

Abstract
Background
Mycoplasma pneumoniae is a common pathogen that causes upper and lower respiratory tract infections in people of all ages, responsible for up to 40 % of community-acquired pneumonias. It also causes a wide array of extrapulmonary infections and autoimmune phenomena. Phylogenetic studies of the organism have been generally restricted to specific genes or regions of the genome, because whole genome sequencing has been completed for only 4 strains. To better understand the physiology and pathogenicity of this important human pathogen, we performed comparative genomic analysis of 15 strains of M. pneumoniae that were isolated between the 1940s to 2009 from respiratory specimens and cerebrospinal fluid originating from the USA, China and England.
Results
Illumina MiSeq whole genome sequencing was performed on the 15 strains and all genome sequences were completed. Results from the comparative genomic analysis indicate that although about 1500 SNP and indel variants exist between type1 and type 2 strains, there is an overall high degree of sequence similarity among the strains (>99 % identical to each other). Within the two subtypes, conservation of most genes, including the CARDS toxin gene and arginine deiminase genes, was observed. The major variation occurs in the P1 and ORF6 genes associated with the adhesin complex. Multiple hsdS genes (encodes S subunit of type I restriction enzyme) with variable tandem repeat copy numbers were found in all 15 genomes.
Conclusions
These data indicate that despite conclusions drawn from 16S rRNA sequences suggesting rapid evolution, the M. pneumoniae genome is extraordinarily stable over time and geographic distance across the globe with a striking lack of evidence of horizontal gene transfer.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1801-0) contains supplementary material, which is available to authorized users.
Keywords: Arginine deiminase, CARDS toxin, Epigenetics
Background
Mycoplasma pneumoniae is a parasitic bacterium belonging to Mollicutes, a class of bacteria lacking cell walls and typically having small genomes under 1,000 kb. It is a common pathogen of the upper and lower respiratory tract of humans in all age groups worldwide. It is also the most comprehensively analyzed species of Mycoplasma, with recent studies characterizing M. pneumoniae’s transcriptome, proteome and metabolome [1–3]. M. pneumoniae causes up to 40 % of community acquired pneumonias [4]. Although the infection is mild in most cases, patients can occasionally develop severe to fatal diseases. In addition to respiratory infections, as many as 25 % of M. pneumoniae infections are accompanied by extrapulmonary complications, which can affect almost any organ system either by direct infection or by infection-associated autoimmune phenomena [4, 5].
The pathogenicity of M. pneumoniae is still under active investigation and several virulence mechanisms have been identified. M. pneumoniae is primarily an extracellular pathogen requiring close association with host cells to survive as its highly reduced genome renders it incapable of de novo synthesis of amino acids, nucleotides, and other essential molecules. Also, the mycoplasmas are unique among bacteria in their growth requirement for host cholesterol. Adherence to the host respiratory epithelium is believed to be the initiating event that facilities local cell injury, tissue disruption, and cytotoxic effects [4]. Several protein components of the adhesin complex have been identified including the P1 protein. The two M. pneumoniae subtypes, type 1 and type 2, were established based on P1 sequence polymorphisms [6]. Hydrogen peroxide and superoxide radicals are known virulence factors of M. pneumoniae [7, 8]. The Community Acquired Respiratory Distress Syndrome (CARDS) toxin, an ADP-ribosylating and vacuolating toxin of M. pneumoniae, is capable of inducing pulmonary inflammation and airway hyperreactivity [9–14]. Inappropriate host immune responses also contribute to the pathogenesis of M. pneumoniae infection. The molecular mimicry by M. pneumoniae adhesin proteins and glycolipids of various host cell components may trigger autoimmune disorders that involve multiple organ systems [4, 15]. M. pneumoniae may also be a facultative intracellular pathogen; viable bacteria have been shown to move into the interior of human cells in vitro [16]. This aspect of the organism’s life cycle and the ability to form biofilms on epithelial tissue likely contribute to the establishment of chronic infection [17].
Whole genome sequencing has greatly facilitated our understanding of M. pneumoniae. At present there are 4 distinct M. pneumoniae strains completely sequenced. The genome of the type 1 strain M129 (ATCC 29342) was sequenced by using a laborious approach involving the construction of an ordered cosmid library. The sequence was reported in 1996 and reannotated in 2000 as having 816,394 bp, 730 genes, and an average GC content of 40 % [14, 18]. The genome sequences of the type 2 strains FH and 309 were completed using next generation sequencing methods (Roche 454 sequencers) [19, 20]. Another strain, M29 was recently submitted (accession number GCA_00733995.1) and has not yet been annotated. A preliminary comparison of the first three genomes indicated that they are very similar, except for variation in a 6-kb insertion region coding lipoproteins [20]. We have resequenced M129 and FH and also have sequenced 13 additional strains obtained from different geographic regions over a period of several decades. For each strain, the sequence was completed to generate a single, circular contig. Analysis of the genomes reveals numerous differences between type 1 and type 2 isolates but a striking degree of homogeneity between strains of the same type, suggesting clonality.
Methods
M. pneumoniae strains
A total of 15 M. pneumoniae strains were sequenced in this study (Table 1), including 11 clinical isolates and 4 reference ATCC strains. These strains were originally isolated over a wide period of time, 1944 to 2009, and geographic range, North America, Asia and Europe. Except for one specimen from cerebrospinal fluid, most of these clinical strains were isolated from respiratory specimens after minimal growth in culture media. There is one macrolide resistant strain from the US (54089). The ATCC strains were purchased from ATCC in 2002, grown in 2005, and the 3rd passage was used in this study.
Table 1.
Summary of the sequenced strains
| Strain | Description | Isolation source | Date collected | Site originated | |
|---|---|---|---|---|---|
| Type 1 | M129 | ATCC 29342 (Reference Strain) | Patient with pneumonia | 1968 | USA/NC |
| 142.8 | ATCC 29085 | Throat | 1960 | USA/MD | |
| 51494 | Clinical isolate | Cerebral spinal fluid | 2006 | USA/CO | |
| 54089 | Macrolide resistant isolate | Throat | 2009 | USA/AL | |
| 54524 | Low passage isolate | Throat | 2009 | USA/AL | |
| 85084 | Respiratory specimen | Prior to 1985 | China | ||
| 85138 | Respiratory specimen | Prior to 1985 | China | ||
| Type 2 | FH | ATCC 15531 (Reference Strain) | Sputum | 1954 | USA/MA |
| 19294 | Low passaged isolate | Throat | 1994 | USA/OH | |
| 39443 | Low passaged isolate | Throat | 1999 | USA/AL | |
| M1139 | Respiratory specimen | 1981 | England | ||
| M2192 | Respiratory specimen | 1982 | England | ||
| M2592 | Respiratory specimen | 1982 | England | ||
| MAC | ATCC 15492 | Human lung tissue | 1944 | USA/CA | |
| UAB PO1 | Low passage isolate | Throat | 1980 | USA/AL | |
Culture and DNA preparation
All strains were grown in 25 ml SP4 medium in T-flasks at 36.5 °C until color change. Non-adherent organisms were discarded. The adherent organisms were gently washed twice with 10 ml PBS (phosphate buffered saline, pH 7.4), scraped from the bottom of the flasks, and suspended in 5 ml PBS. Genomic DNA was purified using the QIAamp DNA Blood Maxi Kit (Qiagen, Valencia, CA) according to manufacturer’s instruction. After determining the DNA concentration (NanoDrop 1000, Wilmington, DE) and quality (0.8 % agarose gel), all DNA products were stored at −80 °C until use.
Next-generation sequencing (NGS)
NGS of all M. pneumoniae strains was performed using the Illumina MiSeq platform in the UAB Heflin Genomic Core. Paired-end 250-bp reads were used.
Genome assembly and annotation
NGS sequencing reads were assembled de novo using ABySS v 1.3.7 [21]. Kmer values were tested iteratively to find the value yielding an assembly with the lowest number of contigs, while retaining a total contig length of approximately 800 kb. The s parameter was changed to double the kmer value, per recommendations by the developer. All other parameters were set to default. The number of contigs generated ranged from 5 to 13 per strain were generated by de novo assembly. These contigs were mapped to the M129 reference genome using BLAT [22] and visualized using IGV [23, 24]. This mapping was used to develop PCR primers to join the contigs. High fidelity PCR reactions and Sanger sequencing were performed using standard methods. Overlapping and joining of the contigs was performed manually with the aid of HVDR fragment merger tool [25] and Audrius Meskauskas’s reverse complement tool [26]. Completed, circularized genomes were annotated using RAST [27, 28], the NCBI prokaryotic pipeline [29] and manual reconciliation.
Single nucleotide polymorphism (SNP) and insertion/deletion (indel) analysis
To call SNPs and indels, completed genomes were first broken into 10-kb “reads” at 1-kb intervals and then aligned to either the M129 or FH reference strains (NCBI accession numbers NC_000912 and NC_017504, respectively) using BWA v0.7.7 [30]. The resulting BAM files were used as input for GATK v3.0-0 [31]. We used GATK’s Unified Genotyper and Haplotype Caller to call SNPs and indels. Because the “reads” came from an assembled genome, a perfect quality score was assigned to each base. This necessitated the use of the allow PotentiallyMisencodedQuals parameter when running GATK’s Realigner Target Creator. Otherwise, GATK was run using standard parameters according to GATK Best Practices recommendations [32, 33]. The effects of the SNPs and indels in the resulting VCF files were evaluated using snpEff v3.3 [34]. Although snpEff does annotate each SNP and indel with the gene that they fall into, we reannotated the VCF files using the latest annotations of the M129 and FH genomes downloaded from NCBI.
Functional annotation
Functional annotation of gene lists was performed using BRITE search from the Kyoto Encyclopedia of Genes and Genomes (KEGG) [35].
Protein sequence analysis
Gene sequences were downloaded from the RAST server after annotation. To identify specific genes, these sequences in FASTA format were compared to the reference sequence as found on NCBI by using BLAST. Genes were translated using the translation tool at Bioinformatics Organization [36], and the protein sequences aligned using CLUSTAL Omega [36].
Comparative genomics
Completed genomes were aligned using BRIG [37] to visualize overall sequence similarity between the strains. The annotated genomes, in the form of GenBank files from RAST, were aligned with MAUVE [38] to identify structural variations and which genes they may affect. For phylogenetic tree generation, completed genomes were aligned using MAFFT [39, 40] via the CIPRES science gateway [41]. We generated phylogenetic trees from the genome alignment using MrBayes [42]. To generate trees for protein sequences, Clustal X [43] was used to align protein sequences and to generated trees. For both genome sequence and protein sequence trees, 1000 iterations of boostraping analysis were used to generate confidence values. Trees were visualized using Dendroscope [44, 45] and FigTree [46]. Tandem repeats across the genome were identified by Tandem Repeat Finder 9 [47].
Results
Genome assembly
We sequenced the 15 M. pneumoniae strains with NGS and computationally de novo assembled them into contigs. The characteristics of these assemblies are found in Additional file 1: Table S1. The resulting contigs were mapped to the M129 reference genome and joined via PCR. All fifteen genomes had all contigs joined to form a single, continuous (circular) contig. Following assembly and editing, the genomes underwent automated gene annotation. Summary statistics for the completed genomes, including submission numbers are found in Table 2. These genomes, having about 40 % of GC and ranging from 816402 to 818633 bp, code for a total of 853 to 870 genes.
Table 2.
Characteristics of the completed genome assemblies
| Strain | Accession | Length | %GC | Genes | ||||
|---|---|---|---|---|---|---|---|---|
| CDS | rRNA | tRNA | Total | |||||
| Type 1 | M129 | CP003913 | 816451 | 0.40038 | 790 | 6 | 74 | 870 |
| 142.8 | CP010538 | 816496 | 0.40011 | 790 | 6 | 74 | 870 | |
| 51494 | CP010541 | 816404 | 0.40005 | 781 | 6 | 74 | 861 | |
| 54089 | CP010542 | 816565 | 0.40010 | 784 | 6 | 74 | 864 | |
| 54524 | CP010543 | 816583 | 0.40009 | 780 | 6 | 74 | 860 | |
| 85084 | CP010544 | 816404 | 0.40011 | 788 | 6 | 74 | 868 | |
| 85138 | CP010545 | 816402 | 0.40011 | 788 | 6 | 74 | 868 | |
| Type 2 | FH | CP010546 | 817207 | 0.39981 | 786 | 6 | 72 | 864 |
| 19294 | CP010539 | 818633 | 0.40001 | 780 | 6 | 72 | 858 | |
| 39443 | CP010540 | 817184 | 0.39979 | 782 | 6 | 72 | 860 | |
| M1139 | CP010547 | 817045 | 0.39977 | 789 | 6 | 72 | 867 | |
| M2192 | CP010548 | 817169 | 0.39978 | 783 | 6 | 72 | 861 | |
| M2592 | CP010549 | 817198 | 0.39981 | 783 | 6 | 72 | 861 | |
| MAC | CP010550 | 817156 | 0.39983 | 788 | 6 | 72 | 866 | |
| PO1 | CP010551 | 817216 | 0.39982 | 788 | 6 | 72 | 866 | |
Genome comparison
The 15 sequenced genomes were aligned using a variety of methods. To determine their overall similarity, the genomes were aligned to the reference M129 genome using BRIG, a BLAST-based alignment method. Overall, the genomes were 99 % to > 99 % identical; the similarity within each subtype group was less than 0.1 % difference among the strains. There was one distinct region where identity dropped to about 95 % in the type 2 strains (Fig. 1). This region corresponds to the P1 gene (Fig. 1). We also aligned the genomes using MAUVE to detect large chromosomal rearrangements, deletions, and duplications (Fig. 2a). MAUVE detected two subtype-specific insertions (Fig. 2b and c): the type 1-specific 557178–560601 (M129 numbering) insertion and type 2-specific 713023–713984 (M129 numbering) insertion. All of the genes affected by these insertions were hypothetical proteins, with the exception of a tRNA gene (MPNt26) in the type 1 specific insertion. MPNt26 codes for a serine TCG tRNA. Both type 1 and type 2 strains have another serine TCG tRNA gene (MPNt25) immediately upstream of the insertion point. The genomes (including M. hominis as an outgroup) were aligned with MAFFT and a phylogenetic tree was generated using MrBayes (Fig. 3). Not surprisingly, the 15 strains fall into 2 groups representing type 1 and type 2.
Fig. 1.
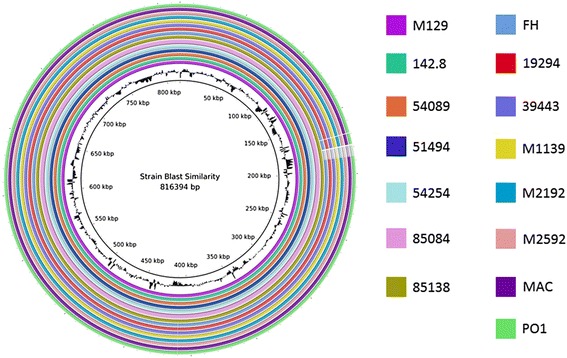
Overall sequence identity of the 15 sequenced strains with the reference M129 genome. BLAST-based similarity of a given strain versus the M129 reference is represented as a colored ring. Colors by strain are indicated to the right. Solid coloration indicates >99 % identity and transparent grey indicates approximately 95 % identity. Location in the reference genome is indicated by numeration on the inside of the ring. GC content in the reference genome is indicated by the black bar graphs between the genomic coordinates and the colored rings (bars pointing toward the outside of the circle indicate high GC content). Note that genomic structural alterations are not visible using this method
Fig. 2.
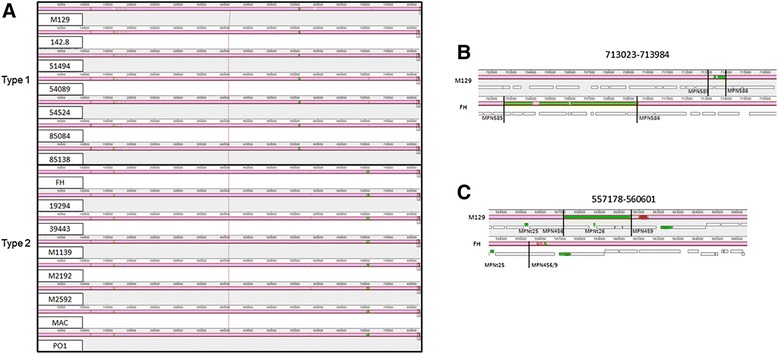
Whole genome alignment of the 15 sequenced strains using MAUVE. Regions colored in mauve are conserved across all strains. Differently colored blocks are conserved in some strains. Blocks that are lower are inverted relative to the other strains. Open boxes indicate the location of genes. tRNA genes are shaded in green and rRNA genes are shaded in red. Genes affected by the indicated variants are labeled. Numbers above intervals indicate locations relative to the M129 strain. a Alignment showing all 15 strains. b Close up of the type 2-specific insertion. M129 and FH are shown and are typical of the other type 1 and 2 strains, respectively. Lines indicate relative point of insertion. c Close up of the type 1-specific insertion. M129 and FH are shown and are typical of the other type 1 and 2 strains, respectively. Lines indicate relative point of insertion
Fig. 3.
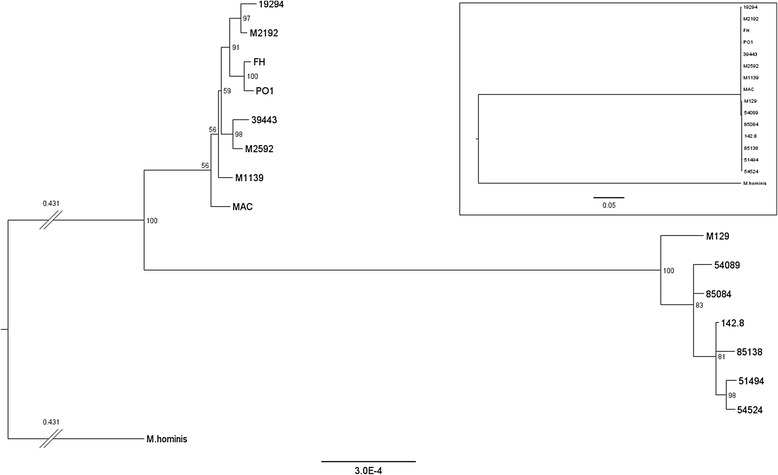
Phylogenetic tree based on whole genome alignment of the 15 sequenced strains. The 15 sequenced M. pneumoniae strains and M. hominis (included as an outgroup) were aligned, and a tree was generated using the alignment. Confidence values, represented as percent of supporting bootstrapping iterations are shown for each node. Scale, in differences per site, is indicated at the bottom. The branches between M. hominis and the M. pneumoniae strains have been truncated (indicated by double slashes), and the branch length (in differences per site) is indicated above the branch. The inset shows the same tree (rescaled, note the new scale bar) without any branches truncated
SNP and indel analysis
SNPs and indels were compared relative to type 1 and type 2 reference strains, M129 and FH, respectively. To our knowledge, there is no whole-genome alignment program that generates an alignment file that can be used with current SNP and indel callers like GATK. Instead of going back to the un-assembled short reads, we broke the completed genomes into 10-kb “reads” in 1-kb intervals, aligned these “reads” to the genome, and used the resulting BAM file as input for GATK. We tested GATK’s UnifiedGenotyper and HaplotypeCaller for genotyping. HaplotypeCaller is newer and calls indels better, but lacks a haploid-genome setting. Overall, the results were highly similar for the two SNP callers. For a subset of genes that were examined, SNP and indel calls were almost perfectly concordant. For indels involving tandem polynucleotide repeats, UnifiedGenotyper missed a few indels found by HaplotypeCaller. We therefore utilized the results from HaplotypeCaller. SNP and indel effects were annotated with snpEff, and we manually re-annotated gene symbols for the SNPs using the latest gene annotations of the M129 and FH reference strains from NCBI.
The SNP and indel counts, by predicted functional effect as annotated by snpEff, relative to the M129 (type 1) reference genome are found in Table 3. As expected, type 1 strains showed fewer variants (235-431) than type 2 strains (1569–1615). As a test of the system, the M129 reference sequence downloaded from NCBI was processed in the same way as the 15 sequenced strains and had SNPs called against itself. No variants were found. SNPs and indels were also called against the FH (type 2) reference genome, and the results were similar with about 1500 variants and 200 variants detected in type 1 and type 2 strains, respectively. No variants were detected when running the FH reference genome against itself. However, the gene annotation for the M129 reference genome appeared to be more complete with fewer unnamed genes. Therefore, all further variant analysis was done using the M129 (type 1) reference genome.
Table 3.
Variants relative to the M129 reference strain
| Strain | Total variants | Non-synonymous genic SNPs | Synonymous genic SNPs | Genic indels | IntragenicSNPs and indels | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Coding | Start lost | Stop gain | Stop lost | In frame | Frameshift | |||||
| Type 1 | M129 | 235 | 113 | 0 | 3 | 0 | 41 | 10 | 36 | 32 |
| 142.8 | 334 | 174 | 1 | 2 | 1 | 67 | 4 | 22 | 63 | |
| 51494 | 382 | 202 | 1 | 3 | 1 | 77 | 7 | 24 | 67 | |
| 54089 | 431 | 223 | 1 | 3 | 1 | 90 | 8 | 36 | 69 | |
| 54524 | 385 | 208 | 1 | 2 | 1 | 76 | 5 | 22 | 70 | |
| 85084 | 404 | 199 | 1 | 2 | 1 | 88 | 8 | 38 | 67 | |
| 85138 | 365 | 183 | 1 | 1 | 1 | 72 | 2 | 25 | 80 | |
| Type 2 | FH | 1581 | 705 | 0 | 8 | 6 | 474 | 35 | 109 | 244 |
| 19294 | 1606 | 705 | 0 | 11 | 6 | 490 | 33 | 110 | 251 | |
| 39443 | 1615 | 713 | 0 | 10 | 6 | 489 | 36 | 110 | 251 | |
| M1139 | 1581 | 687 | 0 | 8 | 6 | 485 | 33 | 114 | 248 | |
| M2192 | 1604 | 705 | 0 | 11 | 6 | 490 | 35 | 109 | 248 | |
| M2592 | 1588 | 697 | 0 | 8 | 6 | 484 | 33 | 111 | 249 | |
| MAC | 1569 | 677 | 0 | 8 | 7 | 477 | 34 | 114 | 252 | |
| PO1 | 1588 | 700 | 0 | 8 | 6 | 482 | 34 | 116 | 242 | |
To test the accuracy of our assemblies, we compared the variants in the resequenced M129 strain relative to the original M129 reference sequence. Of the 203 genic SNPs and indels in the resequenced M129, 101 were found in all other sequenced strains or in all of the sequenced type 1 strains, suggesting that these variants were actually errors in the original M129 sequence. The remaining 102 variants represent about 0.01 % of M129’s genomic sequence. A further 56 were found in at least one other type 1 strain (in almost all cases, the variant was found in all type 2 strains or in type 1 strains 54089 and 85084). Another 10 were found in several type 2 strains, but no type 1 strains. This left 36 variants that are unique to the resequenced M129 strain. These 36 variants, the most likely candidates for sequencing errors, variant miss-calls, or new mutations, represent about 0.004 % of M129’s genomic sequence. Two of these variants were found in the MPN413 gene and the rest were found in MPN489. These two genes code for proteins of unknown function.
To explore the variable and invariable regions of the M. pneumoniae genome, we identified the genes with the most and least non-synonymous variants in type 2 strains compared to the M129 (type 1) reference genome. The top 10 genes with the most non-synonymous variants are found in Table 4. When looking for genes with the fewest variants, we found 182 genes with no variants in any of the sequenced strains. The list of these genes is found in Additional file 2: Table S2. For those genes that could be classified by KEGG, a summary of the functional groups into which these genes fall is found in Table 5.
Table 4.
Type 2 genes with the most variants as compared to M129 (type 1)
| Gene | Function | FH | 19294 | 39443 | M1139 | M2192 | M2592 | MAC | PO1 |
|---|---|---|---|---|---|---|---|---|---|
| MPN457 | Unknown | 165 | 164 | 165 | 165 | 164 | 165 | 165 | 165 |
| MPN141 | P1 | 52 | 67 | 68 | 65 | 66 | 67 | 67 | 66 |
| MPN142 | ORF6 | 17 | 16 | 16 | 16 | 16 | 16 | 16 | 16 |
| MPN286 | Unknown | 13 | 14 | 13 | 13 | 14 | 13 | 13 | 13 |
| MPN205 | Unknown | 12 | 12 | 12 | 12 | 12 | 12 | 13 | 12 |
| MPN503 | Unknown | 10 | 10 | 10 | 11 | 10 | 10 | 12 | 10 |
| MPN439 | Unknown | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| MPN489 | Unknown | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| MPN370 | Unknown | 9 | 11 | 10 | 9 | 10 | 9 | 9 | 9 |
| MPN048 | Unknown | 10 | 9 | 9 | 10 | 9 | 9 | 10 | 10 |
Table 5.
Functional annotation of genes with no variants
| KEGG BRITE hierarchy | Count | Genes | |
|---|---|---|---|
| mpn03100 | Non-coding RNA | 37 | tRNAs, 5S rRNA, 4.5S rRNA, RNaseP RNA |
| mpn01000 | Enzymes | 28 | def, deoC, dhfr, gatB, gcp, gidB, grs1, lacA, lgt, lip2, lspA, nox, pheS, polA, ppnK, pstB, rimK, rnc, udk, upp, yaaC, yacA, ygiH, yjeQ, yjfU, yjfW, MPN047, MPN479 |
| mpn03011 | Ribosome | 25 | 50S and 30S ribosomal proteins, 5S rRNA |
| mpn0200 | Transporters | 9 | amiD, permease, glnQ, dnaK, pstA, pstB, oppB, yjfU |
| mpn03029 | Mitochondrial biogenesis | 9 | grs1, gatB, dnaJ, dnaK, groEL, groES, YidC, rpsP, ssb |
| mpn03009 | Ribosome Biogenesis | 5 | gidB, rnc, rimK, yjeQ, rbfA, spg |
| mpn03016 | Transfer RNA biogenesis | 5 | gidA, grs1, gcp, RNaseP RNA, pheS, yacA |
| mpn03110 | Chaperones and folding catalysts | 4 | dnaJ, dnaK, groEL, groES, trx |
| mpn03036 | Chromosome | 4 | gidA, gidB, rnc, scpB, soj |
| mpn02044 | Secretion system | 4 | yidC, secE, secG, MPN680 |
| mpn03400 | DNA repair and recombination | 4 | polA, recA, rpoE, ssb |
| mpn00194 | Photosynthesis proteins | 3 | atpG, atpF, atpE |
| mpn03012 | Translation factors | 2 | infA, efp |
| mpn01007 | Amino acid related enzymes | 2 | grs1, pheS |
| mpn03032 | DNA replication | 2 | polA, ssb |
| mpn04147 | Exosome | 2 | groEL, dnaK |
| mpn01002 | Peptidases | 2 | lspA, gcp |
| mpn03021 | Transcription machinery | 1 | rpoE |
| mpn01004 | Lipid biosynthesis proteins | 1 | ygiH |
| mpn04090 | Cellular antigens | 1 | cdd |
| mpn04812 | Cytoskeleton proteins | 1 | soj |
Gene specific analysis
The two adjacent genes P1 and ORF6 code for critical components of the M. pneumoniae adhesin complex. These genes are within the largest region of sequence polymorphism between type 1 and type 2 strains (Fig. 1) and are the top two named genes by non-synonymous SNP count. To examine the variance in these genes further, the protein sequences were aligned and found to have identical amino acid sequences for most of their length. However, for both P1 and ORF6, there was one region where type 1 and type 2 strains had virtually no sequence identity. The alignments for the regions of high variation are shown in Fig. 4. Other variants found in these genes not in the large region of variation are listed in Additional file 3: Table S3 and Additional file 4: Table S4. Most of these other variants, like the sequence of the large regions of variation, are subtype specific, rather than strain specific. However, there were also some unique, strain specific variations with potentially significant effects in both P1 and ORF6 (Fig. 5). One of these was a variation in the number of AGT trinucleotide repeats coding for serine in a region between the repetitive elements RepMP4 and RepMP2/3 [48] within the P1 gene. The number of serine repeats ranged from 5–17 with strain 19294 bearing the longest (Fig. 5a).
Fig. 4.

Multiple protein sequence alignments showing the differences in P1 and ORF6 between type 1 and type 2 strains. a The large region of variation in P1. Type1 is representative sequence for all type 1 strains and type2 is representative of all type 2 strains. b The large region of variation in ORF6. Type1 is representative of all type 1 strains, except M129, which is also shown (differences in M129 highlighted in red). Type2 is representative of all type 2 strains except for MAC, which is also shown (differences in MAC highlighted in red)
Fig. 5.

Multiple protein sequence alignments showing strain-specific differences in P1 and ORF6. Type1 and 2 strain names are highlighted in blue and green, respectively. a A poly-serine repeat in P1 with varying lengths in various strains. 19294 has a uniquely long allele of the poly-serine repeat, and the repeat-length in the other strains does not strictly correspond to strain type. b A stop-loss mutation in MAC results in an additional 7 amino acids added to the protein sequence. c A pair of frameshifts results in the truncation of the beginning of ORF6 in MAC. Type1 and Type2 are representative sequences for type 1 and other type 2 strains, respectively. The new protein is predicted to use an alternate start codon by RAST: the starting methionine in MAC is the same codon as that which codes for the leucine in other type 1 and type 2 strains
MPN372 codes for the CARDS toxin, an important virulence factor of M. pneumoniae. There were no non-synonymous variants in any of the type 1 strains, and one non-synonymous SNP common to all of the type 2 strains (T1112G, I371S). Additionally, the FH and M2192 strains each had one unique non-synonymous SNP (FH: C74T, S25L and M2192: G1507A, G503S). The unique SNP in FH was near, but not part of, the sequences comprising the active site of the CARDS toxin.
Our attention was also drawn to the M. pneumoniae arcA gene, an essential component of the arginine deiminase pathway that is thought to be inactive in M. pneumoniae [49]. There are two copies of arcA in M. pneumoniae, MPN304 and MPN560. MPN304 is truncated by a frameshift yielding a premature stop codon and is contiguous to arcC. MPN560 is not truncated but is found in another part of the genome. There were no variants in the coding sequence of either gene (before premature stop for MPN304) in any of the 15 sequenced strains. We also aligned the amino acid sequence of both proteins against the amino acid sequence of ArcA of several other Mycoplasma species. The protein sequence of arginine deiminase from Streptococcus pneumoniae strain R6 (spr0822) was included as an outgroup. MPN304 and MPN560 were more similar to ArcA sequences from other species than they were to each other, with MPN304 being most similar to ArcA from Mycoplasma fermentans and MPN560 being most similar to ArcA from Mycoplasma gallisepticum (Fig. 6).
Fig. 6.

Phylogenetic tree of ArcA. The protein sequences of the two ArcA protein sequences from M. pneumoniae (MPN304 and MPN560), ArcA sequences from other Mycoplasma species, and the protein sequence of arginine deiminase from Streptococcus pneumoniae strain R6 (spr0822_Spn_R6) were aligned and a tree was generated from the alignment. Confidence values, represented as percent of supporting bootstrapping iterations are shown for each node. Scale, in differences per site, is indicated at the bottom
The type I restriction and modification (R-M) enzymes protect bacteria from invading foreign DNA. They are composed of three types of subunits: hsdR encodes the restriction (R) subunit, hsdM encodes the modification (M) subunit and hsdS encodes the DNA sequence specificity (S) subunit [50]. They are pentameric enzymes with two R subunits, two M subunits and one S subunit. The S subunit is composed of two target recognition domains (TRDs) and each TRD comprises a DNA-binding domain and an alpha helical dimerization domain. The M129 genome has 2 hsdM (MPN198 and MPN342) and 10 hsdS genes scattered across the genome (Table 6). The hsdR gene contains frameshift mutations resulting in small ORFs (MPN345, MPN346, and MPN347) that are predicted to be nonfunctional. No prophages, conjugative elements, or genes unique to any one of the genomes were identified from the genome sequences. It is striking that the genome sequences of M. pneumoniae revealed so little evidence of horizontal gene transfer and the absence of an intact hsdR.
Table 6.
hsdS genes in M. pneumoniae strain M129
| Gene | Location (M129) | TR sequence | Amino acid repeat |
|---|---|---|---|
| MPN089 | 111610–112617 | CCGAGCTAAGCG | AELS |
| MPN201 | 244484–245569 | CCGAGCTAAG | AEL |
| MPN285 | 340244–341533 | CCGAGCTAAGTG(A) | A(T)ELS |
| MPN289 | 347169–347732 | CCGAGCTAAGCG | AELS |
| MPN290 | 347871–348308 | ||
| MPN343 | 409562–410863 | CCGAACTAAGCG | AELS |
| MPN365 | 435618–436730 | CCGAGCTAAGCG | AELS |
| MPN507 | 617366–618457 | ||
| MPN615 | 738245–739351 | CCGAGCTAAGCG | AELS |
| MPN638 | 764400–765527 |
The 10 hsdS genes are found in all sequenced strains (Table 7). MPN289 and MPN290 appear to be two truncated subunits derived from an integral hsdS locus that was interrupted by a point mutation resulting in a stop codon. MPN365 and MPN615 in all sequenced type 2 strains are truncated due to a premature stop. MPN285 is also truncated in 3 strains (MAC, PO1, and 142.8) due to frameshifts. Interestingly, a 12-bp tandem repeat (TR) corresponding to a 4-amino acids repeat (AELS or TELS) within the first alpha helical dimerization domain was found in 7 out of the 10 hsdS genes (Table 6). The copy number of this TR varies in 6 out of the 7 hsdS genes among the 15 strains (Table 7). It also varies in the same strains from different passages/laboratory conditions, e.g. in published M129 and FH genomes and our resequenced M129 and FH genomes (Table 7). Because two copies of the hsdS gene (MPN089 and MPN343) are part of two of the strain specific genomic structure variants annotated by MAUVE, we aligned the sequences of these proteins to look at variations in these genes. In both copies of the hsdS, the main source of variation is the TR region of varying length with two different repetitive units (TELS and AELS). The repeat in MPN089 consists only of AELS units, although all strains have one TELS unit, like all other TR-containing hsdS genes (Fig. 7a). The copy number of the repeat varies from 2 to 6 and does not correspond to strain subtype. However, in MPN343, the repeats are much longer in type 1 strains (10 – 16 copies) compared to type 2 strains (1 – 2 copies). Three type 1 strains (51494, 54089, and 54524) have long repeats of mixed TELS and AELS unit (Fig. 7b and Table 7).
Table 7.
Tandem repeat copy numbers of each hsdS gene in 15 M. pneumoniae strains
| Strain | MPN089 | MPN201 | MPN285 | MPN289 | MPN290 | MPN343 | MPN365 | MPN507 | MPN615 | MPN638 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Type 1 | M129 | 5 | 0 | 15 | 4 | 0 | 16 | 1 | 0 | 1 | 0 |
| M129 rs | 3 | 0 | 28 | 3 | 0 | 12 | 1 | 0 | 1 | 0 | |
| 142.8 | 3 | 0 | 21 | 5 | 0 | 12 | 1 | 0 | 5 | 0 | |
| 51494 | 3 | 0 | 24 | 1 | 0 | 10 | 1 | 0 | 4 | 0 | |
| 54089 | 2 | 0 | 33 | 4 | 0 | 13 | 1 | 0 | 4 | 0 | |
| 54524 | 3 | 0 | 31 | 6 | 0 | 11 | 1 | 0 | 5 | 0 | |
| 85084 | 6 | 0 | 16 | 3 | 0 | 13 | 1 | 0 | 4 | 0 | |
| 85138 | 5 | 0 | 14 | 4 | 0 | 13 | 1 | 0 | 5 | 0 | |
| Type 2 | FH | 5 | 0 | 15 | 4 | 0 | 1 | 1 | 0 | 1 | 0 |
| FH rs | 4 | 0 | 17 | 2 | 0 | 1 | 5 | 0 | 4 | 0 | |
| 19294 | 3 | 0 | 26 | 4 | 0 | 1 | 1 | 0 | 4 | 0 | |
| 39443 | 4 | 0 | 15 | 6 | 0 | 1 | 3 | 0 | 1 | 0 | |
| M1139 | 4 | 0 | 7 | 2 | 0 | 1 | 1 | 0 | 2 | 0 | |
| M2192 | 3 | 0 | 13 | 6 | 0 | 1 | 1 | 0 | 4 | 0 | |
| M2592 | 2 | 0 | 20 | 5 | 0 | 1 | 1 | 0 | 3 | 0 | |
| MAC | 4 | 0 | 15 | 3 | 0 | 2 | 3 | 0 | 4 | 0 | |
| PO1 | 3 | 0 | 16 | 6 | 0 | 1 | 4 | 0 | 4 | 0 |
Notes: Strain FH rs and M129 rs are our resequenced strains. Numbers in bold font indicate variations between the published and resequenced genomes
Fig. 7.

Multiple protein sequence of the variable regions in the hsdS genes. Both copies of the hsdS gene had a repetitive region of varying length consisting of TELS and AELS units (highlighted in orange and yellow, respectively). Note that in both copies, the length and composition of the repeat does not correspond to strain subtype. Strain names are to the right and highlighted in blue for type 1 and green for type 2. a Repeat region in the MPN089 copy of the hsdS gene. This is part of the variation in the 108000–126000 region shown in Fig. 2c. b Repeat region in the MPN343 copy of the hsdS gene. This is the variation in the 409700–410900 region shown in Fig. 2c
Macrolide resistance is increasing in M. pneumoniae and is often associated with mutations in 23S rRNA. The macrolide resistant strain 54089 was found to have a point mutation, A2063G (E. coli numbering), in its 23S rRNA gene. This mutation is common among macrolide resistant isolates of M. pneumoniae [51, 52].
Discussion
Quality of genome assembly
We present here 15 M. pneumoniae genomes in single, circular contigs. Our alignments of these genomes did not detect any apparent chromosomal alterations that were not found in other strains. Additionally, our comparison of the resequenced M129 strain with the original M129 reference sequence suggests a low level of assembly errors. Variants conserved in all sequenced strains likely represent errors in the original reference genome but some variants may be mutations that arose in the laboratory in which M129 was first sequenced. Similarly, variants found only in the resequenced M129 genome may be the result of assembly errors or mutations that occurred in our laboratory. Such variants were found in only two genes. These findings suggest that our genome assemblies are high quality and that the resequenced M129 genome is more accurate than the original.
Comparison of the genome assemblies
Overall the similarity between the M. pneumoniae strains was striking with > 99 % sequence identity. The similarity within each subtype group was even stronger with less than 0.1 % difference among strains of the same subtype. The type 1 and type 2 groups of strains appear to be clonal as are some other bacteria species such as Mycobacterium tuberculosis [53]. Despite their geographic separation, the strains may have only recently diverged. The differences between the type 1 and 2 strains were concentrated to specific areas of the genome, rather than being evenly distributed. This suggests the existence of positive selection pressure for some variants, as might be expected for genes coding for proteins that interact with host cells such as the P1 adhesin.
We identified two large regions of genomic structural variation. These intervals were located at 557178–560601 and 713023–713984 in M129, and both were type specific. The type 2-specific insertion (Fig. 2b) contained lipoprotein genes and was identified as an insertion event previously reported only in strain 309 [54]. This block was found in all of our sequenced type 2 strains, including FH, making it a type 2 strain signature, rather than a unique feature of strain 309. The type 1 specific insertion (Fig. 2c) spanned only genes encoding hypothetical proteins.
P1 and ORF6
The tightest clustering of the genomic differences between the type 1 and type 2 strains are found in two contiguous, functionally related genes: P1 and ORF6. Both genes are in the same operon and code for components of the M. pneumoniae adhesin complex, which is necessary for successful colonization of the respiratory tract [55]. The major sequence differences in the P1 and ORF6 genes between type 1 and type 2 strains are localized to specific domains of the proteins, rather than scattered across the protein. The P1 and ORF6 genes each have a long region of divergence between the type 1 and type 2 strains while the rest of the protein sequence is almost completely identical. For each gene, the region of divergence is about 350 bp, across all strains. In the case of P1, this region of variation has been mapped to a known surface-exposed domain [56]. The large regions of variation in both the P1 and ORF6 genes are within RepMP sequences. RepMP sequences are found throughout the M. pneumoniae genome, and previous studies suggest that recombination between RepMP sequences is responsible for antigenic variation [57, 58]. Our data support these prior studies. The within-type consistency between the type 1 and type 2 suggests that such recombination is a rare event. Our data also suggest that while type 1 and type 2 strains diverged via recombination events in the P1 and ORF6 genes (among other loci, Fig. 2), they have been relatively stable evolutionarily at these loci for at least the last six decades. This stability further suggests that the function of P1 and ORF6 is critical for survival of both type 1 and type 2 strains, even though the function of the proteins may be subtly different in each strain subtype. It is also possible that recombination events involving the RepMP sequences regularly occur but that they are immediately out-competed by the superior type 1 and type 2 variants.
Besides the variable RepMP sequences in the P1 gene, an AGT trinucleotide repeat variation was observed in all strains. This variation was previously reported in a total of 85 clinical isolates from China in two studies (repeat 5–16 times) [59, 60]. Serine repeats may form a hinge structure of a protein and hinge bending motions play an important role in catalysis and protein-ligand interactions [61]. The global P1 protein structure contains three domains that are linked by the flexible hinges [56]. The serine repeats are located in conserved domain I, close to but not included in the predicted flexible hinges. Although P1 is a surface antigen clearly related to adherence, no known protein functions such as ligand binding have been clearly predicted or characterized to it [56]. It is possible that the serine repeat variation could potentially affect its interaction with the host. Interestingly, we showed that strain 19294 has a uniquely long expansion of the polyserine repeat, and this strain has unusual morphology, as seen by electron microscopy, compared to most published photomicrographs of M. pneumoniae [62]. The unique expansion of the polyserine repeat may cause this phenotype by changing the way P1 folds or by changing its flexibility, and therefore its interactions with other proteins in the adhesin complex. As protein glycosylation at serine residues has recently been described in mycoplasma, the serine repeat might be a region that is heavily glycosylated with potential consequences on P1 function [63].
The functional effects of these and other variable domains in P1 and ORF6 should be the subject of future studies, but our results already suggest that a large fraction of the functional differences between type 1 and type 2 M. pneumoniae strains lies within one protein complex and two genes coding for components of that complex.
CARDS toxin
The CARDS toxin is an important virulence factor of M. pneumoniae. First identified in 2006 as a surfactant protein A-binding protein, recombinant CARDS toxin induces ADP-ribosylation of multiple cellular proteins and vacuolization of host cells both in vitro and in vivo in rodents and primates [9, 13]. Production of CARDS toxin is upregulated by growth of M. pneumoniae in vivo in mammalian lung [64]. Functional analysis of the recombinant toxin reveals that the ADP-ribosylating activity resides in the N-terminal region of the protein while the cell membrane-binding and vacuolating activities are dependent on the C-terminal region [65]. Unlike P1 and ORF6, the CARDS toxin gene has very little variation among strains. There was only one SNP distinguishing the type 1 and type 2 strains. Two of the type 2 strains bear one unique SNP each, but it is not clear whether these SNPs might affect toxin function. The N-terminal S25L SNP in FH lies between two of the three conserved domains of the pertussis superfamily 1 region but does not appear to be conserved. The G503S SNP in M2192 might affect functions known to be associated with the C-terminal of the protein (the receptor binding/internalization and vacuolating activities) but the structure-function relationships underlying these activities is unknown at present [9, 65]. Additionally, we found no SNPs upstream of the gene that could potentially alter promoter function. These findings suggest that the CARDS toxin is under little selective pressure to vary.
Arginine deiminase
Arginine deiminase activity has never been demonstrated in M. pneumoniae. Enzyme function is thought to be inactive due to a frameshift leading to a premature stop codon in the arcA gene (MPN304). We confirmed the existence of this premature stop in all of the 15 sequenced strains. However, we also observed another, intact copy of arcA (MPN560) in all 15 strains. The prematurely stopped copy of arcA was contiguous to arcC, while the intact copy is in a different part of the genome and appears to have a different origin based on protein sequence alignment (Fig. 6). It is likely that MPN304 was the “original” copy of arcA and that MPN560 was acquired later. What is most striking is that both copies of arcA have no nonsynonymous variants in the entirety of their coding sequences in all of the 15 sequenced strains. The lack of variation in these genes at both loci suggests that the genes are not diverging and hence may be functional. Although previous studies have suggested that the arginine deiminase pathway is inactive in M. pneumoniae [49], ArcA (encoded by MPN560) protein has been detected by proteogenomic assay [66]. ArcA may have a regulated activity that is not observable under prior assay conditions and/or might be involved in cellular function(s) other than arginine metabolism. All of these results warrant further study into the activity and the possible inducibility of the arginine deiminase pathway in M. pneumoniae.
hsdS variation
Each of the strains examined had 10 copies of hsdS scattered throughout the genome, eight of which were identified as transcribed and 5 as translated by transcriptome and proteome analysis of M129 [1, 3, 67]. Multiple copies of hsdS genes are also found in other mycoplasma species, e.g., 9 in M. suis [68] and 21 in M. haemofelis [69]. M. pulmonis has “only” 6 hsdS genes but about 2 dozen hsdS variants, all of which are functional, can be generated by high-frequency DNA inversions with the site-specific recombination sites located within the hsdS coding regions [70]. M. pneumoniae should lack type I restriction endonuclease activity because of the apparent absence of a functional hsdR gene. Nevertheless, the HsdS proteins can combine with the HsdM proteins to form a functional type I modification enzyme. Indeed, the DNA methylome of M. pneumoniae reveals a type I DNA modification enzyme activity that recognizes the target sequence GAN7TAY [67]. None of the hsdS genes of M. pneumoniae should be thought of as orphans because each of the HsdS proteins can complex with any of the HsdM subunits that are available. Hence, a small number of hsdM genes can support a large number of hsdS genes to generate a system in which multiple modification enzymes with differing DNA sequence specificities are active simultaneously.
We observed a 12-bp TR within the dimerization domains in 7 hsdS genes, and the TR copy numbers vary in 6 of hsdS in all strains and in the same strain from different passages. Tandem repeats were also identified in the hsdS genes of M. haemofelis genome [69]. It is known that the length of the alpha helices determines the number of nucleotides in the non-specific spacer of the DNA recognition sequence and thus variations in this domain change the target specificity [50, 71]. Hence, the gain or loss of TR units as would be expected to occur as a result of slipped strand mispairing during DNA replication would alter the DNA methylome. The specificities of the HsdS proteins may vary during infection as has been reported for the type I restriction enzymes in M. pulmonis [72]. Even the truncated copies of hsdS may be functional as have been described for other bacteria [73]. Maintaining so many variable hsdS genes in the genome suggests epigenetic mechanisms for gene regulation [74].
Conclusions
It has been estimated based primarily on rRNA analysis that the mycoplasmas are evolving more rapidly than most bacteria and that M. pneumoniae and closely related species are evolving more rapidly than most other mycoplasmas, suggesting a high mutation rate [75, 76]. Nevertheless, the complete sequence and assembly of 15 M. pneumoniae genomes from isolates collected over the past 6 decades from diverse localities across the globe demonstrate striking conservation of most genes within the two identified subtypes, including the CARDS toxin gene, an important virulence factor. Two genes coding for ArcA, a protein that is integral to the function of arginine deiminase, were also found to be completely conserved both in the apparently prematurely stopped MPN304 as well as the apparently functional MPN560. The major region of variability occurs in the P1 and ORF6 genes associated with the adhesin complex. One isolate with unique cell morphology was found to have an extended polyserine region in P1. Multiple hsdS genes with variable TR numbers were identified in all 15 genomes, suggesting the importance of epigenetics in this species. These data provide the basis for further studies on the evolution and structure-function pathobiology of this highly specialized pathogen.
Acknowledgements
We thank Lynn Duffy for technical assistance. This work was supported by NIH grant number R01AI63909 to KD, internal funding from the UAB Department of Medicine/Division of Clinical Immunology & Rheumatology to TPA, and the National Center for Advancing Translational Sciences of the NIH under award number UL1TR00165. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Additional files
Initial assembly characteristics. (XLSX 19 kb)
Genes with no variants. (XLSX 12 kb)
Variants in the P1 gene excluding the large variation region (nucleotides 181509-181906), relative to the M129 reference strain. (XLSX 13 kb)
Variants in ORF6 excluding the large variation region (nucleotides 186572-187144), relative to the M129 reference strain. (XLSX 11 kb)
Footnotes
Li Xiao and Travis Ptacek contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LX carried out the molecular genetic studies, participated in the sequence alignment and drafted the manuscript. TP assembled the sequence, carried out sequence alignment and drafted the manuscript. JDO participated in sequence assembly and annotation and helped draft the manuscript. DMC grew the strains. WLS participated in the sequence alignment and helped to draft the manuscript. KBW, TPA and EJL participated in the study design and coordination and helped to draft the manuscript. KD conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Li Xiao, Email: lixiao@uab.edu.
Travis Ptacek, Email: tptacek@uab.edu.
John D. Osborne, Email: ozborn@uab.edu
Donna M. Crabb, Email: dmcrabb@uab.edu
Warren L. Simmons, Email: wsimmons@uab.edu
Elliot J. Lefkowitz, Email: ElliotL@uab.edu
Ken B. Waites, Email: waiteskb@uab.edu
T. Prescott Atkinson, Email: PAtkinson@peds.uab.edu.
Kevin Dybvig, Email: dybvig@uab.edu.
References
- 1.Guell M, van Noort V, Yus E, Chen WH, Leigh-Bell J, Michalodimitrakis K, et al. Transcriptome complexity in a genome-reduced bacterium. Science. 2009;326(5957):1268–71. doi: 10.1126/science.1176951. [DOI] [PubMed] [Google Scholar]
- 2.Yus E, Maier T, Michalodimitrakis K, van Noort V, Yamada T, Chen WH, et al. Impact of genome reduction on bacterial metabolism and its regulation. Science. 2009;326(5957):1263–8. doi: 10.1126/science.1177263. [DOI] [PubMed] [Google Scholar]
- 3.Kuhner S, van Noort V, Betts MJ, Leo-Macias A, Batisse C, Rode M, et al. Proteome organization in a genome-reduced bacterium. Science. 2009;326(5957):1235–40. doi: 10.1126/science.1176343. [DOI] [PubMed] [Google Scholar]
- 4.Waites KB, Talkington DF. Mycoplasma pneumoniae and its role as a human pathogen. Clin Microbiol Rev. 2004;17(4):697–728. doi: 10.1128/CMR.17.4.697-728.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Narita M. Pathogenesis of extrapulmonary manifestations of Mycoplasma pneumoniae infection with special reference to pneumonia. J Infect Chemother. 2010;16(3):162–9. doi: 10.1007/s10156-010-0044-X. [DOI] [PubMed] [Google Scholar]
- 6.Sasaki T, Kenri T, Okazaki N, Iseki M, Yamashita R, Shintani M, et al. Epidemiological study of Mycoplasma pneumoniae infections in Japan based on PCR-restriction fragment length polymorphism of the P1 cytadhesin gene. J Clin Microbiol. 1996;34(2):447–9. doi: 10.1128/jcm.34.2.447-449.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Somerson NL, Walls BE, Chanock RM. Hemolysin of Mycoplasma pneumoniae: tentative identification as a peroxide. Science. 1965;150(693):226–8. doi: 10.1126/science.150.3693.226. [DOI] [PubMed] [Google Scholar]
- 8.Almagor M, Kahane I, Yatziv S. Role of superoxide anion in host cell injury induced by Mycoplasma pneumoniae infection. A study in normal and trisomy 21 cells. J Clin Invest. 1984;73(3):842–7. doi: 10.1172/JCI111279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kannan TR, Baseman JB. ADP-ribosylating and vacuolating cytotoxin of Mycoplasma pneumoniae represents unique virulence determinant among bacterial pathogens. Proc Natl Acad Sci U S A. 2006;103(17):6724–9. doi: 10.1073/pnas.0510644103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kannan TR, Provenzano D, Wright JR, Baseman JB. Identification and characterization of human surfactant protein A binding protein of Mycoplasma pneumoniae. Infect Immun. 2005;73(5):2828–34. doi: 10.1128/IAI.73.5.2828-2834.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Medina JL, Coalson JJ, Brooks EG, Le Saux CJ, Winter VT, Chaparro A, et al. Mycoplasma pneumoniae CARDS toxin exacerbates ovalbumin-induced asthma-like inflammation in BALB/c mice. PLoS One. 2014;9(7) doi: 10.1371/journal.pone.0102613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Medina JL, Coalson JJ, Brooks EG, Winter VT, Chaparro A, Principe MF, et al. Mycoplasma pneumoniae CARDS toxin induces pulmonary eosinophilic and lymphocytic inflammation. Am J Respir Cell Mol Biol. 2012;46(6):815–22. doi: 10.1165/rcmb.2011-0135OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hardy RD, Coalson JJ, Peters J, Chaparro A, Techasaensiri C, Cantwell AM, et al. Analysis of pulmonary inflammation and function in the mouse and baboon after exposure to Mycoplasma pneumoniae CARDS toxin. PLoS One. 2009;4(10) doi: 10.1371/journal.pone.0007562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dandekar T, Huynen M, Regula JT, Ueberle B, Zimmermann CU, Andrade MA, et al. Re-annotating the Mycoplasma pneumoniae genome sequence: adding value, function and reading frames. Nucleic Acids Res. 2000;28(17):3278–88. doi: 10.1093/nar/28.17.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma MB, Chaudhry R, Tabassum I, Ahmed NH, Sahu JK, Dhawan B, et al. The presence of Mycoplasma pneumoniae infection and GM1 ganglioside antibodies in Guillain-Barré syndrome. J Infect Dev Ctries. 2011;5(6):459–64. doi: 10.3855/jidc.1508. [DOI] [PubMed] [Google Scholar]
- 16.Dallo SF, Baseman JB. Intracellular DNA replication and long-term survival of pathogenic mycoplasmas. Microb Pathog. 2000;29(5):301–9. doi: 10.1006/mpat.2000.0395. [DOI] [PubMed] [Google Scholar]
- 17.Simmons WL, Daubenspeck JM, Osborne JD, Balish MF, Waites KB, Dybvig K. Type 1 and type 2 strains of Mycoplasma pneumoniae form different biofilms. Microbiology. 2013;159(Pt 4):737–47. doi: 10.1099/mic.0.064782-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li BC, Herrmann R. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 1996;24(22):4420–49. doi: 10.1093/nar/24.22.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krishnakumar R, Assad-Garcia N, Benders GA, Phan Q, Montague MG, Glass JI. Targeted chromosomal knockouts in Mycoplasma pneumoniae. Appl Environ Microbiol. 2010;76(15):5297–9. doi: 10.1128/AEM.00024-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kenri T, Ohya H, Horino A, Shibayama K. Identification of Mycoplasma pneumoniae type 2b variant strains in Japan. J Med Microbiol. 2012;61(Pt 11):1633–5. doi: 10.1099/jmm.0.046441-0. [DOI] [PubMed] [Google Scholar]
- 21.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19(6):1117–23. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12(4):656–64. doi: 10.1101/gr.229202.ArticlepublishedonlinebeforeMarch2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–6. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–92. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.HVDR Online Tools. [http://hvdr.bioinf.wits.ac.za/fmt/]
- 26.Compute reverse complement of the nucleotide sequence. [http://reverse-complement.com/]
- 27.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST) Nucleic Acids Res. 2014;42(Database issue):D206–14. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tatusova T, Ciufo S, Fedorov B, O'Neill K, Tolstoy I. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 2014;42(Database issue):D553–9. doi: 10.1093/nar/gkt1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, et al. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Current Protoc Bioinformatics. 2013;43(11):11.10.11–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.KEGG Mapper. [http://www.genome.jp/kegg/tool/map_brite1.html]
- 36.Ma J, Chen T, Mandelin J, Ceponis A, Miller NE, Hukkanen M, et al. Regulation of macrophage activation. Cell Mol Life Sci. 2003;60:2334–46. doi: 10.1007/s00018-003-3020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5(6) doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–66. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES science gateway for inference of large phylogenetic trees. In: Proceedings for the Gateway Computing Environments Workshop (GCE): 14 Nov. 2010. New Orelans; 2010. p. 1–8.
- 42.MrBayes: Bayesian Inference of Phylogeny. [http://mrbayes.sourceforge.net/]
- 43.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 44.Huson DH, Richter DC, Rausch C, Dezulian T, Franz M, Rupp R. Dendroscope: an interactive viewer for large phylogenetic trees. BMC Bioinformatics. 2007;8:460. doi: 10.1186/1471-2105-8-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huson DH, Scornavacca C. Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst Biol. 2012;61(6):1061–7. doi: 10.1093/sysbio/sys062. [DOI] [PubMed] [Google Scholar]
- 46.FigTree. [http://tree.bio.ed.ac.uk/software/figtree/]
- 47.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–80. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruland K, Wenzel R, Herrmann R. Analysis of three different repeated DNA elements present in the P1 operon of Mycoplasma pneumoniae: size, number and distribution on the genome. Nucleic Acids Res. 1990;18(21):6311–7. doi: 10.1093/nar/18.21.6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rechnitzer H, Rottem S, Herrmann R. Reconstitution of an active arginine deiminase pathway in Mycoplasma pneumoniae M129. Infect Immun. 2013;81(10):3742–9. doi: 10.1128/IAI.00441-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loenen WA, Dryden DT, Raleigh EA, Wilson GG. Type I restriction enzymes and their relatives. Nucleic Acids Res. 2014;42(1):20–44. doi: 10.1093/nar/gkt847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eshaghi A, Memari N, Tang P, Olsha R, Farrell DJ, Low DE, et al. Macrolide-resistant Mycoplasma pneumoniae in humans, Ontario, Canada, 2010–2011. Emerg Infect Dis. 2013;19(9):1525-1527. [DOI] [PMC free article] [PubMed]
- 52.Zhao F, Liu G, Wu J, Cao B, Tao X, He L, et al. Surveillance of macrolide-resistant Mycoplasma pneumoniae in Beijing, China, from 2008 to 2012. Antimicrob Agents Chemother. 2013;57(3):1521–3. doi: 10.1128/AAC.02060-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dos Vultos T, Mestre O, Rauzier J, Golec M, Rastogi N, Rasolofo V, et al. Evolution and diversity of clonal bacteria: the paradigm of Mycobacterium tuberculosis. PLoS One. 2008;3(2) doi: 10.1371/journal.pone.0001538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kenri T, Horino A, Matsui M, Sasaki Y, Suzuki S, Narita M, et al. Complete genome sequence of Mycoplasma pneumoniae type 2a strain 309, isolated in Japan. J Bacteriol. 2012;194(5):1253–4. doi: 10.1128/JB.06553-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Catrein I, Dumke R, Weiner J, 3rd, Jacobs E, Herrmann R. Cross-complementation between the products of the genes P1 and ORF6 of Mycoplasma pneumoniae subtypes 1 and 2. Microbiology. 2004;150(Pt 12):3989–4000. doi: 10.1099/mic.0.27506-0. [DOI] [PubMed] [Google Scholar]
- 56.Nakane D, Adan-Kubo J, Kenri T, Miyata M. Isolation and characterization of P1 adhesin, a leg protein of the gliding bacterium Mycoplasma pneumoniae. J Bacteriol. 2011;193(3):715–22. doi: 10.1128/JB.00796-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dumke R, Schurwanz N, Jacobs E. Characterisation of subtype- and variant-specific antigen regions of the P1 adhesin of Mycoplasma pneumoniae. Int J Med Microbiol. 2008;298(5-6):483–91. doi: 10.1016/j.ijmm.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 58.Kenri T, Taniguchi R, Sasaki Y, Okazaki N, Narita M, Izumikawa K, et al. Identification of a new variable sequence in the P1 cytadhesin gene of Mycoplasma pneumoniae: evidence for the generation of antigenic variation by DNA recombination between repetitive sequences. Infect Immun. 1999;67(9):4557–62. doi: 10.1128/iai.67.9.4557-4562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao F, Cao B, Li J, Song S, Tao X, Yin Y, et al. Sequence analysis of the p1 adhesin gene of Mycoplasma pneumoniae in clinical isolates collected in Beijing in 2008 to 2009. J Clin Microbiol. 2011;49(8):3000–3. doi: 10.1128/JCM.00105-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tian XJ, Dong YQ, Dong XP, Li JY, Li D, Jiang Y, et al. P1 gene of Mycoplasma pneumoniae in clinical isolates collected in Beijing in 2010 and relationship between genotyping and macrolide resistance. Chin Med J (Engl) 2013;126(20):3944–8. [PubMed] [Google Scholar]
- 61.Flores SC, Lu LJ, Yang J, Carriero N, Gerstein MB. Hinge Atlas: relating protein sequence to sites of structural flexibility. BMC Bioinformatics. 2007;8:167. doi: 10.1186/1471-2105-8-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoek KL, Cassell GH, Duffy LB, Atkinson TP. Mycoplasma pneumoniae-induced activation and cytokine production in rodent mast cells. J Allergy Clin Immunol. 2002;109(3):470–6. doi: 10.1067/mai.2002.121951. [DOI] [PubMed] [Google Scholar]
- 63.Jordan DS, Daubenspeck JM, Laube AH, Renfrow MB, Dybvig K. O-linked protein glycosylation in mycoplasma. Mol Microbiol. 2013;90(5):1046–53. doi: 10.1111/mmi.12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kannan TR, Musatovova O, Balasubramanian S, Cagle M, Jordan JL, Krunkosky TM, et al. Mycoplasma pneumoniae Community Acquired Respiratory Distress Syndrome toxin expression reveals growth phase and infection-dependent regulation. Mol Microbiol. 2010;76(5):1127–41. doi: 10.1111/j.1365-2958.2010.07092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kannan TR, Krishnan M, Ramasamy K, Becker A, Pakhomova ON, Hart PJ, et al. Functional mapping of community-acquired respiratory distress syndrome (CARDS) toxin of Mycoplasma pneumoniae defines regions with ADP-ribosyltransferase, vacuolating and receptor-binding activities. Mol Microbiol. 2014;93(3):568–81. doi: 10.1111/mmi.12680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jaffe JD, Berg HC, Church GM. Proteogenomic mapping as a complementary method to perform genome annotation. Proteomics. 2004;4(1):59–77. doi: 10.1002/pmic.200300511. [DOI] [PubMed] [Google Scholar]
- 67.Lluch-Senar M, Luong K, Llorens-Rico V, Delgado J, Fang G, Spittle K, et al. Comprehensive methylome characterization of Mycoplasma genitalium and Mycoplasma pneumoniae at single-base resolution. PLoS Genet. 2013;9(1) doi: 10.1371/journal.pgen.1003191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guimaraes AM, Santos AP, SanMiguel P, Walter T, Timenetsky J, Messick JB. Complete genome sequence of Mycoplasma suis and insights into its biology and adaption to an erythrocyte niche. PLoS One. 2011;6(5) doi: 10.1371/journal.pone.0019574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Santos AP, Guimaraes AM, do Nascimento NC, Sanmiguel PJ, Martin SW, Messick JB. Genome of Mycoplasma haemofelis, unraveling its strategies for survival and persistence. Vet Res. 2011;42:102. doi: 10.1186/1297-9716-42-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dybvig K, Sitaraman R, French CT. A family of phase-variable restriction enzymes with differing specificities generated by high-frequency gene rearrangements. Proc Natl Acad Sci U S A. 1998;95(23):13923–8. doi: 10.1073/pnas.95.23.13923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Price C, Lingner J, Bickle TA, Firman K, Glover SW. Basis for changes in DNA recognition by the EcoR124 and EcoR124/3 type I DNA restriction and modification enzymes. J Mol Biol. 1989;205:115–25. doi: 10.1016/0022-2836(89)90369-0. [DOI] [PubMed] [Google Scholar]
- 72.Gumulak-Smith J, Teachman A, Tu AH, Simecka JW, Lindsey JR, Dybvig K. Variations in the surface proteins and restriction enzyme systems of Mycoplasma pulmonis in the respiratory tract of infected rats. Mol Microbiol. 2001;40(4):1037–44. doi: 10.1046/j.1365-2958.2001.02464.x. [DOI] [PubMed] [Google Scholar]
- 73.Meister J, MacWilliams M, Hubner P, Jutte H, Skrzypek E, Piekarowicz A, et al. Macroevolution by transposition: drastic modification of DNA recognition by a type I restriction enzyme following Tn5 transposition. EMBO J. 1993;12(12):4585–91. doi: 10.1002/j.1460-2075.1993.tb06147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Casadesus J, Low D. Epigenetic gene regulation in the bacterial world. Microbiol Mol Biol Rev. 2006;70(3):830–56. doi: 10.1128/MMBR.00016-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Woese CR. Bacterial evolution. Microbiol Rev. 1987;51(2):221–71. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maniloff J. Phylogeny of mycoplasmas. In: Maniloff J, McElhaney RN, Finch LR, Baseman JB, editors. Mycoplasmas molecular biology and pathogenesis. Washington, D.C.: American Society for Microbiology; 1992. pp. 549–59. [Google Scholar]