

Abstract
Cardiolipin (CL), the signature phospholipid of mitochondrial membranes, is crucial for both mitochondrial function and cellular processes outside of the mitochondria. The importance of CL in cardiovascular health is underscored by the life-threatening genetic disorder Barth syndrome (BTHS), which manifests clinically as cardiomyopathy, skeletal myopathy, neutropenia, and growth retardation. BTHS is caused by mutations in the gene encoding tafazzin, the transacylase that carries out the second CL remodeling step. In addition to BTHS, CL is linked to other cardiovascular diseases (CVDs), including cardiomyopathy, atherosclerosis, myocardial ischemia-reperfusion injury, heart failure, and Tangier disease. The link between CL and CVD may possibly be explained by the physiological roles of CL in pathways that are cardioprotective, including mitochondrial bioenergetics, autophagy/mitophagy, and mitogen activated protein kinase (MAPK) pathways. In this review, we focus on the role of CL in the pathogenesis of CVD as well as the molecular mechanisms that may link CL functions to cardiovascular health.
1. Introduction
Cardiolipin (CL) is the signature lipid of mitochondrial membranes. It contains two phosphatidyl moieties joined by a central glycerol backbone, forming a dimeric structure [1]. Thus, unlike other phospholipids that contain two fatty acyl chains linked by glycerol, CL has four acyl chains. Considering the potential number of combinations of fatty acyl groups, a very large number of CL species may be possible. Interestingly, in most organisms and tissues, the fatty acyl composition of CL is unique and specific. In humans, CL acyl species vary in different tissues, but the most abundant species in the heart is tetralinoleoyl-CL [2]. While CL plays critical roles in mitochondrial biogenesis, fusion and fission, respiration, and protein import [3], it is also involved in various cellular processes outside of the mitochondria. These include, but are not limited to, cell wall biogenesis [4], vacuole homeostasis [5], ageing [6], the cell cycle [7], and apoptosis [8]. In this review, we focus on the role of CL in the pathogenesis of CVD as well as the molecular mechanisms that may link CL functions to cardiovascular health.
2. CL Synthesis
Unlike mitochondrial membrane lipids that are synthesized in the endoplasmic reticulum, de novo synthesis of CL occurs exclusively in the inner membrane of the mitochondria [9], in a series of well-characterized steps that are highly conserved from yeast to higher eukaryotes [10]. As shown in Figure 1, the first step in the CL biosynthetic pathway is the conversion of phosphatidic acid (PA) to CDP-diacylglycerol (CDP-DAG), which is catalyzed in the inner membrane by CDP-DAG synthase encoded by TAM41 [11–13] in yeast. PGS1 encoded phosphatidylglycerolphosphate synthase catalyzes transfer of the phosphatidyl group from CDP-DAG to a glycerol-3-phosphate molecule to generate phosphatidylglycerolphosphate (PGP) [14, 15]. PGP is subsequently dephosphorylated to phosphatidylglycerol (PG) by PGP phosphatase [16, 17], encoded by PTPMT1 in mammals [18, 19] and GEP4 in yeast [20]. The final step in the biosynthetic pathway is carried out by CL synthase, encoded by hCLS1 in human cells [21–23] and by CRD1 in yeast [24–26]. In this step, a second phosphatidyl group is added to PG from another CDP-DAG molecule, generating unremodeled CL [9, 23, 27].
Figure 1.
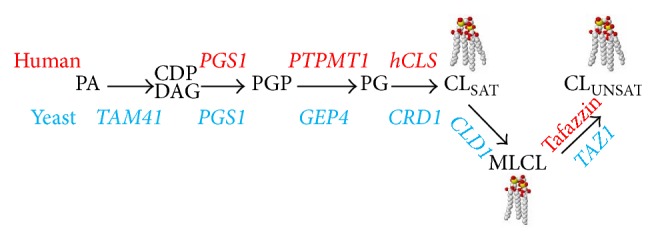
Cardiolipin synthesis and remodeling pathway in humans and yeast. Phosphatidic acid (PA) is converted to CDP-diacylglycerol (CDP-DAG) by CDP-DAG synthase. Phosphatidylglycerolphosphate synthase catalyzes the conversion of CDP-DAG to phosphatidylglycerolphosphate (PGP), which is dephosphorylated to phosphatidylglycerol (PG). PG is converted to unremodeled CL with mostly saturated acyl chains (CLSAT). CLSAT is deacylated to monolyso-CL (MLCL) by phospholipases and MLCL is reacylated to CL with mostly unsaturated acyl chains (CLUNSAT). The genes encoding human enzymes are indicated in red, and genes that encode yeast enzymes are in blue.
The acyl composition of CL varies in different tissues, due primarily to CL remodeling following de novo synthesis. CL remodeling may occur through two mechanisms (Figure 1) [28]. In the two-step mechanism, CL is first deacylated to monolyso-CL (MLCL) by phospholipases [29]. In yeast, the only CL-specific phospholipase is encoded by CLD1 [30] while in mammals, several phospholipases are reported to have CL-hydrolyzing activities, including iPLA2 β, iPLA2 γ, cPLA2, and sPLA2 [31–33]. MLCL is then reacylated to remodeled CL by the transacylase tafazzin, encoded by the tafazzin gene (TAZ/G4.5) located on Xq28 in human cells [34] and by TAZ1 in yeast [35, 36]. Acyltransferases encoded by ALCAT1 [37] and MLCLAT1 [38] have also been described in mammalian cells. In the one-step mechanism, CL remodeling occurs by direct transacylation [39, 40]. Mutations in tafazzin perturb CL remodeling and cause the life-threatening genetic disorder Barth syndrome (BTHS) [41], which is discussed below.
3. Relationship between CL and CVD
3.1. Cardiomyopathy
3.1.1. Barth Syndrome
The most direct link between CVD and CL is seen in Barth syndrome (BTHS), an X-linked genetic disorder of CL remodeling caused by tafazzin mutations. BTHS manifests clinically as cardiomyopathy, skeletal myopathy, neutropenia, and growth retardation [42]. Biochemical phenotypes include decreased levels of CL, increased MLCL, and altered CL fatty acyl composition [43–45]. More than 160 mutations in the tafazzin gene have been identified in BTHS patients [46–48]. Interestingly, there is a wide disparity of clinical phenotypes, even among patients with the same mutation, ranging from being asymptomatic to death of newborns. Thus, some patients with an increased MLCL/CL ratio appear asymptomatic [49]. A study in which mutated BTHS tafazzin proteins were expressed in the yeast taz1Δ mutant reported that 18 of 21 BTHS proteins did not restore MLCL levels to normal, as expected [50]. However, expression of 3 of the 21 BTHS proteins restored MLCL levels in the yeast taz1Δ mutant to normal. In typical cases, total CL is decreased to about 80% in BTHS platelets and skeletal muscle and 20% in cardiac tissue [44]. CL species vary in different tissues. Tetralinoleoyl-CL (L4-CL) is the most abundant CL species in heart, skeletal muscle, and most other tissues, whereas acyl species such as arachidonic and docosahexaenoic acids are found in brain [51, 52]. L4-CL is absent in BTHS, while increases in other CL species are found [43–45]. As mentioned, tafazzin deficiency results in decreased CL, increased MLCL, and altered CL species, any of which may cause the pathology in BTHS. Recent findings in yeast indicate that deletion of Cld1-mediated deacylation rescues growth and lifespan defects in tafazzin-deficient cells [53, 54]. Because the CLD1 mutation restored CL levels without generating remodeled CL, these findings suggest that, at least in yeast, decreased total CL and/or increased MLCL but not decreased remodeled CL leads to defects associated with tafazzin deficiency. If this is true in BTHS cells, inhibiting CL deacylation may, thus, be a novel potential strategy to treat BTHS patients.
3.1.2. Diabetic Cardiomyopathy
Diabetes is a metabolic disease characterized by increased levels of glucose in the blood over a prolonged period. It is due to poor insulin production (type I) or insulin resistance with β-cell dysfunction (type II) [55]. Diabetic complications include a group of diseases derived from microvascular and macrovascular damage, including diabetic cardiomyopathy, myonecrosis, stroke, peripheral vascular disease, nephropathy, retinopathy and encephalopathy [56]. Diabetes doubles the risk of CVD, of which diabetic cardiomyopathy is the leading cause of mortality. Diabetic cardiomyopathy is characterized by altered lipid composition and mitochondrial dysfunction in the diabetic myocardium [57]. In the early stages of pathological development in the type II diabetic mouse model, a sharp decrease in total cardiac CL is observed [58]. In addition to a decrease in the whole cell CL content, there is also a shift from the predominant fatty acyl species, L4-CL (18 : 2), to longer and polyunsaturated fatty acids, due to aberrant CL remodeling [58, 59]. Strikingly, these alterations are similar to changes observed in the type I model of diabetes. In type II diabetic mice treated with the antidiabetic drug rosiglitazone, the wild type CL profile in the heart was restored, as total CL and L4-CL increased, and polyunsaturated CL decreased [60]. Impairment of CL synthesis plays a causal role in mitochondrial dysfunction [61–63], and mitochondrial dysfunction is associated with the pathogenesis of diabetic CVD, especially with the sequential events following silent myocardial ischemia in diabetics [64]. Thus, the sharp decrease in total cardiac CL and the altered CL fatty acyl species in the early stages of diabetic pathogenesis may play a key role in the progression of this disease.
3.2. Myocardial Ischemia-Reperfusion Injury
Myocardial ischemia occurs when the myocardium does not receive sufficient blood flow, resulting in irreversible injury and cell death [65]. Restoration of circulation in the ischemic myocardium leads to reperfusion injury [65]. Ischemia-reperfusion injury causes diverse myocardial dysfunctions, including cardiac contractile abnormalities [66–68], abnormal left-ventricular pressure [69], arrhythmia [70–72], and increased occurrence of ventricular fibrillation [73, 74].
In the early stages of myocardial ischemia, there is an increase in reactive oxygen species (ROS). During and after ischemia-reperfusion, ROS is thought to trigger lipid peroxidation as well as damage to cellular macromolecules and the electron transport chain which, together, lead to apoptosis, necrosis, and tissue damage [75–77]. Unsaturated CL acyl species in the mitochondrial inner membrane that are close to the site of ROS generation are vulnerable to oxidative damage. Consistent with this, total CL was decreased and peroxidized CL was increased in the rat heart during ischemia-reperfusion [78]. A study of ischemia-reperfusion in rabbit heart reported that reduction of total CL was due in large part to a significant decrease in CL in the subsarcolemmal mitochondria, whereas CL in the interfibrillar mitochondria was unchanged [79]. The levels of all other phospholipids remained unaffected. Decreased CL was proposed to be the cause of decreased enzyme activities of electron transport chain complexes I [80], III [81], and IV [78] in the rat heart ischemia-reperfusion model. The enzyme activities were restored by the addition of exogenous CL, but not by other phospholipids or peroxidized CL [78]. In summary, a feedback loop appears to be formed, in which CL is damaged by ischemia-reperfusion-induced ROS, and damaged CL leads to impairment of electron transport chain complexes, resulting in the generation of more ROS. CL also directly binds to cytochrome c (Cytc), and CL-bound Cytc has peroxidase activity that can produce CL hydroperoxides [82]. A known factor that stimulates the activity of this CL/Cytc peroxidase is increased H2O2 [83]. Peroxidized CL has a much lower affinity for Cytc [84]. In addition, several studies show that apoptosis factors, t-Bid and Bax, preferentially localize to the inner and outer membrane contact sites, which are rich in CL [85–87]. t-Bid binding and Bax insertion at the contact sites cause irreversible membrane permeabilization and promotes release of Cytc into the cytosol [87, 88], resulting in apoptosis.
3.3. Atherosclerosis
Atherosclerosis is a form of arteriosclerosis in which an artery wall thickens due to chronic invasion and further accumulation of white blood cells, remnants of dead cells, cholesterol, and triglycerides [89]. Oxidized CL (oxCL) is found to accumulate both in rabbit and human atherosclerotic lesions [90] and in the aortic root of mice fed a high fat diet [91]. Increased anti-oxCL IgG [92–94] and IgM [93, 95] antibodies are associated with atherosclerosis development. oxCL is recognized as a natural antigen that stimulates proinflammatory effects in the artery and promotes formation of atherosclerotic plaques [92, 96]. However, some studies purport that autoantibodies to oxCL may serve a protective role against the onset and development of atherosclerosis [97, 98]. The discrepancies regarding the effects of anti-oxCL antibodies on atherosclerosis may reflect the influence of potential physiological modifiers, including age, gender, and other existing diseases. The anticoagulation protein annexin A5 has been reported to bind to and inhibit the proinflammatory effects of oxCL [99], providing the basis for a potential therapeutic strategy for oxCL positive atherosclerosis.
3.4. Emerging Relationships between CL and Dilated Cardiomyopathy with Ataxia Syndrome (DCMA), Heart Failure (HF), and Tangier Disease
3.4.1. DCMA
Dilated cardiomyopathy with ataxia (DCMA) syndrome is an autosomal recessive genetic disorder that is characterized by early onset dilated cardiomyopathy with conduction defects, nonprogressive cerebellar ataxia, testicular dysgenesis, growth failure, and 3-methylglutaconic aciduria [100]. The clinical manifestations of DCMA are similar to those found in BTHS. Patients with DCMA have a common mutation, a G→C base substitution within a splice site of the DNAJC19 gene [100]. DNAJC19 protein localizes to the mitochondria and shares sequence and location similarity with yeast Tim14, an essential subunit of the TIM23 complex [101, 102]. TIM23 is required for the import of protein precursors from the cytoplasm into the mitochondrial matrix and inner membrane [103]. This suggests that the DCMA phenotype may result from defective mitochondrial protein import. As the loss of CL also leads to defective mitochondrial protein import [104–108], it is interesting to speculate that defective import of specific mitochondrial proteins may be common to DCMA and BTHS.
A recent study suggests that CL may play a role in the pathogenesis of DCMA [109]. DNAJC19 protein is reported to form a PHB/DNAJC19 complex with prohibitin, a ring-like scaffold protein located in the mitochondrial inner membrane. The PHB/DNAJC19 complex modulates CL remodeling by regulating tafazzin activity. siRNA-mediated knockdown of DNAJC19 did not affect CL or MLCL levels but altered the acyl chain composition of CL [109], while knockout of PHB2 resulted in reduced total CL, accumulated MLCL, and altered CL species. These data suggest that the PHB/DNAJC19 complex plays a role in CL synthesis and remodeling. However, whether the cause of DCMA is due to defective protein import, altered CL fatty acyl species that results from loss of DNAJC19, or a combination of the two remains unknown.
3.4.2. HF
Heart failure (HF) results from inability of the heart to pump blood with normal efficiency, resulting in edema, shortness of breath, and lack of energy. HF is usually the end stage of CVD, including cardiomyopathy, heart attack, cardiac valvular disease, atrial fibrillation, and high blood pressure [110]. In both the spontaneously hypertensive HF rat model (SHHF) and human HF patients, decreased tafazzin mRNA levels were observed, concomitant with compensatory increases in the activity of phosphatidylglycerolphosphate synthase and MLCL acyltransferase [111]. However, studies of the CL profile in HF are controversial. While most studies report a significant reduction of total CL and L4-CL in human HF [112–114] and in the rat HF model [112, 115], one study reported an unchanged CL profile in a rat model with intracoronary microembolization-induced HF [116]. It is likely that different HF pathogenesis mechanisms lead to varying degrees of CL profile change and mitochondrial damage.
3.4.3. Tangier Disease
Tangier disease (TD) is a genetic disorder of cholesterol efflux and lipid metabolism characterized by a nearly complete absence of plasma high-density lipoproteins (HDLs), atherosclerosis, peripheral neuropathy, and an increased risk for developing CVD [117, 118]. The genetic cause of TD is the mutation of the ABCA1 gene, which is located on chromosome 9 [119]. ABCA1 encodes a highly conserved ATP-binding cassette transporter. The ABCA subfamily of ABC transporters is involved in lipoprotein metabolism and lipid transport across the plasma membrane [120]. Researchers propose that a physical interaction between apoA-I and ABCA1 results in the formation of a phospholipid-apoA-I complex that promotes cholesterol efflux [121]. Three phospholipids, including CL, lysoCL 1, and 2 (LC1 and LC2), which together contribute only a small fraction of the total cellular phospholipid content, were found to be enriched up to fivefold in TD fibroblasts compared to wild type cells [122]. This finding suggests that phospholipid and cholesterol efflux may be coregulated and, therefore, dually impaired in TD cells. Additionally, it is possible that increased CL may play an as yet uncharacterized regulatory role in cholesterol trafficking and efflux.
4. CL Plays a Role in Cellular Events and Pathways That Are Important for Maintaining Cardiovascular Health
4.1. Mitochondrial Function
4.1.1. Mitochondrial Dysfunction and CVD
To support the normal function of the heart, cardiomyocytes have a high mitochondrial density that comprises about 30% of the total intracellular volume [123]. This allows cardiomyocytes to produce ATP quickly to satisfy the high demand for energy. Even subtle alterations in mitochondrial function or membrane potential can cause a significant change in cardiomyocyte energy production and further harm cardiovascular health.
As discussed in Section 3.4, mitochondrial dysfunction and ROS play a causative role in the pathogenesis of myocardial ischemia-reperfusion injury. Mitochondrial dysfunction and related morphological abnormalities, ROS generation, and altered mitochondrial permeability transition pore and mitochondrial Ca2+ storage also contribute to the development of diabetic cardiomyopathy [124–126], dilated cardiomyopathy [127–129], dystrophic cardiomyopathy [130, 131], and hypertrophic cardiomyopathy [132–134]. Mitochondrial dysfunction is also linked to the development of HF, as demonstrated in the hamster [135]. The role of mitochondrial dysfunction as a cofactor accelerating the progression of existing CVD to HF has been addressed elsewhere [136, 137].
4.1.2. CL Deficiency Leads to Mitochondrial Dysfunction
CL interacts with many inner mitochondrial membrane proteins, including electron transport chain (ETC) complex proteins that are components of complex I [62, 138], complex III [61, 138–140], complex IV [61, 139, 140], complex V [141], cytochrome c [142], and transporter proteins such as the ADP-ATP carrier [143], pyruvate carrier [144], and phosphate carrier [145]. Thus, CL deficiency can negatively impact the activity and efficiency of these proteins. Several studies demonstrate that ROS-induced CL oxidation causes concomitant inactivation of complexes I, III, and IV [146–148]. In vitro studies indicate that adding CL liposomes, but not PE, PC, or oxidized CL liposomes, prevents ETC complex defects caused by CL oxidation [146]. In addition to interactions with single complexes, CL is required for the proper assembly and stability of ETC supercomplexes. In mammalian mitochondria, supercomplexes are comprised of complex I associated with complex III dimers and up to four monomers of complex IV [148]. Yeast mitochondria, which lack complex I, contain small supercomplexes of complex III dimers. Large supercomplexes are characterized by two small supercomplexes associated with complex IV [148]. CL is required for the assembly and stability of these supercomplexes. Supercomplexes of complexes III and IV are destabilized in yeast crd1Δ cells as detected by CN-PAGE [61, 140]. In lymphoblast cells of BTHS patients, complex IV readily dissociates from the supercomplex, and I/III supercomplex levels are decreased [149]. In addition to the impact of CL on the respiratory chain, CL deficiency also leads to other manifestations of mitochondrial dysfunction such as defective protein import and mitophagy, as discussed below.
4.1.3. Mitochondrial Pharmaceutics in CVD
Because mitochondrial dysfunction plays a pivotal role in the pathogenesis and progress of CVD, the field of mitochondrial pharmaceutics is rapidly expanding [150]. Therapeutics that target heart mitochondria, including synthetic peptides (SS peptide family) [151–153], superoxide dismutase mimetics [154], and triphenylphosphonium- (TPP-) ligated antioxidants such as vitamin E [155], ubiquinone [156], and lipoic acid [157], exhibit promise in alleviating mitochondrial damage in CVD. Several of these drugs are currently being tested in clinical trials [150].
4.2. Mitochondrial Protein Import
More than 98% of mitochondrial proteins are encoded in the nucleus, synthesized in the cytosol as precursors, and imported into the mitochondria [158]. Thus, mitochondrial protein import is essential for maintaining normal mitochondrial function. As discussed above, a link between defective mitochondrial protein import and CVD was suggested by mutations in the DNAJC19 gene in DCMA syndrome. Two in vitro studies showed that the unfolding of an artificial mitochondrial protein precursor by CL was required for binding to isolated yeast mitochondrial outer membranes or liposomes. These findings were the first to demonstrate a mechanistic link between CL and protein import [105, 106]. A more direct demonstration of the role of CL in mitochondrial protein import was shown by decreased protein import in the yeast CL mutant crd1Δ [108]. CL was also shown to be involved in the biogenesis of mitochondria outer membrane protein import complexes [104]. Functional assays of precursor binding to the TOM complex, the translocase of the mitochondrial outer membrane, and the SAM complex, the outer membrane sorting and assembly machinery, revealed partially impaired precursor binding in CL mutants [104]. Loss of CL also leads to defective import of mitochondrial ATPase subunit precursors, which are located in the inner membrane or matrix [108].
4.3. Autophagy/Mitophagy
4.3.1. Autophagy/Mitophagy as a Protective Mechanism against Cardiac Aging and Ischemia-Reperfusion
Autophagy refers to the cellular process in which cytoplasmic contents are delivered into the lysosome or vacuole for degradation. Autophagy is further classified as selective and nonselective autophagy [159]. Various types of selective autophagy have been identified, including mitophagy, pexophagy, lipophagy, nucleophagy, lysophagy, reticulophagy/ER-phagy, and ribophagy [160]. Mitophagy is the selective degradation of mitochondria by autophagy [161]. Mitophagy and autophagy are generally not distinguished in studies of CVD and will be discussed together here.
Numerous studies link autophagy to CVD. In the heart, autophagy is an important housekeeping process that is essential for maintaining cardiac health [162]. Deletion of ATG5, the gene encoding a protein that regulates phagophore expansion, is known to result in cardiomyopathy in mice [163]. Autophagic activity declines with age, and decreased or impaired autophagy leads to accumulation of proteins and damaged mitochondria, contributing to cardiac aging [164].
As early as the 1970s, autophagy was shown to be increased during ischemia [165]. After decades of research, the relationship between autophagy and cardiovascular physiology is only partially clear. As discussed above, damage to mitochondria is a hallmark of ischemia. During mild and chronic ischemia, mitophagy is increased as an adaptive and protective strategy to eliminate damaged mitochondria [166–168]. Increased autophagy is accompanied by decreased apoptosis during ischemia, suggesting that autophagy limits apoptotic necrosis of cardiomyocytes [166, 169]. Many studies implicate the involvement of AMPK activation in triggering autophagy/mitophagy during ischemia [170–172], although this is not conclusive [173]. Following reperfusion, autophagy is even more dramatically increased in animal models [166, 174, 175] and primary neonatal cardiomyocytes [169], having a detrimental effect that is at least partially mediated by activation of Beclin-1, the protein required for autophagosome formation [169, 170, 176].
4.3.2. CL Is Needed for Maintaining Normal Mitophagy
CL is reported to externalize the outer mitochondrial membrane as an elimination signal for mitophagy in neuronal cells and to bind the microtubule-associated protein 1 light chain 3 (MAP1LC3/LC3), the marker protein of autophagic membranes. Binding induces recognition of mitochondria as the cargo by the autophagic machinery [177, 178]. The role of CL in mitophagy is supported by the finding that ALCAT1-catalyzed remodeling of CL with aberrant acyl groups leads to defective mitophagy in hepatocytes [179]. Interestingly, the autophagy-related protein Beclin-1 is preferentially enriched in lipid membranes that contain high concentrations of CL [180]. Deletion of ATG5, which is essential for autophagy, results in cardiomyopathy in mice [163]. These findings invite speculation that loss of CL and defective CL remodeling may contribute to the development of cardiomyopathy by a mechanism related to perturbation of mitophagy.
4.4. The PKC Pathway
4.4.1. The Role of PKC in Cardiovascular Health
Protein kinase C (PKC) is a family of protein kinases that regulate the function of other proteins through specific phosphorylation of hydroxyl groups on threonine and serine residues. Human cells have fifteen PKC isozymes [181]. Overstimulation of PKCα, PKCβ, PKCδ, or PKCε results in hypertrophy of cardiomyocytes through activation of the extracellular signal-related kinase (ERK) pathway [182]. However, during ischemia preconditioning, PKCα, PKCδ, PKCε, and PKCη have been shown to translocate to the active membrane pool and perform cardioprotective functions [182]. Activation of PKCδ results in intracellular pH changes and viability protection; activation of PKCη protects against myocardial stunning; activation of both PKCδ and PKCη provides global myocardial protection against necrosis, acidosis, and myocardial stunning [183]. Blocking the phosphatidylinositol-specific phospholipase C- (PI-PLC-) induced translocation of PKCα, PKCε, and PKCη during ischemia impairs myocardial recovery [184]. Therefore, PKC isozymes have dual functions in the pathogenesis and progression of CVD. However, unlike other PKC isozymes that have dual roles in different CVDs, PKCη is mainly reported to play a cardioprotective role during ischemia.
4.4.2. Loss of CL Leads to Defective PKC
During hyperthermia-induced apoptosis, PKCδ phosphorylates phospholipid scramblase 3 (PLS3), which then induces CL translocation from the inner to outer mitochondrial membrane [185–187]. This series of reactions is considered an indicator of both apoptosis and autophagy. The relationship between CL and PKC appears to be interdependent. While CL translocation is regulated by PKCδ, CL may also be a regulator of the PKC pathway. Studies in yeast, which have only one PKC (Pkc1), show that loss of CL may lead to defects in the activation of the PKC pathway [188]. Human PKCη is the only human PKC isozyme that can complement the defects caused by deletion of PKC1 in yeast through activation of the same protein kinase cascade [189]. This suggests that PKCη shares both functional homology and structural homology with Pkc1. Extrapolating from the finding in yeast that CL plays a role in PKC pathway activation, the cardioprotective role of PKCη activation during ischemia preconditioning may be dependent on CL.
5. Conclusion
As discussed above, CL plays important roles in cellular processes and pathways that are crucial for heart function, including mitochondrial function, mitochondrial protein import, autophagy/mitophagy, and the PKC pathway. CL synthesis and remodeling are highly regulated under physiological conditions, and perturbation of this regulation results in aberrant CL profiles in associated cardiac disorders, including cardiomyopathy, myocardial ischemia-reperfusion injury, HF, atherosclerosis, and Tangier disease. However, the mechanisms linking CL to these pathologies remain to be elucidated.
Mechanisms underlying the role of ox-CL in the pathogenesis of myocardial ischemia-reperfusion injury and atherosclerosis have been suggested. Apoptosis and necrosis during ischemia-reperfusion may result from decreased binding of cytochrome c to ox-CL [84], which likely leads to the release of cytochrome c and to increased permeability of the mitochondrial membrane to apoptosis factors [82, 190]. In addition, ox-CL also functions as an antigen to stimulate proinflammatory effects during the formation of atherosclerosis.
The relative contribution of CL/MLCL levels and acyl composition in maintaining respiratory chain function and cardiovascular health is not understood. Many studies have suggested that the lack of unsaturated L4-CL may be the cause of the pathology in BTHS [43, 44]. Consistent with the importance of CL acyl composition, knockdown of DNAJC19 alters the acyl chain composition of CL without influencing the total CL level [109]. However, the finding that growth and respiratory defects of the yeast taz1 mutant are rescued by deletion of CLD1, which restores CL/MLCL levels without generating remodeled CL, suggests that CL/MLCL levels are more important for mitochondrial function than CL acyl composition [53, 54].
In summary, elucidating the mechanisms whereby CL regulates cardiac function remains a vastly unexplored and exciting frontier that holds the promise of potential new therapies to treat cardiac disorders.
Acknowledgments
The Greenberg lab gratefully acknowledges support from the National Institutes of Health R01 HL117880 and from the Barth Syndrome Foundation, Barth Syndrome Foundation of Canada, and Association Barth France.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.De Bruijn J. H. Chemical structure and serological activity of natural and synthetic cardiolipin and related compounds. British Journal of Venereal Diseases. 1966;42(2):125–128. doi: 10.1136/sti.42.2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schlame M., Brody S., Hostetler K. Y. Mitochondrial cardiolipin in diverse eukaryotes. European Journal of Biochemistry. 1993;212(3):727–733. doi: 10.1111/j.1432-1033.1993.tb17711.x. [DOI] [PubMed] [Google Scholar]
- 3.Joshi A. S., Zhou J., Gohil V. M., Chen S., Greenberg M. L. Cellular functions of cardiolipin in yeast. Biochimica et Biophysica Acta. 2009;1793(1):212–218. doi: 10.1016/j.bbamcr.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhong Q., Gvozdenovic-Jeremic J., Webster P., Zhou J., Greenberg M. L. Loss of function of KRE5 suppresses temperature sensitivity of mutants lacking mitochondrial anionic lipids. Molecular Biology of the Cell. 2005;16(2):665–675. doi: 10.1091/mbc.e04-09-0808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen S., Tarsio M., Kane P. M., Greenberg M. L. Cardiolipin mediates cross-talk between mitochondria and the vacuole. Molecular Biology of the Cell. 2008;19(12):5047–5058. doi: 10.1091/mbc.e08-05-0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou J., Zhong Q., Li G., Greenberg M. L. Loss of cardiolipin leads to longevity defects that are alleviated by alterations in stress response signaling. The Journal of Biological Chemistry. 2009;284(27):18106–18114. doi: 10.1074/jbc.m109.003236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen S., Liu D., Finley R. L., Jr., Greenberg M. L. Loss of mitochondrial DNA in the yeast cardiolipin synthase crd1 mutant leads to up-regulation of the protein kinase Swe1p that regulates the G2/M transition. The Journal of Biological Chemistry. 2010;285(14):10397–10407. doi: 10.1074/jbc.m110.100784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X.-X., Tsoi B., Li Y.-F., Kurihara H., He R.-R. Cardiolipin and its different properties in mitophagy and apoptosis. Journal of Histochemistry & Cytochemistry. 2015;63(5):301–311. doi: 10.1369/0022155415574818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hostetler K. Y., van den Bosch H., van Deenen L. L. M. The mechanism of cardiolipin biosynthesis in liver mitochondria. Biochimica et Biophysica Acta. 1972;260(3):507–513. doi: 10.1016/0005-2760(72)90065-3. [DOI] [PubMed] [Google Scholar]
- 10.Tian H.-F., Feng J.-M., Wen J.-F. The evolution of cardiolipin biosynthesis and maturation pathways and its implications for the evolution of eukaryotes. BMC Evolutionary Biology. 2012;12(1, article 32) doi: 10.1186/1471-2148-12-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tamura Y., Harada Y., Nishikawa S.-I., et al. Tam41 is a CDP-diacylglycerol synthase required for cardiolipin biosynthesis in mitochondria. Cell Metabolism. 2013;17(5):709–718. doi: 10.1016/j.cmet.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kutik S., Rissler M., Guan X. L., et al. The translocator maintenance protein Tam41 is required for mitochondrial cardiolipin biosynthesis. Journal of Cell Biology. 2008;183(7):1213–1221. doi: 10.1083/jcb.200806048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steiner M. R., Lester R. L. In vitro studies of phospholipid biosynthesis in Saccharomyces cerevisiae . Biochimica et Biophysica Acta (BBA)—Lipids and Lipid Metabolism. 1972;260(2):222–243. doi: 10.1016/0005-2760(72)90035-5. [DOI] [PubMed] [Google Scholar]
- 14.Chang S.-C., Heacock P. N., Clancey C. J., Dowhan W. The PEL1 gene (renamed PGS1) encodes the phosphatidylglycerophosphate synthase of Saccharomyces cerevisiae . The Journal of Biological Chemistry. 1998;273(16):9829–9836. doi: 10.1074/jbc.273.16.9829. [DOI] [PubMed] [Google Scholar]
- 15.Hirabayashi T., Larson T. J., Dowhan W. Membrane-associated phosphatidylglycerophosphate synthetase from Escherichia coli: purification by substrate affinity chromatography on cytidine 5′-Diphospho-1,2-diacyl-sn-glycerol sepharose. Biochemistry. 1976;15(24):5205–5211. doi: 10.1021/bi00669a002. [DOI] [PubMed] [Google Scholar]
- 16.van den Bosch H., van Golde L. M., van Deenen L. L. Dynamics of phosphoglycerides. Ergebnisse der Physiologie. 1972;13:13–145. doi: 10.1007/3-540-05882-6_2. [DOI] [PubMed] [Google Scholar]
- 17.Kelly B. L., Greenberg M. L. Characterization and regulation of phosphatidylglycerolphosphate phosphatase in Saccharomyces cerevisiae . Biochimica et Biophysica Acta—Lipids and Lipid Metabolism. 1990;1046(2):144–150. doi: 10.1016/0005-2760(90)90181-v. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J., Guan Z., Murphy A. N., et al. Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metabolism. 2011;13(6):690–700. doi: 10.1016/j.cmet.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao J., Engel J. L., Zhang J., Chen M. J., Manning G., Dixon J. E. Structural and functional analysis of PTPMT1, a phosphatase required for cardiolipin synthesis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(29):11860–11865. doi: 10.1073/pnas.1109290108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Osman C., Haag M., Wieland F. T., Brügger B., Langer T. A mitochondrial phosphatase required for cardiolipin biosynthesis: the PGP phosphatase Gep4. The EMBO Journal. 2010;29(12):1976–1987. doi: 10.1038/emboj.2010.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen D., Zhang X.-Y., Shi Y. Identification and functional characterization of hCLS1, a human cardiolipin synthase localized in mitochondria. Biochemical Journal. 2006;398(2):169–176. doi: 10.1042/BJ20060303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu B., Xu F. Y., Jiang Y. J., et al. Cloning and characterization of a cDNA encoding human cardiolipin synthase (hCLS1) Journal of Lipid Research. 2006;47(6):1140–1145. doi: 10.1194/jlr.c600004-jlr200. [DOI] [PubMed] [Google Scholar]
- 23.Houtkooper R. H., Akbari H., van Lenthe H., et al. Identification and characterization of human cardiolipin synthase. FEBS Letters. 2006;580(13):3059–3064. doi: 10.1016/j.febslet.2006.04.054. [DOI] [PubMed] [Google Scholar]
- 24.Tuller G., Hrastnik C., Achleitner G., Schiefthaler U., Klein F., Daum G. YDL142c encodes cardiolipin synthase (Cls1p) and is non-essential for aerobic growth of Saccharomyces cerevisiae . FEBS Letters. 1998;421(1):15–18. doi: 10.1016/s0014-5793(97)01525-1. [DOI] [PubMed] [Google Scholar]
- 25.Jiang F., Rizavi H. S., Greenberg M. L. Cardiolipin is not essential for the growth of Saccharomyces cerevisiae on fermentable or non-fermentable carbon sources. Molecular Microbiology. 1997;26(3):481–491. doi: 10.1046/j.1365-2958.1997.5841950.x. [DOI] [PubMed] [Google Scholar]
- 26.Chang S.-C., Heacock P. N., Mileykovskaya E., Voelker D. R., Dowhan W. Isolation and characterization of the gene (CLS1) encoding cardiolipin synthase in Saccharomyces cerevisiae . Journal of Biological Chemistry. 1998;273(24):14933–14941. doi: 10.1074/jbc.273.24.14933. [DOI] [PubMed] [Google Scholar]
- 27.van den Bosch H. Phosphoglyceride metabolism. Annual Review of Biochemistry. 1974;43:243–277. doi: 10.1146/annurev.bi.43.070174.001331. [DOI] [PubMed] [Google Scholar]
- 28.Ye C., Shen Z., Greenberg M. L. Cardiolipin remodeling: a regulatory hub for modulating cardiolipin metabolism and function. Journal of Bioenergetics and Biomembranes. 2014 doi: 10.1007/s10863-014-9591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lands W. E. M. Metabolism of glycerolipids. II. The enzymatic acylation of lysolecithin. Journal of Biological Chemistry. 1960;235(8):2233–2237. [PubMed] [Google Scholar]
- 30.Beranek A., Rechberger G., Knauer H., Wolinski H., Kohlwein S. D., Leber R. Identification of a cardiolipin-specific phospholipase encoded by the gene CLD1 (YGR110W) in yeast. The Journal of Biological Chemistry. 2009;284(17):11572–11578. doi: 10.1074/jbc.m805511200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buckland A. G., Kinkaid A. R., Wilton D. C. Cardiolipin hydrolysis by human phospholipases A2: the multiple enzymatic activities of human cytosolic phospholipase A2. Biochimica et Biophysica Acta—Lipids and Lipid Metabolism. 1998;1390(1):65–72. doi: 10.1016/s0005-2760(97)00170-7. [DOI] [PubMed] [Google Scholar]
- 32.Hsu Y.-H., Dumlao D. S., Cao J., Dennis E. A. Assessing phospholipase A2 activity toward cardiolipin by mass spectrometry. PLoS ONE. 2013;8(3) doi: 10.1371/journal.pone.0059267.e59267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dennis E. A., Cao J., Hsu Y.-H., Magrioti V., Kokotos G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chemical Reviews. 2011;111(10):6130–6185. doi: 10.1021/cr200085w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bione S., D'Adamo P., Maestrini E., Gedeon A. K., Bolhuis P. A., Toniolo D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nature Genetics. 1996;12(4):385–389. doi: 10.1038/ng0496-385. [DOI] [PubMed] [Google Scholar]
- 35.Gu Z., Valianpour F., Chen S., et al. Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Molecular Microbiology. 2004;51(1):149–158. doi: 10.1046/j.1365-2958.2003.03802.x. [DOI] [PubMed] [Google Scholar]
- 36.Vaz F. M., Houtkooper R. H., Valianpour F., Barth P. G., Wanders R. J. A. Only one splice variant of the human TAZ gene encodes a functional protein with a role in cardiolipin metabolism. Journal of Biological Chemistry. 2003;278(44):43089–43094. doi: 10.1074/jbc.m305956200. [DOI] [PubMed] [Google Scholar]
- 37.Cao J., Liu Y., Lockwood J., Burn P., Shi Y. A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA:lysocardiolipin acyltransferase (ALCAT1) in mouse. The Journal of Biological Chemistry. 2004;279(30):31727–31734. doi: 10.1074/jbc.m402930200. [DOI] [PubMed] [Google Scholar]
- 38.Taylor W. A., Hatch G. M. Identification of the human mitochondrial linoleoyl-coenzyme a monolysocardiolipin acyltransferase (MLCL AT-1) The Journal of Biological Chemistry. 2009;284(44):30360–30371. doi: 10.1074/jbc.m109.048322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu Y., Kelley R. I., Blanck T. J. J., Schlame M. Remodeling of cardiolipin by phospholipid transacylation. Journal of Biological Chemistry. 2003;278(51):51380–51385. doi: 10.1074/jbc.m307382200. [DOI] [PubMed] [Google Scholar]
- 40.Yamashita A., Sugiura T., Waku K. Acyltransferases and transacylases involved in fatty acid remodeling of phospholipids and metabolism of bioactive lipids in mammalian cells. Journal of Biochemistry. 1997;122(1):1–16. doi: 10.1093/oxfordjournals.jbchem.a021715. [DOI] [PubMed] [Google Scholar]
- 41.Barth P. G., Scholte H. R., Berden J. A., et al. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. Journal of the Neurological Sciences. 1983;62(1–3):327–355. doi: 10.1016/0022-510x(83)90209-5. [DOI] [PubMed] [Google Scholar]
- 42.Barth P. G., Wanders R. J. A., Vreken P. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome)—MIM 302060. Journal of Pediatrics. 1999;135(3):273–276. doi: 10.1016/s0022-3476(99)70118-6. [DOI] [PubMed] [Google Scholar]
- 43.Valianpour F., Wanders R. J. A., Overmars H., et al. Cardiolipin deficiency in X-linked cardioskeletal myopathy and neutropenia (Barth syndrome, MIM 302060): a study in cultured skin fibroblasts. The Journal of Pediatrics. 2002;141(5):729–733. doi: 10.1067/mpd.2002.129174. [DOI] [PubMed] [Google Scholar]
- 44.Schlame M., Towbin J. A., Heerdt P. M., Jehle R., DiMauro S., Blanck T. J. J. Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Annals of Neurology. 2002;51(5):634–637. doi: 10.1002/ana.10176. [DOI] [PubMed] [Google Scholar]
- 45.Schlame M., Kelley R. I., Feigenbaum A., et al. Phospholipid abnormalities in children with Barth syndrome. Journal of the American College of Cardiology. 2003;42(11):1994–1999. doi: 10.1016/j.jacc.2003.06.015. [DOI] [PubMed] [Google Scholar]
- 46.Chen R., Tsuji T., Ichida F., et al. Mutation analysis of the G4.5 gene in patients with isolated left ventricular noncompaction. Molecular Genetics and Metabolism. 2002;77(4):319–325. doi: 10.1016/s1096-7192(02)00195-6. [DOI] [PubMed] [Google Scholar]
- 47.D'Adamo P., Fassone L., Gedeon A., et al. The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. American Journal of Human Genetics. 1997;61(4):862–867. doi: 10.1086/514886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hijikata A., Yura K., Ohara O., Go M. Structural and functional analyses of Barth syndrome-causing mutations and alternative splicing in the tafazzin acyltransferase domain. Meta Gene. 2015;4:92–106. doi: 10.1016/j.mgene.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bowron A., Honeychurch J., Williams M., et al. Barth syndrome without tetralinoleoyl cardiolipin deficiency: a possible ameliorated phenotype. Journal of Inherited Metabolic Disease. 2015;38(2):279–286. doi: 10.1007/s10545-014-9747-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Claypool S. M., Whited K., Srijumnong S., Han X., Koehler C. M. Barth syndrome mutations that cause tafazzin complex lability. Journal of Cell Biology. 2011;192(3):447–462. doi: 10.1083/jcb.201008177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Houtkooper R. H., Vaz F. M. Cardiolipin, the heart of mitochondrial metabolism. Cellular and Molecular Life Sciences. 2008;65(16):2493–2506. doi: 10.1007/s00018-008-8030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheng H., Mancuso D. J., Jiang X., et al. Shotgun lipidomics reveals the temporally dependent, highly diversified cardiolipin profile in the mammalian brain: temporally coordinated postnatal diversification of cardiolipin molecular species with neuronal remodeling. Biochemistry. 2008;47(21):5869–5880. doi: 10.1021/bi7023282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baile M. G., Sathappa M., Lu Y.-W., et al. Unremodeled and remodeled cardiolipin are functionally indistinguishable in yeast. The Journal of Biological Chemistry. 2014;289(3):1768–1778. doi: 10.1074/jbc.m113.525733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ye C., Lou W., Li Y., et al. Deletion of the Cardiolipin-specific Phospholipase Cld1 rescues growth and life span defects in the Tafazzin Mutant: implications for Barth Syndrome. The Journal of Biological Chemistry. 2014;289(6):3114–3125. doi: 10.1074/jbc.m113.529487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gardner D. G., Shoback D. Greenspan's Basic and Clinical Endocrinology. 9th. chapter 17. New York, NY, USA: McGraw-Hill; 2011. [Google Scholar]
- 56.Nathan D. M. Long-term complications of diabetes mellitus. The New England Journal of Medicine. 1993;328(23):1676–1685. doi: 10.1056/nejm199306103282306. [DOI] [PubMed] [Google Scholar]
- 57.Han X., Abendschein D. R., Kelley J. G., Gross R. W. Diabetes-induced changes in specific lipid molecular species in rat myocardium. Biochemical Journal. 2000;352(1):79–89. doi: 10.1042/0264-6021:3520079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han X., Yang J., Cheng H., Yang K., Abendschein D. R., Gross R. W. Shotgun lipidomics identifies cardiolipin depletion in diabetic myocardium linking altered substrate utilization with mitochondrial dysfunction. Biochemistry. 2005;44(50):16684–16694. doi: 10.1021/bi051908a. [DOI] [PubMed] [Google Scholar]
- 59.Han X., Yang J., Yang K., Zhongdan Z., Abendschein D. R., Gross R. W. Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: a shotgun lipidomics study. Biochemistry. 2007;46(21):6417–6428. doi: 10.1021/bi7004015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pan H.-J., Lin Y., Chen Y. E., Vance D. E., Leiter E. H. Adverse hepatic and cardiac responses to rosiglitazone in a new mouse model of type 2 diabetes: relation to dysregulated phosphatidylcholine metabolism. Vascular Pharmacology. 2006;45(1):65–71. doi: 10.1016/j.vph.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 61.Pfeiffer K., Gohil V., Stuart R. A., et al. Cardiolipin stabilizes respiratory chain supercomplexes. The Journal of Biological Chemistry. 2003;278(52):52873–52880. doi: 10.1074/jbc.m308366200. [DOI] [PubMed] [Google Scholar]
- 62.Mileykovskaya E., Dowhan W. Cardiolipin-dependent formation of mitochondrial respiratory supercomplexes. Chemistry and Physics of Lipids. 2014;179:42–48. doi: 10.1016/j.chemphyslip.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koshkin V., Greenberg M. L. Cardiolipin prevents rate-dependent uncoupling and provides osmotic stability in yeast mitochondria. Biochemical Journal. 2002;364(1):317–322. doi: 10.1042/bj3640317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sack M. N. Type 2 diabetes, mitochondrial biology and the heart. Journal of Molecular and Cellular Cardiology. 2009;46(6):842–849. doi: 10.1016/j.yjmcc.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carden D. L., Granger D. N. Pathophysiology of ischaemia-reperfusion injury. The Journal of Pathology. 2000;190(3):255–266. doi: 10.1002/(sici)1096-9896(200002)190:3lt;255::aid-path526>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 66.Ganote C. E. Contraction band necrosis and irreversible myocardial injury. Journal of Molecular and Cellular Cardiology. 1983;15(2):67–73. doi: 10.1016/0022-2828(83)90283-3. [DOI] [PubMed] [Google Scholar]
- 67.Ruiz-Meana M., Garcia-Dorado D., Hofstaetter B., Piper H. M., Soler-Soler J. Propagation of cardiomyocyte hypercontracture by passage of Na+ through gap junctions. Circulation Research. 1999;85(3):280–287. doi: 10.1161/01.res.85.3.280. [DOI] [PubMed] [Google Scholar]
- 68.Allen D. G., Orchard C. H. Myocardial contractile function during ischemia and hypoxia. Circulation Research. 1987;60(2):153–168. doi: 10.1161/01.RES.60.2.153. [DOI] [PubMed] [Google Scholar]
- 69.Chandrashekhar Y., Prahash A. J., Sen S., Gupta S., Anand I. S. Cardiomyocytes from hearts with left ventricular dysfunction after ischemia-reperfusion do not manifest contractile abnormalities. Journal of the American College of Cardiology. 1999;34(2):594–602. doi: 10.1016/S0735-1097(99)00222-3. [DOI] [PubMed] [Google Scholar]
- 70.Wit A. L., Janse M. J. Reperfusion arrhythmias and sudden cardiac death: a century of progress toward an understanding of the mechanisms. Circulation Research. 2001;89(9):741–743. [PubMed] [Google Scholar]
- 71.Cascio W. E., Yang H., Muller-Borer B. J., Johnson T. A. Ischemia-induced arrhythmia: the role of connexins, gap junctions, and attendant changes in impulse propagation. Journal of Electrocardiology. 2005;38(4, supplement):55–59. doi: 10.1016/j.jelectrocard.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 72.Ghuran A. V., Camm A. J. Ischaemic heart disease presenting as arrhythmias. British Medical Bulletin. 2001;59(1):193–210. doi: 10.1093/bmb/59.1.193. [DOI] [PubMed] [Google Scholar]
- 73.Luqman N., Sung R. J., Wang C.-L., Kuo C.-T. Myocardial ischemia and ventricular fibrillation: pathophysiology and clinical implications. International Journal of Cardiology. 2007;119(3):283–290. doi: 10.1016/j.ijcard.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 74.Kazbanov I. V., Clayton R. H., Nash M. P., et al. Effect of global cardiac ischemia on human ventricular fibrillation: insights from a multi-scale mechanistic model of the human heart. PLoS Computational Biology. 2014;10(11) doi: 10.1371/journal.pcbi.1003891.e1003891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ferrari R., Ceconi C., Curello S., et al. Role of oxygen free radicals in ischemic and reperfused myocardium. The American Journal of Clinical Nutrition. 1991;53(1):215S–222S. doi: 10.1093/ajcn/53.1.215S. [DOI] [PubMed] [Google Scholar]
- 76.Kloner R. A., Przyklenk K., Whittaker P. Deleterious effects of oxygen radicals in ischemia/reperfusion. Resolved and unresolved issues. Circulation. 1989;80(5):1115–1127. doi: 10.1161/01.cir.80.5.1115. [DOI] [PubMed] [Google Scholar]
- 77.Kalogeris T., Bao Y., Korthuis R. J. Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biology. 2014;2(1):702–714. doi: 10.1016/j.redox.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Paradies G., Petrosillo G., Pistolese M., Di Venosa N., Serena D., Ruggiero F. M. Lipid peroxidation and alterations to oxidative metabolism in mitochondria isolated from rat heart subjected to ischemia and reperfusion. Free Radical Biology and Medicine. 1999;27(1-2):42–50. doi: 10.1016/S0891-5849(99)00032-5. [DOI] [PubMed] [Google Scholar]
- 79.Lesnefsky E. J., Slabe T. J., Stoll M. S. K., Minkler P. E., Hoppel C. L. Myocardial ischemia selectively depletes cardiolipin in rabbit heart subsarcolemmal mitochondria. The American Journal of Physiology—Heart and Circulatory Physiology. 2001;280(6):H2770–H2778. doi: 10.1152/ajpheart.2001.280.6.H2770. [DOI] [PubMed] [Google Scholar]
- 80.Paradies G., Petrosillo G., Pistolese M., Di Venosa N., Federici A., Ruggiero F. M. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circulation Research. 2004;94(1):53–59. doi: 10.1161/01.res.0000109416.56608.64. [DOI] [PubMed] [Google Scholar]
- 81.Petrosillo G., Ruggiero F. M., Di Venosa N., Paradies G. Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: role of reactive oxygen species and cardiolipin. The FASEB Journal. 2003;17(6):714–716. doi: 10.1096/fj.02-0729fje. [DOI] [PubMed] [Google Scholar]
- 82.Kagan V. E., Tyurin V. A., Jiang J., et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nature Chemical Biology. 2005;1(4):223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 83.Vladimirov Y. A., Proskurnina E. V., Izmailov D. Y., et al. Cardiolipin activates cytochrome c peroxidase activity since it facilitates H2O2 access to heme. Biochemistry. 2006;71(9):998–1005. doi: 10.1134/s0006297906090082. [DOI] [PubMed] [Google Scholar]
- 84.Nomura K., Imai H., Koumura T., Kobayashi T., Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochemical Journal. 2000;351(1):183–193. doi: 10.1042/0264-6021:3510183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lutter M., Perkins G. A., Wang X. The pro-apoptotic Bcl-2 family member tBid localizes to mitochondrial contact sites. BMC Cell Biology. 2001;2, article 22 doi: 10.1186/1471-2121-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lucken-Ardjomande S., Montessuit S., Martinou J.-C. Contributions to Bax insertion and oligomerization of lipids of the mitochondrial outer membrane. Cell Death and Differentiation. 2008;15(5):929–937. doi: 10.1038/cdd.2008.9. [DOI] [PubMed] [Google Scholar]
- 87.Sani M.-A., Dufourc E. J., Gröbner G. How does the Bax-α1 targeting sequence interact with mitochondrial membranes? The role of cardiolipin. Biochimica et Biophysica Acta: Biomembranes. 2009;1788(3):623–631. doi: 10.1016/j.bbamem.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 88.Epand R. F., Martinou J.-C., Fornallaz-Mulhauser M., Hughes D. W., Epand R. M. The apoptotic protein tBid promotes leakage by altering membrane curvature. The Journal of Biological Chemistry. 2002;277(36):32632–32639. doi: 10.1074/jbc.m202396200. [DOI] [PubMed] [Google Scholar]
- 89.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362(6423):801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 90.Tuominen A., Miller Y. I., Hansen L. F., Kesäniemi Y. A., Witztum J. L., Hörkkö S. A natural antibody to oxidized cardiolipin binds to oxidized low-density lipoprotein, apoptotic cells, and atherosclerotic lesions. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(9):2096–2102. doi: 10.1161/01.ATV.0000233333.07991.4a. [DOI] [PubMed] [Google Scholar]
- 91.Zhong H., Lu J., Xia L., Zhu M., Yin H. Formation of electrophilic oxidation products from mitochondrial cardiolipin in vitro and in vivo in the context of apoptosis and atherosclerosis. Redox Biology. 2014;2:878–883. doi: 10.1016/j.redox.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Marai I., Shechter M., Langevitz P., et al. Anti-cardiolipin antibodies and endothelial function in patients with coronary artery disease. The American Journal of Cardiology. 2008;101(8):1094–1097. doi: 10.1016/j.amjcard.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 93.Türkoğlu O., Bariş N., Kütükçüler N., Şenarslan Ö., Güneri S., Atilla G. Evaluation of serum anti-cardiolipin and oxidized low-density lipoprotein levels in chronic periodontitis patients with essential hypertension. Journal of Periodontology. 2008;79(2):332–340. doi: 10.1902/jop.2008.070321. [DOI] [PubMed] [Google Scholar]
- 94.Lopez D., Kobayashi K., Merrill J. T., Matsuura E., Lopez L. R. IgG autoantibodies against β 2-glycoprotein I complexed with a lipid ligand derived from oxidized low-density lipoprotein are associated with arterial thrombosis in antiphospholipid syndrome. Clinical and Developmental Immunology. 2003;10(2–4):203–211. doi: 10.1080/10446670310001642113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Su J., Frostegård A. G., Hua X., et al. Low levels of antibodies against oxidized but not nonoxidized cardiolipin and phosphatidylserine are associated with atherosclerotic plaques in systemic lupus erythematosus. Journal of Rheumatology. 2013;40(11):1856–1864. doi: 10.3899/jrheum.121173. [DOI] [PubMed] [Google Scholar]
- 96.Bochkov V. N., Oskolkova O. V., Birukov K. G., Levonen A.-L., Binder C. J., Stöckl J. Generation and biological activities of oxidized phospholipids. Antioxidants & Redox Signaling. 2010;12(8):1009–1059. doi: 10.1089/ars.2009.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Frostegård A. G., Su J., Hua X., Vikström M., De Faire U., Frostegård J. Antibodies against native and oxidized cardiolipin and phosphatidylserine and phosphorylcholine in atherosclerosis development. PLoS ONE. 2014;9(12) doi: 10.1371/journal.pone.0111764.e111764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Su J., Hua X., Vikström M., et al. Low levels of IgM antibodies to oxidized cardiolipin increase and high levels decrease risk of cardiovascular disease among 60-year olds: a prospective study. BMC Cardiovascular Disorders. 2013;13, article 1 doi: 10.1186/1471-2261-13-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wan M., Hua X., Su J., et al. Oxidized but not native cardiolipin has pro-inflammatory effects, which are inhibited by Annexin A5. Atherosclerosis. 2014;235(2):592–598. doi: 10.1016/j.atherosclerosis.2014.05.913. [DOI] [PubMed] [Google Scholar]
- 100.Davey K. M., Parboosingh J. S., McLeod D. R., et al. Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. Journal of Medical Genetics. 2006;43(5):385–393. doi: 10.1136/jmg.2005.036657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.D'Silva P. D., Schilke B., Walter W., Andrew A., Craig E. A. J protein cochaperone of the mitochondrial inner membrane required for protein import into the mitochondrial matrix. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(2):13839–13844. doi: 10.1073/pnas.1936150100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mokranjac D., Sichting M., Neupert W., Hell K. Tim14, a novel key component of the import motor of the TIM23 protein translocase of mitochondria. The EMBO Journal. 2003;22(19):4945–4956. doi: 10.1093/emboj/cdg485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rehling P., Pfanner N., Meisinger C. Insertion of hydrophobic membrane proteins into the inner mitochondrial membrane—a guided tour. Journal of Molecular Biology. 2003;326(3):639–657. doi: 10.1016/s0022-2836(02)01440-7. [DOI] [PubMed] [Google Scholar]
- 104.Gebert N., Joshi A. S., Kutik S., et al. Mitochondrial cardiolipin involved in outer membrane protein biogenesis: implications for Barth syndrome. Current Biology. 2009;19(24):2133–2139. doi: 10.1016/j.cub.2009.10.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Endo T., Eilers M., Schatz G. Binding of a tightly folded artificial mitochondrial precursor protein to the mitochondrial outer membrane involves a lipid-mediated conformational change. The Journal of Biological Chemistry. 1989;264(5):2951–2956. [PubMed] [Google Scholar]
- 106.Endo T., Schatz G. Latent membrane perturbation activity of a mitochondrial precursor protein is exposed by unfolding. The EMBO Journal. 1988;7(4):1153–1158. doi: 10.1002/j.1460-2075.1988.tb02925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Eilers M., Endo T., Schatz G. Adriamycin, a drug interacting with acidic phospholipids, blocks import of precursor proteins by isolated yeast mitochondria. The Journal of Biological Chemistry. 1989;264(5):2945–2950. [PubMed] [Google Scholar]
- 108.Jiang F., Ryan M. T., Schlame M., et al. Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. The Journal of Biological Chemistry. 2000;275(29):22387–22394. doi: 10.1074/jbc.m909868199. [DOI] [PubMed] [Google Scholar]
- 109.Richter-Dennerlein R., Korwitz A., Haag M., et al. DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metabolism. 2014;20(1):158–171. doi: 10.1016/j.cmet.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 110.McMurray J. J. V., Pfeffer M. A. Heart failure. The Lancet. 2005;365(9474):1877–1889. doi: 10.1016/s0140-6736(05)66621-4. [DOI] [PubMed] [Google Scholar]
- 111.Saini-Chohan H. K., Holmes M. G., Chicco A. J., et al. Cardiolipin biosynthesis and remodeling enzymes are altered during development of heart failure. Journal of Lipid Research. 2009;50(8):1600–1608. doi: 10.1194/jlr.m800561-jlr200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sparagna G. C., Chicco A. J., Murphy R. C., et al. Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. Journal of Lipid Research. 2007;48(7):1559–1570. doi: 10.1194/jlr.M600551-JLR200. [DOI] [PubMed] [Google Scholar]
- 113.Chatfield K. C., Sparagna G. C., Sucharov C. C., et al. Dysregulation of cardiolipin biosynthesis in pediatric heart failure. Journal of Molecular and Cellular Cardiology. 2014;74:251–259. doi: 10.1016/j.yjmcc.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Le C. H., Mulligan C. M., Routh M. A., et al. Delta-6-desaturase links polyunsaturated fatty acid metabolism with phospholipid remodeling and disease progression in heart failure. Circulation: Heart Failure. 2014;7(1):172–183. doi: 10.1161/circheartfailure.113.000744. [DOI] [PubMed] [Google Scholar]
- 115.Reibel D. K., O'Rourke B., Foster K. A., Hutchinson H., Uboh C. E., Kent R. L. Altered phospholipid metabolism in pressure-overload hypertrophied hearts. The American Journal of Physiology—Heart and Circulatory Physiology. 1986;250(1):H1–H6. doi: 10.1152/ajpheart.1986.250.1.H1. [DOI] [PubMed] [Google Scholar]
- 116.Rosca M., Minkler P., Hoppel C. L. Cardiac mitochondria in heart failure: normal cardiolipin profile and increased threonine phosphorylation of complex IV. Biochimica et Biophysica Acta: Bioenergetics. 2011;1807(11):1373–1382. doi: 10.1016/j.bbabio.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 117.Fredrickson D. S. The inheritance of high density lipoprotein deficiency (Tangier disease) The Journal of Clinical Investigation. 1964;43:228–236. doi: 10.1172/jci104907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Oram J. F. Tangier disease and ABCA1. Biochimica et Biophysica Acta (BBA)—Molecular and Cell Biology of Lipids. 2000;1529(1–3):321–330. doi: 10.1016/s1388-1981(00)00157-8. [DOI] [PubMed] [Google Scholar]
- 119.Rust S., Rosier M., Funke H., et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nature Genetics. 1999;22(4):352–355. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- 120.Knight B. L. ATP-binding cassette transporter A1: regulation of cholesterol efflux. Biochemical Society Transactions. 2004;32(1):124–127. doi: 10.1042/bst0320124. [DOI] [PubMed] [Google Scholar]
- 121.Wang N., Silver D. L., Thiele C., Tall A. R. ATP-binding cassette transporter A1 (ABCA1) functions as a cholesterol efflux regulatory protein. The Journal of Biological Chemistry. 2001;276(26):23742–23747. doi: 10.1074/jbc.m102348200. [DOI] [PubMed] [Google Scholar]
- 122.Fobker M., Voss R., Reinecke H., Crone C., Assmann G., Walter M. Accumulation of cardiolipin and lysocardiolipin in fibroblasts from Tangier disease subjects. FEBS Letters. 2001;500(3):157–162. doi: 10.1016/S0014-5793(01)02578-9. [DOI] [PubMed] [Google Scholar]
- 123.Maack C., O'Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Research in Cardiology. 2007;102(5):369–392. doi: 10.1007/s00395-007-0666-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Beaudoin M.-S., Perry C. G. R., Arkell A. M., et al. Impairments in mitochondrial palmitoyl-CoA respiratory kinetics that precede development of diabetic cardiomyopathy are prevented by resveratrol in ZDF rats. The Journal of Physiology. 2014;592(12):2519–2533. doi: 10.1113/jphysiol.2013.270538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Montaigne D., Marechal X., Coisne A., et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation. 2014;130(7):554–564. doi: 10.1161/circulationaha.113.008476. [DOI] [PubMed] [Google Scholar]
- 126.Dhalla N. S., Rangi S., Zieroth S., Xu Y.-J. Alterations in sarcoplasmic reticulum and mitochondrial functions in diabetic cardiomyopathy. Experimental & Clinical Cardiology. 2012;17(3):115–120. [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang M., Wei J., Shan H., et al. Calreticulin-STAT3 signaling pathway modulates mitochondrial function in a rat model of furazolidone-induced dilated cardiomyopathy. PLoS ONE. 2013;8(6) doi: 10.1371/journal.pone.0066779.e66779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang X., Bathina M., Lynch J., et al. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes & Development. 2013;27(12):1351–1364. doi: 10.1101/gad.215855.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hayashi D., Ohshima S., Isobe S., et al. Increased 99mTc-sestamibi washout reflects impaired myocardial contractile and relaxation reserve during dobutamine stress due to mitochondrial dysfunction in dilated cardiomyopathy patients. Journal of the American College of Cardiology. 2013;61(19):2007–2017. doi: 10.1016/j.jacc.2013.01.074. [DOI] [PubMed] [Google Scholar]
- 130.Jung C., Martins A. S., Niggli E., Shirokova N. Dystrophic cardiomyopathy: amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovascular Research. 2008;77(4):766–773. doi: 10.1093/cvr/cvm089. [DOI] [PubMed] [Google Scholar]
- 131.Burelle Y., Khairallah M., Ascah A., et al. Alterations in mitochondrial function as a harbinger of cardiomyopathy: lessons from the dystrophic heart. Journal of Molecular and Cellular Cardiology. 2010;48(2):310–321. doi: 10.1016/j.yjmcc.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chouchani E. T., Methner C., Buonincontri G., et al. Complex I deficiency due to selective loss of Ndufs4 in the mouse heart results in severe hypertrophic cardiomyopathy. PLoS ONE. 2014;9(4) doi: 10.1371/journal.pone.0094157.e94157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hagen C. M., Aidt F. H., Hedley P. L., et al. Mitochondrial haplogroups modify the risk of developing hypertrophic cardiomyopathy in a Danish population. PLoS ONE. 2013;8(8) doi: 10.1371/journal.pone.0071904.e71904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Liu X., Ye B., Miller S., et al. Ablation of ALCAT1 mitigates hypertrophic cardiomyopathy through effects on oxidative stress and mitophagy. Molecular and Cellular Biology. 2012;32(21):4493–4504. doi: 10.1128/mcb.01092-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lin L., Sharma V. K., Sheu S.-S. Mechanisms of reduced mitochondrial Ca2+ accumulation in failing hamster heart. Pflügers Archi—European Journal of Physiology. 2007;454(3):395–402. doi: 10.1007/s00424-007-0257-8. [DOI] [PubMed] [Google Scholar]
- 136.Karamanlidis G., Lee C. F., Garcia-Menendez L., et al. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metabolism. 2013;18(2):239–250. doi: 10.1016/j.cmet.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Boudina S., Abel E. D. Diabetic cardiomyopathy revisited. Circulation. 2007;115(25):3213–3223. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 138.Fry M., Green D. E. Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. The Journal of Biological Chemistry. 1981;256(4):1874–1880. [PubMed] [Google Scholar]
- 139.Zhang M., Mileykovskaya E., Dowhan W. Gluing the respiratory chain together: cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. The Journal of Biological Chemistry. 2002;277(46):43553–43556. doi: 10.1074/jbc.c200551200. [DOI] [PubMed] [Google Scholar]
- 140.Zhang M., Mileykovskaya E., Dowhan W. Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. The Journal of Biological Chemistry. 2005;280(33):29403–29408. doi: 10.1074/jbc.m504955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gohil V. M., Hayes P., Matsuyama S., Schägger H., Schlame M., Greenberg M. L. Cardiolipin biosynthesis and mitochondrial respiratory chain function are interdependent. The Journal of Biological Chemistry. 2004;279(41):42612–42618. doi: 10.1074/jbc.m402545200. [DOI] [PubMed] [Google Scholar]
- 142.Ranieri A., Millo D., Di Rocco G., et al. Immobilized cytochrome c bound to cardiolipin exhibits peculiar oxidation state-dependent axial heme ligation and catalytically reduces dioxygen. Journal of Biological Inorganic Chemistry. 2015;20(3):531–540. doi: 10.1007/s00775-015-1238-6. [DOI] [PubMed] [Google Scholar]
- 143.Beyer K., Klingenberg M. ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by 31P nuclear magnetic resonance. Biochemistry. 1985;24(15):3821–3826. doi: 10.1021/bi00336a001. [DOI] [PubMed] [Google Scholar]
- 144.Paradies G., Ruggiero F. M. The effect of doxorubicin on the transport of pyruvate in rat-heart mitochondria. Biochemical and Biophysical Research Communications. 1988;156(3):1302–1307. doi: 10.1016/s0006-291x(88)80774-5. [DOI] [PubMed] [Google Scholar]
- 145.Kadenbach B., Mende P., Kolbe H. V. J., Stipani I., Palmieri F. The mitochondrial phosphate carrier has an essential requirement for cardiolipin. FEBS Letters. 1982;139(1):109–112. doi: 10.1016/0014-5793(82)80498-5. [DOI] [PubMed] [Google Scholar]
- 146.Paradies G., Petrosillo G., Pistolese M., Ruggiero F. M. The effect of reactive oxygen species generated from the mitochondrial electron transport chain on the cytochrome c oxidase activity and on the cardiolipin content in bovine heart submitochondrial particles. FEBS Letters. 2000;466(2-3):323–326. doi: 10.1016/S0014-5793(00)01082-6. [DOI] [PubMed] [Google Scholar]
- 147.Musatov A. Contribution of peroxidized cardiolipin to inactivation of bovine heart cytochrome c oxidase. Free Radical Biology and Medicine. 2006;41(2):238–246. doi: 10.1016/j.freeradbiomed.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 148.Schägger H., Pfeiffer K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. The EMBO Journal. 2000;19(8):1777–1783. doi: 10.1093/emboj/19.8.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.McKenzie M., Lazarou M., Thorburn D. R., Ryan M. T. Mitochondrial respiratory chain supercomplexes are destabilized in barth syndrome patients. Journal of Molecular Biology. 2006;361(3):462–469. doi: 10.1016/j.jmb.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 150.Ajith T. A., Jayakumar T. G. Mitochondria-targeted agents: future perspectives of mitochondrial pharmaceutics in cardiovascular diseases. World Journal of Cardiology. 2014;6(10):1091–1099. doi: 10.4330/wjc.v6.i10.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhao K., Zhao G.-M., Wu D., et al. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. Journal of Biological Chemistry. 2004;279(33):34682–34690. doi: 10.1074/jbc.m402999200. [DOI] [PubMed] [Google Scholar]
- 152.Cho J., Won K., Wu D., et al. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coronary Artery Disease. 2007;18(3):215–220. doi: 10.1097/01.mca.0000236285.71683.b6. [DOI] [PubMed] [Google Scholar]
- 153.Szeto H. H. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxidants & Redox Signaling. 2008;10(3):601–619. doi: 10.1089/ars.2007.1892. [DOI] [PubMed] [Google Scholar]
- 154.Kelso G. F., Maroz A., Cochemé H. M., et al. A mitochondria-targeted macrocyclic Mn(II) superoxide dismutase mimetic. Chemistry & Biology. 2012;19(10):1237–1246. doi: 10.1016/j.chembiol.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 155.Smith R. A. J., Porteous C. M., Coulter C. V., Murphy M. P. Selective targeting of an antioxidant to mitochondria. European Journal of Biochemistry. 1999;263(3):709–716. doi: 10.1046/j.1432-1327.1999.00543.x. [DOI] [PubMed] [Google Scholar]
- 156.Kelso G. F., Porteous C. M., Coulter C. V., et al. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. The Journal of Biological Chemistry. 2001;276(7):4588–4596. doi: 10.1074/jbc.m009093200. [DOI] [PubMed] [Google Scholar]
- 157.Brown S. E., Ross M. F., Sanjuan-Pla A., Manas A.-R. B., Smith R. A. J., Murphy M. P. Targeting lipoic acid to mitochondria: synthesis and characterization of a triphenylphosphonium-conjugated α-lipoyl derivative. Free Radical Biology and Medicine. 2007;42(12):1766–1780. doi: 10.1016/j.freeradbiomed.2007.02.033. [DOI] [PubMed] [Google Scholar]
- 158.Sickmann A., Reinders J., Wagner Y., et al. The proteome of Saccharomyces cerevisiae mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(23):13207–13212. doi: 10.1073/pnas.2135385100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nair U., Klionsky D. J. Molecular mechanisms and regulation of specific and nonspecific autophagy pathways in yeast. The Journal of Biological Chemistry. 2005;280(51):41785–41788. doi: 10.1074/jbc.r500016200. [DOI] [PubMed] [Google Scholar]
- 160.Okamoto K. Organellophagy: eliminating cellular building blocks via selective autophagy. The Journal of Cell Biology. 2014;205(4):435–445. doi: 10.1083/jcb.201402054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wang K., Klionsky D. J. Mitochondria removal by autophagy. Autophagy. 2011;7(3):297–300. doi: 10.4161/auto.7.3.14502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Moyzis A. G., Sadoshima J., Gustafsson Å. B. Mending a broken heart: the role of mitophagy in cardioprotection. The American Journal of Physiology—Heart and Circulatory Physiology. 2015;308(3):H183–H192. doi: 10.1152/ajpheart.00708.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nakai A., Yamaguchi O., Takeda T., et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nature Medicine. 2007;13(5):619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 164.Linton P.-J., Gurney M., Sengstock D., Mentzer R. M., Jr., Gottlieb R. A. This old heart: cardiac aging and autophagy. Journal of Molecular and Cellular Cardiology. 2015;83:44–54. doi: 10.1016/j.yjmcc.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sybers H. D., Ingwall J., DeLuca M. Autophagy in cardiac myocytes. Recent Advances in Studies on Cardiac Structure and Metabolism. 1976;26-29(12):453–463. [PubMed] [Google Scholar]
- 166.Yan L., Vatner D. E., Kim S.-J., et al. Autophagy in chronically ischemic myocardium. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(39):13807–13812. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hamacher-Brady A., Brady N. R., Logue S. E., et al. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death and Differentiation. 2007;14(1):146–157. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- 168.Huang C., Yitzhaki S., Perry C. N., et al. Autophagy induced by ischemic preconditioning is essential for cardioprotection. Journal of Cardiovascular Translational Research. 2010;3(4):365–373. doi: 10.1007/s12265-010-9189-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Valentim L., Laurence K. M., Townsend P. A., et al. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. Journal of Molecular and Cellular Cardiology. 2006;40(6):846–852. doi: 10.1016/j.yjmcc.2006.03.428. [DOI] [PubMed] [Google Scholar]
- 170.Matsui Y., Takagi H., Qu X., et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and beclin 1 in mediating autophagy. Circulation Research. 2007;100(6):914–922. doi: 10.1161/01.res.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 171.Wang L.-T., Chen B.-L., Wu C.-T., Huang K.-H., Chiang C.-K., Liu S. H. Protective role of AMP-activated protein kinase-evoked autophagy on an in vitro model of ischemia/reperfusion-induced renal tubular cell injury. PLoS ONE. 2013;8(11) doi: 10.1371/journal.pone.0079814.e79814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Guo R., Ren J. Deficiency in AMPK attenuates ethanol-induced cardiac contractile dysfunction through inhibition of autophagosome formation. Cardiovascular Research. 2012;94(3):480–491. doi: 10.1093/cvr/cvs127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Paiva M. A., Rutter-Locher Z., Gonçalves L. M., et al. Enhancing AMPK activation during ischemia protects the diabetic heart against reperfusion injury. The American Journal of Physiology—Heart and Circulatory Physiology. 2011;300(6):H2123–H2134. doi: 10.1152/ajpheart.00707.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Huang C., Liu W., Perry C. N., et al. Autophagy and protein kinase C are required for cardioprotection by sulfaphenazole. The American Journal of Physiology—Heart and Circulatory Physiology. 2010;298(2):H570–H579. doi: 10.1152/ajpheart.00716.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Decker R. S., Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. The American Journal of Pathology. 1980;98(2):425–444. [PMC free article] [PubMed] [Google Scholar]
- 176.Ma X., Liu H., Foyil S. R., et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125(25):3170–3181. doi: 10.1161/circulationaha.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chu C. T., Ji J., Dagda R. K., et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nature Cell Biology. 2013;15(10):1197–1205. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chu C. T., Bayir H., Kagan V. E. LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: implications for Parkinson disease. Autophagy. 2014;10(2):376–378. doi: 10.4161/auto.27191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wang L., Liu X., Nie J., et al. ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to steatosis. Hepatology. 2015;61(2):486–496. doi: 10.1002/hep.27420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Huang W., Choi W., Hu W., et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Research. 2012;22(3):473–489. doi: 10.1038/cr.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mellor H., Parker P. J. The extended protein kinase C superfamily. Biochemical Journal. 1998;332, part 2:281–292. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Marso S. P., Stern D. M. Diabetes and Cardiovascular Disease: Integrating Science and Clinical Medicine. Lippincott Williams & Wilkins; 2003. [Google Scholar]
- 183.Meldrum D. R., Cleveland J. C., Jr., Meng X., et al. Protein kinase C isoform diversity in preconditioning. Journal of Surgical Research. 1997;69(1):183–187. doi: 10.1006/jsre.1997.5072. [DOI] [PubMed] [Google Scholar]
- 184.Munakata M., Stamm C., Friehs I., et al. Protective effects of protein kinase C during myocardial ischemia require activation of phosphatidyl-inositol specific phospholipase C. Annals of Thoracic Surgery. 2002;73(4):1236–1245. doi: 10.1016/S0003-4975(01)03594-9. [DOI] [PubMed] [Google Scholar]
- 185.Jiang W., Bian L., Ma L.-J., Tang R.-Z., Xun S., He Y.-W. Hyperthermia-induced apoptosis in Tca8113 cells is inhibited by heat shock protein 27 through blocking phospholipid scramblase 3 phosphorylation. International Journal of Hyperthermia. 2010;26(6):523–537. doi: 10.3109/02656731003793393. [DOI] [PubMed] [Google Scholar]
- 186.He Y., Liu J., Grossman D., et al. Phosphorylation of mitochondrial phospholipid scramblase 3 by protein kinase C-δ induces its activation and facilitates mitochondrial targeting of tBid. Journal of Cellular Biochemistry. 2007;101(5):1210–1221. doi: 10.1002/jcb.21243. [DOI] [PubMed] [Google Scholar]
- 187.Kowalczyk J. E., Beresewicz M., Gajkowska B., Zabłocka B. Association of protein kinase C delta and phospholipid scramblase 3 in hippocampal mitochondria correlates with neuronal vulnerability to brain ischemia. Neurochemistry International. 2009;55(1–3):157–163. doi: 10.1016/j.neuint.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 188.Zhong Q., Li G., Gvozdenovic-Jeremic J., Greenberg M. L. Up-regulation of the cell integrity pathway in Saccharomyces cerevisiae suppresses temperature sensitivity of the pgs1Δ mutant. Journal of Biological Chemistry. 2007;282(22):15946–15953. doi: 10.1074/jbc.m701055200. [DOI] [PubMed] [Google Scholar]
- 189.Nomoto S., Watanabe Y., Ninomiya-Tsuji J., et al. Functional analyses of mammalian protein kinase C isozymes in budding yeast and mammalian fibroblasts. Genes to Cells. 1997;2(10):601–614. doi: 10.1046/j.1365-2443.1997.1470346.x. [DOI] [PubMed] [Google Scholar]
- 190.Korytowski W., Basova L. V., Pilat A., Kernstock R. M., Girotti A. W. Permeabilization of the mitochondrial outer membrane by bax/truncated Bid (tBid) proteins as sensitized by cardiolipin hydroperoxide translocation: mechanistic implications for the intrinsic pathway of oxidative apoptosis. The Journal of Biological Chemistry. 2011;286(30):26334–26343. doi: 10.1074/jbc.m110.188516. [DOI] [PMC free article] [PubMed] [Google Scholar]