

Abstract
The toxicity of pharmacological ascorbate is mediated by the generation of H2O2 via the oxidation of ascorbate. Since pancreatic cancer cells are sensitive to H2O2 generated by ascorbate they would also be expected to become sensitized to agents that increase oxidative damage such as ionizing radiation. The current study demonstrates that pharmacological ascorbate enhances the cytotoxic effects of ionizing radiation as seen by decreased cell viability and clonogenic survival in all pancreatic cancer cell lines examined, but not in non-tumorigenic pancreatic ductal epithelial cells. Ascorbate radiosensitization was associated with an increase in oxidative stress-induced DNA damage, which was reversed by catalase. In mice with established heterotopic and orthotopic pancreatic tumor xenografts, pharmacological ascorbate combined with ionizing radiation decreased tumor growth and increased survival, without damaging the gastrointestinal tract or increasing systemic changes in parameters indicative of oxidative stress. Our results demonstrate the potential clinical utility of pharmacological ascorbate as a radiosensitizer in the treatment of pancreatic cancer.
Introduction
Pharmacological ascorbate induces cytotoxicity and oxidative stress in pancreatic cancer cells, compared to normal cells (1). In the extracellular environment, pharmacological concentrations of ascorbate can oxidize to form hydrogen peroxide (H2O2) (1, 2, 3, 4). H2O2 will diffuse readily across the cell membrane causing oxidative damage to cellular proteins, lipids, and DNA. The generation of H2O2 correlates with the concentration of ascorbate in both a time- and dose-dependent manner. Ascorbate has been shown to decrease viability in all pancreatic cancer cell lines studied, but has no effect on non-tumorigenic pancreatic ductal epithelial cells (1) and cytotoxicity was reversed with scavengers of H2O2. Furthermore, in vivo treatment with pharmacological ascorbate inhibited tumor growth and prolonged survival. Thus, ascorbate has been hypothesized to be a “pro-drug” for formation of H2O2 in pancreatic cancer xenografts (1, 3). Therapeutic interventions designed to increase oxidant stress (such as ionizing radiation) in combination with pharmacological ascorbate would be predicted to preferentially sensitize tumor cells vs. normal cells via metabolic oxidative stress (1, 5).
Ionizing radiation (IR) has long been known to induce DNA damage. In addition to direct damage, IR generates reactive oxygen species (ROS) that can damage proteins, lipids, and DNA, inducing both single- and double-strand DNA breaks (6). Formation of double-strand breaks results in the rapid phosphorylation of histone H2AX (7). Mammalian phosphorylated H2AX (γ-H2AX) is believed to facilitate the recruitment and retention of DNA repair and checkpoint proteins (8, 9). Radiosensitive tumor cells have been shown to retain γ-H2AX for a longer duration after IR than radio-resistant cells.
Pharmacological ascorbate-mediated H2O2 formation also causes DNA damage, which involves transition metal ions such as Fe2+ associated with DNA (10). Fe2+ reacts with H2O2, producing site-specific hydroxyl radical (HO•), damaging DNA bases as well as the sugar/phosphate backbone of DNA (11). The base excision repair pathway is the major system for repair of oxidative-induced DNA damage (12). Thus, DNA damage can be assessed by measuring γ-H2AX which is upregulated in the presence of double-strand breaks.
Because both IR and pharmacological ascorbate initiate DNA damage, we hypothesize that pharmacological ascorbate has potential as a radiosensitizer in pancreatic cancer. Here we demonstrate that pharmacological ascorbate is a selective radio-sensitizer in pancreatic cancer vs. normal non-tumorigenic pancreatic ductal epithelial cells. Pharmacological ascorbate also enhanced IR-induced DNA damage in pancreatic cancer cells as well as enhancing IR-induced inhibition of tumor growth in established human pancreatic xenografts without causing systemic changes in parameters of parameters indicative of oxidative stress or enhancing normal tissue injury to the gut epithelium. These results demonstrate the potential utility of pharmacological ascorbate as an adjuvant to pancreatic cancer radiotherapy.
Methods
Cell culture
MIA PaCa-2, AsPC-1, and PANC-1 human pancreatic adenocarcinoma cells were obtained from American Type Culture Collection (Manassas, VA) and passaged for fewer than six months after receipt. MIA PaCa-2 cells were maintained in Dulbecco’s modified Eagle media (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. AsPC-1 cells were maintained in RPMI 1640 with 10% FBS and 1% penicillin-streptomycin. PANC-1 cells were cultured in DMEM supplemented with 10% FBS. Also, patient-derived pancreatic cancer cell lines (403 and 339s) from the Medical College of Wisconsin surgical oncology tissue bank (13, 14) were cultured in Dulbecco’s Modified Eagle’s Media Nutrient Mixture F-12 with Penicillin/Streptomycin, human recombinant EGF, Bovine Pituitary Extract, hydrocortisone, and human recombinant insulin. In addition to the pancreatic cancer cell lines, we also used the non-tumorigenic HPV16-E6E7 immortalized cell line derived from normal pancreatic ductal epithelium (H6c7) with near normal genotype and phenotype of pancreatic duct epithelial cells (15). H6c7 cells were maintained in keratinocyte serum-free media supplemented with epidermal growth factor (5 ng/mL) and bovine pituitary extract (50 μg/mL). The H6c7 cells were characterized by IDEXX-RADIL. Profile data was not available for comparison purposes for H6c7 cells. However, the genetic profile for H6c7 cells was compared to the cell line genetic profiles available in the DSMZ STR database and did not match any other reported profiles in the DSMZ database. The H6c7 cells alone do not form colonies so feeder cells were used as described (154).
L-ascorbic acid was purchased from Macron Chemicals (Center Valley, PA). Stock solutions of ascorbate (1.0 M, pH 7.0) were made as previously described (1). For ascorbate treatments, cells were placed in fresh media and treated with ascorbate for 1 h at 37 ° C. To determine clonogenic survival, cells were treated, trypsinized, counted, diluted, and plated for clonogenic cell survival assay as previously described (1). Surviving colonies were fixed and stained with Coomassie blue after 10–14 days and counted under an inverted light microscope. As another indicator of cell viability an assay monitoring the reduction of MTT (3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide) was used. Cells were seeded in 60 mm2 dishes at 3 x 105 cells/well and allowed to attach for 24 h. Next, cells were irradiated with cesium-137 source, doses ranging from 0–10 Gy. Immediately prior and during IR, cells were treated with ascorbate (0.25 mM) in DMEM with 10% FBS for 1 h. After treatment, the media were changed, and cells were allowed to recover for 72 h. The media were removed, and cells were then incubated in a solution of MTT, 1 mg/mL (Sigma-Aldrich, St. Louis, MO), dissolved in serum-free DMEM at 37 °C for 3 h in the dark. At the end of the incubation time, the media were removed, and DMSO was added to each plate to dissolve the precipitate. Samples were transferred to a 96-well plate for plate reader analysis and read at 590 nm on a Tecan SpectraFluor Plus plate reader (Tecan, Research Triangle Park, NC).
Catalase treatment
To determine whether H2O2 was responsible for the cytotoxic effects of ascorbate and radiation, cells were treated with various forms of catalase including adenovirus catalase (AdCAT), bovine catalase (100 U/mL) or catalase-polyethylene glycol (PEG-CAT) (200 U/mL). Catalase and PEG-catalase were purchased from Sigma-Aldrich (St. Louis, MO). The AdCAT construct used was a replication-defective, E1- and partial E3 deleted recombinant adenovirus (16). Inserted into the E1 region of the adenovirus genome is the human catalase gene, which is driven by a cytomegalovirus promoter. For the adenovirus experiments approximately 106 cells were plated in 10 mL of complete media in a 100 mm2 tissue culture dish and allowed to attach for 24 h. Cells were then washed 3 times in serum- and antibiotic-free media. The adenovirus constructs were applied to cells in 4 mL of serum-and antibiotic-free media. Control cells were treated with the empty adenovirus (AdEmpty) construct. Cells were incubated with the adenovirus constructs for 24 h. Media was replaced with 4 mL of complete media for an additional 24 h before cells were harvested for Western blot or treated for clonogenic assay.
Cell cycle analysis
Cell viability was measured by flow cytometry using propidium iodide (PI). After treatment, cells were collected by trypsinization, centrifuged at 500 g and resuspended in 500 μL of HBSS. After addition of 5 μL of PI (50 μg/mL), cells were incubated in the dark at room temperature for 5 min. PI fluorescence was analyzed by flow cytometry (excitation at 488 nm, emission at > 550 nm). To analyze alterations in cell cycle by quantitation of DNA content, cells were collected and fixed in suspension with 70% ethanol for 4 h at 4 °C. Cells were washed with 1 mL PBS, centrifuged, and resuspended in 100 μL RNase A (1 mg/mL in PBS). After 30-min incubation at room temperature, 500 μL PI (35 μg/mL in PBS) were added to each sample. After 1-h incubation in the dark at room temperature, PI fluorescence was analyzed by flow cytometry.
Determination of intracellular hydrogen peroxide
Intracellular H2O2 concentrations were determined by analysis of the rate of aminotriazole-mediated inactivation of endogenous catalase activity (17). Catalase is irreversibly inactivated by aminotriazole (3-AT, 3-amino-1,2,4-triazole, Sigma-Aldrich, St. Louis, MO) in the presence of H2O2. Cells grown in 150 mm2 culture dishes were irradiated at 3 Gy and then treated with ascorbate (20 mM) in the presence of 3-AT (20 mM) for 0, 5, 10, 20, 30, 60 and 120 min at 37 °C. Cells were washed with ice-cold PBS, harvested, and lysed by freeze/thaw at −80 °C followed by room temperature. To determine the amount of fully active cellular catalase, cell lysates (2 mL) were introduced into the reaction chamber of an oxygen monitor (YSI model 5300, YSI Inc., Yellow Springs, OH). Then 333 μM of H2O2 was injected into the reaction chamber, and the rate of production of oxygen was continuously recorded for 5 min or until the curve reached a plateau. The rate of appearance of dioxygen reflects the amount of active catalase in the cell lysate. Cell protein was determined by Bio-Rad DC protein assay. The intracellular steady-state concentration of H2O2 was calculated from the equation [H2O2]ss = kinactivation / k1, where kinactivation is the experimental pseudo first-order rate constant of catalase inactivation (s−1) and k1 = 1.7 x 107 M−1 s−1, i.e. the rate constant for the formation of catalase compound I. The rate of extracellular production of H2O2 was determined with a Clark electrode as in (1, 5).
Intracellular GSH and GSSG measurement
Cells grown in 100 mm2 dishes were treated with IR, ascorbate, or in combination. After treatments, cells were washed with PBS and harvested by trypsinization (floating cells in culture media and PBS were collected and combined with trypsinized cells). After centrifugation, cell pellets were suspended in 100 μL PCA buffer (5% perchloric acid with 100 μM DETAPAC (diethylenetriaminepentaacetic acid); Sigma-Aldrich Chemical Co., St. Louis, MO) to precipitate protein. Samples were sonicated and centrifuged at 18,500 g for 5 min at 4 ° C using an Eppendorf Microcentrifuge. Supernatants were collected and stored at −80 ° C or analyzed immediately using HPLC (ESA CoulArray, Dionex/Thermo Scientific, Sunnyvale, CA) with electrochemical detection (ECD) following the protocol described in Park et al. (18). In addition, in separate groups of treated mice, red blood cells (RBCs) were harvested and centrifuged (500 g for 5 min). The plasma was removed from the sample and the remaining pellet of intact RBCs was washed twice with cold isotonic saline. An aliquot was removed for counting by a hemocytometer. From the RBC pellet, 100 μL of RBCs were lysed with 300 μL of PCA buffer and then centrifuged to pellet the protein (500 g, 5 min). The clean supernatant was stored at −80 ° C or immediately analyzed for total glutathione (tGSH) using a plate-reader based assay (19).
Immunoblot analysis
Protein (10–40 μg) was electrophoresed in a 4–20% Bio-Rad ready gel then electrotransferred to an Immobilon PVDF membrane (EMD Millipore, Billerica, MA). Membranes were blocked in 5% nonfat milk for 1 h, then treated with anti-γ-H2AX (Ser 139) antibody (1:1000; EMD Millipore, Billerica, MA), or with anti-catalase antibody (1:5000 dilution; Cell Signaling Technology, Danvers, MA). Horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse (1:50,000; Chemicon International, Temecula, CA) was used as a secondary antibody. Anti-GAPDH (1:1000; EMD Millipore, Billerica, MA) or anti-actin (1:4000; Sigma-Aldrich, St. Louis, MO) were used as a loading control. Blots were treated with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Rockford, IL) and exposed to autoradiography film.
Immunohistochemistry
MIA PaCa-2 cells were seeded in 8-well (0.8 cm2/well) glass Lab-Tek Chamber slides at a density of 30,000 cells/well. After 48 h, cells were treated with ascorbate prior to IR treatment. Cells were fixed in 3% paraformaldehyde and rehydrated in Dulbecco’s PBS (pH 7.4) 30 min after treatment and then incubated in RNAse (0.5 μg/mL) at 37 °C for 30 min, and 10% normal goat serum was applied overnight for blocking. Cells were incubated in anti-γ-H2AX primary antibody (1:200, Calbiochem) for 1.5 h. A goat anti-rabbit antibody conjugated to Alexa Fluor® 488 (1:500) was used as a secondary antibody to fluorescently tag γ-H2AX. Slides were mounted in Vectashield® with DAPI as a nuclear counterstain. Slides were imaged on a Bio-Rad Radiance 2100 confocal multi-photon microscope with laser sharp software. Z-plane images of each treatment were captured, and the intensity of fluorescence was quantified on ImageJ® using a maximum intensity Z-projection with identical threshold values to calculate the mean fluorescence intensity for individual cells.
Animal experiments
Thirty-day-old athymic nude mice were obtained from Harlan Sprague-Dawley (Indianapolis, IN). The nude mice protocol was reviewed and approved by the Animal Care and Use Committee of The University of Iowa. The animals were housed four to a cage and fed a sterile commercial stock diet and tap water, ad libitum. Animals were allowed to acclimate in the unit for one week before any manipulations were performed. Each experimental group consisted of nine to twelve mice. MIA PaCa-2 or PANC-1 human pancreatic tumor cells (2 x 106) were delivered subcutaneously into the flank region of nude mice with a 1-mL tuberculin syringe equipped with a 25-gauge needle. The tumors were allowed to grow until they reached between 3 mm and 4 mm in greatest dimension (2 weeks), at which time the mice were randomized and treatment was initiated. This was defined as day 1 of the experiment. Before IR, the animals were anesthetized with 80–100 mg/kg ketamine and 10 mg/kg xylazine i.p. and shielded in a lead block such that only the tumor-bearing right hind flank was irradiated. Tumor size was measured twice a week using a digital caliper, and tumor volume was estimated according to the following formula: tumor volume = π/6 x L x W2, where L is the greatest dimension of the tumor, and W is the dimension of the tumor in the perpendicular direction (20). Animals were euthanized by CO2 asphyxiation when the tumors reached a predetermined size of 1000 mm3. In the orthotopic pancreatic cancer model, 6-week-old athymic nude mice were injected with the human pancreatic cancer cell line MIA PaCa-2, which had previously been stably transfected with firefly luciferase. Ultrasound guidance was used to identify the pancreas as described (21) and 400,000 cells suspended in 20 μL of a 1:1 mixture of PBS and matrigel were injected into the pancreas under direct visualization. Bioluminescence-imaging microscopy was used to identify mice with established tumors within the pancreas and tumor-containing mice were randomly assigned and treatment was initiated. Tumor growth was monitored using bioluminescence-imaging microscopy to determine the average radiance (photons/s−1/cm2) of the tumors.
In separate groups of treated animals, mouse tumors were excised and processed as described to determine 4-hydroxy-2-nonenal-(4HNE)-modified proteins (22). Briefly, tissues were homogenized and 25 μg of protein was blotted onto PVDF membranes. Blots were incubated with the primary antibody recognizing the Michael addition product of 4HNE-modified cellular proteins (23) diluted 1:2,000 overnight at 4 °C, followed by 2 h in secondary antibody, HRP-conjugated goat anti-rabbit polyclonal antibody (1:20,000), and chemiluminescence detection with X-ray film. Immunoreactive protein on the dot blot was analyzed using integrated densities determined with ImageJ® software.
In separate groups of mice, a crypt cell assay was performed. Mice were exposed to 10 Gy or 13 Gy of total abdominal radiation with and without ascorbate (4 g/kg/d). After 48 h, each animal was sacrificed and sections were made of the jejunum and then viewed under light microscopy and expressed as surviving cells/circumference. Additionally, blood samples for TNF-α were collected into 4.0 mL heparinized vial from control mice and mice treated with radiation (13 Gy) and/or ascorbate (4g/kg). Samples were centrifuged at 200 g for 20 min. Serum samples were collected from each tube and stored at −80 °C. TNF-α levels in the serum were measured using a Quantikine ELISA kit (RnDSystem, Minneapolis, MN).
Statistical analysis
A single factor ANOVA followed by Tukey’s post-hoc test was used to determine statistical differences between means for multiple comparisons, or a student’s t-test was used when only 2 comparisons. All means were calculated from three experiments, with error bars representing the standard error of the mean (SEM). All Western blots were repeated at least twice. All data are expressed as means ± SEM. A dose-modifying factor (DMF) was calculated for each cell line based on clonogenic surviving fraction. For the in vivo studies, the statistical analyses focused on the effects of different treatments on tumor progression. The primary outcomes of interest were time to death and tumor growth over time. The log-rank test was used to compare the survival times between treatment groups. Kaplan-Meier survival plots were constructed to estimate survival. Linear mixed effects regression models were used to estimate and compare group-specific tumor growth curves. Tests of statistical significance were two-sided and performed using the Systat (Evanston, IL) and SAS (Cary, NC) software.
Results
Ascorbate enhances IR-induced cytotoxicity
Previous studies from our laboratory have demonstrated that pharmacological ascorbate is cytotoxic to pancreatic cancer cells while normal cells are resistant (1). To test the effect of ascorbate on radiation response in cancer vs. non-cancerous cell lines, we performed clonogenic (Figure 1A – D) and cell viability assays (Figure 1E, F) using three pancreatic ductal adenocarcinomas (MIA PaCa-2, PANC-1 (Figure A, B) and AsPC-1 (Figure 1C), compared to normal non-tumorigenic H6c7 pancreatic ductal epithelial cells (Figure 1D). The radiation survival curves were normalized to ascorbate- or sham-treated controls and fit with a linear quadratic model. Pharmacological ascorbate (0.25 mM) enhanced IR-induced decreases in clonogenic survival. The pancreatic cancer cell lines showed dose modification factors at 10% iso-survival of 2.5 (MIA PaCa-2) and 2.2 (PANC-1) (Figure 1A, B) and 1.25 (AsPC-1, Figure 1C). In contrast no radio-sensitization was noted with ascorbate in the normal non-tumorigenic H6c7 pancreatic ductal epithelial cells (Figure 1D). Similar results were seen using cell viability where there was radiosensitization in the MIA PaCa-2 cell line (Figure 1E) but no radiosensitization in the H6c7 cell line (Figure 1F). To determine if ascorbate radiosensitization occurs in the physiologically relevant environment seen in pancreatic cancer, we treated patient derived pancreatic cancer cell lines, 339 (Figure 1G) and 403 (Figure 1H) (13, 14) in 4% O2. Again we demonstrated ascorbate radiosensitization in the 339 line with a DMF of 1.4 at 40% iso-survival and a DMF of 1.6 in the 403 cell line at 40% iso-survival. The timing for the administration of pharmacological ascorbate is also important. Treating cells with ascorbate for 1 h prior to IR or 1 h immediately after IR demonstrated a similar decrease in clonogenic survival when compared to IR or ascorbate alone. However, when ascorbate was administered 6 h after IR, the decrease in clonogenic survival was decreased (Supplemental Information Figure 1). These data strongly support the hypothesis that pharmacological ascorbate is a selective radio-sensitizer in pancreatic cancer cells vs. normal non-tumorigenic pancreatic ductal epithelial cells.
Figure 1. Ascorbate radiosensitizes pancreatic cancer cells.

A. MIA PaCa-2 pancreatic cancer cells were irradiated (0 – 3 Gy) with and without ascorbate (0.25 mM) and clonogenic survival determined. The resulting dose modification factor at 10% iso-survival was 2.5 indicating enhanced radiosensitivity (Means ± SEM, n = 3). Pharmacological ascorbate (0.25 mM) produces a flux of H2O2 of 550 amol−1 cell−1 s−1 under our experimental conditions. This rate is considerably greater than the rate of cellular oxygen consumption by these cells, 57 amol−1 cell−1 s−1 (29).
B. PANC-1 pancreatic cancer cells were irradiated (0–10 Gy) and treated with ascorbate (0.25 mM) in a similar fashion. Clonogenic survival yielded a DMF of 2.2 indicating enhanced radiosensitivity. (Means ± SEM, n = 3).
C. AsPC-1 human pancreatic cancer cells were irradiated (0 – 10 Gy) with and without ascorbate (0.25 mM) and clonogenic survival determined. The resulting dose modification factor at 10% iso-survival was 1.25 indicating enhanced radiosensitivity. (Means ± SEM, n = 3).
D. H6c7 immortalized human pancreatic ductal epithelial cells were irradiated (0–6 Gy) and treated with ascorbate (0.25 mM) to determine clonogenic survival. The clonogenic survival assay demonstrates no radiosensitization after IR with or without ascorbate. (Means ± SEM, n = 3).
E. MIA PaCa-2 pancreatic cancer cells were irradiated (0–4 Gy) and treated with ascorbate (0.25 mM) to determine viability using the MTT assay and demonstrated radiosensitization. Data were normalized to drug or vehicle control. (Means ± SEM, n = 3).
F. H6c7 immortalized human pancreatic ductal epithelial cells were irradiated (0–10 Gy) and treated with ascorbate (0.25 mM) to determine cell viability. The MTT assay demonstrates minimal changes in cell viability after IR with or without ascorbate. (Means ± SEM, n = 3).
G. Patient derived pancreatic cancer cells (339) in 4% O2 were irradiated (0 – 6 Gy) with and without ascorbate (0.5 mM) and clonogenic survival determined. The resulting dose modification factor at 40% iso-survival was 1.4 indicating enhanced radiosensitivity. (Means ± SEM, n =3).
H. Patient derived pancreatic cancer cells (403) in 4% O2 were irradiated (0 – 6 Gy) with and without ascorbate (0.5 mM) and clonogenic survival determined. The resulting dose modification factor at 40% iso-survival was 1.6 indicating enhanced radiosensitivity. (Means ± SEM, n =3).
Cell cycle analysis of ascorbate-induced radiosensitization
Hydrogen peroxide generation has been reported to induce cell cycle arrest at G1 and G2 (24). IR-induced DNA damage also triggers G1 or G2 arrest, allowing time for cells to repair DNA damage (25). In MIA PaCa-2 cells treated with ascorbate (2 mM) or 2 Gy alone or combination for 6, 24, 48 and 72 h, there was no significant change in cell cycle distribution. However, the sub-G1 population increased in cells treated with ascorbate (2 mM) or combination of 2 Gy IR and ascorbate (2 mM) at 6 and 24 h. At 24 h, there was a significant sub-G1 fraction in the combination group compared to the control, IR alone, and ascorbate alone groups (Supplemental Information, Figure 2). In addition, the PI viability assay at 24 h after treatment demonstrated that there was no significant change in viability with IR (2 Gy) alone while pharmacological ascorbate alone induced an 89 ± 1% sub-G1 fraction that was increased to 96 ± 2% with the combination treatment. These data taken together with the clonogenic survival and cell viability assays, strongly support the hypothesis that pharmacological ascorbate is a radio-sensitizer in pancreatic cancer cells.
Ascorbate radiosensitization is mediated by H2O2
Ascorbate-induced cytotoxicity is mediated by the formation of H2O2 during the oxidation of ascorbate. Similarly, ionizing radiation generates numerous ROS, including H2O2 and hydroxyl radical from the radiolysis of water. We hypothesized that ascorbate-induced radiosensitization is mediated by H2O2. To determine if intracellular levels of H2O2 change upon exposure to pharmacological ascorbate, intracellular concentrations of H2O2 were determined by the rate of aminotriazole-mediated inactivation of endogenous catalase activity. Cells were treated with IR (3 Gy), ascorbate (20 mM), or in combination and catalase activity was assayed in cells treated with 3-aminotriazole (3-AT). Using the stoichiometric inactivation of catalase in the presence of H2O2 and excess 3-AT, baseline intracellular concentration of H2O2 6 h after treatment was 53 ± 6 pM in pancreas cancer cells; this increased to 71 ± 9 pM with exposure to pharmacological ascorbate, and 79 ± 14 pM following IR. However, the combination of IR + ascorbate increased the concentration of intracellular H2O2 to 105 ± 2 pM (Figure 2A).
Figure 2. Ascorbate radiosensitization is mediated by H2O2.

A. MIA PaCa-2 cells were treated with 3-amino-1,2,4-triazole (3-AT) to determine intracellular H2O2. Cells treated with IR + ascorbate (20 mM) showed an increased concentration of intracellular H2O2 (n = 3, means ± SEM, P < 0.01). Pharmacological ascorbate at 20 mM produces H2O2 at a flux of 6200 amol−1 cell−1 s−1 in our experimental conditions.
B. Ascorbate (2 mM) radiosensitization depletes GSH and increases the half-cell reduction potential (Ehc). Ascorbate at 2 mM produces H2O2 at a flux of 1400 amol−1 cell−1 s−1 in our experimental conditions. (Means ± SEM, n =3).
C. Catalase partially rescues ascorbate (2 mM) radiosensitization. Western blot demonstrates increased catalase immunoreactive protein in cells treated with the AdCAT vector compared to AdEmpty treated cells.
D. MIA PaCa-2 cells were transfected with AdEmpty (50 MOI) or AdCAT (50 MOI) for 48 h, then subjected to ionizing radiation (2 Gy), ascorbate (2 mM) or the combined treatment. Ascorbate radiosensitization was reversed with catalase overexpression. (n = 3, means ± SEM, P < 0.01).
Under steady-state conditions, intracellular GSH is maintained at millimolar concentrations, which keeps cells in a reduced environment and serves as the principal intracellular redox buffer when cells are subjected to an oxidative stressor including H2O2 (26). Glutathione peroxidase (GPx) activity catalyzes the reduction of H2O2 to water with the conversion of GSH to glutathione disulfide (GSSG). Under steady-state conditions, GSSG is recycled back to GSH by glutathione disulfide reductase using reducing equivalents from NADPH. However, under conditions of increased H2O2 flux, this recycling mechanism may become overwhelmed leading to a depletion of intracellular GSH (27, 28).
Because ascorbate combined with radiation significantly increases the intracellular H2O2, we next examined the effects of the combination of ascorbate and IR on the GSH/GSSG intracellular redox couple. MIA PaCa-2 cells were treated with IR (2 Gy) and ascorbate (2 mM) and harvested at various time points (1–24 h). Cells treated with IR + ascorbate showed a significant decrease in intracellular GSH concentration at 6 h compared to untreated cells (2.5 ± 0.1 mM vs. 2.1 ± 0.1 mM respectively, Means ± S.D., n = 3, P < 0.05) and failed to recover after 24 h (Figure 2B). The half-cell reduction potential (Ehc) of the GSSG/2GSH couple, which is a marker for overall redox environment of the cell (26), was calculated using the Nernst equation. Ehc increased (more positive) indicating a more oxidized intracellular redox environment. This increase mirrored the depletion of GSH (Figure 2B). MIA PaCa-2 cells have a baseline oxygen consumption rate of 57 amol cell−1 s−1 (29); assuming 1% efficiency this corresponds to a metabolic rate for the production of H2O2 of less than 1 amol cell−1 s−1. The flux of H2O2 in these experiments was 1400 amol cell−1 s−1, considerably greater than the metabolic flux, thus recycling of GSSG to GSH is rate-limiting and depletion of GSH and an increase (more positive) in Ehc would be expected (28). Changes in Ehc have been shown to correlate with biological status of the cell with a change from −240 mV to −170 mV as cells shift from the state of proliferation to the onset of cell death (26). These results indicate that ascorbate radiosensitization can create an overwhelming oxidative stress to pancreatic cancer cells resulting in oxidation/depletion of the GSH intracellular redox buffer, resulting in cell death.
To further determine whether H2O2 was responsible for the cytotoxic effects of ascorbate + radiation, cells were pre-treated with either AdEmpty (50 MOI) or AdCAT (50 MOI) adenoviral vectors. Figure 2C demonstrates that there was robust expression of immunoreactive intracellular catalase in MIA PaCa-2 cells treated with the AdCAT vector when compared to control or AdEmpty treated cells. IR decreased clonogenic survival to 50% of AdEmpty alone values while ascorbate (2 mM) decreased clonogenic survival to 26 ± 2% (Figure 2D). The combination of 2 Gy IR and ascorbate (2 mM) further decreased clonogenic survival to 6 ± 1%. This decrease in clonogenic survival was significantly reversed with overexpression of intracellular catalase resulting in clonogenic survival of 41 ± 1% which was not significantly different from IR, suggesting that H2O2 mediates ascorbate radiosensitization (Figure 2D).
Ascorbate radiosensitization induces DNA damage
γ-H2AX is a sensor of DNA strand breaks (30) and promotes efficient double strand break repair (31). As shown in Figure 3A, 2 Gy IR and ascorbate (2 mM) induced formation of γ-H2AX in MIA PaCa-2 cells. However, the combination of IR and ascorbate increased formation of γ-H2AX, indicating a greater number of double strand breaks. Previous studies have demonstrated that ascorbate-induced cytotoxicity is mediated by generation of H2O2 (1, 2, 3). To determine if H2O2 mediates the increase in γ-H2AX during ascorbate-induced radiosensitization, cells were treated with extracellular catalase. Figure 3B demonstrates that the combination treatment of IR and ascorbate induced increased formation of γ-H2AX in AsPC-1 cells compared to either treatment alone. The increase in γ-H2AX with the combination treatment was inhibited with catalase pretreatment suggesting that H2O2 mediates the increase in γ-H2AX during ascorbate-induced radiosensitization. In addition to Western blot, we demonstrated similar increases in double strand DNA breaks as seen by γ-H2AX on immunohistochemistry (Figure 3C). In cells treated with ascorbate (1 mM), IR alone, or the combination of the two treatments, the increase in fluorescence was reversed with catalase pretreatment. Once again, quantification of these images demonstrated significant increases in fluorescence after ascorbate treatment and after IR + ascorbate combination treatment that was ameliorated with catalase pretreatment (Figure 3D). These experiments were repeated with varying doses of pharmacological ascorbate and varying doses of IR with similar results (Supplemental Information, Figure 3). Thus, H2O2 significantly contributes to the DNA damage observed upon exposure to pharmacological ascorbate and IR, as seen by γ-H2AX.
Figure 3. Ascorbate radiosensitization induces DNA damage.

A. In MIA PaCa-2 pancreatic cancer cells, both IR (2 Gy) and ascorbate (2 mM) induced the formation of γ-H2AX as determined by Western blot, while the combination treatment further enhanced γ-H2AX formation.
B. In AsPC-1 pancreatic cancer cells, ascorbate (2 mM) radiosensitization increased γH2AX immunoreactive protein, which was reversed with catalase pretreatment.
C. γ-H2AX immunohistochemistry demonstrated increased γH2AX formation with the combination treatment, which was reversed with catalase pretreatment. Cells were treated with ascorbate (1 mM) and IR (1 Gy) or catalase (100 U/mL). Ascorbate at 1 mM produces H2O2 at a flux of 690 amol−1 cell−1 s−1 under our experimental conditions.
D. Quantification of γ-H2AX immunohistochemistry in MIA PaCa-2 cells treated with ascorbate (1 mM) and IR (1 Gy) demonstrating increased induction of γ-H2AX during ascorbate radiosensitization which was reversed with catalase pretreatment. (n = 3, means ± SEM, P < 0.01).
Ascorbate-induced radiosensitization in vivo
We have previously demonstrated that ascorbate alone inhibits tumor growth in a mouse xenograft model with intraperitoneal administration of 4 g/kg twice daily (1). Pre-established MIA PaCa-2 tumors in nude mice were treated with either: saline (1 M NaCl i.p. daily); IR (7.5 Gy on days 5 and 8 and 1 M NaCl i.p. daily); ascorbate (4 g/kg i.p. daily); or ascorbate + IR. Table 1 in supplementary information provides statistical summaries of tumor volumes used in the mixed linear regression analysis of growth curves. The sample sizes (N) given in the table are the total number of measurements available within each group. Pairwise comparisons were carried out to assess group differences. Treatment with the combination of ascorbate + IR significantly delayed tumor growth compared to controls or ascorbate alone (Figure 4A). Summary statistics for the survival analysis are presented in Table 2 of supplementary information. The estimated survival curve for the treatment groups are given by the Kaplan-Meier plots in Figure 4B. Ascorbate + IR also significantly increased overall survival compared to controls, IR alone or ascorbate alone (Figure 4B). Most notably, 54% of mice treated with the combination of IR + ascorbate had no measurable tumors (Table 2 supplementary information). Treatment was well tolerated as evidenced by similar weight gain patterns in all groups of mice (Supplementary Figure 4).
Figure 4. Pharmacological ascorbate radiosensitization in vivo.

A. Linear mixed effects regression models were used to estimate and compare group-specific tumor growth curves. Tumor growth was significantly inhibited with the ascorbate + IR treatment compared to control animals or animals that received ascorbate alone. Control was saline (1 M NaCl i.p. daily, 22.7 μL g−1); IR (7.5 Gy on days 5 and 8 and 1 M NaCl i.p. daily); ascorbate (4 g kg−1 i.p. daily); or ascorbate + IR. (Means ± SEM, n = 9–11 animals/group).
B. Kaplan-Meier survival plots demonstrating survival as a function of time. The log-rank test was used for pairwise treatment group comparisons of survival between treatment groups demonstrating significantly increased overall survival of animals receiving pharmacological ascorbate and IR.
C. Pharmacological ascorbate alters the status of the GSH redox buffer of RBCs. Blood was collected from separate groups of mice after treatments and assayed for the intracellular concentration of GSH in the RBCs. Both IR and ascorbate alone decreased intracellular GSH compared to controls. The combination of ascorbate and IR did not further decrease GSH when compared to IR alone.
D. Complete blood counts and differential in control mice and those treated with ascorbate, IR and IR + ascorbate. White blood cell counts were decreased with IR which was unchanged when ascorbate was added to the treatment regimen.
E. Initially, purified bovine serum albumin was reacted with excess purified 4-HNE to create a positively labelled protein control which is seen in the upper panel. The negative control is purified bovine serum albumin without purified 4-HNE. Then to determine 4-HNE modified proteins in separate groups of mice, dot blots for 4-HNE modified proteins in cardiac muscle showed no changes in immunoreactive protein after any of the treatments compared to controls.
F. Dot blots for 4-HNE modified proteins in heart, kidney and liver demonstrated no changes in immuno-reactive protein after any of the treatments compared to controls.
Oxidative stress indicators
Glutathione is a measurable marker indicative of the oxidation state of the thiol redox buffer in cells. In severe systemic oxidative stress, the GSSG/2GSH couple may become oxidized, i.e. the concentration of GSH decreases and GSSG may increase because the capacity to recycle GSSG to GSH becomes rate-limiting. As seen in Figure 4C, intracellular concentrations of GSH in red blood cells of mice after ascorbate, IR, and ascorbate + IR treatments decreased. However, the addition of pharmacological ascorbate with IR did not significantly reduce GSH levels compared to 5 Gy IR alone. This suggests that the very high levels of pharmacological ascorbate in these experiments may have a pro-oxidant toward red blood cells as seen by a decrease in the capacity of the intracellular redox buffer. To further determine the effect of these treatments on hematological parameters, complete blood counts were determined in separate groups of mice. Although ascorbate and IR decreased intracellular GSH in red blood cells, there were no significant changes in hemoglobin and/or hematocrits between groups of mice. As seen in Figure 4D, mice that received IR alone or IR + ascorbate had a significant decrease in white blood cells suggesting some immunosuppression with IR. However, ascorbate did not add to further decreases in white blood cells and the percentage of neutrophils and lymphocyte counts were unchanged between groups.
Oxidation of lipids generates highly reactive aldehydes such as 4-hydroxynonenal (4-HNE), which can be used as a marker of oxidative damage [32, 33]. To further examine systemic oxidative stress, we harvested heart, kidney and liver from mice treated with the various combinations to determine 4-HNE modified proteins. As seen in Figure 4E, there were no differences in 4-HNE modified proteins in heart tissue in mice treated with the various combinations. In addition, dot blot analysis of the other organs demonstrated no differences in 4-HNE proteins after the various treatments (Figure 4F). These data support the hypothesis that ascorbate radiosensitization does not cause an increase in oxidative damage from lipid-derived aldehydes to other organs.
Orthotopic tumors were established using ultrasound guidance as described (21) (Figure 5A). After bioluminescence-imaging microscopy identified mice with established tumors within the pancreas (Figure 5B), mice were randomly assigned to one of four treatment groups: controls received NaCl (1 M) i.p. daily; ascorbate 4 g/kg i.p. daily; NaCl i.p. daily and two separate fractions of 5 Gy IR one week apart on days 4 and 11; a group that received both IR and ascorbate. Tumor sizes were measured weekly throughout the experiments, resulting in repeated measurements for each mouse. Linear mixed effects regression models were used to estimate and compare group-specific tumor growth curves. Table 3 in supplemental information provides statistical summaries of tumor volumes used in the linear mixed effects regression analysis of growth curves. The sample sizes (N) given in the table are the total number of measurements available within each group. Pairwise comparisons were carried out to assess group differences. P-values for the comparisons are listed in Table 3 (Supplemental information). Tumor growth as determined by bioluminescence-imaging microscopy (mean radiance photons/s−1/cm2) demonstrated a decrease in tumor growth of animals receiving both IR and ascorbate compared to controls and compared to IR alone (*P < 0.05 control vs. Ascorbate + IR; *#P < 0.05 Ascorbate + IR vs. IR alone. (Figure 5C, Supplemental Figure 5). As seen in the previous heterotopic study, weights of the animals did not change during the study (data not shown).
Figure 5. Pharmacologic ascorbate radiosensitization in an orthotopic model.
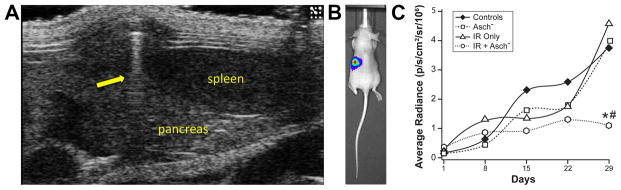
A. Ultrasound guided pancreatic injections. 4 x 105 MIA PaCa-2 cells suspended in a 20 μL 1:1 mixture of PBS and Matrigel were injected into athymic nude mice who were sedated with Isoflurane. Under ultrasound guidance, injections were performed directly into the pancreas just medial and inferior to the spleen. The spleen and pancreas are labeled. The yellow arrow indicates the shadow of the needle during injection.
B. Bioluminescence imaging microscopy. Four days after the initial injections, mice were imaged using the Xenogen IVIS 200 microscope 10 minutes after injection with 200 μL of 15 mg/mL luciferin. Initial exposure time was 1 minute. Displayed is a color scale of photons/second emitted superimposed over a photograph of the mouse as viewed in the prone position. The bioluminescence is localized to the left quadrant central quadrant as would be expected for a pancreatic tumor.
C. Linear mixed effects regression models demonstrated significant inhibition in tumor growth with the ascorbate + IR treatment compared to control animals or animals that received IR alone. Control was saline (1 M NaCl i.p. daily, 22.7 μL g−1); IR (7.5 Gy on days 4 and 11 and 1 M NaCl i.p. daily); ascorbate (4 g kg−1 i.p. daily); or ascorbate + IR. (Means, n = 7 – 12 animals/group). *P < 0.05 control vs. Ascorbate + IR; #P < 0.05 Ascorbate + IR vs. IR alone.
Radiation-induced gastrointestinal toxicity is highly relevant to the treatment of pancreatic cancer with radiation. To determine if pharmacological ascorbate changes the response of the gastrointestinal tract following radiation in a clinically meaningful way, a crypt cell assay was performed (Figure 6). Control animals and those treated with pharmacological ascorbate (4 g/kg) had similar degrees of crypt cell generation. IR alone (10 Gy) decreased crypts to 28% of control values, while 13 Gy decreased crypts to 44% of control values. Addition of pharmacological ascorbate partially reversed the decreases in both the 10 Gy and 13 Gy groups of mice suggesting that ascorbate may protect the gastrointestinal tract from the damaging effects of IR. Additionally, previous studies have demonstrated that radiation-induced jejunal toxicity was accompanied by increases in serum tumor necrosis factor-α (TNF-α) (34). TNF-α was 7.5 ± 2.2 pg/ml in controls and was increased to 25.0 ± 1.5 pg/ml after 13 Gy. Treatment with pharmacological ascorbate combined with IR, decreased TNF-α to 12.5 ± 0.7 pg/ml (Means ± SEM, n = 3, P < 0.05 IR alone vs. ascorbate + IR). Taken together, these data suggest that pharmacological ascorbate may protect the gut locally by decreasing IR-induced damage to the crypt cells, and systemically, by ameliorating increases in TNF-α
Figure 6. Pharmacological ascorbate partially reverses IR-induced jejunal damage.

A. Crypt cell assay from control mice. Sections were made of the jejunum and then viewed under light microscopy and expressed as surviving cells/circumference.
B. Mice were treated with ascorbate 4 g/kg I.P. for 5 days and then sacrificed.
C. Mice were exposed to 10 Gy of total abdominal radiation. After 48 h, each animal was sacrificed and sections were made of the jejunum as described.
D. Mice treated with pharmacological ascorbate (4 g/kg for 5 days with 10 Gy total abdominal radiation on day 3 and then sacrificed on day 5.
E. Mice exposed to 13 Gy of total abdominal radiation and sacrificed 48 h later. Note marked decrease in number of regenerating crypts/circumference of the sectioned jejunum.
F. Mice treated with pharmacological ascorbate (4 g/kg for 5 days with 13 Gy total abdominal radiation on day 3 and then sacrificed on day 5. Note increase in regenerating crypts compared to panel E.
G. Quantification of crypt cell assay. Means ± SEM, n = 8–10 samples/group. * P < 0,01 vs. Controls and ascorbate treated animals. #P < 0.05 vs. Control and ascorbate treated animals.
The in vivo animal experiments were repeated using PANC-1 tumors. In these sets of experiments we added gemcitabine to the IR group as done in clinical practice (35). The mice were divided into four groups and treated with either: saline (1 M NaCl i.p. daily); IR/ gemcitabine (5 Gy on days 4 and 8 with gemcitabine 60 mg/kg I.P. every 4th day for two weeks and 1 M NaCl i.p. daily); ascorbate (4 g/kg i.p. daily); or ascorbate + IR/gemcitabine. As seen in Figure 7A treatment with IR/gemcitabine delayed tumor growth compared to ascorbate alone (P < 0.05). However, the combination of ascorbate + IR/gemcitabine further delayed tumor growth compared to ascorbate alone (P < 0.001). Summary statistics for the survival analysis are presented in Table 4 of supplementary information. The estimated survival curve for the treatment groups are given by the Kaplan-Meier plots in Figure 7B. Ascorbate + IR/gemcitabine also significantly increased overall survival compared to ascorbate alone (Figure 7B). Most notably, 100% of the mice on day-43 that were treated with the combination of ascorbate + IR/gemcitabine were surviving (Figure 7B). Treatment was well tolerated as evident by similar weight gain patterns in all groups of mice (Figure 7C).
Figure 7. Pharmacological ascorbate radiosensitization in vivo.

A. Linear mixed effects regression models were used to estimate and compare group-specific tumor growth curves. Tumor growth was significantly inhibited in mice with pancreatic tumor xenografts treated with IR/gemcitabine compared to controls and ascorbate alone. Tumor growth was further inhibited in mice that received the ascorbate + IR/gemcitabine treatment compared to control animals or animals that received ascorbate alone. (Means ± SEM, n = 11–12 mice/group).
B. Kaplan-Meier survival plots demonstrating survival as a function of time. The log-rank test was used for pairwise treatment group comparisons of survival between treatment groups demonstrating significantly increased overall survival animals receiving pharmacological ascorbate and IR/gemcitabine treatment on day 43 after the initiation of treatment.
C. Weight changes of mice during treatment periods in mice during treatments from day 1 through day 15.
Discussion
Our current study demonstrates the potential for pharmacological ascorbate as a radiosensitizer in the treatment of pancreatic cancer. IR is the standard of care in pancreatic cancer in a variety of clinical scenarios including: 1) locally advanced pancreatic cancer; 2) positive margins or positive lymph nodes after pancreatic resection; or 3) large tumors which obstruct the duodenum (36). Chemotherapy combined with radiotherapy has also shown to improve survival in pancreatic cancer when compared with single-modality therapy (37). In addition, radiotherapy can also provide palliation to patients with locally advanced disease (38). Thus, pharmacological ascorbate-induced radiosensitization may have clinical benefits.
In our present study we have shown that pharmacological ascorbate significantly decreases clonogenic survival and inhibits the growth of all pancreatic cancer cell lines as a single agent, as well as sensitizes cancer cells to IR. This corresponds well with other reports demonstrating that pharmacological ascorbate enhances IR-induced cell killing and DNA fragmentation leading to induction of apoptosis in HL60 leukemia cells (39). In addition, Hurst et al. demonstrated that pharmacological ascorbate combined with IR leads to increased numbers of double-strand DNA breaks and cell cycle arrest when compared to either treatment alone (40). Our previous studies demonstrated that pharmacological ascorbate could serve as a “pro-drug” for the delivery of H2O2 to tumors (1, 2, 3, 4). There was both a time and dose-dependent increase in measured H2O2 production with increased concentrations of ascorbate. Others have demonstrated increased levels of double-strand breaks with pharmacological ascorbate and H2O2 treatment to tumor cells (41, 42). In addition, the double-strand breaks induced by H2O2 were more slowly repaired.
Previously we demonstrated that the immortalized, non-tumorigenic pancreatic ductal epithelial cells H6c7 are resistant to ascorbate-induced toxicity (1). Consistent with this study, our current study demonstrates that H6c7 cells are also resistant to the combination treatment of IR and ascorbate. The selective ascorbate-induced cytotoxicity may be due to the combination of low levels of antioxidant enzymes and high endogenous levels of ROS in cancer cells (43, 44, 45). Moreover, tumor cells are often defective in DNA repair, whereas normal cells are proficient (46). The combination of ascorbate and IR provide two distinct mechanisms of action: ascorbate-induced toxicity due to extracellular production of H2O2 that then diffuses into cells and causes damage to DNA, protein, and lipids; and radiation-induced toxicity as a result of ROS-induced damage to DNA. In addition, redox metal metals like Fe2+ may play an important role in ascorbate-induced cytotoxicity. By catalyzing the oxidation of ascorbate, labile iron can enhance the rate of formation of H2O2; labile iron can also react with H2O2. Recently our group has demonstrated that pharmacological ascorbate and IR increase the labile iron in tumor homogenates from this murine model of pancreatic cancer (47). In our present study using a pancreatic cancer xenograft model, we demonstrated that ascorbate or IR alone decreased tumor growth, but the combination treatment further inhibited tumor growth, indicating that pharmacological ascorbate is an effective radiosensitizer in vivo. In addition, all groups of mice had similar weight gain during treatment suggesting lack of normal tissue toxicity with the combination treatment.
That approximately 30% of pancreatic patients receive a diagnosis of advanced locoregional disease underscores the importance of radiotherapy for local disease control (48). Our preclinical model shows that the combination of IR and ascorbate enhances toxicity in pancreatic cancer cells compared to either treatment alone. In a Phase I clinical trial that investigated the use of pharmacological ascorbate as an adjuvant to gemcitabine, the standard of care, in the treatment of pancreatic cancer it was well-tolerated with few dose-limiting toxicities (49). The data reported here support testing of pharmacological ascorbate as an adjuvant treatment for radiotherapy in pancreatic cancer patients after surgical resection or with locally advanced disease. Indeed, the pre-clinical translational studies in this current study have led us to develop and implement a phase I clinical trial (www.clinicaltrials.gov; NCT01852890, Cullen PI) with FDA approval (IND 105715, Cullen sponsor-investigator).
Supplementary Material
Acknowledgments
We thank The University of Iowa Central Microscopy Research Facility for their guidance and technical advice. We thank Dr. Luke I. Szweda from the Medical Research Foundation, Oklahoma City, Oklahoma. The University of Iowa ESR Facility and the Radiation and Free Radical Research Core in the Holden Comprehensive Cancer Center provided invaluable assistance in the execution of these studies.
Supported by NIH grants RO1 CA184051, RO1 CA169046, P30 CA086862, P42 ES013661, RO1 CA111365, RO1 CA182804, T32 CA078586, T32 CA148062, the Medical Research Service, an ASTRO Career Development Award JF2014-1, and the Department of Veterans Affairs 1I01BX001318-01A2.
Footnotes
There are no conflicts of interest
References
- 1.Du J, Martin SM, Levine M, Wagner BA, Buettner GR, Wang SH, et al. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin Cancer Res. 2010;16:509–20. doi: 10.1158/1078-0432.CCR-09-1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Q, Espey MG, Krishna MC, Mitchell JB, Corpe CP, Buettner GR, et al. Ascorbic acid at pharmacologic concentrations selectively kills cancer cells: ascorbic acid as a pro-drug for hydrogen peroxide delivery to tissues. Proc Natl Acad Sci USA. 2005;102:13604–09. doi: 10.1073/pnas.0506390102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Q, Espey MG, Sun AY, Lee JH, Krishna MC, Shacter E, et al. Ascorbic acid in pharmacologic concentrations: a pro-drug for selective delivery of ascorbate radical and hydrogen peroxide to extracellular fluid in vivo. Proc Natl Acad Sci USA. 2007;104:8749–54. doi: 10.1073/pnas.0702854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochimica et Biophysica Acta – Reviews on Cancer. 2012;1826:443–457. doi: 10.1016/j.bbcan.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rawal M, Schroeder SR, Wagner BA, Cushing CM, Welsh J, Button AM, Du J, Sibenaller ZA, Buettner GR, Cullen JJ. Manganoporphyrins increase ascorbate-induced cytotoxicity by enhancing H2O2 generation. Cancer Res. 2013;73(16):5232–5241. doi: 10.1158/0008-5472.CAN-13-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kobayashi J, Iwabuchi K, Miyagawa K, Sonoda E, Suzuki K, Takata M, et al. Current topics in DNA double-strand break repair. J Radiat Res (Tokyo) 2008;49:93–103. doi: 10.1269/jrr.07130. [DOI] [PubMed] [Google Scholar]
- 7.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on Serine 139. J Biol Chem. 1998;273(10):5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 8.Lowndes NF, Toh GWL. DNA repair: the importance of phosphorylating histone H2AX. Current Biology. 2005;15(3):R99–R102. doi: 10.1016/j.cub.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 9.Taneja N, Davis M, Choy JS, Beckett MA, Singh R, Kron SJ, et al. Histone H2AX phosphorylation as a predictor of radiosensitivity and target for radiotherapy. J Biol Chem. 2004;279(3):2273–80. doi: 10.1074/jbc.M310030200. [DOI] [PubMed] [Google Scholar]
- 10.Duarte TL, Almeida GM, Jones GD. Investigation of the role of extracellular H2O2 and transition metal ions in the genotoxic action of ascorbic acid in cell culture models. Toxicol Lett. 2007;170:57–65. doi: 10.1016/j.toxlet.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Henle ES, Linn S. Formation, prevention, and repair of DNA damage by iron/hydrogen peroxide. J Biol Chem. 1997;272(31):19095–98. doi: 10.1074/jbc.272.31.19095. [DOI] [PubMed] [Google Scholar]
- 12.Storr SJ, Woolston CM, Martin SG. Base excision repair, the redox environment and therapeutic implications. Curr Mol Pharmacol. 2012;5:88–101. [PubMed] [Google Scholar]
- 13.Roy I, Zimmerman NP, Mackinnon AC, Tsai S, Evans DB, Dwinell MB. CXCL12 chemokine expression suppresses human pancreatic cancer growth and metastasis. PLOS ONE. 2014;9(3):1–13. doi: 10.1371/journal.pone.0090400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, Gallick GE. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nature Protocols. 2009;4(11):1670–1680. doi: 10.1038/nprot.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qian J, Niu J, Li M, Chiao PJ, Tsao MS. In vitro modeling of human pancreatic duct epithelial cell transformation defines gene expression changes induced by K-ras oncogenic activation in pancreatic carcinogenesis. Cancer Res. 2005;65:5045–53. doi: 10.1158/0008-5472.CAN-04-3208. [DOI] [PubMed] [Google Scholar]
- 16.Du J, Daniels DH, Asbury C, Venkataraman S, Liu J, Spitz DR, et al. Mitochondrial production of reactive oxygen species mediate dicumarol-induced cytotoxicity in cancer cells. J Biol Chem. 2006;281(49):37416–26. doi: 10.1074/jbc.M605063200. [DOI] [PubMed] [Google Scholar]
- 17.Olney KE, Du J, van ‘t Erve TJ, Witmer JR, Sibenaller ZA, Wagner BA, et al. Inhibitors of hydroperoxide metabolism enhance ascorbate-induced cytotoxicity. Free Radical Research. 2013;47:154–63. doi: 10.3109/10715762.2012.755263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park HJ, Mah E, Bruno RS. Validation of high performance liquid chromatography-boron-doped diamond detection for assessing hepatic glutathione redox status. Analytical Biochemistry. 2010;407:151–159. doi: 10.1016/j.ab.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 19.Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nature protocols. 2006;1(6):3159–3165. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- 20.Euhus DM, Hudd C, LaRegina MC, Johnson FE. Tumor measurement in the nude mouse. J Surg Oncol. 1986;31(4):229–34. doi: 10.1002/jso.2930310402. [DOI] [PubMed] [Google Scholar]
- 21.Huynh AS, Abrahams DF, Torres MS, Baldwin MK, Gillies RJ, Morse DL. Development of an orthopic human pancreatic cancer xenograft model using ultrasound guided injection of cells. 2011;6(5) doi: 10.1371/journal.pone.0020330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allen BG, Bhatia SK, Buatti JM, et al. Ketogenic diets enhance oxidative stress and radio-chemo-therapy responses in lung cancer xenografts. Clin Cancer Res. 2013;19:3905–3913. doi: 10.1158/1078-0432.CCR-12-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohn JA, Tsai L, Friquet b, Szweda LI. Chemical characterization of a protein-4-hydroxy-2-nonenal cross-link: Immunochemical detection in mitochondria exposed to oxidative stress. Arch Biochem Biophys. 1996;328:158–164. doi: 10.1006/abbi.1996.0156. [DOI] [PubMed] [Google Scholar]
- 24.Oyama K, Takahashi K, Sakurai K. Hydrogen peroxide induces cell cycle arrest in cardiomyoblast H9c2 cells, which is related to hypertrophy. Biol Pharm Bull. 2011;34:501–06. doi: 10.1248/bpb.34.501. [DOI] [PubMed] [Google Scholar]
- 25.Samuel T, Weber HO, Funk JO. Link DNA damage to cell cycle checkpoints. Cell Cycle. 2002;1:162–68. [PubMed] [Google Scholar]
- 26.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 27.Sies H. Glutathione and its role in cellular functions. Free Radic Biol Med. 1999;27(9–10):916–21. doi: 10.1016/s0891-5849(99)00177-x. [DOI] [PubMed] [Google Scholar]
- 28.Ng CF, Schafer FQ, Buettner GR, Rodgers VGJ. The rate of cellular hydrogen peroxide removal shows dependency on GSH: Mathematical insight into in vivo H2O2 and GPx concentrations. Free Rad Res. 2007;41:1201–1211. doi: 10.1080/10715760701625075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagner BA, Venkataraman S, Buettner GR. The rate of oxygen utilization by cells. Free Radic Biol Med. 2011;51:700–712. doi: 10.1016/j.freeradbiomed.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finn K, Lowndes NF, Grenon M. Eukaryotic DNA damage checkpoint activation in response to double-strand breaks. Cell Mol Life Sci. 2012;69:1447–73. doi: 10.1007/s00018-011-0875-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Redon CE, Nakamura AJ, Zhang YW, Ji JJ, Bonner WM, Kinders RJ, et al. Histone γH2AX and poly(ADP-Ribose) as clinical pharmacodynamic biomarkers. Clin Cancer Res. 2010;16(18):4532–42. doi: 10.1158/1078-0432.CCR-10-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robinson CE, Keshavarzian A, Pasco DS, Frommel TO, Winship DH, Holmes EW. Determination of protein carbonyl groups by immunoblotting. Anal Biochem. 1999;266(1):48–57. doi: 10.1006/abio.1998.2932. [DOI] [PubMed] [Google Scholar]
- 33.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42(4):318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 34.Ito Y, Kinoshita M, Yamamoto T, Sato T, Obara T, Saitoh D, Seki S, Takahashi Y. A combination of pre- and post-exposure ascorbic acid rescues mice from radiation-induced lethal gastrointestinal damage. Int J Mol Sci. 2013;14:19618–19635. doi: 10.3390/ijms141019618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loehrer PJ, Feng Y, Cardenes H, Wagner L, Brell JM, Cella D, Flynn P, Ramanathan RK, Crane CH, Alberts SR, Benson AB. Gemcitabine Alone Versus Gemcitabine Plus Radiotherapy in Patients With Locally Advanced Pancreatic Cancer: An Eastern Cooperative Oncology Group Trial. Journal of Clinical Oncology. 2011;29(31):4105–4112. doi: 10.1200/JCO.2011.34.8904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hazard L, Tward JD, Szabo A, Shrieve DC. Radiation therapy is associated with improved survival in patients with pancreatic adenocarcinoma: Results of a study from the surveillance, epidemiology, and end results (SEER) registry data. Cancer. 2007;110(10):2191–201. doi: 10.1002/cncr.23047. [DOI] [PubMed] [Google Scholar]
- 37.Chang DT, Schellenberg D, Shen J, Kim J, Goodman KA, Fisher GA, et al. Stereotactic radiotherapy for unresectable adenocarcinoma of the pancreas. Cancer. 2009;115(3):665–72. doi: 10.1002/cncr.24059. [DOI] [PubMed] [Google Scholar]
- 38.Minsky BD, Hilaris B, Fuks Z. The role of radiation therapy in the control of pain from pancreatic carcinoma. J Pain Symptom Manage. 1988;3(4):199–205. doi: 10.1016/0885-3924(88)90031-0. [DOI] [PubMed] [Google Scholar]
- 39.Shinozaki K, Hosokawa Y, Hazawa M, Kashiwakura I, Okumura K, Kaku T, et al. Ascorbic acid enhances radiation-induced apoptosis in an HL60 human leukemia cell line. J Radiat Res. 2011;52(2):229–37. doi: 10.1269/jrr.10089. [DOI] [PubMed] [Google Scholar]
- 40.Herst PM, Broadley KW, Harper JL, McConnell MJ. Pharmacological concentrations of ascorbate radiosensitize glioblastoma multiforme primary cells by increasing oxidative DNA damage and inhibiting G2/M arrest. Free Radic Biol Med. 2012;52(8):1486–93. doi: 10.1016/j.freeradbiomed.2012.01.021. [DOI] [PubMed] [Google Scholar]
- 41.Driessens N, Versteyhe S, Ghaddhab C, Bumiat A, De Deken X, Van Sande J, et al. Hydrogen peroxide induces DNA single- and double-strand breaks in thyroid cells and is therefore a potential mutagen for this organ. Endocrine Related Cancer. 2009;16:845–56. doi: 10.1677/ERC-09-0020. [DOI] [PubMed] [Google Scholar]
- 42.Ma Y, Chapman J, Levine M, Polireddy K, Drisko J, Chen Q. High-dose parental ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci Transl Med. 2014;6:1–10. doi: 10.1126/scitranslmed.3007154. [DOI] [PubMed] [Google Scholar]
- 43.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51(3):794–98. [PubMed] [Google Scholar]
- 44.Oberley LW. Mechanism of the tumor suppressive effect of MnSOD overexpression. Biomed Pharmacother. 2005;59:143–48. doi: 10.1016/j.biopha.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 45.Du J, Nelson ES, Simons AL, Olney KE, Moser JC, Schrock HE, et al. Regulation of pancreatic cancer growth by superoxide. Molecular Carcinogenesis. 2013;52:555–67. doi: 10.1002/mc.21891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 2011;11(4):239–53. doi: 10.1038/nrc3007. [DOI] [PubMed] [Google Scholar]
- 47.Moser JC, Rawal M, Wagner BA, Du J, Cullen JJ, Buettner GR. Pharmacologic ascorbate and ionizing radiation (IR) increase labile iron in pancreatic cancer. Redox Biology. 2014;2:22–27. doi: 10.1016/j.redox.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hidalgo M. Pancreatic Cancer. New Engl J Med. 2010;362(17):1650–17. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 49.Welsh JL, Wagner BA, van ’t Erve TJ, Zehr PS, Berg DJ, Halfdanarson TR, Yee NS, Bodeker KL, Du J, Roberts LJ, 2nd, Levine M, Buettner GR, Cullen JJ. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): Results from a phase I clinical trial. Cancer Chemotherapy and Pharmacology. 2013;71(3):765–75. doi: 10.1007/s00280-013-2070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.