

Summary
Molecular dynamics simulations are an effective tool to study the structure, dynamics, and thermodynamics of carbohydrates and proteins. However, the simulations of heterogeneous glycoprotein systems have been limited due to the lack of appropriate molecular force field parameters describing the linkage between the carbohydrate and the protein regions as well as the tools to prepare these systems for modeling studies. In this work we outline the recent developments in the CHARMM carbohydrate force field to treat glycoproteins and describe in detail the step-by-step procedures involved in building glycoprotein geometries using the recently developed CHARMM-GUI Glycan Reader.
Keywords: N-glycosylation, O-glycosylation, carbohydrates, empirical force field, molecular dynamics simulations
1. Introduction
Glycosylation is a common posttranslational modification of proteins that involves the covalent attachment of carbohydrates to the side chains of the amino acids asparagine (Asn) (N-linked) or serine (Ser)/threonine (Thr) (O-linked). [1] Glycans attached to proteins are shown to have a wide variety of roles in processes ranging from protein folding, immune recognition, and developmental regulation. [2,3] The N-glycosidic linkage generally occurs between a β-N-acetylglucosamine (GlcNAc) carbohydrate moiety and the side chain of Asn, wherein Asn is embedded in the consensus tripeptide sequence Asn-Xxx-Ser/Thr, Xxx being any amino acid but proline. [4,5] The O-glycosidic linkage on the other hand has been found to occur between different carbohydrate moieties (α-N-acetylgalactosamine (GalNAc), [6] β-N-acetylglucosamine (GlcNAc), [7] α-L-fucose (Fuc), [8] β-D-glucose (Glc), [9] and α-mannose (Man)[10]) and the side chains of Ser/Thr. Contrary to Asn involved in the N-linkages, Ser/Thr involved in O-linkages do not show any particular amino acid sequence preference. [11]
Structural studies of glycoproteins are essential to understanding the carbohydrate-protein interactions that are responsible for their functional activity. However, structural studies of glycoproteins are complicated by the inherent flexibility of carbohydrates, which hinders their crystallization, [12,13] as well as by the heterogeneous nature of the glycans themselves. For example, nearly 70% of all proteins deposited in sequence databases show potential N-glycosylation sites; [14] however only 7% of PDB (Protein Data Bank)[15] entries contain carbohydrate residues. [16] Moreover, these PDB entries were found to contain a high rate of error, which lead to a remediation of the PDB database[17] and the development of tools to aid researchers in the validation of carbohydrate residues in PDB entries. [18,19]
To this end, molecular modeling and dynamics (MD) studies using accurate force fields (FFs) have the potential to provide insights into the structure, dynamics and functional properties of biomolecular systems. [20] Classical FF development efforts aimed at enabling accurate modeling of carbohydrates have been ongoing for over a decade. [21–26] While successful for carbohydrates, these FFs were found to be limited when attempting to model heterogeneous biomolecular systems containing proteins, lipids, and/or nucleic acids. This was due to the fact that much of the parameter development work was not done in the wider context of a comprehensive biomolecular FF. This issue has been addressed by recent efforts to revise and make the resulting carbohydrate parameters compatible with the related family of FFs, with two examples being the revised GROMOS and GLYCAM FFs. [23,24,27]
While the other FFs required reparameterization, the CHARMM carbohydrate FF was developed ground up to be consistent with the other components of the CHARMM additive all-atom biomolecular FF, which includes proteins, [28,29] nucleic acids, [30,31] lipids, [32–35] and drug-like small molecules, [36] thereby allowing for the modeling of heterogeneous biomolecular systems. To date parameters for the additive all-atom CHARMM carbohydrate FF have been optimized and validated for pyranose and furanose monosaccharides, [37,38] aldose and ketose linear carbohydrates, and their reduced counterparts, the sugar alcohols. [39] Towards studying heterogeneous systems parameters have been presented for glycosidic linkages involving both pyranoses and furanoses, [40,41] deoxy, oxidized, or N- methylamine monosaccharide derivatives as well as covalent N- and O- linkages to proteins. [42–44] These parameters are available for download at http://mackerell.umaryland.edu.
Generation of inputs for the bio-molecular simulation program CHARMM [45] is now facilitated by the web-based graphical user interface CHARMM-GUI (http://www.charmm-gui.org). [46] During a web session this interface allows users to build and validate a molecular system in an interactive fashion and provides users with input files that can be used to setup the equilibration and production MD simulations as well as a range of MD related calculations. In an attempt to simplify glycan modeling, the newly developed CHARMM carbohydrate FF parameters have been incorporated with the CHARMM-GUI resulting in the development of the Glycan Reader and its web-based interface (http://www.charmm-gui.org/input/glycan). [47]
The aim of this work is to provide the reader with an overview of the parameter development protocol and the subsequent use of these parameters in the context of glycoprotein modeling. In the Theory section, the parametrization protocol used to generate the patch residues for N- and O-linkages to proteins is briefly outlined, especially highlighting the selection of model compounds to treat these linkages. In the Methods section, the glycoprotein building procedures using scripts generated by Glycan Reader are described. In the Notes section, we discuss a number of issues that users should consider when performing simulations of glycoproteins.
2. Theory
The potential energy function that, along with the parameters described below, comprises the CHARMM additive FF and is described as
| (1) |
The first five sums in Eqn. 1 account for bonded interactions, which can be combined and termed as the internal potential energy. In these sums, Kb, Kθ, KS, Kχ and Kϕ are force constant parameters for bond, valence angle, Urey-Bradley angle, dihedral angle, and improper dihedral angle, respectively. b, θ, S, χ and ϕ are the bond distance, valence angle, Urey-Bradley 1,3-distance, dihedral angle, and improper dihedral angle values. The subscript 0 indicates an equilibrium value parameter. Additionally, for the dihedral term, n is the multiplicity and δ is the phase angle as in a cosine series. The next two terms in Eqn. 1 sum over nonbonded pairs ij which includes a Lennard-Jones (LJ) 6–12 term to account for dispersion and Pauli exclusion and a Coulomb term to account for electrostatic interactions. These two sums combined together are termed as the external potential energy. εij is the LJ well depth, Rmin,ij is the interatomic distance at the LJ energy minimum, qi and qj are the partial atomic charges, and rij is the distance between atoms i and j. The energy function represented in Eqn. 1 is referred to as an additive FF, as the charges are static such that the total electrostatic energy of a system is simply the sum of all the atom pair-wise electrostatic interactions. Recently, non-additive or polarizable models have started to be developed in which the charge distribution responds to the surrounding electric field. [48–50] While such FFs represent the next generation of tools for simulations of biomolecular systems the present manuscript focuses on glycoprotein simulations using the additive CHARMM FF.
The aim of a parametrization effort is to optimize the above mentioned parameters to allow the resulting FF to reproduce a variety of target properties, like molecular geometries and vibrations, free energies of solvation and other condensed phase properties. There are a number of excellent reviews detailing the overall parameterization procedure used in the CHARMM FF. [20,28,51,52] A general flow diagram of this procedure is presented in Figure 1. The strategy involves the selection of small model compounds, which may ultimately be combined to describe the larger biomolecular systems of interest. The model compounds are generally chosen in accordance with the availability of experimental data, like geometries (high resolution X-ray structures), vibrations (IR data), and conformational information (NMR data), which is used for the validation of the parameters. In the absence of experimental data, small model compounds may be subjected to ab initio quantum mechanical (QM) calculations to generate additional target data, like optimized geometries, vibrational information, and conformational energies, which is also used to drive the parametrization process. Here it is important to note that sole reliance on QM methods is inappropriate. This is particularly important when optimizing nonbonded parameters relevant to the external potential energy (i.e., LJ and electrostatic terms in Eqn. 1), where dispersion interactions are important. It is also important to highlight that a lot of the parametrization procedure relies on the assumption that parameters from the smaller model compounds are transferable to macromolecules. Importantly, for the development of a comprehensive additive biomolecular FF it is essential that all new parameters are developed to be consistent with the pre-existing components of the FF. This caveat was followed in the development of the CHARMM carbohydrate FF, making it compatible with the remainder of the CHARMM additive all-atom biomolecular FF. [28–36] While necessary for development of a consistent heterogeneous FF, this approach allows for the transfer of parameters, both internal and external, from the existing FF to the new entities to initiate the parameter optimization process.
Figure 1.
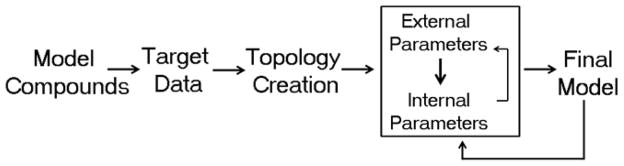
Parametrization flow chart.
2.1 Model Compounds
The model compound selection strategy for the O- and N-linkages is presented in Figure 2. For the O-linkages, the initial transfer of bonded and non-bonded parameters from the existing FFs left only those parameters associated with the glycosidic torsions about the C1O1 bond (O5C1O1Cβ and C2C1O1Cβ), O1Cβ bond (C1O1CβCα and additionally C1O1CβCγ in the Thr-linked analogs) and the CβCα bond (O1CβCαN and O1CβCαC), as targets for parametrization. Since these dihedrals span both the carbohydrate and the protein regions, the complete dipeptides were chosen as the model compounds as depicted in Figure 2a. In contrast, in the N-linkages the presence of an additional –CH2– spacer between the carbohydrate and protein regions allowed the selection of smaller model compounds. The presence of available parameters in the CHARMM carbohydrate FF for the N-acetylamine substitution at the C2 position, as developed for N-acetylglucosamine (GlcNAc) and N-acetylgalactosamine (GalNAc), allowed for the transfer of parameter for the N-acetylamine substitution at the C1 position, which formed the first set of model compounds. Since N-glycosylation commonly involves the linkage of GlcNAc to the side chain of Asn, parameters were required for the dihedral angle between the nitrogens of the N-acetlyamine groups at positions C1 (anomeric carbon) and C2 of GlcNAc involved in such a linkage. To parametrize this dihedral, tetrahydropyrans with N-acetlyamine substitutions at both the C1 and C2 positions were chosen as the second set of model compounds as depicted in Figure 2b. The target data for parameter optimization included full two-dimensional energy surfaces defined by the glycosidic dihedral angle pairs for the O-linkages and one-dimensional energy surfaces for the N-linkages, and these energy surfaces were determined by QM MP2/cc-pVTZ single point energies on MP2/6-31+G(d) optimized structures. Parameters were validated in the context of crystals of relevant monosaccharides, as well as NMR and/or X-ray crystallographic data on larger systems including O- and N-linked glycopeptides. For complete details of the parametrization procedure and the accompanying validations the readers are referred to the relevant publications. [42,43]
Figure 2.
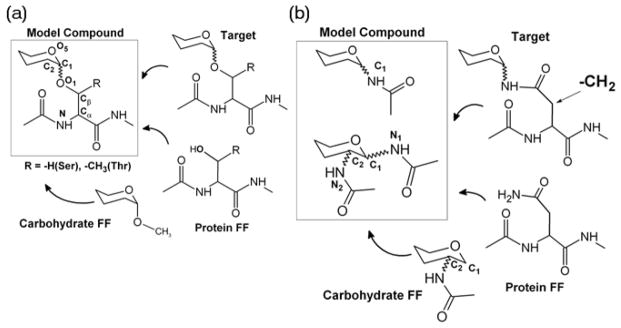
Model compound selection strategy for (a) O-linkages and (b) N-linkages.
2.2 Patch Residues
Patch residues are an easy way of manipulating and connecting generated segments within CHARMM. [45] The patch command allows, for instance, the addition of disulfide bridges, changing the protonation state of a titratible residue or to make a histidine to heme crosslink. The development of parameters for the model compounds resulted in the creation of patches that can be used to setup the glycosidic linkages between carbohydrates and proteins. Two patches were created for the O-linkages to Ser (SGPA and SGPB) and Thr (TGPA and TGPB), wherein the SGPA and TGPA patches link Ser and Thr to the carbohydrate with an α conformation at the anomeric center, while SGPB and TGPB link Ser and Thr to the carbohydrate with a β conformation at the anomeric center. Similarly NGLA and NGLB link Asn to the carbohydrate in the α and β conformations at the anomeric center, respectively. These patches and the related parameters are available as “toppar_all36_glycopeptide.str” in the CHARMM carbohydrate FF distribution for download at http://mackerell.umaryland.edu. For all the patch residues the syntax, as shown in Scheme 1, requires the protein information first followed by the carbohydrate information.
Scheme 1.
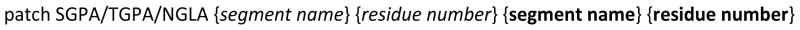
Description of the CHARMM commands for use of the carbohydrate-protein glycosidic patches. The first {segment name} and {residue number} information corresponds to the protein segment, and the subsequent information {segment name} and {residue number} corresponds to the carbohydrate segment.
Below in Scheme 2 the topology information for the α conformation patch residues for each of the amino acids Ser, Thr and Asn, is presented highlighting the changes that are made to each residue upon applying the respective patches.
Scheme 2.


Details of the CHARMM patches to create the carbohydrate-protein glycosidic linkages.
These patches are also included in the automated CHARMM-GUI Glycan Reader (http://www.charmm-gui.org/input/glycan), which can be used to interactively setup the glycoprotein system.
3. Methods
The crystal structure of glucoamylase from Aspergillus awamori var. X100 (PDB id: 3GLY) [53] is used as an example in this study to describe the procedure of setting up a glycoprotein system using CHARMM-GUI. This system was selected as it contains all three types of linkages, O-linkages to Ser and Thr and N-linkages to Asn. A complete example file including scripts from CHARMM-GUI used to setup this system is available for download at http://mackerell.umaryland.edu. The example file contains inputs for each step of the system setup and the subsequent MD process. In Scheme 3 below we present an overview of the example files describing the entire process;
Scheme 3.

Description of the example file and the system setup procedure.
3.1 Generation of the glycoprotein system (Step 1)
The first step involves setting up the glycoprotein systems and the generation of the PSF (Protein Structure File) in the Glycan Reader in preparation for the subsequent steps. Since most of the PDB files do not have hydrogen positions the Reduce software was used to place the missing hydrogens and choose optimal Asn and Gln side chain amide and His side chain ring orientations. [54] Based on the positions of the hydrogen, HIS residue is renamed to HSD or HSE for neutral His and HSP for protonated His. The PDB files generated after this initial assignment were used as inputs to the Glycan Reader interface. Each segment of the whole system is generated independently and patch residues in CHARMM are subsequently used to link them together, as shown in Scheme 4. To do this various regions of the structure (e.g. protein, glycan, or water) are identified and separated based on the “ATOM” and “HETATM” records present in the PDB file.
Scheme 4.



Description of the CHARMM commands to generate and link the carbohydrate-protein system.
The generation step is shown in detail in Scheme 4 to highlight the intricacies involved in the glycoprotein setup procedure. It is to be noted that while Glycan Reader identifies the linkages and assigns the patches involved, errors due to incorrect assignment of bonds in glycan chains could interfere with glycosidic linkage detection. Thus, users of Glycan Reader and CHARMM-GUI must make sure that the input is correct, with regard to the patches being applied, and the output is as intended. [47] In case of polysaccharides one needs to make sure that the correct sugar units are identified and the appropriate patches are used to generate the polysaccharides. While a lot of monosaccharide derivatives have been parameterized for the CHARMM carbohydrate FF, [42–44] there still may be instances wherein parameters for monosaccharide derivatives may be missing in the FF. In such cases the Glycan Reader would fail to identify the carbohydrate and ignore it completely. [47] In many cases the coordinates of alcoholic side chains are missing in the PDB file and these need to be built before the simulation. To do this we use the internal coordinates (IC) information in the topology file of the CHARMM FF as described in Scheme 5.
Scheme 5.

Description of the CHARMM IC commands to build missing coordinates.
3.2 Solvation and equilibration (Step 2)
Once the glycoprotein is properly generated (Figure 3) it is immersed in a pre-equilibrated water box that extends at least 10 Å beyond the non-hydrogen atoms of the protein structure, resulting in a simulation box of size 90 × 90 × 90 Å3 in the case of PDB:3GLY. Water molecules with the oxygen overlapping with the non-hydrogen solute atoms within a distance of 2.8 Å are deleted. Based on the overall charge of the system the appropriate number of ions, 29 potassium ions for 3GLY, are added to neutralize the system. For all of the subsequent minimization and MD simulation steps, periodic boundary conditions are employed using the CRYSTAL module implemented in the CHARMM program. Before performing an equilibration, the water molecules are allowed to rearrange around the fixed solute atoms by a short minimization cycle of 50 steepest descent (SD) steps followed by 50 adopted-basis Newton-Raphson (ABNR) steps. Next, with a mass-weighted harmonic restraint of 1.0 kcal/mol/Å on the non-hydrogen atoms of the glycopeptides, the system is subjected to a 50-step SD minimization followed by a 50-step ABNR minimization cycle. This is followed by a 100 ps simulation in the constant-volume, constant-temperature (NVT) ensemble with the same harmonic restraints to equilibrate the solvent molecules around the glycoprotein. A 200 ps constant-pressure, constant-temperature NPT simulation at 1 atm and 298 K follows the NVT simulation, wherein all the previous restraints are removed. In the NPT simulation the center of mass of the glycoprotein is restrained near the origin by using the MMFP module[55] in CHARMM using a harmonic restraint of 1.0 kcal/mol/Å applied to the center of mass of the glycoprotein. This is done to keep the glycoprotein from drifting out of the simulation box. The electrostatic interactions are treated via the particle mesh Ewald method with a real-space cutoff of 12 Å, a kappa value of 0.34 Å−1, and a sixth-order spline. [56] Nonbond interaction lists are updated heuristically out to 16 Å with a force switch smoothing function from 10 to 12 Å used for the Lennard-Jones interactions. [57] An integration time step of 2 fs is used with the Leapfrog integrator, while the SHAKE algorithm was used to constrain all covalent bonds involving hydrogen atoms. [58] The temperature was maintained at 298 K by a Nose-Hoover heat bath with a thermal piston parameter of 2000 kcal mol−1 ps2. [59] Constant pressure of 1 atm was controlled using the Langevin piston with a mass of 8767 amu (i.e., Pmass = integer(system mass/50.0)). [60]
Figure 3.

Structure of glucoamylase from Aspergillus awamori var. X100 (PDB id: 3GLY). The protein is shown as a cartoon drawing in cyan. The Ser-O-linked monosaccharides are shown as licorice drawing in red, while the Thr-O-linked monosaccharides are shown in green. The N-linked polysaccharides are shown as licorice drawing in yellow. The figure was prepared using VMD. [69]
3.3 Production (Step 3)
Production simulations may be performed with CHARMM, NAMD, or any other simulation package. Glycan Reader and the CHARMM-GUI currently produce both standard CHARMM and XPLOR[61] format PSFs, with the later allowing for MD simulations with the program NAMD, [62] which was used for the production simulation in the present study. The CHARMM commands used to generate the XPLOR format PSF, along with the PDB format coordinate file to initiate the simulation in NAMD are shown in Scheme 6.
Scheme 6.

CHARMM commands to generate a PDB format coordinate file and a XPLOR format PSF.
The equilibrated structure obtained from the calculations in CHARMM described above is used to initiate the production simulations, which presently involved a 16-ns simulation performed using NAMD version 2.7b1. [62] The CHARMM FF is supported within NAMD using the following keywords shown in Scheme 7.
Scheme 7.

NAMD commands required to access the CHARMM parameter files.
A Langevin coupling coefficient of 1 ps−1 with a temperature bath of 298 K is applied to all atoms to achieve constant pressure. A piston oscillation period of 200 fs and a barostat damping time scale of 100 fs are used to maintain a piston pressure of 1 atm.
3.4 Analysis
Proper analysis of the MD simulation is necessary to assure that the simulation itself was performed correctly as well as to extract the relevant structural and dynamic information required to understand the properties of the system and relate them to the relevant chemistry or biology. Confirmation of the quality of the simulation includes monitoring the potential energy of the system versus time as well as other properties such at the volume of the simulation cell or the change in the structure of the glycoprotein via root-mean-square difference (RMSD) analysis. While validating the quality of the simulation such analysis is important to judge the convergence of the simulation as typically there is an initial relaxation of the glycoprotein and surrounding environment followed by fluctuations around the equilibrium structure. Such equilibration often takes a nanosecond or more, which is typically discarded from subsequent analysis. Details of the analysis of the 3GLY simulation are described in the remainder of this section.
Analysis of the 3GLY trajectory revealed that the glycan portions of the glycoprotein exhibited greater conformational variability than the protein portions. The overall RMSD of the complete glycoprotein remained lower than 3 Å for the entire simulation length (Figure 4a). On decomposing the overall RMSD into carbohydrate and protein components, the carbohydrate regions were found to be more flexible with the RMSD as large as 3.5 Å, while the underlying protein remains more stable, with the RMSD lower than 2 Å. The high RMSD for the carbohydrate regions is consistent with the high flexibility of carbohydrates, as observed in both NMR and crystallographic studies (B-factors). [12,13]
Figure 4.
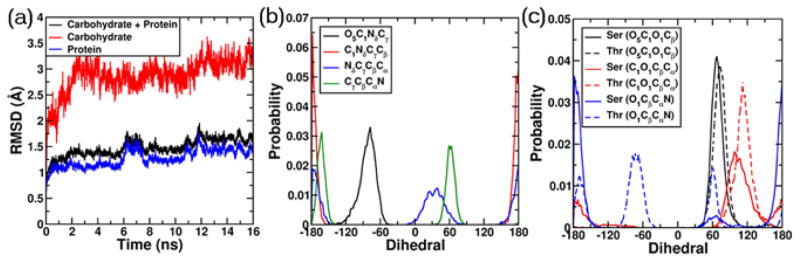
(a) RMSD analysis for the protein 3GLY. RMSD values are for all non-hydrogen atoms following RMS alignment with the crystallographic structure. Color code: black (RMSD for all carbohydrate-protein heavy atoms), red (RMSD for carbohydrate heavy atoms only), blue (RMSD for protein heavy atoms only). (b) Dihedral probability distributions for all the dihedrals involved in the N-linkages from the last 10 ns of the simulation trajectory. Color code: black (O5C1NδCγ), red (C1NδCγCβ), blue (NδCγCβCα), green (CγCβCαN). (c) Dihedral probability distributions for all the dihedrals involved in the O-linkages from the last 10 ns of the simulation trajectory. The distributions from the Ser-O-linkages are presented as solid lines, while the distributions from Thr-O-linkages are presented as broken lines. Color code: black (O5C1O1Cβ), red (C1O1CβCα), blue (O1CβCαN).
Pooled data from the last 10 ns of the simulation trajectory was used to assess the flexibility of the N- and O-linkages. In Figure 4b the probability distributions associated with the O5C1NδCγ, C1NδCγCβ, NδCγCβCα and CγCβCαN dihedrals involved in the N-linkages are presented. It was found that O5C1NδCγ and C1NδCγCβ exhibited unimodal distributions around −75º and ±180º, while NδCγCβCα and CγCβCαN exhibited bimodal distributions around (+30º, ±180º) and (+60º, −160º). These distributions are in agreement with the survey of over 500 N-linked glycans in the PDB. [63] Consistent with the experimental observation it was found that there was greater flexibility associated with the dihedral atoms exclusively in the Asn side chain (NδCγCβCα and CγCβCαN) as compared to the dihedral atoms involved in the glycosidic linkage (O5C1NδCγ and C1NδCγCβ). Presented in Figure 4c are the probability distributions associated with the O5C1O1Cβ, C1O1CβCα and O1CβCαN dihedrals in the O-linkages. It was found that O5C1O1Cβ dihedral samples in the region of +60º consistent with the exoanomeric effect observed in sugars. [64] The C1O1CβCα dihedral was found to be more flexible in the Ser O-linkages when compared to the Thr O-linkages consistent with previous results, [42,43] while the O1CβCαN dihedral was found to adopt conformations with values around −60º, ±180º, and +60º, which correspond to the g+, anti, and g+ conformational states, again consistent with previous studies. [42,43] It is to be noted that these dihedral distributions are important when setting up glycosidic linkages in the absence of experimental geometry information.
In Table 1 we summarize the bridge water occupancies for water bridges between carbohydrates and proteins (carbohydrate-H2O-protein) from the MD simulation for both the O- and N-linkages. We use a sequential numbering scheme to describe the O-linkages; thus 1-Man describes the α-O-Man linkage at protein residue 443 and subsequent linkages from 2-Man to 10-Man describe O-Man linkages at protein reisudes 444, 452, 453, 455, 457, 459, 460, 462, and 464, respectively. Results show that the carbohydrates closer to the terminal, 4-Man to 8-Man, are involved in various bridging interactions with the protein residues. 8-Man (0.710), 4-Man (0.608), and 6-Man (0.431) form strong bridges with protein residues Val-461, Tyr-458, and Ser-455, respectively. Four of the five water bridges detected by the MD simulation were present in the crystallographic structure, suggesting that these bridging water molecules stabilize the relative orientation of the carbohydrate with respect to the protein.
Table 1.
Significant Carbohydrate-H2O-Protein bridge water occupancies occurring in the 3GLY MD simulation. Also presented are the distances between the heavy atoms involved in the bridge-water interactions from the crystal structure.
| O-linkages
| |||
|---|---|---|---|
| Carbohydrate-H2O-Protein
| |||
| Carbohydrate | Protein | Occupancy | dA-H2O, dB-H2O (crys)a |
|
| |||
| Man-8 (O3/HO3) | Val-461 (O) | 0.710 | 2.84, 2.66 |
| Man-4 (O3/HO3) | Tyr-458 (HN) | 0.608 | 2.71, 3.09 |
| Man-6 (O2/HO2) | Ser-455 (O) | 0.431 | --- |
| Man-4 (O2/HO2) | Ser-99 (O) | 0.378 | 3.34, 2.50 |
| Man-8 (O2/HO2) | Ile-87 (O) | 0.279 | 2.81, 3.15 |
| N-linkages
| |||
|---|---|---|---|
| Carbohydrate-H2O-Protein
| |||
| Carbohydrate | Protein | Occupancy | dA-H2O, dB-H2O (crys)a |
|
| |||
| Asn-171 | |||
|
| |||
| β-GlcNAc-1 (O6/HO6) | Gln-219 (O) | 0.803 | 2.48, 2.97 |
| β-GlcNAc-1 (O) | Ser-184 (O/OH) | 0.776 | 2.58, 2.65 |
| β-GlcNAc-1 (O6/HO6) | Ser-226 (O) | 0.775 | 2.85, 3.75 |
| β-GlcNAc-1 (O6/HO6) | Ala-450 (O) | 0.655 | 2.48, 2.96 |
| β-GlcNAc-1 (O3/HO3) | Tyr-223 (O/OH) | 0.565 | 2.87, 2.89 |
| β-GlcNAc-1 (O) | Trp-170 (O) | 0.525 | 2.58, 2.81 |
| β-GlcNAc-2 (O6/HO6) | Asp-238 (Oδ1/Oδ2) | 0.513 | 3.08, 4.16 |
| Man-5 (O2/HO2) | Arg-241 (O) | 0.354 | 4.17, 2.63 |
| Man-5 (O3/HO3) | Arg-241 (O) | 0.216 | 3.51, 2.63 |
|
| |||
| Asn-395 | |||
|
| |||
| Man-7 (O3/HO3) | Ser-399 (O/OH) | 0.876 | 2.63,2.81 |
| Man-7 (O3/HO3) | Ser-411 (O) | 0.805 | 2.63,2.79 |
| β-GlcNAc-1 (O6/HO6) | Asn-395 (O) | 0.629 | --- |
| β-GlcNAc-1 (O) | Asp-414 (Oδ1/Oδ2) | 0.922 | --- |
| Man-7 (O4/HO4) | Arg-413 (NH2) | 0.500 | 3.11,3.22 |
| Man-7 (O4/HO4) | Thr-43 (O/OH) | 0.493 | 3.11,2.89 |
| Man-3 (O6/HO6) | Trp-28 (O) | 0.494 | 2.66,2.52 |
| Man-2 (O6/HO6) | Trp-28 (O) | 0.454 | 2.84,2.52 |
all distances are in Å.
This effect was even more pronounced for the N-linkages at Asn-171 and Asn-395. The sequential numbering used to describe the polysaccharide at Asn-171 is presented below,
while that for the branched polysaccharide at Asn-395 is as follows.
Analysis shows the polysaccharides to be involved in a higher number of multiple water bridges, which span the entire length of the polysaccharide. These water bridges were also observed in the crystal structure as summarized by the distances also presented in Table 1. These results suggest that the solvent mediated interactions between the carbohydrate and protein may be involved in the overall stabilization of the glycoprotein systems and they need to be described accurately by the FF.
4. Notes
Following are a number of items users should consider when performing MD simulations of glycoproteins and several of the items are appropriate for simulations of any system.
It is preferable to use high-resolution structures where available.
-
NMR J-coupling information can be used to determine the conformational state of the O1CβCαN dihedral, which can adopt the g+, anti, and g+ conformational states (−60 º, ±180º, and +60º). The initial geometry of these dihedrals can be set to the particular conformational state using the IC edit command in CHARMM.
ic edit dihe PEPT 1 N PEPT 1 CA PEPT 1 CB PEPT 1 OG -60/180/60 end In this case PEPT is the {segment name} and 1 is the {residue number}. O- and N-linkages are known to be involved in the formation of water mediated hydrogen bonds. [65,66] Thus it is important to allow for additional equilibration with restraints on the glycoprotein structure allowing waters in the vicinity of the carbohydrate-protein interface to relax.
The length of the simulation has to be decided depending on the questions being addressed by the simulation. Convergence is a major issue when working with carbohydrate structures and needs to be addressed accordingly. For example, ring puckering of carbohydrates has been shown to require microsecond length simulations and cannot be addressed by nanoscecond length simulations. [67]
Conformational sampling of glycosidic linkages can be improved by using sampling techniques like temperature or Hamiltonian replica exchange method. [68] An example of the application of such methods to glycopeptides has recently been reported. [43]
-
The residue names in the CHARMM carbohydrate FF in some cases can be up to six characters long eg; BGLCNA (β-N-acetylglucosamine). In such cases the coordinate information must be read in from a CHARMM formatted coordinate file (.crd) and not a PDB (.pdb) file as the PDB format allows only up to three characters for the residue name. The Glycan Reader by default converts the PDB coordinates information into a CHARMM formatted coordinate information for all the HETATM records in the PDB file. In the example described above the protein coordinates are read from a PDB file while the carbohydrate coordinates are read from a CHARMM formatted coordinate file.
! Read PROA open read card unit 10 name 3gly_proa.pdb ! Reading the protein coordinates !Read CARA open read card unit 10 name 3gly_cara.crd !Reading the carbohydrate coordinates read coor card unit 10 append
In case of missing parameters and topology information the Glycan Reader allows the user to upload a user defined topology and parameter file. In the above example we encountered this with the sulfate ions present in the crystal structure for which the topology information was not present in the CHARMM FF, while the parameters were already present. A user defined topology file (so4.rtf) was uploaded to treat the sulfate ions in the system.
The user needs to remove the ATOMS record in the CHARMM parameter files to be able to use the CHARMM FF parameters files with NAMD. Samples of such modified parameters files have been included in the toppar_namd directory in the example file.
One consistent trend in the parametrization of the carbohydrate FF was the overestimation of crystal volumes for neutral compounds.28–36 One possible explanation is the highly directional hydrogen bonding in the crystal environment that is not accounted for in the parametrization protocol for hydroxyl groups, which targeted the molecular volumes and heats of vaporization of neat alcohols. Current work on introducing electronic polarizability into the molecular mechanics framework may help to alleviate this limitation. We refer the reader to the parametrization manuscripts for in-depth details and limitation of the FF.28–36
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grant GM070855 (to ADM) and the University of Kansas General Research Fund allocation #2301388-003 (to WI).
References
- 1.Varki A. Essentials of glycobiology. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 2009. [PubMed] [Google Scholar]
- 2.Dwek RA. Glycobiology: Toward Understanding the Function of Sugars. Chem Rev. 1996;96 (2):683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 3.Lis H, Sharon N. Protein glycosylation. Structural and functional aspects. Eur J Biochem. 1993;218 (1):1–27. doi: 10.1111/j.1432-1033.1993.tb18347.x. [DOI] [PubMed] [Google Scholar]
- 4.Hart GW, Brew K, Grant GA, Bradshaw RA, Lennarz WJ. Primary structural requirements for the enzymatic formation of the N-glycosidic bond in glycoproteins. Studies with natural and synthetic peptides. J Biol Chem. 1979;254 (19):9747–9753. [PubMed] [Google Scholar]
- 5.Bause E. Structural requirements of N-glycosylation of proteins. Studies with proline peptides as conformational probes. Biochem J. 1983;209 (2):331–336. doi: 10.1042/bj2090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strous GJ, Dekker J. Mucin-type glycoproteins. Crit Rev Biochem Mol Biol. 1992;27 (1–2):57–92. doi: 10.3109/10409239209082559. [DOI] [PubMed] [Google Scholar]
- 7.Wells L, Kreppel LK, Comer FI, Wadzinski BE, Hart GW. O-GlcNAc transferase is in a functional complex with protein phosphatase 1 catalytic subunits. J Biol Chem. 2004;279 (37):38466–38470. doi: 10.1074/jbc.M406481200. [DOI] [PubMed] [Google Scholar]
- 8.Haltiwanger RS. Regulation of signal transduction pathways in development by glycosylation. Curr Opin Struct Biol. 2002;12 (5):593–598. doi: 10.1016/s0959-440x(02)00371-8. [DOI] [PubMed] [Google Scholar]
- 9.Shao L, Luo Y, Moloney DJ, Haltiwanger R. O-glycosylation of EGF repeats: identification and initial characterization of a UDP-glucose: protein O-glucosyltransferase. Glycobiology. 2002;12 (11):763–770. doi: 10.1093/glycob/cwf085. [DOI] [PubMed] [Google Scholar]
- 10.Strahl-Bolsinger S, Gentzsch M, Tanner W. Protein O-mannosylation. Biochim Biophys Acta. 1999;1426 (2):297–307. doi: 10.1016/s0304-4165(98)00131-7. [DOI] [PubMed] [Google Scholar]
- 11.Van den Steen P, Rudd PM, Dwek RA, Opdenakker G. Concepts and principles of O-linked glycosylation. Crit Rev Biochem Mol Biol. 1998;33 (3):151–208. doi: 10.1080/10409239891204198. [DOI] [PubMed] [Google Scholar]
- 12.Crispin M, Stuart DI, Jones EY. Building meaningful models of glycoproteins. Nat Struct Mol Biol. 2007;14 (5):354. doi: 10.1038/nsmb0507-354a. discussion 354–355. [DOI] [PubMed] [Google Scholar]
- 13.Read RJ, Adams PD, Arendall WB, 3rd, Brunger AT, Emsley P, Joosten RP, Kleywegt GJ, Krissinel EB, Lutteke T, Otwinowski Z, Perrakis A, Richardson JS, Sheffler WH, Smith JL, Tickle IJ, Vriend G, Zwart PH. A new generation of crystallographic validation tools for the protein data bank. Structure. 19(10):1395–1412. doi: 10.1016/j.str.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta. 1999;1473 (1):4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 15.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucl Acids Res. 2000;28 (1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lutteke T. Analysis and validation of carbohydrate three-dimensional structures. Acta Cryst. 2009;D65(2):156–168. doi: 10.1107/S0907444909001905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henrick K, Feng Z, Bluhm WF, Dimitropoulos D, Doreleijers JF, Dutta S, Flippen-Anderson JL, Ionides J, Kamada C, Krissinel E, Lawson CL, Markley JL, Nakamura H, Newman R, Shimizu Y, Swaminathan J, Velankar S, Ory J, Ulrich EL, Vranken W, Westbrook J, Yamashita R, Yang H, Young J, Yousufuddin M, Berman HM. Remediation of the protein data bank archive. Nucl Acids Res. 2008;36 (suppl 1):D426–D433. doi: 10.1093/nar/gkm937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lutteke T, von der Lieth CW. pdb-care (PDB carbohydrate residue check): a program to support annotation of complex carbohydrate structures in PDB files. BMC Bioinformatics. 2004;5:69. doi: 10.1186/1471-2105-5-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakahara T, Hashimoto R, Nakagawa H, Monde K, Miura N, Nishimura S-I. Glycoconjugate Data Bank:Structures-an annotated glycan structure database and N-glycan primary structure verification service. Nucl Acids Res. 2008;36 (suppl 1):D368–D371. doi: 10.1093/nar/gkm833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacKerell AD., Jr Empirical force fields for biological macromolecules: Overview and issues. J Comput Chem. 2004;25 (13):1584–1604. doi: 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- 21.Momany FA, Willett JL. Computational studies on carbohydrates: solvation studies on maltose and cyclomaltooligosaccharides (cyclodextrins) using a DFT/ab initio-derived empirical force field, AMB99C. Carbohydr Res. 2000;326 (3):210–226. doi: 10.1016/s0008-6215(00)00043-4. [DOI] [PubMed] [Google Scholar]
- 22.Momany FA, Willett JL. Computational studies on carbohydrates: in vacuo studies using a revised AMBER force field, AMB99C, designed for alpha-(1-->4) linkages. Carbohydr Res. 2000;326 (3):194–209. doi: 10.1016/s0008-6215(00)00042-2. [DOI] [PubMed] [Google Scholar]
- 23.Kirschner KN, Yongye AB, Tschampel SM, Gonzalez-Outeirino J, Daniels CR, Foley BL, Woods RJ. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J Comput Chem. 2008;29 (4):622–655. doi: 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lins RD, Hunenberger PH. A new GROMOS force field for hexopyranose-based carbohydrates. J Comput Chem. 2005;26 (13):1400–1412. doi: 10.1002/jcc.20275. [DOI] [PubMed] [Google Scholar]
- 25.Kony D, Damm W, Stoll S, Van Gunsteren WF. An improved OPLS–AA force field for carbohydrates. J Comput Chem. 2002;23 (15):1416–1429. doi: 10.1002/jcc.10139. [DOI] [PubMed] [Google Scholar]
- 26.Woods RJ, Dwek RA, Edge CJ, Fraser-Reid B. Molecular Mechanical and Molecular Dynamic Simulations of Glycoproteins and Oligosaccharides. 1. GLYCAM_93 Parameter Development. J Phys Chem. 1995;99 (11):3832–3846. [Google Scholar]
- 27.Hansen HS, Hünenberger PH. A reoptimized GROMOS force field for hexopyranose-based carbohydrates accounting for the relative free energies of ring conformers, anomers, epimers, hydroxymethyl rotamers, and glycosidic linkage conformers. J Comput Chem. 2011;32 (6):998–1032. doi: 10.1002/jcc.21675. [DOI] [PubMed] [Google Scholar]
- 28.MacKerell AD, Jr, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102 (18):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 29.MacKerell AD, Jr, Feig M, Brooks CL. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25 (11):1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 30.Foloppe N, MacKerell AD., Jr All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J Comput Chem. 2000;21 (2):86–104. [Google Scholar]
- 31.MacKerell AD, Jr, Banavali NK. All-atom empirical force field for nucleic acids: II. Application to molecular dynamics simulations of DNA and RNA in solution. J Comput Chem. 2000;21 (2):105–120. [Google Scholar]
- 32.Klauda JB, Brooks BR, MacKerell AD, Jr, Venable RM, Pastor RW. An ab Initio Study on the Torsional Surface of Alkanes and Its Effect on Molecular Simulations of Alkanes and a DPPC Bilayer. J Phys Chem B. 2005;109 (11):5300–5311. doi: 10.1021/jp0468096. [DOI] [PubMed] [Google Scholar]
- 33.Feller SE, Gawrisch K, MacKerell AD., Jr Polyunsaturated Fatty Acids in Lipid Bilayers: Intrinsic and Environmental Contributions to Their Unique Physical Properties. J Am Chem Soc. 2001;124 (2):318–326. doi: 10.1021/ja0118340. [DOI] [PubMed] [Google Scholar]
- 34.Feller SE, MacKerell AD., Jr An Improved Empirical Potential Energy Function for Molecular Simulations of Phospholipids. J Phys Chem B. 2000;104 (31):7510–7515. [Google Scholar]
- 35.Yin D, MacKerell AD., Jr Combined ab initio/empirical approach for optimization of Lennard–Jones parameters. J Comput Chem. 1998;19 (3):334–348. doi: 10.1002/jcc.1166. [DOI] [PubMed] [Google Scholar]
- 36.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, MacKerell AD., Jr CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. 2010;31 (4):671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guvench O, Greene SN, Kamath G, Brady JW, Venable RM, Pastor RW, MacKerell AD., Jr Additive empirical force field for hexopyranose monosaccharides. J Comput Chem. 2008;29 (15):2543–2564. doi: 10.1002/jcc.21004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hatcher E, Guvench O, MacKerell AD., Jr CHARMM additive all-atom force field for aldopentofuranoses, methyl-aldopentofuranosides, and fructofuranose. J Phys Chem B. 2009;113 (37):12466–12476. doi: 10.1021/jp905496e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hatcher E, Guvench O, MacKerell AD., Jr CHARMM Additive All-Atom Force Field for Acyclic Polyalcohols, Acyclic Carbohydrates and Inositol. J Chem Theory Comput. 2009;5 (5):1315–1327. doi: 10.1021/ct9000608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guvench O, Hatcher ER, Venable RM, Pastor RW, MacKerell AD., Jr CHARMM Additive All-Atom Force Field for Glycosidic Linkages between Hexopyranoses. J Chem Theory Comput. 2009;5 (9):2353–2370. doi: 10.1021/ct900242e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raman EP, Guvench O, MacKerell AD., Jr CHARMM additive all-atom force field for glycosidic linkages in carbohydrates involving furanoses. J Phys Chem B. 2010;114 (40):12981–12994. doi: 10.1021/jp105758h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guvench O, Mallajosyula SS, Raman EP, Hatcher E, Vanommeslaeghe K, Foster TJ, Jamison FW, MacKerell AD., Jr CHARMM Additive All-Atom Force Field for Carbohydrate Derivatives and Its Utility in Polysaccharide and Carbohydrate-Protein Modeling. J Chem Theory Comput. 2011;7 (10):3162–3180. doi: 10.1021/ct200328p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mallajosyula SS, MacKerell AD., Jr Influence of Solvent and Intramolecular Hydrogen Bonding on the Conformational Properties of O-Linked Glycopeptides. J Phys Chem B. 2011;115 (38):11215–11229. doi: 10.1021/jp203695t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mallajosyula SS, Guvench O, Hatcher E, MacKerell AD., Jr CHARMM Additive All-Atom Force Field for Phosphate and Sulfate Linked to Carbohydrates. J Chem Theory Comput. 2012 doi: 10.1021/ct200792v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brooks BR, Brooks CL, 3rd, MacKerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30 (10):1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J Comput Chem. 2008;29 (11):1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 47.Jo S, Song KC, Desaire H, MacKerell AD, Jr, Im W. Glycan Reader: Automated Sugar Identification and Simulation Preparation for Carbohydrates and Glycoproteins. J Comput Chem. 2011;32 (14):3135–3141. doi: 10.1002/jcc.21886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Halgren TA, Damm W. Polarizable force fields. Curr Opin Struct Biol. 2001;11 (2):236–242. doi: 10.1016/s0959-440x(00)00196-2. [DOI] [PubMed] [Google Scholar]
- 49.Ponder JW, Wu C, Ren P, Pande VS, Chodera JD, Schnieders MJ, Haque I, Mobley DL, Lambrecht DS, DiStasio RA, Jr, Head-Gordon M, Clark GN, Johnson ME, Head-Gordon T. Current status of the AMOEBA polarizable force field. J Phys Chem B. 2010;114 (8):2549–2564. doi: 10.1021/jp910674d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lopes P, Roux B, MacKerell A. Molecular modeling and dynamics studies with explicit inclusion of electronic polarizability: theory and applications. Theoretica Chimica Acta. 2009;124 (1):11–28. doi: 10.1007/s00214-009-0617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Becker OMMJ, AD, Roux B, Watanabe M, editors. Computational Biochemistry and Biophysics. Marcel-Dekker, Inc; New York: 2001. [Google Scholar]
- 52.Zhu X, Lopes PEM, MacKerell AD., Jr Recent developments and applications of the CHARMM force fields. WIREs Comput Mol Sci. 2(1):167–185. doi: 10.1002/wcms.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aleshin AE, Hoffman C, Firsov LM, Honzatko RB. Refined Crystal Structures of Glucoamylase from Aspergillus awamori var. X100. J Mol Biol. 1994;238 (4):575–591. doi: 10.1006/jmbi.1994.1316. [DOI] [PubMed] [Google Scholar]
- 54.Word JM, Lovell SC, Richardson JS, Richardson DC. Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol. 1999;285 (4):1735–1747. doi: 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- 55.Beglov D, Roux B. An Integral Equation To Describe the Solvation of Polar Molecules in Liquid Water. J Phys Chem B. 1997;101 (39):7821–7826. [Google Scholar]
- 56.Darden T, York D, Pedersen L. Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98 (12):10089–10092. [Google Scholar]
- 57.Steinbach PJ, Brooks BR. New spherical-cutoff methods for long-range forces in macromolecular simulation. J Comput Chem. 1994;15 (7):667–683. [Google Scholar]
- 58.Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23 (3):327–341. [Google Scholar]
- 59.Nose S. A unified formulation of the constant temperature molecular dynamics methods. J Chem Phys. 1984;81 (1):511–519. [Google Scholar]
- 60.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: The Langevin piston method. J Chem Phys. 1995;103 (11):4613–4621. [Google Scholar]
- 61.Brünger AT. X-PLOR, Version 3.1, A System for X-ray Crystallography and NMR. Yale University Press; New Haven, CT: 1992. [Google Scholar]
- 62.Kalé L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, Schulten K. NAMD2: Greater Scalability for Parallel Molecular Dynamics. J Comput Phys. 1999;151 (1):283–312. [Google Scholar]
- 63.Petrescu AJ, Milac AL, Petrescu SM, Dwek RA, Wormald MR. Statistical analysis of the protein environment of N-glycosylation sites: implications for occupancy, structure, and folding. Glycobiology. 2004;14 (2):103–114. doi: 10.1093/glycob/cwh008. [DOI] [PubMed] [Google Scholar]
- 64.Rao VSR, Qasba PK, Balaji PV, Chandrasekaran R. Conformation of Carbohydrates. Harwood Academic Publishers; Amsterdam: 1998. [Google Scholar]
- 65.Tachibana Y, Fletcher GL, Fujitani N, Tsuda S, Monde K, Nishimura S-I. Antifreeze Glycoproteins: Elucidation of the Structural Motifs That Are Essential for Antifreeze Activity. Angew Chem Int Ed Engl. 2004;43(7):856–862. doi: 10.1002/anie.200353110. [DOI] [PubMed] [Google Scholar]
- 66.Stanca-Kaposta EC, Gamblin DP, Cocinero EJ, Frey J, Kroemer RT, Fairbanks AJ, Davis BG, Simons JP. Solvent Interactions and Conformational Choice in a Core N-Glycan Segment: Gas Phase Conformation of the Central, Branching Trimannose Unit and its Singly Hydrated Complex. J Am Chem Soc. 2008;130 (32):10691–10696. doi: 10.1021/ja801892h. [DOI] [PubMed] [Google Scholar]
- 67.Sattelle BM, Hansen SU, Gardiner J, Almond A. Free Energy Landscapes of Iduronic Acid and Related Monosaccharides. J Am Chem Soc. 132(38):13132–13134. doi: 10.1021/ja1054143. [DOI] [PubMed] [Google Scholar]
- 68.Gnanakaran S, Nymeyer H, Portman J, Sanbonmatsu KY, García AE. Peptide folding simulations. Curr Opin Struct Biol. 2003;13 (2):168–174. doi: 10.1016/s0959-440x(03)00040-x. [DOI] [PubMed] [Google Scholar]
- 69.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14 (1):33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]