

Abstract
Background
The National Birth Defects Prevention Study (NBDPS) contains a wealth of information on affected and unaffected family triads, and thus provides numerous opportunities to study gene-environment interactions (GxE) in the etiology of birth defect outcomes. Depending on the research objective, several analytic options exist to estimate GxE effects that utilize varying combinations of individuals drawn from available triads.
Methods
In this paper we discuss several considerations in the collection of genetic data and environmental exposures. We will also present several population- and family-based approaches that can be applied to data from the NBDPS including case-control, case-only, family-based trio, and maternal versus fetal effects. For each, we describe the data requirements, applicable statistical methods, advantages and disadvantages.
Discussion
A range of approaches can be used to evaluate potentially important GxE effects in the NBDPS. Investigators should be aware of the limitations inherent to each approach when choosing a study design and interpreting results.
Keywords: Gene-environment interaction, genetic epidemiology, birth defects, family-based study, neural tube defects, congenital heart defects, National Birth Defects Prevention Study, case-only, transmission disequilibrium test
Introduction
Abnormal embryonic development likely depends on both inherited genetic risk factors that reflect interactions between maternal and paternal genetics and on maternal environmental influences at specific gestational times. Such gene-environment interactions (GxE) have been reported for several congenital abnormalities, including neural tube defects (NTDs) (Etheredge et al., 2012), congenital heart defects (CHD) (Hobbs et al., 2010), and oral–facial clefts (Murray, 2002). Nevertheless, the biological mechanisms underlying such interactions generally remain somewhat unclear, and public health recommendations have only begun to consider how combinations of genetic and environmental effects could interact to influence the risk of birth defects (Sharp and Barrett, 2000).
Often GxE studies build upon existing knowledge about the individual main effects of genetic or environmental factors. For example, animal studies, observational research, and clinical trials determined the existence of a link between folate and the occurrence of NTDs (Nelson et al., 1952; Laurence et al., 1981; Mulinare et al., 1988; Bower and Stanley, 1989; Mills et al., 1989; Milunsky et al., 1989; MRC Vitamin Study Research Group, 1991). Later, Christensen et al. (1999) proposed that there was also a gene-nutrient interaction between a folate related gene, methylenetetrahydrofolate reductase (MTHFR), and maternal folate levels for NTDs. Furthermore, we are often interested in scenarios where the GxE effect is greater than the simple combination of the genetic or environmental factor alone. Before widespread availability of genotyped genetic data, family history was used as a proxy measurement for inherited genetic risk. For example, Honein et al. (2000) conducted a case-control study for clubfoot that found the joint effect of smoking and family history (odds ratio (OR) = 20.30, 95% confidence interval (95%CI): 7.90 to 52.17) to be greater than the individual or multiplicative effects of either smoking (OR = 1.34, 95%CI: 1.04, 1.72) or family history (OR = 6.52, 95% CI: 2.95, 14.41).
In GxE studies of birth defects, it is important to recognize that multiple interaction effects may occur simultaneously. Any maternal environmental exposure may interact with either the mother’s genes or the infant’s genes to alter risks for birth defects, resulting in coexisting infant GxE and maternal GxE effects. Additionally, there could be interactions between infants’ genotype and their mothers’ genotype, yielding maternal-fetal gene-gene interactions (GxG).
Researchers can investigate such interactions in the National Birth Defects Prevention Study (NBDPS), a population based case-control study with 10 recruitment centers across the country (Arkansas, California, Georgia, Iowa, Massachusetts, New Jersey, New York, North Carolina, Texas, and Utah). The NBDPS collected DNA samples from over 19,000 stillborn and live-born case infants with birth defects from a pre-specified list of over 30 conditions. Also available is information on the mothers of these case infants regarding genetic, lifestyle and environmental exposures, as well as genetic data on fathers who provided DNA samples (Yoon et al., 2001; Webber et al., 2015). The same information was collected from over 6,000 population-based unmatched control infants without birth defects and their parents (Cogswell et al., 2009; Webber et al., 2015).
The NBDPS has detected a number of associations, including between birth defects and maternal “environmental” factors such as obesity (Waller et al., 2007) and diet quality (Carmichael et al., 2012) or with mothers’ pharmaceutical drug usage of antidepressants (Alwan et al., 2007) and nitrosatable drugs (Brender et al., 2011). Investigators have also found associations between candidate genes and birth defects including genes in folate-related pathways for CHD (Hobbs et al., 2014) and glucose homeostasis genes for NTDs (Lupo et al., 2012). Hobbs et al. (2010) also identified multiple gene-environment interactions between folate-related genes and maternal smoking or alcohol intake for CHDs. These examples represent only a handful of the numerous etiologic questions that either have or could be investigated in the NBDPS.
There are many considerations for epidemiologic studies in which the main research objective is to interrogate GxE associations for birth defects. These include choices in study design, type of genetic information to collect, and statistical approaches to use. Here we present and discuss some of the current study designs for, analytic methods used in, and challenges to identifying GxE effects in studies of birth defects.
Methods
Measuring Genes and Environment
Genes
To date, most studies of GxE for birth defects have evaluated single-nucleotide polymorphisms (SNPs) from candidate genes or loci. SNPs are generally chosen based on some prior biological rationale or from main effect findings from candidate gene studies. For example, in gene-nutrient studies, only genes from a pathway involved in the transport or metabolism of that nutrient may be considered.
The focus on candidate genes has given way to genome-wide assessments of GxE, on account of the increasing affordability of high-resolution genotyping arrays that are commonly used in genome wide association studies (GWAS). Genome-wide interrogations of GxE effects, termed gene-by-environment wide interaction studies (GEWIS) (Khoury and Wacholder, 2008) often begin with individually testing each SNP for its association with the outcome of interest, and then the most strongly associated SNPs are evaluated for their interactions with environmental exposures. Murcray et al. (2008) developed a method to extend GWAS analyses to include a GEWIS analysis that uses a two-step approach. The first step serves as a screening phase and employs a test similar to the case-only approach (described below). In the second step, only SNPs that are significant in the first step are tested for GxE using the traditional case-control approach.
In many two-step approaches investigators must optimize filtering significance (i.e., p-values) thresholds in their selection of SNPs for analysis in the second step. Alternatively, one can use two-stage data collection with high-density genotyping and analysis in the first stage followed by genotyping of only the most statistically significant SNPs using a custom array in the second stage (Satagopan et al., 2002; Thomas, 2010). However the cost differential between high-density and targeted arrays has become small enough that most studies simply run a high-density array on all study subjects. Several Bayesian approaches are also available that average results from both case-only and case-control approaches to estimate GxE effects (Thomas, 2010). Specific pathway based analyses combine the strengths of candidate gene and GWAS approaches; inter-related genes that play a part in the same biological pathway are considered while using genetic data from genome-wide genotyping arrays. This approach has only been recently applied to GxE studies (Wei et al., 2012).
One major point of contention in all GxE studies is whether detection of a main genetic effect is necessary prior to testing interaction effects. In birth defects research, GxE effects are typically assessed only for variants that have already shown evidence of a main genetic effect. Recent methods, however, suggest that this approach may overlook important findings. For example, some epidemiologic studies may have been unable to detect a clear association between the C677T allele in the MTHFR gene, which plays a role in folate metabolism, and risk of NTDs due to a lack of varying levels of folate intake in some study populations, especially post fortification. It was only after a meta-analysis was conducted using 17 studies from North America and Europe that the link was finally confirmed (Botto and Yang, 2000). Therefore, SNPs with a GxE effect may not make it to the list of candidate SNPs due to a diminished genetic effect in the absence of an environmental exposure that also contributes to the outcome, as was the case with the MTHFR variant and folate intake for NTDs (Daly et al., 1995; Crider et al., 2014). Some SNPs may only be associated with an outcome in the presence of an environmental factor. In such a case, the main effect for a SNP will not be found for a causal reason—because both the genetic factor and the environmental factor must be present to observe the outcome. Etheredge et al. (2012) reported that the risk of NTDs was only slightly elevated for infants who possessed a risk SNP, rs11627387, in another folate-related gene MTHFD1 (OR=1.11, 95%CI: 0.87 to 1.41), but for infants who also had low folate intake the risk of NTDs increased four-fold (OR=4.25, 95%CI: 2.33 to 7.75). If there is a true etiologic mechanism by which the genetic and environmental factors interact to produce the outcome, then a main genetic effect should not be required to investigate the presence of a GxE effect.
Environment
There are unique considerations in studying maternal exposures just prior to and during early pregnancy. In the data collection phase, maternal environmental exposure ascertainment is challenging because mothers often underreport behaviors or lifestyle choices that are known to potentially cause harm to their fetuses. A study in New Zealand found that maternal smoking was underreported for half of all mothers based on a comparison between self-reported smoking and levels of serum cotinine, a nicotine metabolite (Ford et al., 1997). Additionally, mothers that did not respond to the lifestyle questionnaire were more likely to be heavy smokers in the first trimester (40%) than mothers who did respond (16%) (Ford et al., 1997). Similar underreporting has also been found for medication use in the United States (Newport et al., 2008). In addition, other environmental exposures may be difficult to accurately measure, such as pollutants or industrial byproducts (e.g., Bisphenol A), although for many of these, any biased recall may not be differ between cases and controls. Since many single birth defect phenotypes are rare, most research participants are identified after the outcome has occurred and are asked to retrospectively recall their exposures during narrow windows of time. At times women may be asked about exposures that occurred many months or years earlier.
For main environmental effects, if the level of misclassification is similar (non-differential) among cases and controls, then estimates for the environmental factor will be biased towards the null, making it more difficult to detect any true effects. If the misclassification is more (or less) prevalent among cases than controls (differential), then estimates can be biased in either direction. These issues can become more complex in the context of GxE estimates and are less often discussed (Greenland, 1993). The direction of for a GxE effect estimate may be predicted when there is no true association between the genotype and environmental exposure among the controls, and when misclassification of the environmental exposure is non-differential between genotypes (García-Closas et al., 1998). Under these conditions, in the presence of a true multiplicative interaction, differential misclassification of the environmental exposure biases the GxE effect estimate toward the null. In the absence of multiplicative interaction, differential misclassification of the environmental exposure does not bias the GxE effect estimate. Importantly, the first of these conditions could be tested in the NBDPS data via a test of independence of the environmental and genotype data in the controls.
Design and Statistical Analysis for Gene-Environment Interactions
The following discussions of study designs and analyses will be limited to dichotomous phenotypes since most birth defect phenotypes are defined as binary. Similarly we will assume a qualitative (binary) environmental exposure but many approaches can be extended to quantitative (continuous) environmental exposures.
Case-control studies of unrelated individuals are commonly used for evaluating rare diseases such as birth defects. However, case-only (i.e., affected infants), case-parent dyad (i.e., affected infants and their mothers), and triad (i.e., affected infants and both of their parents) designs can also be used to evaluate GxE. Note that the case-sibling study design in which an affected individual is matched to his or her sibling can be more efficient for detecting GxE than case-control or triad designs especially for rare SNPs (Witte et al., 1999; Chatterjee et al., 2005). We will not expand on the discordant sibship model because our goal here is to describe the possible approaches that could be applied to data from the NBDPS, which contains affected and unaffected family dyads and triads, and does not include siblings.
In general, sample sizes required to detect GxE can be substantially larger than those required for detecting main genetic or environmental effects. They are influenced by: 1) the magnitude of the GxE effect; 2) the allele frequency of the causal SNP; 3) the prevalence of the environmental exposure in the study population; 4) the model of inheritance assumed; and 5) the strength of correlation between causal and tag SNPs. Strong main genetic effects could also affect the ability to detect GxE, although the extent of this depends on the causal structure of the GxE relationship (Thomas, 2010). To get a sense of the sample sizes required, assuming a true GxE odds ratio of 1.25, an environmental exposures with 40% prevalence, a minor allele frequency of 0.3, a dominant mode of inheritance and directly measuring the causal SNPs requires over 10,000 case-control pairs to achieve 80% power (Hein et al., 2008). In contrast, if the GxE odds ratio is 2.0, fewer than 1,600 pairs would be required, holding everything else constant. In many power and sample size calculations, a larger GxE effect is typically expected. For example, an assumption of at least a 3-fold increased risk under a gene-environment interaction model is consistent with, and is in fact less than, what others have used as a guide (Hwang et al., 1994). Although Hein et al. (2008) found that sample size requirements did not differ greatly based on the assumed inheritance model (e.g., recessive, additive, or dominant), typically recessive models require greater sample sizes to achieve the same level of power than dominant models in case-only (Clarke and Morris, 2010) and case-control (Palmer and Cardon, 2005) studies. Since the causal SNP involved in the GxE effect is generally not directly genotyped but rather assayed by a tag SNP, the degree of linkage disequilibrium and concordance of allele frequencies between the causal and tag SNPs among the controls also impact sample size requirements. There is an inverse relationship between the causal and tag SNP correlation and the sample size required to detect GxE effects would increase (Hein et al., 2008).
There is specialized statistical software available for GxE power calculations, including Quanto (Gauderman, 2002) for matched case-control, case-sibling, case-parent, and case-only study designs, PBAT (Lange et al., 2004) or the R implementation, pbatR, (Hoffmann and Lange, 2006) for family-based study designs, and the R package, trio, (Schwender et al., 2014) specifically for trios. A set-based approach is implemented in SBERIA, which weights main effects of SNPs within a gene region using a genetic risk score that is included in interaction tests (Jiao et al., 2013).
Results
Case-Control Study
The case-control approach utilizes information from affected and unaffected individuals who are unrelated as depicted in Figure 1. To evaluate GxE in a case-control study, the traditional approach is to fit a logistic regression model with terms that include the genetic effect, environmental effect, GxE effect, and any covariates:
| (1) |
Figure 1. Possible Approaches for Family Trio Data.
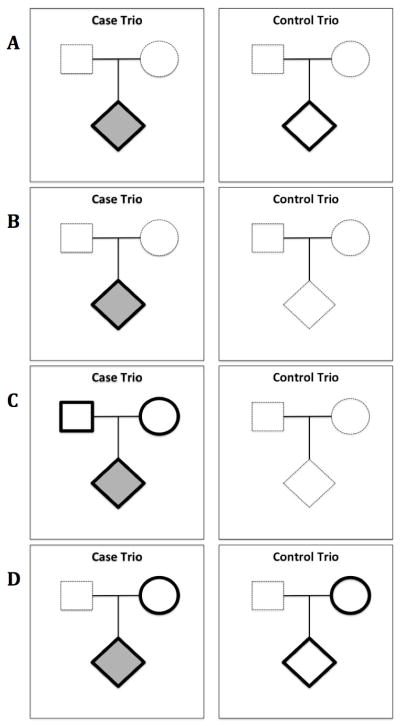
Required data from individuals indicated by bolded black outline. Affected individuals noted by shaded shapes. Optional or not required individuals noted by dashed outline.
Panel A: Case-Control Study
Requires genotypes of unrelated affected and unaffected individuals. In a hypothetical sample of 100 case trios and 100 control trios, there would be 200 individuals in the study, 100 affected individuals and 100 unaffected individuals.
Panel B: Case-Only Study
Requires genotypes of only affected individuals. In a hypothetical sample of 100 case trios and 100 control trios, there would be 100 affected individuals in the study for this approach.
Panel C: Family-Based Trio Study
Requires genotypes of affected individuals and both their parents. In a hypothetical sample of 100 case trios and 100 control trios, there would be 300 individuals in the study for this approach, 100 affected infants and 200 parents.
Panel D: Maternal vs. Fetal Effects
Requires genotypes of affected and unaffected individuals and their mothers. In a hypothetical sample of 100 case trios and 100 control trios, there would be 400 individuals in the study, 100 affected individuals their 100 mothers, 100 unaffected individuals, and their 100 mothers.
The vector Y represents the binary outcome with possible values for affected (1) and unaffected individuals (0). The model intercept is given by β0, while G represents the genotype of the affected or unaffected individuals following an additive, dominant or recessive model and its effects on Y are estimated by βG. The parameter E is the maternal environmental exposure of the affected or unaffected individual and its effect is estimated by βE. The GxE effect is estimated by βGxE. Therefore evaluating the presence of an interaction between the genetic and environmental factors requires testing the null hypothesis that βGxE = 0 (i.e., no interaction), while βGxE ≠ 0 indicates interaction is present. A joint two-degrees of freedom test of both the main genetic (βG) and GxE (βGxE) effects can provide greater power than standard case-control and case-only approaches when the causal structure describing genetic, environmental, and GxE effects on the outcome is not well known (Kraft, 2007). The case-control approach is common in genetic association studies. As an example of a basic GxE test, in a population-based case-control study of 69 infants with cleft palate and 284 controls with non-cleft birth defects, Hwang et al. (1994) observed a GxE effect between maternal smoking and the infant’s genotype for the transforming grown factor alpha (TGFA) locus.
The case-control design is susceptible to population stratification bias, especially when the study includes individuals with varying genetic ancestries, as is the case with the NBDPS. The issue arises if both the frequency of genetic variants and the risk of birth defects vary among the ancestrally different populations. This can result in a non-causal association between genetic ancestry and the individual’s outcome even in the absence of a true association between genotypes and birth defect phenotypes due to confounding. This is generally adjusted for in the logistic regression model by including covariates (C) as a vector of eigenvectors, which represent the principal components that identify distinct subpopulations or by including ancestry informative markers. Their effects are then estimated by βC. Commonly used methods to generate these principal components include EIGENSTRAT using the EIGENSOFT package (Patterson et al., 2006; Price et al., 2006) and multi-dimensional scaling using PLINK (Purcell et al. 2007). The case-control logistic regression model can be fit in any standard statistical software package, such as in R using the glm function, with the CGEN package (Bhattacharjee et al., 2012) or in PLINK (Purcell et al., 2007) as described in Table 1.
Table 1.
Comparison of Different Gene-environment Interaction Approaches
| Approach | Advantages | Disadvantages | Software Implementation | Effect Estimated |
|---|---|---|---|---|
| Case-control study |
|
|
|
|
| Case-only study |
|
|
|
|
| Family-based trio study |
|
|
|
|
| Maternal vs. Fetal Effects |
|
|
|
|
Case-Only Study
This design requires only affected individuals (Figure 1). The case-only approach can be powerful for detecting GxE, and useful when identifying and recruiting control subjects is problematic or not feasible. However, the approach assumes that the genetic and environmental factors are independent in the study’s source population (Piegorsch et al., 1994). One can estimate the association between gene and environment among cases from the model using logistic regression:
| (2) |
where G is the genotype (estimated for an dominant or recessive model in this case, but could be coded for an additive model using ordinal or multinomial logistic models (Clarke and Morris 2010)), E is the environmental exposure, β0 is the model intercept and βGxE is the ln(OR) for the association between environment and genotype (i.e., the estimate of the GxE effect). This OR among cases is equivalent to the synergy index calculated from a case-control design, which is the OR of the GxE effect divided by the product of the OR of the genetic effect and the OR of the environmental exposure (Khoury and Flanders, 1996). A synergy index greater than one implies the presence of multiplicative GxE. This approach could be especially useful in re-analyzing existing data using only the cases of prior case-control studies. As an example of this study design, Zeiger et al. (2005) implemented a case-only pooled meta-analysis using 335 cleft palate cases from five previously published studies and found a GxE effect between the TGFA Taq1 C2 allele genotype and maternal smoking.
The case-only analysis can be more powerful (and potentially, financially much less costly if data collection of controls is required) than a traditional case-control study, as the assumption of gene-environment independence reduces the standard error of the estimate of GxE interaction (Khoury and Flanders, 1996; Albert, 2001). Violations of the independence assumption, however, would preclude using the case-only design to assess GxE (Albert, 2001). The independence assumption may not hold when behavior is affected by genetic factors (Gatto, 2004). For example, those who possess the risk allele in the D2 dopamine receptor gene (DRD2) may experience increased dopamine levels when consuming nicotine and thus more likely to be cigarette smokers (Noble et al., 1994). A case-only study of the interaction between DRD2 and smoking in lung cancer could be problematic if this relationship between DRD2 and smoking existed in the population. Note that many of the family-based methods (discussed below) also require that gene and environment are independent within families (Umbach and Weinberg, 2000; Chatterjee et al., 2005).
Potential drawbacks to the case-only approach are that main effects (genetic or environmental) cannot be estimated, and it is vulnerable to bias due to population stratification (Wang and Lee, 2008) (Table 1). However, if truly causal, the presence of both genetic and environmental factors is more likely to occur in cases than controls, which makes this approach appealing for studying rare genes or environmental exposures. The case-only logistic regression analysis can be implemented using standard statistical software.
Family-Based Trio Study
GxE can also be evaluated with a study of parents and an affected infant (Figure 1). The most common approach here is the transmission-disequilibrium test (TDT). The affected infant is the case (with genotypes transmitted from the parents), and is matched to three pseudo-controls (with the non-transmitted parental genotypes). One then conducts what is essentially a matched 1:3 case-control analysis where each family serves as a matching stratum (Spielman et al., 1993; Neumann et al., 2014). This approach requires genetic information from both affected individual’s parents; incomplete trios cannot be used in this analysis without prior imputation for any missing parental genotypes (Table 1).
Using a TDT, the main effect of the environmental factor cannot be directly tested as all four ‘individuals’ (the case and three ‘pseudo-controls’) would be exposed to the same maternal environment. Nevertheless, the GxE effect can be estimated without the environmental main effect with the following conditional logistic regression model:
| (3) |
where Y represents the presence (1) or absence (0) of a birth defect in the infant, G represents the genotypes for the affected infant and its three pseudo-controls following an additive, dominant, or recessive model, E is the maternal environmental exposure, βi is the model intercept which is unique to each family strata (i), βG is the infant’s genetic main effect, and βGxE is the GxE effect. As an example, Beaty (2011) used the TDT in a study of 550 family trios with non-syndromic cleft palate that identified evidence of a GxE effect between maternal alcohol intake and infant genotype (in the MLLT3 and SMC2 genes), and between maternal smoking and infant genotype (in the TBK1 and ZNF236 genes).
Family-based designs such as the TDT avoid confounding due to population stratification because the analysis is conducted within families who share the same genetic ancestry (Gauderman et al., 1999; Witte et al., 1999; Thomas and Witte, 2002). However, a major drawback is that collecting DNA samples from both parents and the affected infant can be difficult. The issue is particularly problematic for late-onset diseases since parents may no longer be alive. This concern is more easily overcome in the study of birth defects since sample collection would be expected to occur near the time of the infant’s birth when both parents are likely still alive and available to provide DNA.
Conducting a genotypic TDT analysis for GxE can be implemented using the R-package trio and the colGxE function developed by Schwender et al. (2014), which is conveniently able to read in .ped and binary files created from PLINK software. Extensions of this approach to other family structures also exist (e.g., sibships, combinations of sibships and trios) (Lake and Laird, 2004; Dudbridge, 2008; Vansteelandt et al., 2008; Hoffmann et al., 2009, 2011; Moerkerke et al., 2010) which can be tested using the PBAT software (Lange et al., 2004) and in R using the pbatR package (Hoffmann and Lange, 2006).
Maternal versus Fetal Effects
As noted above, it may be important to evaluate GxE not only in the affected infant, but also in their mothers as well as any potential maternal-fetal GxG. For example, using data from the NBDPS, Lupo et al. (2014) found suggestive evidence that maternal genes related to metabolic conditions interacted with fetal genes related to glucose homeostasis to increase the risk of NTDs. Such interactions can be disentangled using log-linear models that adjust for maternal genetics when estimating infants’ genetic effects and that adjust for infants’ genetics when estimating maternal genetic effects (Umbach and Weinberg, 1997; 2000). This approach requires genetic information from affected and unaffected infant-mother dyads (Figure 1) but can also incorporate genotype information from fathers (Ainsworth et al., 2011). To evaluate GxE, we can first model main genetic effects within strata of the environmental factor then test for heterogeneity of the stratum-specific estimates. Note that a similar approach could be used for the case-control and trio designs. Hobbs et al. (2014) used an extended log-linear model to evaluate GxE effects on 616 case trios and 1645 control trios; with this approach, they found 19 maternal SNPs and 9 fetal SNPs with evidence of GxE between the genotype and maternal folate supplementation.
The log-linear approach uses a multinomial model to calculate risk ratios of genetic effects by estimating the penetrance for all possible combinations of the genotypes of the infant and the mother under a multiplicative risk model. In particular, the model estimates the effect of the infant or mother carrying one or two copies of a genetic variant on the infants risk. Then several maternal-fetal GxG (γij) parameters given by γ11, γ12, γ21, and γ22 describe the interaction between the mother’s genotype and the infant’s genotype in which i (j) indicates the number of copies of the risk allele that the mother (infant) possesses. For example, γ12 is the interaction between mother and infant genotypes when the mother possesses one copy and the infant possesses two copies.
The log-linear models must be fit within homogenous genetic populations and bias due to population stratification is possible if ancestrally varying populations are used. Implementation of this approach is easily conducted using the PREMIM and EMIM software (Howey and Cordell, 2012). To extend this genetic model to test for GxE, the log ORs and their standard errors provided in the output of the EMIM software can be read into any standard statistical software with the ability to conduct a test of heterogeneity such as the R package, rmeta using the meta.summaries function. The test of heterogeneity is essentially testing whether the stratum-specific estimates for the genetic effect are different between the two strata of the binary environmental factor. Unfortunately, this only provides a p-value rather than a direct estimate of the GxE and is limited to binary environmental exposures but it has the advantage of discriminating between maternal GxE and infant GxE effects (Table 1).
Discussion
By collecting both affected and unaffected infant/mother/father triads, the NBDPS allows for flexibility in the choice of analytic approach and resulting estimates of genetic, environmental and GxE effects. The approach used should reflect the research question of interest. For example, all described approaches can estimate the GxE effect contributed by the infant’s genotype, but only the log-linear model also allows for estimation of the maternal GxE. Additionally, some causal assumptions limit the use of certain approaches. If, for instance, the genetic and environmental factors may not be independent in the study population, then the case-only approach is not appropriate. As another example, if genetic ancestry cannot be determined accurately, a genotypic TDT approach may be best since it controls for potential population stratification.
Another important point for GxE studies is how to appropriately control for potential confounding. Instead of only including the main effect of the corresponding covariate, one may also need to include interaction terms between the confounder and each of the genetic and environmental factors (Keller, 2014). The model would then control for any covariates that may confound the GxE effect, either by being correlated with the genotype or with the environmental factor. This type of adjustment is not yet common practice and so the degree of confounding due to improper adjustment is largely unknown, but it is an important consideration when assessing GxE effects (Keller, 2014).
When answering etiologic questions about interactions, an important consideration is whether the presence of interaction is assessed on the additive or multiplicative scale. All approaches presented in this paper assume a multiplicative scale, but it may be important to also consider additive interaction since the interpretation of the GxE effect, which can have public health implications, may differ based on scale (Ottman, 1996). Some have criticized case-only designs for being only relevant to the multiplicative scale, but if certain assumptions can be made, the case-only approach also allows inference about mechanistic interactions that are typically only identified on the additive scale (VanderWeele et al., 2010).
Many challenges still exist when testing for GxE. Typically in GWAS, independent replication is expected for validation of observed associations. The standards for GxE studies are not as well established and thus replication of GxE associations is much less common. This may in part reflect the generally reduced power to detect interactions of GxE (in contrast with main genetic or environmental effects). Increased focus on meta-analyses could theoretically address this issue, but lack of standardization in environmental exposure definitions can make this difficult in practice. And as there are numerous combinations of genetic and environmental factors, it would be rare that the same SNP and environmental exposure are analyzed for GxE effects in multiple studies. Although for some commonly known GxE effects such as folate-related genes (i.e. MTHFR) and folate intake, this may be possible. Improving comparability across studies will require increased sharing of unpublished results and prior coordination among different studies to standardize collection of genetic and environmental data (Colhoun et al., 2003; Hunter, 2005; Cornelis et al., 2010). Lack of standardization presents a bigger challenge if investigators wish to estimate the effect of environmental factors on a quantitative rather than a qualitative (binary) scale. For example, should smoking be measured in cigarettes per day or total number of cigarettes smoked in a narrow gestational period of relevance?
In summary, the NBDPS provides an important resource for evaluating the genetic and environmental basis of birth defects. A key component of such work is determining whether these factors work in concert to increase risk beyond their individual effects. A unique aspect of the NBDPS is the collection of samples in a manner that allow for numerous different analytic approaches. Understanding the assumptions, strengths and weaknesses of each such approach will allow researchers to further decipher factors that increase risk of birth defects.
Acknowledgments
Funding Information: This work supported by NIH grants: R25CA112355, R01CA088164, and R01HD039054.
Footnotes
Conflicts of Interest: None to report.
References
- Ainsworth HF, Unwin J, Jamison DL, Cordell HJ. Investigation of maternal effects, maternal-fetal interactions and parent-of-origin effects (imprinting), using mothers and their offspring. Genet Epidemiol. 2011;35:19–45. doi: 10.1002/gepi.20547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert PS. Limitations of the Case-only Design for Identifying Gene-Environment Interactions. Am J Epidemiol. 2001;154:687–693. doi: 10.1093/aje/154.8.687. [DOI] [PubMed] [Google Scholar]
- Alwan S, Reefhuis J, Rasmussen SA, Olney RS, Friedman JM National Birth Defects Prevention Study. Use of selective serotonin-reuptake inhibitors in pregnancy and the risk of birth defects. N Engl J Med. 2007;356:2684–2692. doi: 10.1056/NEJMoa066584. [DOI] [PubMed] [Google Scholar]
- Beaty TH, Ruczinski I, Murray JC, Marazita ML, Munger RG, Hetmanski JB, Murray T, Redett RJ, Fallin MD, Liang KY, et al. Evidence for gene-environment interaction in a genome wide study of nonsyndromic cleft palate. Genet Epidemiol. 2011;35:469–478. doi: 10.1002/gepi.20595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee S, Chatterjee N, Han S, Wheeler W. R Package Version 3.0.0. 2012. CGEN: An R package for analysis of case-control studies in genetic epidemiology. [Google Scholar]
- Botto LD, Yang Q. 5,10-Methylenetetrahydrofolate reductase gene variants and congenital anomalies: a HuGE review. Am J Epidemiol. 2000;151:862–877. doi: 10.1093/oxfordjournals.aje.a010290. [DOI] [PubMed] [Google Scholar]
- Bower C, Stanley FJ. Dietary folate as a risk factor for neural-tube defects: evidence from a case-control study in Western Australia. Med J Aust. 1989;150:613–619. doi: 10.5694/j.1326-5377.1989.tb136723.x. [DOI] [PubMed] [Google Scholar]
- Brender JD, Werler MM, Kelley KE, Vuong AM, Shinde MU, Zheng Q, Huber JC, Sharkey JR, Griesenbeck JS, Romitti PA, et al. Nitrosatable Drug Exposure During Early Pregnancy and Neural Tube Defects in Offspring: National Birth Defects Prevention Study. Am J Epidemiol. 2011;174:1286–1295. doi: 10.1093/aje/kwr254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael SL, Yang W, Feldkamp ML, Munger RG, Siega-Riz AM, Botto LD, Shaw G National Birth Defects Prevention Study. Reduced risks of neural tube defects and orofacial clefts with higher diet quality. Arch Pediatr Adolesc Med. 2012;166:121–126. doi: 10.1001/archpediatrics.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee N, Kalaylioglu Z, Carroll RJ. Exploiting gene-environment independence in family-based case-control studies: Increased power for detecting associations, interactions and joint effects. Genet Epidemiol. 2005;28:138–156. doi: 10.1002/gepi.20049. [DOI] [PubMed] [Google Scholar]
- Christensen B, Arbour L, Tran P, Leclerc D, Sabbaghian N, Platt R, Gilfix BM, Rosenblatt DS, Gravel RA, Forbes P, Rozen R. Genetic polymorphisms in methylenetetrahydrofolate reductase and methionine synthase, folate levels in red blood cells, and risk of neural tube defects. Am J Med Genet. 1999;4:151–157. doi: 10.1002/(sici)1096-8628(19990521)84:2<151::aid-ajmg12>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Cogswell ME, Bitsko RH, Anderka M, Caton AR, Feldkamp ML, Sherlock SMH, Meyer RE, Ramadhani T, Robbins JM, Shaw GM, et al. Control Selection and Participation in an Ongoing, Population-based, Case-Control Study of Birth Defects: The National Birth Defects Prevention Study. Am J Epidemiol. 2009;170:975–985. doi: 10.1093/aje/kwp226. [DOI] [PubMed] [Google Scholar]
- Clarke GM, Morris AP. A Comparison of Sample Size and Power in Case-Only Association Studies of Gene-Environment Interaction. American Journal of Epidemiology. 2010;171:498–505. doi: 10.1093/aje/kwp398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colhoun HM, McKeigue PM, Davey Smith G. Problems of reporting genetic associations with complex outcomes. Lancet. 2003;361:865–872. doi: 10.1016/s0140-6736(03)12715-8. [DOI] [PubMed] [Google Scholar]
- Cornelis MC, Agrawal A, Cole JW, Hansel NN, Barnes KC, Beaty TH, Bennett SN, Bierut LJ, Boerwinkle E, Doheny KF, et al. The gene, environment association studies consortium (GENEVA): maximizing the knowledge obtained from GWAS by collaboration across studies of multiple conditions. Genet Epidemiol. 2010;34:364–372. doi: 10.1002/gepi.20492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crider KS, Devine O, Hao L, Dowling NF, Li S, Molloy AM, Li Z, Zhu J, Berry RJ. Population Red Blood Cell Folate Concentrations for Prevention of Neural Tube Defects: Bayesian Model. BMJ. 2014;349:4554. doi: 10.1136/bmj.g4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly LE, Kirke PN, Molloy A, Weir DG, Scott JM. Folate levels and neural tube defects. Implications for prevention. JAMA. 1995;274:1698–1702. doi: 10.1001/jama.1995.03530210052030. [DOI] [PubMed] [Google Scholar]
- Dudbridge F. Likelihood-based association analysis for nuclear families and unrelated subjects with missing genotype data. Hum Hered. 2008;66:87–98. doi: 10.1159/000119108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etheredge AJ, Finnell RH, Carmichael SL, Lammer EJ, Zhu H, Mitchell LE, Shaw GM. Maternal and infant gene-folate interactions and the risk of neural tube defects. Am J Med Genet A. 2012;158A:2439–2446. doi: 10.1002/ajmg.a.35552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford RP, Tappin DM, Schluter PJ, Wild CJ. Smoking during pregnancy: how reliable are maternal self reports in New Zealand? J Epidemiol Community Health. 1997;51:246–251. doi: 10.1136/jech.51.3.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Closas M, Thompson WD, Robins JM. Differential misclassification and the assessment of gene-environment interactions in case-control studies. Am J Epidemiol. 1998;147:426–433. doi: 10.1093/oxfordjournals.aje.a009467. [DOI] [PubMed] [Google Scholar]
- Gatto NM. Further development of the case-only design for assessing gene-environment interaction: evaluation of and adjustment for bias. Int J Epidemiol. 2004;33:1014–1024. doi: 10.1093/ije/dyh306. [DOI] [PubMed] [Google Scholar]
- Gauderman WJ, Witte JS, Thomas DC. Family-based association studies. J Natl Cancer Inst Monogr. 1999:31–37. doi: 10.1093/oxfordjournals.jncimonographs.a024223. [DOI] [PubMed] [Google Scholar]
- Gauderman WJ. Sample size requirements for association studies of gene-gene interaction. Am J Epidemiol. 2002;155:478–484. doi: 10.1093/aje/155.5.478. [DOI] [PubMed] [Google Scholar]
- Greenland S. Basic problems in interaction assessment. Environ Health Perspect. 1993;101:59–66. doi: 10.1289/ehp.93101s459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein R, Beckmann L, Chang-Claude J. Sample size requirements for indirect association studies of gene-environment interactions (G x E) Genet Epidemiol. 2008;32:235–245. doi: 10.1002/gepi.20298. [DOI] [PubMed] [Google Scholar]
- Hobbs CA, Cleves MA, Karim MA, Zhao W, MacLeod SL. Maternal Folate-Related Gene Environment Interactions and Congenital Heart Defects. Obstet Gynecol. 2010;116:316–322. doi: 10.1097/AOG.0b013e3181e80979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs CA, Cleves MA, MacLeod SL, Erickson SW, Tang X, Li J, Li M, Nick T, Malik S the National Birth Defects Prevention Study. Conotruncal heart defects and common variants in maternal and fetal genes in folate, homocysteine, and transsulfuration pathways. Birt Defects Res A Clin Mol Teratol. 2014;100:116–126. doi: 10.1002/bdra.23225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann T, Lange C. P2BAT: a massive parallel implementation of PBAT for genome-wide association studies in R. Bioinforma Oxf Engl. 2006;22:3103–3105. doi: 10.1093/bioinformatics/btl507. [DOI] [PubMed] [Google Scholar]
- Hoffmann TJ, Lange C, Vansteelandt S, Laird NM. Gene-environment interaction tests for dichotomous traits in trios and sibships. Genet Epidemiol. 2009;33:691–699. doi: 10.1002/gepi.20421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann TJ, Vansteelandt S, Lange C, Silverman EK, DeMeo DL, Laird NM. Combining disease models to test for gene-environment interaction in nuclear families. Biometrics. 2011;67:1260–1270. doi: 10.1111/j.1541-0420.2011.01581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honein MA, Paulozzi LJ, Moore CA. Family history, maternal smoking, and clubfoot: an indication of a gene-environment interaction. Am J Epidemiol. 2000;152:658–665. doi: 10.1093/aje/152.7.658. [DOI] [PubMed] [Google Scholar]
- Howey R, Cordell HJ. PREMIM and EMIM: tools for estimation of maternal, imprinting and interaction effects using multinomial modelling. BMC Bioinformatics. 2012;13:149. doi: 10.1186/1471-2105-13-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter DJ. Gene–environment interactions in human diseases. Nat Rev Genet. 2005;6:287–298. doi: 10.1038/nrg1578. [DOI] [PubMed] [Google Scholar]
- Hwang SJ, Beaty TH, Panny SR, Street NA, Joseph JM, Gordon S, McIntosh I, Francomano CA. Association study of transforming growth factor alpha (TGF alpha) TaqI polymorphism and oral clefts: indication of gene-environment interaction in a population-based sample of infants with birth defects. Am J Epidemiol. 1995;141:629–636. doi: 10.1093/oxfordjournals.aje.a117478. [DOI] [PubMed] [Google Scholar]
- Jiao S, Hsu L, Bézieau S, Brenner H, Chan AT, Chang-Claude J, Le Marchand L, Lemire M, Newcomb PA, Slattery ML, Peters U. SBERIA: set-based gene-environment interaction test for rare and common variants in complex diseases. Genet Epidemiol. 2013;37:452–64. doi: 10.1002/gepi.21735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller MC. Gene × environment interaction studies have not properly controlled for potential confounders: the problem and the (simple) solution. Biol Psychiatry. 2014;75:18–24. doi: 10.1016/j.biopsych.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury MJ, Flanders WD. Nontraditional epidemiologic approaches in the analysis of gene-environment interaction: case-control studies with no controls! Am J Epidemiol. 1996;144:207–213. doi: 10.1093/oxfordjournals.aje.a008915. [DOI] [PubMed] [Google Scholar]
- Khoury MJ, Wacholder S. Invited Commentary: From Genome-Wide Association Studies to Gene-Environment-Wide Interaction Studies--Challenges and Opportunities. Am J Epidemiol. 2008;169:227–230. doi: 10.1093/aje/kwn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft P, Yen Y-C, Stram DO, Morrison J, Gauderman WJ. Exploiting gene-environment interaction to detect genetic associations. Hum Hered. 2007;63:111–119. doi: 10.1159/000099183. [DOI] [PubMed] [Google Scholar]
- Lake SL, Laird NM. Tests of gene-environment interaction for case-parent triads with general environmental exposures. Ann Hum Genet. 2004;68:55–64. doi: 10.1046/j.1529-8817.2003.00073.x. [DOI] [PubMed] [Google Scholar]
- Lange C, DeMeo D, Silverman EK, Weiss ST, Laird NM. PBAT: Tools for Family-Based Association Studies. Am J Hum Genet. 2004;74:367–369. doi: 10.1086/381563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurence KM, James N, Miller MH, Tennant GB, Campbell H. Double-blind randomised controlled trial of folate treatment before conception to prevent recurrence of neural-tube defects. Br Med J Clin Res Ed. 1981;282:1509–1511. doi: 10.1136/bmj.282.6275.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupo PJ, Canfield MA, Chapa C, Lu W, Agopian AJ, Mitchell LE, Shaw GM, Waller DK, Olshan AF, Finnell RH, et al. Diabetes and Obesity-Related Genes and the Risk of Neural Tube Defects in the National Birth Defects Prevention Study. Am J Epidemiol. 2012;176:1101–1109. doi: 10.1093/aje/kws190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupo PJ, Mitchell LE, Canfield MA, Shaw GM, Olshan AF, Finnell RH, Zhu H. Maternal–fetal metabolic gene–gene interactions and risk of neural tube defects. Mol Genet Metab. 2014;111:46–51. doi: 10.1016/j.ymgme.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JL, Rhoads GG, Simpson JL, Cunningham GC, Conley MR, Lassman MR, Walden ME, Depp OR, Hoffman HJ. The absence of a relation between the periconceptional use of vitamins and neural-tube defects. National Institute of Child Health and Human Development Neural Tube Defects Study Group. N Engl J Med. 1989;321:430–435. doi: 10.1056/NEJM198908173210704. [DOI] [PubMed] [Google Scholar]
- Milunsky A, Jick H, Jick SS, Bruell CL, MacLaughlin DS, Rothman KJ, Willett W. Multivitamin/folic acid supplementation in early pregnancy reduces the prevalence of neural tube defects. JAMA. 1989;262:2847–2852. doi: 10.1001/jama.262.20.2847. [DOI] [PubMed] [Google Scholar]
- Moerkerke B, Vansteelandt S, Lange C. A doubly robust test for gene-environment interaction in family-based studies of affected offspring. Biostat Oxf Engl. 2010;11:213–225. doi: 10.1093/biostatistics/kxp061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MRC Vitamin Study Research Group. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet. 1991;338:131–137. [PubMed] [Google Scholar]
- Mulinare J, Cordero JF, Erickson JD, Berry RJ. Periconceptional use of multivitamins and the occurrence of neural tube defects. JAMA. 1988;260:3141–3145. [PubMed] [Google Scholar]
- Murcray CE, Lewinger JP, Gauderman WJ. Gene-Environment Interaction in Genome-Wide Association Studies. Am J Epidemiol. 2008;169:219–226. doi: 10.1093/aje/kwn353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray J. Gene/environment causes of cleft lip and/or palate. Clin Genet. 2002;61:248–256. doi: 10.1034/j.1399-0004.2002.610402.x. [DOI] [PubMed] [Google Scholar]
- Nelson M, Asling CW, Evans HM. Production of multiple congenital abnormalities in young by maternal pteroylglutamic acid deficiency during gestation. J Nutr. 1952;48:61–79. doi: 10.1093/jn/48.1.61. [DOI] [PubMed] [Google Scholar]
- Neumann C, Taub MA, Younkin SG, Beaty TH, Ruczinski I, Schwender H. Analytic power and sample size calculation for the genotypic transmission/disequilibrium test in case-parent trio studies: Power and sample size calculation. Biom J. 2014;56:1076–1092. doi: 10.1002/bimj.201300148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newport D, Brennan P, Green P, Ilardi D, Whitfield T, Morris N, Knight B, Stowe Z. Maternal depression and medication exposure during pregnancy: comparison of maternal retrospective recall to prospective documentation. BJOG. 2008;115:681–688. doi: 10.1111/j.1471-0528.2008.01701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble EP, Jeor STS, Ritchie T, Syndulko K, Jeor SCS, Fitch RJ, Brunner RL, Sparkes RS. D2 dopamine receptor gene and cigarette smoking: A reward gene? Med Hypotheses. 1994;42:257–260. doi: 10.1016/0306-9877(94)90127-9. [DOI] [PubMed] [Google Scholar]
- Ottman R. Gene–environment interaction: definitions and study designs. Prev Med. 1996;25:764. doi: 10.1006/pmed.1996.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LJ, Cardon LR. Shaking the tree: mapping complex disease genes with linkage disequilibrium. Lancet. 2005;366:1223–1234. doi: 10.1016/S0140-6736(05)67485-5. [DOI] [PubMed] [Google Scholar]
- Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piegorsch WW, Weinberg CR, Taylor JA. Non-hierarchical logistic models and case-only designs for assessing susceptibility in population-based case-control studies. Stat Med. 1994;13:153–162. doi: 10.1002/sim.4780130206. [DOI] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satagopan JM, Verbel DA, Venkatraman ES, Offit KE, Begg CB. Two-stage designs for gene-disease association studies. Biometrics. 2002;58:163–170. doi: 10.1111/j.0006-341x.2002.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwender H, Li Q, Neumann C, Taub M, Ruczinski I. R Package Version 3.4.0. 2014. trio: Testing of SNPs and SNP Interactions in Case-Parent Trio Studies. [Google Scholar]
- Sharp RR, Barrett JC. The environmental genome project: ethical, legal, and social implications. Environ Health Perspect. 2000;108:279–281. doi: 10.1289/ehp.00108279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielman RS, McGinnis RE, Ewens WJ. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM) Am J Hum Genet. 1993;52:506–516. [PMC free article] [PubMed] [Google Scholar]
- Thomas D. Gene–environment-wide association studies: emerging approaches. Nat Rev Genet. 2010;11:259–272. doi: 10.1038/nrg2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DC, Witte JS. Point: population stratification: a problem for case-control studies of candidate-gene associations? Cancer Epidemiol Biomark Prev. 2002;11:505–512. [PubMed] [Google Scholar]
- Umbach DM, Weinberg CR. Designing and analysing case-control studies to exploit independence of genotype and exposure. Stat Med. 1997;16:1731–1743. doi: 10.1002/(sici)1097-0258(19970815)16:15<1731::aid-sim595>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Umbach DM, Weinberg CR. The use of case-parent triads to study joint effects of genotype and exposure. Am J Hum Genet. 2000;66:251–261. doi: 10.1086/302707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderWeele TJ, Hernández-Díaz S, Hernán MA. Case-only gene-environment interaction studies: when does association imply mechanistic interaction? Genet Epidemiol. 2010;34:327–334. doi: 10.1002/gepi.20484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vansteelandt S, Demeo DL, Lasky-Su J, Smoller JW, Murphy AJ, McQueen M, Schneiter K, Celedon JC, Weiss ST, Silverman EK, et al. Testing and estimating gene-environment interactions in family-based association studies. Biometrics. 2008;64:458–467. doi: 10.1111/j.1541-0420.2007.00925.x. [DOI] [PubMed] [Google Scholar]
- Waller DK, Shaw GM, Rasmussen SA, Hobbs CA, Canfield MA, Siega-Riz A-M, Gallaway MS, Correa A National Birth Defects Prevention Study. Prepregnancy obesity as a risk factor for structural birth defects. Arch Pediatr Adolesc Med. 2007;161:745–750. doi: 10.1001/archpedi.161.8.745. [DOI] [PubMed] [Google Scholar]
- Wang L-Y, Lee W-C. Population Stratification Bias in the Case-Only Study for Gene-Environment Interactions. Am J Epidemiol. 2008;168:197–201. doi: 10.1093/aje/kwn130. [DOI] [PubMed] [Google Scholar]
- Webber DM, Bamshad MJ, Shaw GM, Finnell RH, Shete SS, Witte JS, MacLeod SL, Erickson SW, Murphy LD, Hobbs C. Developments in our understanding of the genetic basis of birth defects. Birt Defects Res A Clin Mol Teratol. 2015;X:XX–XX. doi: 10.1002/bdra.23385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S, Wang L-E, McHugh MK, Han Y, Xiong M, Amos CI, Spitz MR, Wei QW. Genome-wide gene-environment interaction analysis for asbestos exposure in lung cancer susceptibility. Carcinogenesis. 2012;33:1531–1537. doi: 10.1093/carcin/bgs188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte JS, Gauderman WJ, Thomas DC. Asymptotic bias and efficiency in case-control studies of candidate genes and gene-environment interactions: basic family designs. Am J Epidemiol. 1999;149:693–705. doi: 10.1093/oxfordjournals.aje.a009877. [DOI] [PubMed] [Google Scholar]
- Yoon PW, Rasmussen SA, Lynberg MC, Moore CA, Anderka M, Carmichael SL, Costa P, Druschel C, Hobbs CA, Romitti PA, et al. The National Birth Defects Prevention Study. Public Health Rep. 2001;116:32–40. doi: 10.1093/phr/116.S1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeiger JS, Beaty TH, Liang KY. Oral clefts, maternal smoking, and TGFA: a meta-analysis of gene-environment interaction. Cleft Palate Craniofac J. 2005;42:58–63. doi: 10.1597/02-128.1. [DOI] [PubMed] [Google Scholar]