

Abstract
Background
The CHL1 gene codes for a member of the L1 family of neural cell adhesion molecules. It is highly expressed in the central and peripheral nervous system playing an important role in the building and functioning on the brain. CHL1 proteins are also involved in axonal migration, synaptic formation and plasticity. In mice, functional studies showed that the haploinsufficiency of Chl1 gene in the developing brain results in cognitive deficits suggesting that the CHL1 gene at 3p26.3 is a candidate for an autosomal form of intellectual disability. Furthermore, in humans deletions of CHL1 have been described in patients with neurodevelopmental delay characterized by learning and language difficulties, seizures. Less is known about the potential effect of CHL1 overexpression, and microduplications of CHL1 have been rarely identified.
Case presentation
In this report, we describe a male patient with a phenotype characterized by developmental delay, symptoms of hyperactivity, short attention span and speech delay. In addition, minor facial dysmorphic features have been observed. Chromosomal microarray analysis revealed a rare de novo 0.85 Mb microduplication on the short arm (p26.3) of chromosome 3, encompassing a single gene, CHL1. To the best of our knowledge, duplication of chromosome 3p26.3, including only the CHL1 gene, has been described in only one intellectually disabled girl with epilepsy. The duplication described here is the smallest reported so far. In addition, this is the first report describing a patient in which the CHL1 duplication is a de novo event.
Conclusions
The clinical and molecular findings reported here are useful to provide further evidence that CHL1 is a dosage sensitive gene suggesting that not only the deletion but also its duplication can cause non-syndromic neurodevelopmental phenotypes.
Background
Cell adhesion molecules mediate various interactions between cells and also between cells and the extracellular matrix in developing and mature brain. Thus, they are intimately involved in the regulation of brain development and function. The cell adhesion molecule L1-like (CHL1) gene, located at the chromosomal sub-band 3p26.3, codes for a cell adhesion molecule of the immunoglobulin superfamily due to the presence of six Ig-like domains in its extracellular part. CHL1 is highly expressed in neurons but is also detectable in astrocytes, oligodendrocytes, and Schwann cells [1]. In the developing brain, CHL1 regulates neurite outgrowth [1] and neuronal migration [2], while in mature neurons it accumulates in the axonal membrane and regulates synapse function [3]. Deletions or mutations of CHL1 have been associated with learning and language difficulties while Chl1+/− and Chl1−/− knockout mouse models have provided evidence that Chl1 may contribute to mental impairment associated with “3p-syndrome” [4, 5]. Less is known about the clinical consequences due to the reciprocal microduplications. Up to date, only one patient, a female, carrier of a microduplication in the CHL1 gene, inherited from her healthy father, has been reported in literature [6]. The proband showed significant ID, marked speech development delay, generalized tonic-clonic seizures. Being the first patient reported in literature carrier of a duplication encompassing only the CHL1 gene, and since the duplication was inherited from a healthy parent, it remained unclear whether or not CHL1 was responsible for the clinical phenotype observed. Here, we report another patient with developmental delay (DD), symptoms of hyperactivity, short attention span and speech delay who has a de novo duplicated region, less than 1 megabase in size, encompassing only CHL1.
We review the clinical and molecular features of CHL1 gene duplication cases discussing the function of the gene and its role in the etiology of the observed phenotypes.
Case presentation
Case report
The patient is the first child of healthy, non-consanguineous parents. He was born at term after a normal pregnancy by cesarean section. Karyotype was normal male. His younger brothers had normal development and schooling. No family history of congenital anomalies or DD/ID was referred. Development was normal during the neonatal period, no feeding problems were reported. He managed to walk unsupported at the age of 1 year showing, since the first year of life, language problems.
Medical Geneticist first clinically assessed the patient at 2 years and 3 months. His height was 96 cm (90–97th centile), weight was 15 kg (75–90th centile) and head circumference was 52 cm (50–75th centile). Physical examination revealed minor dysmorphic facial features consisting of mild hypertelorism, down-slanting long palpebral fissures with eversion of lateral third of lower eyelids, long philtrum, thin upper lip, mildly prominent ear lobes (Fig. 1). Sleeping and feeding have been considered normal, as well as brain MRI, EEG, ECG, ocular and audiological assessment, abdomen echography, carpal bone X-rays. Routine blood exams, aminoacidemia/aminoaciduria, urinary organic acids’ panel and acyl-carnitine blood spot were all in normal ranges.
Fig. 1.
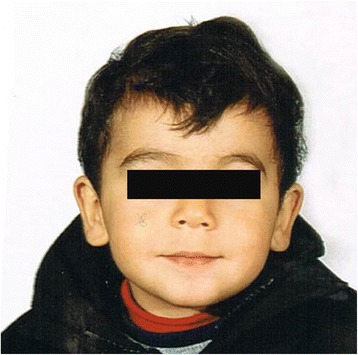
Face of the proband at age of 2 years and 3 months showing mild facial dysmorphic features listed in the text
Although the patient’s behavior was friendly and sociable, he showed symptoms of hyperactivity and his attention span was short, but he was too young to confirm a diagnosis of attention deficit hyperactivity disorder (ADHD). Language development was delayed, and he was able to say few words, incorrectly.
At his most recent clinical evaluation, at the age of 3 years and 3 months, the patient had difficulties to focus and sustain his attention. In addition, he shoved hyperactivity and severe speech delay. The family stimulated spontaneous speech associated with few unarticulated words. A neuropediatric examination ruled out a neurological defect. Fragile X screening was unremarkable.
Results
Chromosomal microarray analysis (CMA), performed by high resolution SNP-array, showed an approximately 850 kb duplication on the short arm of chromosome 3 spanning 1,005 probes: arr[hg19]3p26.3(125,931–975,649)x3, encompassing a single coding gene, CHL1. The adjacent probes of normal copy number are at position 125,742 distally, and 978,510 proximally, for a maximum duplication size of 852 kb. The duplication was not found in the parents and was thus de novo (Fig. 2). We carefully evaluated the microarray results and we excluded the presence of other significant genomic imbalance. The duplication was confirmed by an independent array (data not showed).
Fig. 2.
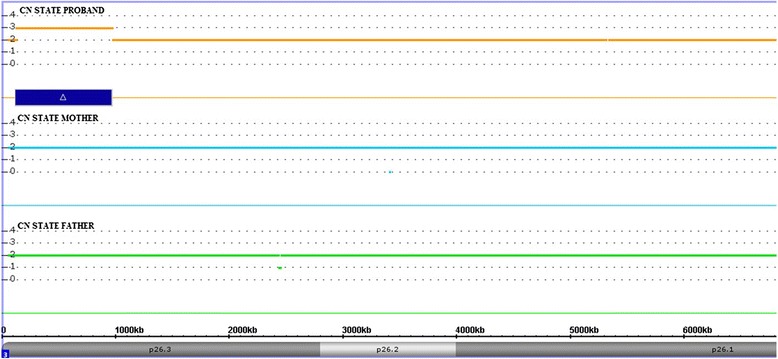
Chromosome microarray analysis performed with the Affymetrix CytoScan HD array and visualized using the Affymetrix Chromosome Analysis Suite version 3.0. Copy number state of each probe is drawn along chromosome 3 from 1 to 6.800.000 bp. The upper panel represents the copy number state of the proband, the middle panel the mother and the lower panel the father. Values of Y-axis indicate the inferred copy number according to probe intensities. Blue bar is the duplicated region identified in the patient
Discussion
In this report, we present the first patient to date reported with a de novo whole gene duplication of CHL1 in 3p26.3 chromosomal region. This is the only duplication encompassing CHL1 identified in our database of over 3,000 individuals referred for copy number variation (CNV) analysis. In addition, duplications of CHL1 have not been reported in healthy individuals suggesting a causative role for duplication of this gene in our patient’s abnormal phenotype.
CHL1 encodes for a 1224-amino acid cell adhesion protein that belongs to the immunoglobulin superfamily due to the presence of six Ig-like domains in its extracellular part. It is highly expressed in neurons but is also detectable in astrocytes, oligodendrocytes, and Schwann cells. Recent work suggests that CHL1 functions as a regulator of synaptic chaperones and vesicle exocytosis. Studies of Chl1-deficient mice have shown impaired sensorimotor gating and neuronal connectivity [7, 8].
In humans, deletions affecting CHL1 underline a spectrum of neurodevelopmental disorders. To date four familial cases presenting deletions of chromosome 3p26.3 confined to CHL1 gene have been described. Pohjola and collaborators [9] described a patient showing mild learning problems, microcephaly and growth retardation carrier of a CHL1 microdeletion inherited from his normal mother; later, a pair of siblings has been reported with features including microcephaly, learning difficulties, and ID who inherited a deletion encompassing only the CHL1 gene from their asymptomatic father [10]. More recently, Tassano et al. [11] described a maternally inherited 0.95 Mb deletion on the 3p26.3, which removed only the CHL1 gene, in a male with microcephaly, short stature, mild ID, learning and language delay, and strabismus.
Since the shared deleted region between the reported cases encompassed only the CHL1 gene, and this latter is highly expressed in the brain, the authors proposed that although the deletion may have incomplete penetrance, the haploinsufficiency of the CHL1 gene could be the main factor contributing to neurodevelopmental delay observed in these patients.
Of note, a balanced translocation [46,Y,t(X;3)(p22.1;p26.3)] with a breakpoint within intron five of the CHL1 gene has previously been linked to ID, and an animal model showed that Chl1+/− mice have a phenotype spectrum ranging from wild type to behavioral abnormalities [5].
Taken together, these clinical, molecular and functional data corroborate the hypothesis that CHL1 is the candidate gene for the cognitive impairments in these patients.
Less is known about the potential effect of CHL1 overexpression, and microduplications of CHL1 have been rarely identified.
Until now, only another patient carrier of a CNV similar for size and chromosomal location to that identified in our patient, has been reported in medical literature by Shoukier et al. [6]. The clinical manifestations of the present case along with the patient reported by Shoukier et al. are listed in Table 1 while the molecular data of the two patients are presented in Fig. 3. A review of clinical features in these two patients revealed overlapping phenotypes, namely, ID/DD and speech delay. Since CHL1 has important regulatory functions both in developing brain and in mature neurons [1–3], we conclude that CHL1 duplication is likely responsible for the patient’s phenotype. Our observation is also corroborated by the fact that the duplication reported in the present case is the first described to date as de novo event in a patient with non-syndromic developmental delay. In addition, copy number variations of genes encoding for neural cell adhesion molecules (NCAM), have been recently published in the literature as responsible for neurodevelopmental disorders further strengthening the hypothesis that chromosomal alteration affecting this family of genes can cause neurodevelopmental disorders [12].
Table 1.
Summary of the clinical features and molecular data of the reported patients with 3p26.3 microduplication encompassing only the CHL1 gene
| Features | Present case | Shoukier et al. [6] |
|---|---|---|
| Dup. size | 0.85 Mb | 1.0 Mb |
| Coordinates (hg19) | 125,931–975,649 | 48,914–1,054,209 |
| Inheritance | de novo | maternal |
| Sex and age at diagnosis | M, 2.3 years | F, 16 years |
| Weight | 15 Kg (75–90th centile) | 57 Kg (50th centile) |
| Height | 96 cm (90–97th centile) | 157 cm (25th centile) |
| Head circumference | 52 cm (50–75th centile) | 53.4 cm (25th centile) |
| DD/ID | + | + |
| Language delay | + | + |
| Seizure | - | + |
| Hyperactivity/attention deficit | + | - |
| Dysmorphisms | + | - |
| Delivery | at term | at term |
| Age at walking | 12 months | 15 months |
| Age at first words | 20 months | 24 months |
Dup duplication, M male, F female, + present, − absent, DD developmental delay, ID intellectual disability, NR not reported
Fig. 3.
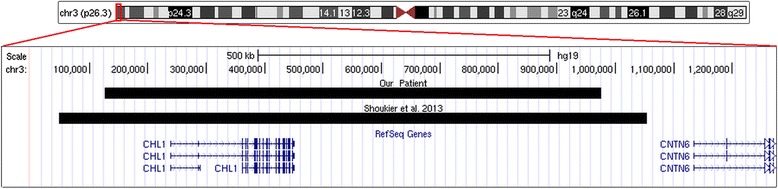
Snapshot of the 3p26.3 region displayed using the UCSC Genome browser [GRCh37/hg19 assembly; http://genome.ucsc.edu] showing the CHL1 duplications seen in the present patient and in the first patient reported in the literature
Interestingly, epileptic seizures were reported in the patient described by Shoukier et al. [6], whereas symptoms of hyperactivity and short attention span were observed in our. Given the young age of ours patient, we cannot exclude that he will manifest later signs of epilepsy. In addition, since in one of the patients described by Cuoco et al. [10], carrier of a CHL1 microdeletion, seizure has been observed, we suggest a clinical surveillance for this feature in the patients with CNVs encompassing the CHL1 gene.
Finally, it is possible that the genes located in the neighboring regions of the duplication have a role in the etiology of the clinical phenotype reported in the patient. In fact, chromosomal rearrangements frequently lead to alteration of the genes’ environment and this may be reflected in a change of expression, referred as a position effect [13]. Of note, located at ~159 kb of the centromeric breakpoint of the 3p26.3 duplication identified in the patient, there is contactin 6 (CNTN6) gene (Fig. 2), which could be affected by the rearrangement and related to the phenotype. CNTN6 is a neuronal membrane protein acting as a cell adhesion molecule involved in the formation of axon connection in the developing nervous system. As well as for other members of this protein family, CNTN6 has been suggested as a disease-causing gene in neurodevelopmental disorders [14]. In mice, CNTN6 participates in embryonic development and postnatal brain maturation and its deficiency causes profound motor coordination abnormalities and learning difficulties [15]. In humans, microdeletions and microduplications of CNTN6 have been reported in patients with DD, ID, speech and language delays, atypical autism suggesting that under- and overexpression of this gene is responsible for the observed phenotypes [16]. In agreement with Shoukier et al. [6], we cannot exclude the existence of a position effect on the CNTN6 gene, as already reported for others copy number variations [17, 18]. Obviously, this observation need to be elucidated by further gene expression studies either on experimental in vivo animal models or on diagnostic material.
Conclusions
In conclusion, to our knowledge this is the first reported case of an isolated de novo CHL1 duplication in a patient with a non-syndromic clinical phenotype characterized by developmental and speech delays, hyperactivity and short attention span. Our data are useful to better understand the role that the duplicated gene play in the clinical outcome, corroborating the hypothesis that not only the deletion but also the duplication of CHL1 is associated with non-syndromic forms of DD/ID.
Materials and methods
Snp array analysis
We extracted DNA from the lymphocytes of patient and his parents using BioRobot EZ1 (Qiagen, Solna, Sweden). Genomic screening for copy number variations (CNVs) was carried out using the CytoScan HD array platform (Affymetrix, Santa Clara, CA) as previously described [19]. The array contains more than 2,600,000 CNV markers across the genome, including 750,000 genotype-able single nucleotide polymorphism (SNP) markers. Data analysis were performed using the Chromosome Analysis Suite Software version 3.0: (1) the raw data file (CEL) was normalized using the default options; (2) an unpaired analysis was performed using as baseline 270 HapMap samples in order to obtain Copy numbers value from. CEL files while the amplified and/or deleted regions were detected using a standard Hidden Markov Model (HMM) method. A copy number variation was validated if an abnormal log2 ratio was obtained for at least 25 contiguous probes. DNA sequence information refer to the public UCSC database hg19 assembly (Build GRCh37, February 2009) while molecular karyotype was designated according to ISCN 2013.
Consent
A copy of the written consent is available for review by the Editor of this journal.
Acknowledgements
This study was supported by grants of the Italian Ministry of Health (Ricerca Corrente 2015 and RF2011-02350693) to MC, and partially funded by the “Progetto Operativo Nazionale”, PON 2007–2013 LAB GTP (PON02_00619). We are grateful to the patient and his family for agreeing to take part in this study.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RF, FN, FP and LZ carried out the clinical genetic diagnosis; OP and PP provided the SNP array analysis and the interpretation of results; OP wrote the manuscript; MC supervised the study and reviewed the paper. All authors read and approved the final manuscript.
Contributor Information
Orazio Palumbo, Email: o.palumbo@operapadrepio.it.
Rita Fischetto, Email: rfischetto@libero.it.
Pietro Palumbo, Email: p.palumbo@operapadrepio.it.
Francesco Nicastro, Email: nicastrofrancesco@yahoo.it.
Francesco Papadia, Email: papadiaf@gmail.com.
Leopoldo Zelante, Email: zelante@operapadrepio.it.
Massimo Carella, Email: m.carella@operapadrepio.it.
References
- 1.Hillenbrand R, Molthagen M, Montag D, Schachner M. The close homologue of the neural adhesion molecule L1 (CHL1). Patterns of expression and promotion of neurite outgrowth by heterophilic interactions. Eur J Neurosci. 1999;11:813–26. doi: 10.1046/j.1460-9568.1999.00496.x. [DOI] [PubMed] [Google Scholar]
- 2.Buhusi M, Midkiff BR, Gates AM, Richter M, Schachner M, Maness PF. Close homolog of L1 is an enhancer of integrin-mediated cell migration. J Biol Chem. 2003;278:25024–31. doi: 10.1074/jbc.M303084200. [DOI] [PubMed] [Google Scholar]
- 3.Leshchyns’ka I, Sytnyk V, Richter M, Andreyeva A, Puchkov D, Schachner M. The adhesion molecule CHL1 regulates uncoating of clathrin-coated synaptic vesicles. Neuron. 2006;52:1011–25. doi: 10.1016/j.neuron.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 4.Angeloni D, Lindor NM, Pack S, Latif F, Wei MH, Lerman MI. CALL gene is haploinsufficient in a 3p- syndrome patient. Am J Med Genet. 1999;86:482–5. doi: 10.1002/(SICI)1096-8628(19991029)86:5<482::AID-AJMG15>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 5.Frints SG, Marynen P, Hartmann D, Fryns JP, Steyaert J, Schachner M, et al. CALL interrupted in a patient with non-specific mental retardation: gene dosage-dependent alteration of murine brain development and behavior. Hum Mol Genet. 2003;12:1463–74. doi: 10.1093/hmg/ddg165. [DOI] [PubMed] [Google Scholar]
- 6.Shoukier M, Fuchs S, Schwaibold E, Lingen M, Gärtner J, Brockmann K, et al. Microduplication of 3p26.3 in nonsyndromic intellectual disability indicates an important role of CHL1 for normal cognitive function. Neuropediatrics. 2013;44:268–71. doi: 10.1055/s-0033-1333874. [DOI] [PubMed] [Google Scholar]
- 7.Montag-Sallaz M, Baarke A, Montag D. Aberrant neuronal connectivity in CHL1-deficient mice is associated with altered information processing-related immediate early gene expression. J Neurobiol. 2003;57:67–80. doi: 10.1002/neu.10254. [DOI] [PubMed] [Google Scholar]
- 8.Irintchev A, Koch M, Needham LK, Maness P, Schachner M. Impairment of sensorimotor gating in mice deficient in the cell adhesion molecule L1 or its close homologue, CHL1. Brain Res. 2004;1029:131–4. doi: 10.1016/j.brainres.2004.09.042. [DOI] [PubMed] [Google Scholar]
- 9.Pohjola P, de Leeuw N, Penttinen M, Kääriäinen H. Terminal 3p deletions in two families--correlation between molecular karyotype and phenotype. Am J Med Genet A. 2010;152A:441–6. doi: 10.1002/ajmg.a.33215. [DOI] [PubMed] [Google Scholar]
- 10.Cuoco C, Ronchetto P, Gimelli S, Béna F, Divizia MT, Lerone M, et al. Microarray based analysis of an inherited terminal 3p26.3 deletion, containing only the CHL1 gene, from a normal father to his two affected children. Orphanet J Rare Dis. 2011;6:12. doi: 10.1186/1750-1172-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tassano E, Biancheri R, Denegri L, Porta S, Novara F, Zuffardi O, et al. Heterozygous deletion of CHL1 gene: detailed array-CGH and clinical characterization of a new case and review of the literature. Eur J Med Genet. 2014;57:626–9. doi: 10.1016/j.ejmg.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 12.Petit F, Plessis G, Decamp M, Cuisset JM, Blyth M, Pendlebury M, et al. 21q21 deletion involving NCAM2: report of 3 cases with neurodevelopmental disorders. Eur J Med Genet. 2015;58:44–6. doi: 10.1016/j.ejmg.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Kleinjan DJ, van Heyningen V. Position effect in human genetic disease. Hum Mol Genet. 1998;7:1611–8. doi: 10.1093/hmg/7.10.1611. [DOI] [PubMed] [Google Scholar]
- 14.Zuko A, Kleijer KT, Oguro-Ando A, Kas MJ, van Daalen E, van der Zwaag B, et al. Contactins in the neurobiology of autism. Eur J Pharmacol. 2013;719:63–74. doi: 10.1016/j.ejphar.2013.07.016. [DOI] [PubMed] [Google Scholar]
- 15.Sakurai K, Toyoshima M, Ueda H, Matsubara K, Takeda Y, Karagogeos D, et al. Contribution of the neural cell recognition molecule NB-3 to synapse formation between parallel fibers and Purkinje cells in mouse. Dev Neurobiol. 2009;69:811–24. doi: 10.1002/dneu.20742. [DOI] [PubMed] [Google Scholar]
- 16.Kashevarova AA, Nazarenko LP, Schultz-Pedersen S, Skryabin NA, Salyukova OA, Chechetkina NN, et al. Single gene microdeletions and microduplication of 3p26.3 in three unrelated families: CNTN6 as a new candidate gene for intellectual disability. Mol Cytogenet. 2014;7:97. doi: 10.1186/s13039-014-0097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurth I, Klopocki E, Stricker S, van Oosterwijk J, Vanek S, Altmann J, et al. Duplications of noncoding elements 5' of SOX9 are associated with brachydactyly-anonychia. Nat Genet. 2009;41:862–3. [DOI] [PubMed]
- 18.Palumbo O, Palumbo P, Delvecchio M, Palladino T, Stallone R, Crisetti M, et al. Microdeletion of 12q24.31: report of a girl with intellectual disability, stereotypies, seizures and facial dysmorphisms. Am J Med Genet A. 2015;167A:438–44. doi: 10.1002/ajmg.a.36872. [DOI] [PubMed] [Google Scholar]
- 19.Palumbo O, Fichera M, Palumbo P, Rizzo R, Mazzolla E, Cocuzza DM, et al. TBR1 is the candidate gene for intellectual disability in patients with a 2q24.2 interstitial deletion. Am J Med Genet A. 2014;164A:828–33. doi: 10.1002/ajmg.a.36363. [DOI] [PubMed] [Google Scholar]