed] [Google Scholar]
- Jin XL, Guo H, Mao C, Atkins N, Wang H, Avasthi PP, Tu YT, Li Y. 2000. Emx1-specific expression of foreign genes using “knock-in” approach. Biochem Biophys Res Commun. 270:978–982. [DOI] [PubMed] [Google Scholar]
- Keeble TR, Halford MM, Seaman C, Kee N, Macheda M, Anderson RB, Stacker SA, Cooper HM. 2006. The Wnt receptor Ryk is required for Wnt5a-mediated axon guidance on the contralateral side of the corpus callosum. J Neurosci. 26:5840–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koester SE, O'Leary DD. 1994. Axons of early generated neurons in cingulate cortex pioneer the corpus callosum. J Neurosci. 14:6608–6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusek GK, Wahlsten D, Herron BJ, Bolivar VJ, Flaherty L. 2007. Localization of two new X-linked quantitative trait loci controlling corpus callosum size in the mouse. Genes Brain Behav. 6:359–363. [DOI] [PubMed] [Google Scholar]
- Levitt P, Rakic P. 1980. Immunoperoxidase localization of glial fibrillary acidic protein in radial glial cells and astrocytes of the developing rhesus monkey brain. J Comp Neur. 193:815–840. [DOI] [PubMed] [Google Scholar]
- Livy DJ, Wahlsten D. 1997. Retarded formation of the hippocampal commissure in embryos from mouse strains lacking a corpus callosum. Hippocampus. 7:2–14. [DOI] [PubMed] [Google Scholar]
- Magara F, Muller U, Li ZW, Lipp HP, Weissmann C, Stagljar M, Wolfer DP. 1999. Genetic background changes the pattern of forebrain commissure defects in transgenic mice underexpressing the beta-amyloid-precursor protein. Proc Natl Acad Sci U S A. 96:4656–4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangale VS, Hirokawa KE, Satyaki PR, Gokulchandran N, Chikbire S, Subramanian L, Shetty AS, Martynoga B, Paul J, Mai MV, et al. 2008. Lhx2 selector activity specifies cortical identity and suppresses hippocampal organizer fate. Science. 319:304–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekada K, Abe K, Murakami A, Nakamura S, Nakata H, Moriwaki K, Obata Y, Yoshiki A. 2009. Genetic differences among C57BL/6 substrains. Exp Anim. 58:141–149. [DOI] [PubMed] [Google Scholar]
- Miyama S, Takahashi T, Nowakowski RS, Caviness VS., Jr 1997. A gradient in the duration of the G1 phase in the murine neocortical proliferative epithelium. Cereb Cortex. 7:678–689. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Fame RM, MacDonald JL, MacQuarrie KL, Macklis JD. 2009. Novel subtype-specific genes identify distinct subpopulations of callosal projection neurons. J Neurosci. 29:12343–12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Hirata T, Hibi M, Macklis JD. 2005. Fezl is required for the birth and specification of corticospinal motor neurons. Neuron. 47:817–831. [DOI] [PubMed] [Google Scholar]
- Monuki ES, Porter FD, Walsh CA. 2001. Patterning of the dorsal telencephalon and cerebral cortex by a roof plate-Lhx2 pathway. Neuron. 32:591–604. [DOI] [PubMed] [Google Scholar]
- Mutch CA, Funatsu N, Monuki ES, Chenn A. 2009. Beta-catenin signaling levels in progenitors influence the laminar cell fates of projection neurons. J Neurosci. 29:13710–13719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niquille M, Garel S, Mann F, Hornung JP, Otsmane B, Chevalley S, Parras C, Guillemot F, Gaspar P, Yanagawa Y, et al. 2009. Transient neuronal populations are required to guide callosal axons: a role for semaphorin 3C. PLoS Biol. 7:e1000230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki HS, Murakami TH, Toyoshima T, Shimada M. 1987. The fibers which leave the Probst’s longitudinal bundle seen in the brain of an acallosal mouse: a study with the horseradish peroxidase technique. Brain Res. 400:239–246. [DOI] [PubMed] [Google Scholar]
- Ozaki HS, Wahlsten D. 1992. Prenatal formation of the normal mouse corpus callosum: a quantitative study with carbocyanine dyes. J Comp Neurol. 323:81–90. [DOI] [PubMed] [Google Scholar]
- Paul LK, Brown WS, Adolphs R, Tyszka JM, Richards LJ, Mukherjee P, Sherr EH. 2007. Agenesis of the corpus callosum: genetic, developmental and functional aspects of connectivity. Nat Rev Neurosci. 8:287–299. [DOI] [PubMed] [Google Scholar]
- Piper M, Moldrich RX, Lindwall C, Little E, Barry G, Mason S, Sunn N, Kurniawan ND, Gronostajski RM, Richards LJ. 2009. Multiple non-cell-autonomous defects underlie neocortical callosal dysgenesis in Nfib-deficient mice. Neural Dev. 4:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper M, Plachez C, Zalucki O, Fothergill T, Goudreau G, Erzurumlu R, Gu C, Richards LJ. 2009. Neuropilin 1-Sema signaling regulates crossing of cingulate pioneering axons during development of the corpus callosum. Cereb Cortex. 19(Suppl 1):i11–i21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter FD, Drago J, Xu Y, Cheema SS, Wassif C, Huang SP, Lee E, Grinberg A, Massalas JS, Bodine D, et al. 1997. Lhx2, a LIM homeobox gene, is required for eye, forebrain, and definitive erythrocyte development. Development. 124:2935–2944. [DOI] [PubMed] [Google Scholar]
- Rash BG, Richards LJ. 2001. A role for cingulate pioneering axons in the development of the corpus callosum. J Comp Neurol. 434:147–157. [DOI] [PubMed] [Google Scholar]
- Ren T, Zhang J, Plachez C, Mori S, Richards LJ. 2007. Diffusion tensor magnetic resonance imaging and tract-tracing analysis of Probst bundle structure in Netrin1- and DCC-deficient mice. J Neurosci. 27:10345–10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson MS, Schoderbek WE, Tremml G, Maurer RA. 1994. Activation of the glycoprotein hormone alpha-subunit promoter by a LIM-homeodomain transcription factor. Mol Cell Biol. 14:2985–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Nakatsuji N. 2001. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev Biol. 240:237–246. [DOI] [PubMed] [Google Scholar]
- Sanchez-Camacho C, Ortega JA, Ocana I, Alcantara S, Bovolenta P. 2011. Appropriate Bmp7 levels are required for the differentiation of midline guidepost cells involved in corpus callosum formation. Dev Neurobiol. 71:337–350. [DOI] [PubMed] [Google Scholar]
- Serafini T, Colamarino SA, Leonardo ED, Wang H, Beddington R, Skarnes WC, Tessier-Lavigne M. 1996. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell. 87:1001–1014. [DOI] [PubMed] [Google Scholar]
- Seth A, Culverwell J, Walkowicz M, Toro S, Rick JM, Neuhauss SC, Varga ZM, Karlstrom RO. 2006. belladonna/(Ihx2) is required for neural patterning and midline axon guidance in the zebrafish forebrain. Development. 133:725–735. [DOI] [PubMed] [Google Scholar]
- Shu T, Butz KG, Plachez C, Gronostajski RM, Richards LJ. 2003. Abnormal development of forebrain midline glia and commissural projections in Nfia knock-out mice. J Neurosci. 23:203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu T, Puche AC, Richards LJ. 2003. Development of midline glial populations at the corticoseptal boundary. J Neurobiol. 57:81–94. [DOI] [PubMed] [Google Scholar]
- Shu T, Richards LJ. 2001. Cortical axon guidance by the glial wedge during the development of the corpus callosum. J Neurosci. 21:2749–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu T, Sundaresan V, McCarthy MM, Richards LJ. 2003. Slit2 guides both precrossing and postcrossing callosal axons at the midline in vivo. J Neurosci. 23:8176–8184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, Edwards MA, Levitt P. 1993. Immunocytochemical demonstration of early appearing astroglial structures that form boundaries and pathways along axon tracts in the fetal brain. J Comp Neurol. 328:415–436. [DOI] [PubMed] [Google Scholar]
- Silver J, Lorenz SE, Wahlsten D, Coughlin J. 1982. Axonal guidance during development of the great cerebral commissures: descriptive and experimental studies, in vivo, on the role of preformed glial pathways. J Comp Neurol. 210:10–29. [DOI] [PubMed] [Google Scholar]
- Smith KM, Ohkubo Y, Maragnoli ME, Rasin MR, Schwartz ML, Sestan N, Vaccarino FM. 2006. Midline radial glia translocation and corpus callosum formation require FGF signaling. Nat Neurosci. 9:787–797. [DOI] [PubMed] [Google Scholar]
- Soriano P. 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 21:70–71. [DOI] [PubMed] [Google Scholar]
- Subramanian L, Sarkar A, Shetty AS, Muralidharan B, Padmanabhan H, Piper M, Monuki ES, Bach I, Gronostajski RM, Richards LJ, et al. 2011. Transcription factor Lhx2 is necessary and sufficient to suppress astrogliogenesis and promote neurogenesis in the developing hippocampus. Proc Natl Acad Sci U S A. 108:E265–E274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh H, Consiglio A, Ray J, Sawai T, D'Amour KA, Gage FH. 2007. In vivo fate analysis reveals the multipotent and self-renewal capacities of Sox2+ neural stem cells in the adult hippocampus. Cell Stem Cell. 1:515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter B, Nowakowski RS, Bhide PG, Caviness VS. 2007. Navigating neocortical neurogenesis and neuronal specification: a positional information system encoded by neurogenetic gradients. J Neurosci. 27:10777–10784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS., Jr 1995. The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J Neurosci. 15:6046–6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tole S, Gutin G, Bhatnagar L, Remedios R, Hebert JM. 2006. Development of midline cell types and commissural axon tracts requires Fgfr1 in the cerebrum. Dev Biol. 289:141–151. [DOI] [PubMed] [Google Scholar]
- Tomasch J. 1954. Size, distribution, and number of fibres in the human corpus callosum. Anat Rec. 119:119–135. [DOI] [PubMed] [Google Scholar]
- Toye AA, Lippiat JD, Proks P, Shimomura K, Bentley L, Hugill A, Mijat V, Goldsworthy M, Moir L, Haynes A, et al. 2005. A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia. 48:675–686. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. 1999. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 23:99–103. [DOI] [PubMed] [Google Scholar]
- Wahlsten D. 1981. Prenatal schedule of appearance of mouse brain commissures. Brain Res. 227:461–473. [DOI] [PubMed] [Google Scholar]
- Wahlsten D. 1982. Deficiency of corpus callosum varies with strain and supplier of the mice. Brain Res. 239:329–347. [DOI] [PubMed] [Google Scholar]
- Wahlsten D, Bishop KM, Ozaki HS. 2006. Recombinant inbreeding in mice reveals thresholds in embryonic corpus callosum development. Genes Brain Behav. 5:170–188. [DOI] [PubMed] [Google Scholar]
- Wang LW, Huang CC, Yeh TF. 2004. Major brain lesions detected on sonographic screening of apparently normal term neonates. Neuroradiology. 46:368–373. [DOI] [PubMed] [Google Scholar]
- Zimmerman L, Parr B, Lendahl U, Cunningham M, McKay R, Gavin B, Mann J, Vassileva G, McMahon A. 1994. Independent regulatory elements in the nestin gene direct transgene expression to neural stem cells or muscle precursors. Neuron. 12:11–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}