

Abstract
We discuss how principles of nuclear architecture drive typical gene rearrangements in B lymphocytes, whereas translocation hotspots and recurrent lesions reflect the extent of AID-mediated DNA damage and selection.
Chromosome rearrangements are essential for human development. At immunoglobulin (Ig) gene loci in bone marrow B cells, the recombination activating gene (RAG) proteins mediate joining of V, D, and J segments to create a diverse array of antibodies. In the periphery, the activation-induced cytidine deaminase AID initiates class switch recombination (CSR) of Igh constant domains, leading to different antibody isotypes. These tightly regulated processes lie at the heart of the adaptive immune response.
Non-targeted rearrangements are also important for cellular homeostasis and genomic integrity. Spontaneous DNA double-strand breaks (DSBs) are usually repaired in cis by non-homologous end joining (NHEJ), or in trans by homologous recombination (HR). Although largely beneficial, these processes occasionally generate cancer-causing translocations that juxtapose oncogenes (e.g. myc) to potent Ig enhancers.
We review three principles of nuclear architecture that influence patterns of recombination in B cells: polymer folding, looping between convergent CTCF motifs, and A–B compartmentalization. While Ig gene recombination evolved to exploit nuclear architecture, DNA damage and selection drive the recurrent, pathological rearrangements in B cell tumorigenesis.
The folding of chromatin polymers and its role in CSR
A simple estimate for the rearrangement frequency between two loci, A and B, is the probability that they are in close spatial proximity within the cell nucleus. In the absence of local folding features (such as loops), this probability ought to decline monotonically as A and B are positioned further apart along the contour of the genome. Such a decline is readily evident using Hi-C experiments (Lieberman-Aiden et al., 2009). The decline passes through multiple scaling regimes. For inter-locus distances between 500kb and 7Mb, early Hi-C experiments showed that the frequency of contact is related to the distance by a power law with an exponent of approximately −1.0.
Polymer theory provides a rationale for these observations. In such studies, theoretical and physical simulations of condensed chromatin are used to deduce the relationship between 1D proximity-in-sequence and 3D proximity-in-space. Presently, the most commonly employed model is the fractal globule, which predicts that the frequency of contact (or recombination) between two loci scales roughly as the reciprocal of the distance between them (Lieberman-Aiden et al., 2009). The resulting predictions closely match the empirical values obtained by Hi-C.
Many studies have confirmed the power of polymer models in predicting recombination profiles. The simplest prediction, that most DSBs ought to be resolved in cis, was confirmed by studies in which breaks were induced by RAGs or by ionizing radiation. More quantitative predictions have been made by using next-generation sequencing to map rearrangements. These methods rely on the insertion of restriction sites for I-SceI, a yeast homing endonuclease with a rare, 18bp long motif, at loci of interest such as Igh and myc. By transducing activated B cells with I-SceI, DSBs form at the targeted loci, and the resulting rearrangements can be profiled in a high-throughput fashion (Chiarle et al., 2011; Klein et al., 2011). The monotonic declines in I-SceI recombination frequency were characterized by power law scalings (−1.3) resembling those estimated from Hi-C and polymer modeling. Thus, mammalian genomes rearrange in cis with a profile that mimics the polymer behavior of chromatin. (Note that recent improvements in Hi-C maps are likely to lead to significant improvements in the underlying polymer models.)
CSR clearly benefits from this propensity. During CSR, activated B cells replace the IgM constant domain (Cμ) with that of a downstream isotype (Cγ, Cα, or Cε). In the mouse, CHs are confined to a relatively small region (160Kb) within the vast Igh locus (2.8Mb, Figure 1A). Recombination is facilitated by transcription of switch (S) regions upstream of each CH domain, which imparts accessibility to AID and leads to DSBs. In experiments where switch regions were replaced by I-SceI sites (Gostissa et al., 2014; Zarrin et al., 2007), the induction of I-SceI breaks promoted CSR. Proximal I-SceI breaks recombined at higher frequencies than distant ones, consistent with the idea that, at least in part, the monotonic decline of Igh interactions influences these rearrangements. Thus, CSR appears to exploit the polymer behavior of chromosomes, bringing about recombination events between switch regions through the repair of DSBs that come into spatial proximity.
Figure 1. Patterns of rearrangement often reflect principles of nuclear architecture.
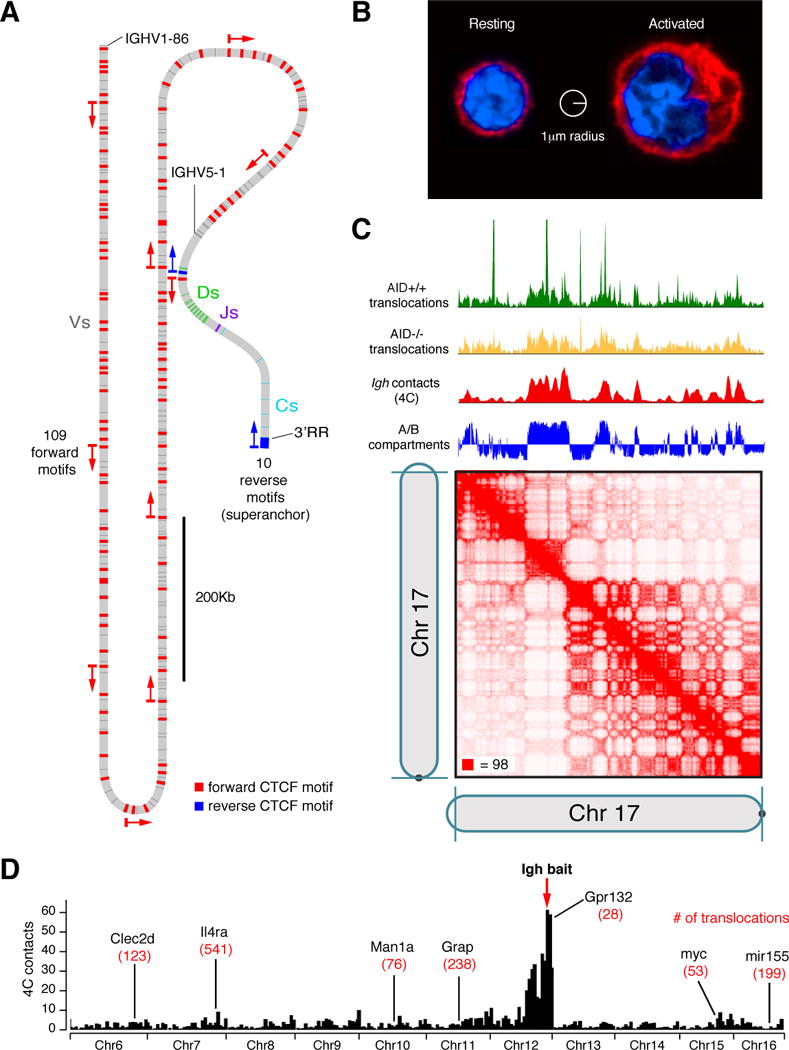
(A) Distribution of CTCF binding motifs (red and blue stripes) at the Igh locus. The position of each site is derived from a list of ChIP-Seq peaks (Choi et al., 2013). The raw signal was weak at several annotated peaks, which were therefore removed by hand. Arrows and colors denote the orientation of the CTCF motifs (forward=red, reverse=blue). The position of CTCF sites and V, D, and J segments in the locus is drawn to scale. Strikingly, the V region contains 109 CTCF motifs, all in the forward orientation. These all face a single reverse motif near the D region. The figure shows an example of a possible loop between a forward CTCF motif and the reverse motif. Note that, in vivo, such looping would be expected occur in the presence of a rearranged DJH, but the locus is shown in the unrearranged state, reflecting the state of most cells during the actual experiment. Notably, a superanchor comprising 10 CTCF motifs in the reverse orientation lies the 3′RR enhancer; these motifs face a single forward CTCF, also at the D locus. (B) Confocal micrograph comparing a resting to a 72h-activated B cell. Samples were stained with anti-α-tubulin (red) and DAPI (blue). Nuclei are much larger after activation. (C) Correlation between A–B compartmentalization (defined by Hi-C eigenvector, blue), interactions with Igh as measured by 4C (red), incidental chromosomal translocations involving Igh and chromosome 17 in the absence of AID (yellow) and in its presence (green). In AID+/+ cells, recurrent hotspots translocation are seen. A Hi-C map is also shown, at 250Kb resolution. Data derived from (Hakim et al., 2012) and (Rao et al., 2014). (D) Igh 4C-Seq profile. Seven (of 236) AID target genes, including Il4ra and Gpr132, are highlighted. The total number of Igh translocations at each gene, as defined by TC-Seq (Klein et al., 2011) is shown.
Long-range CTCF looping facilitates V(D)J recombination and ensures antibody diversity
V(D)J recombination occurs over a broader range of distances than CSR. The first recombination event joins D and JH segments separated by a maximum distance of 100Kb (in the mouse genome, Figure 1A). In contrast, VH-DJH recombination deletes at least 45–150Kb for the most proximal V segment (IghV5-1) and up to ~2.6Mb for the most distal one (IghV1-86, Figure 1A). Thus, if VH-DJH recombination relied exclusively on the monotonic behavior of chromatin polymers, proximal VH segments would dominate the mature Ig repertoire, drastically curtailing antibody diversity. However, microscopy studies have shown that the entire Igh locus undergoes conformational changes during V(D)J recombination (Jhunjhunwala et al., 2008). In particular, the entire VH domain associates with D-JH segments in cells undergoing VH-DJH recombination (reviewed by (Subrahmanyam and Sen, 2012)). Clearly, mechanisms beyond local polymer folding are at work.
A recent Hi-C map of human B lymphoblastoid cells shed light on these mechanisms. The study revealed ~10,000 loops between pairs of CTCF sites whose binding motifs lie in the convergent orientation (i.e. facing one another, (Rao et al., 2014)). The Igh locus is a dramatic illustration of this principle. Its VH region contains >100 CTCF sites, all pointing downstream. They face a single CTCF site, pointing upstream, that is situated at the 5′ end of the D region. (Figure 1A). This configuration is consistent with a model in which CTCF looping facilitates recombination between distant VHs and rearranged DJHs. (Similar configurations are seen at other Ig loci.) In support of this, deletion of CTCF binding sites at the D region decreased recombination with distal VHs (Guo et al., 2011; Lin et al., 2015). Thus, V(D)J recombination may be distinct from CSR insofar as it relies on CTCF-mediated looping rather than polymer folding alone. Besides CTCF, other factors have also been implicated in Igh locus contraction, including E2A, Pax5, cohesin, Brg1, and YY-1 (reviewed by (Alt et al., 2013)). How these factors complement CTCF-mediated looping remains unclear.
A–B compartmentalization shapes incidental rearrangements
The DSBs that form during Ig gene recombination are not always properly repaired in cis, and can instead lead to incidental rearrangements, both in cis and especially in trans. In B cell tumors from humans and other species, chromosomal translocations frequently join oncogenes to potent Ig enhancers that deregulate their expression. The origin of these translocations has been debated for several decades; proximity, patterns of DSB formation, and selection have all been proposed as the driving mechanism.
At first, the relationship between nuclear architecture and translocations was explored using microscopy. These studies tested the hypothesis that oncogenes and Ig loci translocate frequently because they preferentially associate in B cell nuclei. Fluorescence in-situ hybridization (FISH) showed that Igh and myc are within 1μm of each other in a fraction of activated B cells, but the overlap frequency varied greatly across studies. In reports where B cells were analyzed shortly after activation (<15 minutes), Igh and myc appeared to overlap in more than 20% of lymphocytes (Osborne et al., 2007), while at later stages of activation (72h), or in germinal center B cells, values were in the 3–5% range (Gramlich et al., 2012; Wang et al., 2009). These experiments were interpreted to suggest that, at least under some conditions, Igh and myc preferentially associate in B cells.
However, the studies did not consider the frequency at which gene loci overlap at random. For 3D-FISH in a diploid nucleus using a 1μm overlap threshold, the random overlap frequency is given by the volume of two spheres of radius 1μm (~8.4μm3) divided by the total volume of the nucleus. For B cells at rest or shortly after activation (15 minutes), a volume of 55μm3 is typical (Figure 1B), implying that two genes overlap at random in 15% of the cells. In contrast, the average volume of activated B cells >24h post-activation is ~250μm3 (Figure 1B), resulting in a random overlap frequency of roughly 3.5%. (For 2D FISH, the probabilities are 14% before activation and 5% afterwards.) These considerations suggest that the reported differences in Igh-myc overlap may be explained by differences in nuclear volume, rather than by preferential association.
Recent contact mapping experiments have provided more definitive results. In particular, kilobase resolution Hi-C mapping did not reveal peaks of contact frequency between pairs of loci lying in trans (Rao et al., 2014). Similarly, a 4C study found that Igh and myc do not form preferential locus-specific associations in trans (Hakim et al., 2012). Crucially, the observation that there are no biases in trans between individual gene loci does not imply that there are no biases in trans whatsoever. The existence of chromosome territories, and the preferential association between them, is well known; as is the fact that A–B compartmentalization of chromatin leads to spatial segregation of open and closed genomic domains (Lieberman-Aiden et al., 2009; (de Laat and Grosveld, 2007). Consequently, Igh and myc, located on chromosomes 12 and 15 respectively, spatially associate with transcriptionally active domains genome-wide at similar frequencies (Figure 1C, (Hakim et al., 2012)). Consistent with this view, high-throughput FISH experiments show that the frequency of overlap (<1μm) in B cells between Igh and a typical active gene is 3–5%, whereas the frequency of overlap between Igh and any silent locus is 1–2% (Hakim et al., 2012).
These broad patterns of spatial proximity influence rearrangement patterns. In AID−/− B cells or in irradiated pro-B cells – where the distribution of DNA breaks is relatively uniform across the genome – translocation patterns reflect both chromosome territories and A–B compartmentalization (Hakim et al., 2012). Presumably, higher resolution rearrangement data would reflect the 6 chromatin subcompartments (A1/A2 and B1/B2/B3/B4) that subdivide the original A and B compartments, and which are seen in the highest resolution Hi-C maps (Rao et al., 2014). Thus, in the absence of recurrent DNA damage, incidental rearrangements – those that do not arise due to targeted mechanisms such as VDJ recombination – mostly follow the broad contours of nuclear compartmentalization.
Patterns of DNA Damage and selective pressure shape patterns of recurrent rearrangements
The findings discussed so far highlight the significant influence of nuclear architecture on patterns of rearrangement. Yet architecture is not the only factor that influences rearrangement. Another key feature is the frequency of DNA DSBs at particular loci. In the presence of DNA damaging enzymes such as RAGs and AID, the rate of formation of such lesions is not constant across the genome, but is instead targeted to specific sites. In cancer, yet another factor must be considered: tumorigenesis may select for or against particular rearrangements.
Hotspots of AID-mediated DNA damage become hotspots of rearrangement
A primary role of AID-mediated deamination is to promote the formation of DSBs during CSR. However, AID is also a significant driver of rearrangement hotspots. This tendency has been clearly observed in activated and germinal center B cells. In addition to the rearrangements seen in the AID−/− background, activated B cells with intact AID exhibit translocation hotspots at myc and other oncogenes implicated in B cell transformation. By monitoring the accumulation of the repair factors RPA and Rad51 at resected DNA breaks, these hotspots were confirmed to be sites of recurrent AID-induced DNA damage (Hakim et al., 2012). Indeed, there is a direct proportionality between the extent of AID-mediated DSB formation at hotspots and the absolute number of Igh translocations. These translocation frequencies dramatically exceed predictions based on nuclear architecture alone. For instance, Igh is 20-fold more likely to spatially co-locate with Gpr132, which lies 400kb away on chromosome 12, than with Il4ra, which lies, in trans, on chromosome 7 (Figure 1D). But Igh is 20-fold more likely to rearrange with Il4ra than with Gpr132. Similarly, myc is not particularly likely to spatially co-locate with Igh: over 2,000 genes are more likely to do so. Yet myc is one of a relatively small fraction of genes that recurrently rearrange to Igh (Hakim et al., 2012).
Thus, patterns of AID-mediated DSBs are a principal factor in determining sites of recurrent translocation in B cells, irrespective of topology. This also applies to translocations induced by other forms of recurrent DNA damage, such as Rag1/2 activity or the CRISPR-Cas9 system (Frock et al., 2015). These arguments highlight the need to understand the factors that make loci vulnerable to each damage modality. In the case of AID, several proposals have been made, including the presence of super-enhancers, convergent gene transcription, and high interconnectivity between regulatory elements (reviewed by (Alinikula and Schatz, 2014)).
Architecture constrains selection of cancer-causing rearrangements
In tumors, the rate at which a particular rearrangement is found does not reflect its rate of formation in the tumor’s cell of origin. Instead, there is strong positive selection for incidental translocations that deregulate oncogene expression. For instance, Il4ra translocations form 10-fold more frequently than myc translocations in activated B cells (Figure 1D), but – unlike myc – Il4ra translocations are yet to be reported in B cell tumors.
In Ig translocations, oncogene deregulation tends to be the result of the potent Igκ, Igλ, or Igh enhancers. Because these enhancers can work at a distance, they are believed to rely on spatial proximity to drive their biological function. Yet this same architecture imposes constraints on their activity. For instance, when murine B cells lacking AID-mediated damage were cultured under non-selective conditions, incidental translocations placed the Igh 3′ enhancer, known as 3′RR, at a wide range of distances from myc (Kovalchuk et al., 2012). Following selection (during mouse plasmacytomagenesis), the same element was found no more than 500kb from myc. Beyond this point, no local changes were observed in the level of PolII recruitment, gene expression, or in the presence of various epigenetic modifications. The ability of 3′RR to activate translocated genes that lie far away along the contour of the genome has also been observed in other mature B cell tumors (Gostissa et al., 2009). Given the above-noted findings about CTCF orientation, it is likely that the transformational potency of the 3′RR enhancer is related to the fact that it lies adjacent to what we dub a CTCF “superanchor”: a series of 10 CTCF sites all of which point in the same direction. The key implication is that the functional reach of 3′RR and its superanchor – at least in the setting of myc translocations – is approximately half a megabase.
Thus, nuclear architecture not only influences the location and frequency of rearrangements, but it also influences which of these events are selectively favored during transformation. This observation may have important implications for tumors that are not derived from B and T cells and that typically lack targeted, recurrent DNA damage.
In the past 5 years, technological advances have helped unravel many longstanding issues relating to both physiological and pathological rearrangement in B lymphocytes. As the field of 3D genomics advances, we anticipate that it will continue to illuminate these mechanisms and their implications for tumorigenesis, both within the immune system and beyond.
Acknowledgments
We thank Wouter de Laat, Neva Durand, Suhas Rao, and Adrian Sanborn for helpful discussions, and Wolfgang Resch, Marei Dose, Suhas Rao, and Sigrid Knemeyer for assistance in designing the figure.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alinikula J, Schatz DG. Cell. 2014;159:1490–1492. doi: 10.1016/j.cell.2014.12.007. [DOI] [PubMed] [Google Scholar]
- Alt FW, Zhang Y, Meng FL, Guo C, Schwer B. Cell. 2013;152:417–429. doi: 10.1016/j.cell.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, et al. Cell. 2011;147:107–119. doi: 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi NM, Loguercio S, Verma-Gaur J, Degner SC, Torkamani A, Su AI, Oltz EM, Artyomov M, Feeney AJ. J Immunol. 2013;191:2393–2402. doi: 10.4049/jimmunol.1301279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Laat W, Grosveld F. Curr Opin Genet Dev. 2007;17:456–464. doi: 10.1016/j.gde.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Frock RL, Hu J, Meyers RM, Ho YJ, Kii E, Alt FW. Nat Biotechnol. 2015;33:179–186. doi: 10.1038/nbt.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gostissa M, Schwer B, Chang A, Dong J, Meyers RM, Marecki GT, Choi VW, Chiarle R, Zarrin AA, Alt FW. Proc Natl Acad Sci U S A. 2014;111:2644–2649. doi: 10.1073/pnas.1324176111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gostissa M, Yan CT, Bianco JM, Cogne M, Pinaud E, Alt FW. Nature. 2009;462:803–807. doi: 10.1038/nature08633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramlich HS, Reisbig T, Schatz DG. PLoS One. 2012;7:e39601. doi: 10.1371/journal.pone.0039601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Yoon HS, Franklin A, Jain S, Ebert A, Cheng HL, Hansen E, Despo O, Bossen C, Vettermann C, et al. Nature. 2011;477:424–430. doi: 10.1038/nature10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakim O, Resch W, Yamane A, Klein I, Kieffer-Kwon K-R, Jankovic M, Oliveira T, Bothmer A, Voss TC, Ansarah-Sobrinho C, et al. Nature. 2012;484:69–74. doi: 10.1038/nature10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhunjhunwala S, van Zelm MC, Peak MM, Cutchin S, Riblet R, van Dongen JJ, Grosveld FG, Knoch TA, Murre C. Cell. 2008;133:265–279. doi: 10.1016/j.cell.2008.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, Di Virgilio M, Bothmer A, Nussenzweig A, Robbiani DF, et al. Cell. 2011;147:95–106. doi: 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalchuk AL, Ansarah-Sobrinho C, Hakim O, Resch W, Tolarova H, Dubois W, Yamane A, Takizawa M, Klein I, Hager GL, et al. Proc Natl Acad Sci U S A. 2012;109:10972–10977. doi: 10.1073/pnas.1200106109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SG, Guo C, Su A, Zhang Y, Alt FW. Proc Natl Acad Sci U S A. 2015;112:1815–1820. doi: 10.1073/pnas.1424936112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne CS, Chakalova L, Mitchell JA, Horton A, Wood AL, Bolland DJ, Corcoran AE, Fraser P. PLoS Biol. 2007;5:e192. doi: 10.1371/journal.pbio.0050192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subrahmanyam R, Sen R. Curr Top Microbiol Immunol. 2012;356:39–63. doi: 10.1007/82_2011_153. [DOI] [PubMed] [Google Scholar]
- Wang JH, Gostissa M, Yan CT, Goff P, Hickernell T, Hansen E, Difilippantonio S, Wesemann DR, Zarrin AA, Rajewsky K, et al. Nature. 2009;460:231–236. doi: 10.1038/nature08159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrin AA, Del Vecchio C, Tseng E, Gleason M, Zarin P, Tian M, Alt FW. Science. 2007;315:377–381. doi: 10.1126/science.1136386. [DOI] [PubMed] [Google Scholar]