

Abstract
Rates of the most common gynecologic cancer, endometrioid adenocarcinoma (EAC), continue to rise, mirroring the global epidemic of obesity, a well-known EAC risk factor. Thus, identifying novel molecular targets to prevent and/or mitigate EAC is imperative. The prevalent Type 1 EAC commonly harbors loss of the tumor suppressor, Pten, leading to AKT activation. The major endoplasmic reticulum (ER) chaperone, GRP78, is a potent pro-survival protein to maintain ER homeostasis, and as a cell surface protein, is known to regulate the PI3K/AKT pathway. To determine whether targeting GRP78 could suppress EAC development, we created a conditional knockout mouse model utilizing progesterone receptor (PR)-Cre-recombinase to achieve Pten and Grp78 (cPtenf/fGrp78f/f) deletion in the endometrial epithelium. Mice with a single Pten (cPtenf/f) deletion developed well-differentiated EAC by 4 weeks. In contrast, no cPtenf/fGrp78f/f mice developed EAC, even after more than 8 months of observation. Histologic examination of uteri from cPtenf/fGrp78f/f mice also revealed no complex atypical hyperplasia (CAH), a well-established EAC precursor. These histologic observations among the cPtenf/fGrp78f/f murine uteri also corresponded to abrogation of AKT activation within the endometrium. We further observed that GRP78 co-localized with activated AKT on the surface of EAC thus providing an opportunity for therapeutic targeting. Consistent with previous findings that cell surface GRP78 is an upstream regulator of PI3K/AKT signaling, we show here that in vivo short-term systemic treatment with a highly specific monoclonal antibody against GRP78 suppressed AKT activation and increased apoptosis in the cPtenf/f tumors. Collectively, these findings present GRP78-targeting therapy as an efficacious therapeutic option for EAC.
Keywords: endometrial cancer, glucose regulated protein 78 (GRP78), AKT, PTEN, mouse model
INTRODUCTION
Endometrioid adenocarcinoma (EAC) is the most prevalent gynecologic cancer in the United States (US) and accounts for nearly 50,000 incident cases annually.1 Contemporary molecular characterization of EAC has unearthed additional deleterious mutations and mechanisms responsible for tumor development.2 One of the most common mutations detected in the more prevalent Type 1 EAC is the loss of the tumor suppressor gene, Pten (phosphatase and tensin homolog) and its phosphatase protein product.3,4 Pten mutations resulting in PTEN loss are involved in a wide variety of human cancers, including >60% of endometrioid adenocarcinomas of the endometrium.3,4 The deleterious phenotype resulting from Pten-loss has also been observed in in vitro and in vivo tumor models.5–9 While constitutive deletion of Pten results in embryonic loss, conditional deletion of Pten in target cells has permitted exploration of spontaneous tumorigenesis in various tissues.10–12 For EAC a conditional Pten deletion within the endometrial epithelium leads to development of endometrial hyperplasia and Type I EAC in female mice.5 Furthermore, the knockout of Pten by the progesterone receptor (PR)-driven Cre-recombinase progresses along the histologic continuum of complex atypical endometrial hyperplasia (AEH) to EAC, thereby facilitating specific interrogation of Pten-driven EAC development, and exploring strategies to therapeutically block EAC development.5
The glucose regulated protein-78 (GRP78) is a key member of the heat shock protein-70 (HSP70) family and an important endoplasmic reticulum (ER) chaperone protein.13,14 The ER is an essential organelle required for the synthesis and maturation of membrane-associated and secretory proteins. Pathophysiologic conditions that disrupt the ER result in ER stress, activation of the unfolded protein response (UPR), and marked upregulation of GRP78.15 ER stress has been reported to be transmissible to the tumor microenvironment16 and is activated in EAC.17 The role of GRP78 in human cancers appears to be multifaceted, and its overexpression has been cited in a wide variety of human cancers, including breast and gynecologic cancers.18–20
EAC development has been linked to obesity,20 and increased secretory activities associated with obesity lead to ER stress, UPR activation, and GRP78 upregulation.21 Within the uterus, GRP78 overexpression is more frequently apparent in EAC than in normal endometrium.17,19,22 Although benign endometrial epithelial cells will have some basal expression of GRP78, women with EAC whose carcinoma cells exhibit even higher levels of GRP78 expression tend to fare worse, clinically.22 Furthermore, GRP78 expression levels in visceral adipocytes adjacent to the uterine corpus are also higher in women with EAC who have more worrisome clinical features (e.g., myometrial invasion, advanced stage).22 While GRP78 is known to be an upstream regulator of the PI3K/AKT pathway, GRP78 has also been shown to be a downstream target of AKT, an important signal transduction pathway in EAC.19 Additionally, GRP78 is important for the optimal activation of AKT in response to cisplatin in EAC cell lines.19 Despite these observations, the key question of whether GRP78 is required for EAC development or progression is still not known.
Traditionally, GRP78 is regarded as an ER luminal protein critical for protein folding and processing, degrading malfolded ER proteins, and maintaining the transmembrane ER stress sensors and pro-apoptotic components in their inactive forms.15,23–25 Recent discovery of GRP78 localized on the plasma membrane of cancer cells, has led to the further characterization that ER stress, as seen in cancer cells due to both intrinsic and extrinsic factors, can actively promote the surface expression of GRP78.26,27 At the cell surface, GRP78 forms complexes with PI3K and enhances PI(3,4,5)P3 production, consistent with its novel role as a regulator of the PI3K/AKT signaling pathway, which promotes cell proliferation, survival, metastasis, and chemoresistance.28 Furthermore, the preferential presence of GRP78 on the surface of cancer but not normal cells in vivo provides a potential opportunity for highly specific therapeutic intervention.26,29–32 Recently, a high-affinity, highly specific monoclonal antibody (MAb159) against GRP78 has been identified and has shown therapeutic efficacy in reducing tumor growth in vivo.32 In this study, we used a combined approach of genetic knockout and antibody targeting of GRP78 to provide direct evidence that GRP78 is critical and necessary for the development of Pten-driven EAC, and may represent a new therapeutic opportunity for EAC.
RESULTS
Creation of biallelic deletion of Grp78 and Pten in the mouse uterus
Across successive breeding generations, PCR analysis of female pups at 10 days confirmed the generation of the distinct genotypes used throughout these studies: cPtenf/fGrp78+/+, cPtenf/fGrp78f/+, and cPtenf/fGrp78f/f with mice lacking Cre expression serving as wild-type (WT) mice. Mouse tail genomic DNA was used for genotyping and the status of Pten and Grp78 alleles in the uterus was confirmed by PCR of uterine DNA analyzed at 8 weeks (Figure 1a).
Figure 1.

Generation of mice with concurrent Pten and Grp78 ablation in uteri. (a) Representative PCR Pten, Grp78, and Cre genotyping results of mouse uteri DNA from WT, cPtenf/fGrp78+/+, cPtenf/fGrp78f/+ and cPtenf/fGrp78f/f at 8 weeks. Mice without Cre serve as WT controls. (b) Expression of progesterone receptor (PR), PTEN, and GRP78 in uteri of the indicated genotypes (8 weeks). Arrows indicate PR-expressing cells. (c) Western blot analysis confirms substantial reduction of GRP78 and PTEN in uterine tissue lysates from cPtenf/fGrp78f/f mice at 4- and 20-weeks. Vinculin serves as a loading control. Numbers represent relative change in GRP78 expression relative to WT control mice. (d) Immunohistochemistry confirms substantial reduction of GRP78 expression in murine uteri (4- and 8-weeks). Black scale bar, 100 μm. Red scale bar, 25 μm.
Immunohistochemical staining of uterine cross-sections first showed progesterone receptor (PR) primarily localized in the endometrium (Figure 1b). Loss of expression of the targeted genes within the endometrium was then confirmed by immunohistochemical analysis (Figure 1b). GRP78 and PTEN protein expression was detected in the uteri of WT mice, while expression of both proteins was substantially reduced in the endometria from cPtenf/fGrp78f/f mice (Figure 1b).
To assess the level and durability of PTEN and GRP78 loss, Western blot analysis of tissue lysate from the uteri at 4- and 20-weeks was performed. Reduction or loss of PTEN expression was confirmed at each time point. Similarly, GRP78 expression in the uterus declined significantly in mice homozygous for the Grp78 floxed alleles compared to the uteri from WT mice (Figure 1c). Interestingly, we noted that for the cPtenf/fGrp78f/+ mice, the expression level of GRP78 was only modestly reduced at 4 weeks and by 20 weeks, its level was similar to that of WT, thereby suggesting a compensatory response in the heterozygous mice to restore normal levels of GRP78 (Figure 1c). Immunohistochemical evaluation of GRP78 expression in FFPE uterine sections further confirmed durable and near absent GRP78 expression within the endometrial epithelial cells of cPtenf/fGrp78f/f uteri at both 4- and 8-weeks (Figure 1d).
Conditional Grp78 deletion from the endometrium blocks endometrial cancer development
To determine if anatomic differences existed in the murine uteri from different genotypes, biometric data were taken from euthanized mice (Table 1). The mean uterine weights between cPtenf/fGrp78f/f and WT mice were not statistically different at 10 days and 4 weeks (Table 1). However, by 4 weeks, the mean uterine weights of cPtenf/fGrp78+/+ mice were significantly greater than that of both WT and cPtenf/fGrp78f/f mice. cPtenf/fGrp78+/+ mice at 8 weeks showed the greatest mean uterine weight compared to cPtenf/fGrp78f/+ and cPtenf/fGrp78f/f mice (Figure 2). There was no statistically significant difference in mean uterine weights between cPtenf/fGrp78+/+ and cPtenf/fGrp78f/+ mice at both 4- and 8-weeks (Table 1).
Table 1.
Comparative mouse and uterine weights at 10-days, 4-weeks, and 8-weeks.
| Genotype | ||||
|---|---|---|---|---|
|
| ||||
| WT | cPtenf/fGrp78+/+ | cPtenf/fGrp78f/+ | cPtenf/fGrp78f/f | |
| 10 days | n=16 | n=8 | n=7 | n=7 |
|
| ||||
| Mean body weight (g) ±SD | 6.66±0.99 | 7.62±1.12 | 7.49±0.42 | 7.27±1.43 |
| Mean uterine weight (g) ±SD | 0.025±0.017 | 0.040±0.032 | 0.031±0.033 | 0.029±0.026 |
|
| ||||
| 4 weeks | n=10 | n=8 | n=8 | n=8 |
|
| ||||
| Mean body weight (g) ±SD | 16.61±0.13 | 18.67±0.50 | 14.94±2.24 | 14.86±3.50 |
| Mean uterine weight (g) ±SD | 0.071±0.027 | 0.22±0.15*# | 0.18±0.081 | 0.054±0.028± |
|
| ||||
| 8 weeks | n=11 | n=8 | n=8 | n=8 |
|
| ||||
| Mean body weight (g) ±SD | 20.87±2.39 | 21.17±0.98 | 21.90±1.82 | 20.80±1.63 |
| Mean uterine weight (g) ±SD | 0.095±0.085 | 0.76±0.082ξ | 0.59±0.12 | 0.054±0.062δ |
p=0.001 (vs. 4-week WT).
p<0.001 (vs. 4-week cPtenf/fGrp78f/f).
p=0.001 (vs. 8-week cPtenf/fGrp78f/f).
p<0.01 (vs. 4- and 8-week WT, respectively).
Figure 2.

Effect of Grp78 deficiency on the uterine weight of mouse cohorts in Pten-null mediated endometrial cancer model. Individual uterine weights (g) and ratios of uterus/total body weight of mice from each genotype at 10 days, 4 weeks, and 8 weeks. Red bar indicates mean values. **p<0.01.
To account for any variability in constitutive differences in mouse size, uterine weights were individually normalized against the total body weights (TBW). There was no statistically significant difference in mean TBW or normalized uterine weights across the cPtenf/fGrp78+/+, cPtenf/fGrp78f/+, cPtenf/fGrp78f/f mice at 10 days regardless of their Grp78 status (p-values >0.7, Figure 2). However, at 4- and 8-weeks, both cPtenf/fGrp78+/+ and cPtenf/fGrp78f/+ mice had significantly greater normalized uterine weights compared to WT and cPtenf/fGrp78f/f mice (both p<0.01, Figure 2). While the normalized uterine weights for 4 week-old WT and cPtenf/fGrp78f/f mice were similar, interestingly, normalized uterine weights from cPtenf/fGrp78f/f mice at 8 weeks were also significantly smaller than that from WT mice (p =0.01, Figure 2).
Endometrioid adenocarcinoma is histologically undetectable after conditional Grp78 deletion from the endometrium
To determine whether GRP78 blocked or delayed EAC development, the uteri from all mice were anatomically and histologically evaluated from 10 days onwards by a gynecologic pathologist (PMF). Anatomic inspection of uteri at 4- and 8-weeks showed no morphologic differences between cPtenf/fGrp78f/f and WT mice (Figure 3a). Histologically, uteri from cPtenf/fGrp78+/+ mice as early as 10 days showed complex atypical hyperplasia (CAH), and well-differentiated (i.e., Grade 1) EAC was observed as early as 11 days (Figure 3b). EAC was seen in 82% of cPtenf/fGrp78+/+ mice, and 92% of cPtenf/fGrp78f/+ mice by 4 weeks (Figure 3c). At 8 weeks, 80% of cPtenf/fGrp78+/+ mice and 94% of cPtenf/fGrp78f/+ mice demonstrated EAC. Remarkably, however, at 4- and 8-weeks, no EAC was observed in the cPtenf/fGrp78f/f mice (all p-values <0.001). (Figure 3c).
Figure 3.

Grp78 ablation durably blocks Pten-null mediated endometrial tumorigenesis. (a) Representative gross morphology of mouse uteri from the indicated genotypes at 4- and 8-weeks. Scale bar represents 1 cm. (b) Representative histologic sections (10x) of complex atypical hyperplasia (CAH) of the endometrium and EAC detectable at 10 days. Scale bar represents 200 μm. (c) Representative hematoxylin & eosin (H&E) microscopy cross-sections (40x) of murine uteri from indicated genotypes at 10 days, 4- and 8-weeks. Scale bar represents 50 μm.
To characterize whether EAC development was halted or delayed, we performed necropsies of the cPtenf/fGrp78f/f mice at 4- and 5-months. At both time points, no cases of EAC were identified (Figure 4). In contrast, 80% and 100% of cPtenf/fGrp78+/+ and cPtenf/fGrp78f/+ mice, respectively, developed EAC at 4 months. Mice with both Pten and Grp78 inactivated (cPtenf/fGrp78f/f) failed to develop any EAC even after 8 months, findings similar to that seen in WT mice (log-rank p=1.0, Figure 4). Collectively, our data demonstrated that homozygous Grp78 deletion completely blocked development of both Pten-mediated EAC and its precursor, CAH, while heterozygous Grp78 deletion could significantly delay EAC development (log-rank p=0.03, Figure 4) but was insufficient to prevent tumor development altogether (log-rank p<0.0001, Figure 4).
Figure 4.
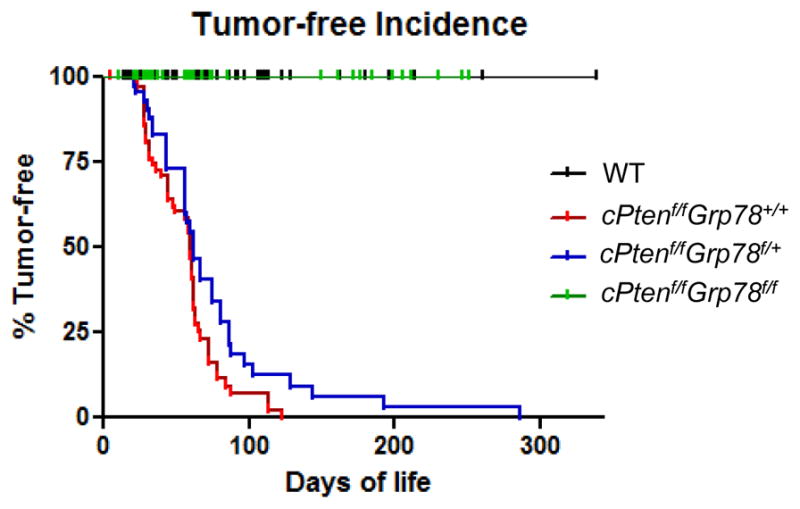
Homozygous deletion of Grp78 durably inhibits PTEN null-mediated endometrial cancer development. Tumor-free incidences of the indicated mouse genotypes plotted against the days of life. WT mice lacking Cre-recombinase served as WT controls.
GRP78 deficiency inhibits AKT activation in Pten-null endometrium
AKT activation is an important mechanism in the development and progression of many solid tumors, including this Pten-null EAC model. Recent in vitro evidence shows that GRP78 is needed for optimal AKT activation in EAC19; therefore, we sought to determine whether genetic deletion of Grp78 from the endometrial epithelia could block AKT activation in cPtenf/fGrp78f/f uteri. As demonstrated by immunofluorescent staining followed by confocal microscopy, endometrial tumors harvested from necropsy showed robust expression of AKT and GRP78 in cPtenf/fGrp78+/+ endometrial epithelial cells (Figure 5a). Although AKT was activated in the cPtenf/fGrp78+/+ uteri as evidenced by serine 473 phosphorylation, this activation was markedly suppressed in the cPtenf/fGrp78f/f uteri, correlating with the lack of GRP78 expression (Figure 5a). This result was confirmed in Western blots using lysates from WT, cPtenf/fGrp78+/+ and cPtenf/fGrp78f/f uteri (Figure 5b). We further determined that S6 phosphorylation (serine 235/236), which is downstream of AKT activation was also suppressed in cPtenf/fGrp78f/f uteri (Figure 5c). Dual immunofluorescent staining of pAKT and GRP78 in 4 week cPtenf/fGrp78+/+ uteri further demonstrated that GRP78 colocalized with pAKT at the cell surface (Figure 5d).
Figure 5.

Pten-null mediated AKT activation was suppressed in uteri of cPtenf/fGrp78f/f mice. (a) Immunofluorescent staining for pAKT (Ser473) (red) and GRP78 (green) in cPtenf/fGrp78+/+ and cPtenf/fGrp78f/f mice uteri (4 weeks). Nuclei were stained with DAPI (blue). Scale bar, 50 μm. (b) Western blot analysis of pAKT and total AKT from uteri of the indicated genotypes. (c) Immunofluorescent staining of phospho-S6 (Ser235/236) (green) and S6 (red) on uteri of indicated genotypes at 4 weeks. Nuclei were stained with DAPI (blue). Scale bar, 200 μm. (d) Co-immunofluorescent staining for pAKT (red) and GRP78 (green) in 4 week cPtenf/fGrp78+/+ mice uteri. Nuclei were stained with DAPI (blue). Yellow represents the colocalization of pAKT and GRP78. Negative control (Neg ctrl) is without primary antibodies. Each row represents one individual mouse uterus. Scale bar, 20 μm.
UPR activation is often associated with tumorigenesis.15 In the Pten-null EAC model, eIF2α phosphorylation downstream of PERK signaling was observed in the cancerous cPtenf/fGrp78+/+ uteri but not in the WT (Figure 6). Phosphorylation of eIF2α was also detected in the cPtenf/fGrp78f/f uteri, consistent with previous reports of eIF2α activation in GRP78 knockdown in other cell types (Figure 6).33 In contrast, the expression of CHOP, a pro-apoptotic UPR marker downstream of PERK signaling, was detected in only a few cells in the cPtenf/fGrp78+/+ uteri and not detected in the WT or the cPtenf/fGrp78f/f uteri (Figure 6).
Figure 6.

Analysis of UPR markers in the mouse uteri. Immunohistochemical staining of p-eIF2α, eIF2α and CHOP on uteri of indicated genotypes at 4 weeks. Examples of CHOP-positive cells are indicated by arrows and the insert showed an enlarged image of a CHOP-positive cell. Scale bar, 100 μm.
Therapeutic targeting of cell surface GRP78 induces endometrial cancer regression
The detection of GRP78 at the surface of EAC cells presents a key opportunity for therapeutic targeting.34 To test this, 3-week-old cPtenf/fGrp78+/+ mice were randomized to receive either IgG-vehicle or the highly specific anti-GRP78 antibody, MAb159 via biweekly intraperitoneal (i.p.) injections for 4 weeks. Uteri were collected at necropsy and examined with a gynecologic pathologist (PMF) blinded to the treatment group. Uteri in both treatment groups were morphologically abnormal with indurated uterine horns; however, MAb159-treated uteri were generally smaller (about 40%) than IgG-treated uteri (Figure 7a). After 4 weeks of treatment, uterine cross-sections from the IgG group showed well-differentiated endometrioid adenocarcinoma defined as and as evidenced by back-to-back endometrial glands exhibiting hyperchromatic nuclei, and prominent nucleoli (Figure 7b). There are also focal areas with superficial myometrial invasion. Uteri from the MAb159-treated group also showed similar morphology as those in the IgG-treated group, but MAb159-treated uteri also showed larger areas of necrosis than their IgG counterparts (Figure 7b). To determine whether or not GRP78-targeting therapy is capable of suppressing AKT and S6 activation, we analyzed the uteri from cPtenf/fGrp78+/+ mice for pAKT and pS6. Both AKT and S6 activation were observed in established cPtenf/fGrp78+/+ EAC tumors at 7 weeks (Figure 7c). Therapeutic delivery of MAb159 against cell surface GRP78 mitigates AKT and S6 activation and results in decreased expression of pro-tumorigenic pAKT and pS6. Furthermore, TUNEL staining revealed an increase in apoptotic cells after MAb159-treatment compared to IgG-vehicle treated cells (Figure 7c), which, taken together, provides additional support for GRP78 as a therapeutic target for EAC treatment by suppressing AKT activation and increasing apoptosis.
Figure 7.

Monoclonal antibody targeting of GRP78 suppressed tumor growth and AKT activation, while increasing apoptosis. (a) Left: representative gross; Right: the relative uterine size in the two treatment groups were graphed (*p<0.05). (b) H&E, (c) pAKT, pS6, and TUNEL stained uterine sections (20x) from cPtenf/fGrp78+/+ mice treated from week 3 to week 7 with control IgG versus MAb159 (10 mg/kg intraperitoneal twice weekly x 4 weeks). In control IgG treated uterus, nests of tumor demonstrate strong expression for pAKT and pS6 but less apoptosis, whereas pAKT and pS6 expression is significantly decreased while apoptosis is increased in uterine tumors after MAb159 treatment. Quantification data are presented as mean ± SEM (n=3 per treatment group). Signals were quantified with Image J. *p<0.02; **p<0.002, as determined by an unpaired 2-tail student t-test. Scale bar, (a) 1 cm, (b) 200 μm, (c) 100 μm. SEM: standard error of the mean.
DISCUSSION
We use a conditional knockout mouse model of EAC to demonstrate that the specific ablation of GRP78 from the endometrium inhibits AKT activation while durably preventing PTEN-mediated EAC development even beyond 300 days of life. Another key finding of particular importance in our work was the prevention of the EAC precursor, complex atypical hyperplasia, after tissue-specific GRP78 knockout. Furthermore, we also show that homozygous Grp78 deletion in the endometrial epithelial cells not only prevents EAC development, it also interrupts and attenuates AKT activation mediated by PTEN loss. Interestingly, re-expression of GRP78 to the WT level was observed in mice still harboring a single functional Grp78 allele, suggesting some compensatory response to restore normal GRP78 levels. While the EAC tumor formation was delayed in these mice, they eventually developed EAC, similar to the cPtenf/fGrp78+/+ mice. Importantly, with GRP78 detectable on the surface of the EAC cells, we show that GRP78 is therapeutically targetable with a monoclonal antibody, and that GRP78-targeted therapy results in reduction of AKT activation and increased areas of tumoral apoptosis.
The critical role of GRP78 in the development of several human cancers and the manifestation of chemo- or hormone-resistant phenotypes is well-documented.18,35,36 Higher GRP78 expression levels in tumors have been correlated with more aggressive tumor characteristics (e.g., invasion, lymph node involvement) and worse clinical outcome (e.g., survival).14 The level of GRP78 expression in clinical samples of invasive breast carcinoma has been shown to predict responsiveness to certain cytotoxic chemotherapies, such as anthracyclines and alkylating agents.18,37 Meanwhile, in cell lines, high GRP78 levels appear to be associated with aggressive phenotypes, including increased growth, invasiveness, decreased apoptosis and increased survival.34,38 GRP78 also plays an integral role in several critical cell signaling pathways, many of which (e.g., PI3K/AKT), figure prominently in human cancers.39
In vitro and in vivo cancer models support the relationship between GRP78 and AKT activation.14,27,32,40,41 For example, in PTEN loss driven prostate cancer and leukemia, a partial reduction of GRP78 suppresses AKT activation.6,7 But, how GRP78 affects AKT activation could be multifactorial. Although GRP78 is generally regarded as an ER resident protein, a subfraction of GRP78 localizes at the cell surface in selected cell types, notably cancer cells.27,41 Cell surface GRP78, through its interaction with different ligands and cell surface proteins, can mediate important signal transduction pathways including PI3K/AKT and this could play a major role since MAb159 targeting cell surface GRP78 is highly effective in suppressing AKT activation in a variety of tumors, including EAC. However, it is also possible that, as a major molecular chaperone in the ER, GRP78 may be required for processing important growth factors and the cell surface expression of their corresponding receptors, which might regulate AKT activation.
One proposed mechanism in which cell surface GRP78 activates AKT is by complexing with PI3K to promote PIP3 formation.28 Consistent with this notion, colocalization of GRP78 with pAKT on the cell surface in Pten-null EAC was detected, similar to the phenomena seen in pancreatic adenocarcinoma.42 Collectively, we report here for the first time that, in this murine Pten-null EAC, endometrium-specific GRP78 deletion is able to suppress AKT and S6 activation. Our targeted genetic deletion of GRP78 using floxed murine models provides compelling evidence to explore and identify pharmacologic means to block GRP78 and further represents a therapeutic opportunity to exploit this target.14,30
In endometrial cancer, perturbation of cell surface GRP78 with a polyclonal antibody directed against its C-terminus induced apoptosis in AN3CA cells in vitro in association with reduced AKT phosphorylation.34 We have recently identified a highly specific monoclonal antibody against GRP78 that is capable of inhibiting PI3K/AKT activation and suppressing tumorigenesis, thus, representing a new anti-cancer therapeutic strategy.32 In the current study, monoclonal antibody targeting of GRP78 in vivo not only resulted in decreased tumor with increased tumoral apoptosis, but also in the suppression of AKT activation, further supporting the interplay between GRP78 and AKT during EAC development and growth. Given the pleiotropic role of GRP78 in tumorigenesis and progression, additional mechanisms for this anti-tumor phenotype towards interruption of endometrial epithelial cell growth and maturation, merits further exploration.
In the decades since identifying the link between EAC, obesity and hyperestrogenism, significant progress has been made to better understand the molecular and genetic underpinnings of endometrial carcinoma.43 The whole genome characterization of endometrial carcinomas by The Cancer Genome Atlas (TCGA) further implicates PTEN mutations and co-existing AKT pathway alterations as common genomic events early in the development of EAC.2 Endometrioid adenocarcinomas are clearly the more prevalent form of EAC, accounting for >65% of all EACs. This type of EAC also has a recognizable precancerous state, complex atypical hyperplasia, which is blocked experimentally by tissue-specific GRP78 deletion. Given the pervasiveness of EAC due to the obesity epidemic, preventive and therapeutic strategies to block obesity-related CAH and EAC carry significant potential to deliver a tremendous public health impact.
MATERIALS AND METHODS
Mouse models
The parental male mouse PRCre/+ Ptenf/f, on a C57BL6/129SV genetic background5 was mated with parental female mouse Ptenf/fGrp78f/f on a C57/BL6xDBA2/129 genetic background.6 These mice were then crossed over successive generations to yield, first, PRCre/+Ptenf/fGrp78f/+ mice, then PRCre/+Ptenf/fGrp78f/f (cPtenf/fGrp78f/f) mice (Supplementary Figure 1). Mice without the PRCre/+ allele served as the phenotypic wild-type controls. All animal experiments were conducted in concordance with the U.S. Public Health Service Policy on Human Care and Use of Laboratory Animals after protocol approval from the USC Institutional Animal Care and Use Committee (IACUC). Genotyping of tail or uteri genomic DNA were performed using PCR primers (Supplementary Table 1) as previously described.5,6 At various time points, all uteri were harvested, weighed, and measured at the time of necropsy, just prior to snap freezing for tissue lysate or fixation in 10% buffered formalin for microscopic examination. Tissue morphology was examined after hematoxylin-eosin (H&E) staining of 5 μm thick formalin-fixed paraffin-embedded (FFPE) sections. All histologic diagnoses were rendered by a gynecologic pathologist with expertise in gynecologic cancers (PMF).
Immunohistochemical staining
Antibodies against GRP78 (Abcam, Cambridge, MA; 1:1,000), progesterone receptor (PgR 636, Dako, Carpinteria, CA; 1:50), PTEN (D4.3 XP, Cell Signaling, Danvers, MA; 1:2,000), p-eIF2α (Ser51, D9G8, Cell Signaling Danvers, MA; 1:50), eIF2α (Cell Signaling, Danvers, MA; 1:200), and CHOP (Santa Cruz Biotechnology, Dallas, TX; 1:50) were used for immunohistochemistry on 5 μm thick FFPE slides as previously described.6 Briefly, after deparaffinization and rehydration, standard antigen retrieval (Retrievagen A, BD Biosciences, San Jose, CA) and blocking of endogenous peroxidases (3% H2O2/PBS) and nonspecific epitopes (5% normal horse serum) were performed. Sections were incubated with the appropriate primary antibody overnight at 4°C with the appropriate negative control. Detection was performed using the antibody-specific Vectastain Elite® Avidin-Biotin Complex (ABC) kit (Vector Laboratories, Burlingame, CA), and visualization was achieved with 3,3′-diaminobenzidine (DAB, BD Biosciences) and Gill’s No. 3 hematoxylin (Sigma-Aldrich, St. Louis, MO) counterstaining.
Phospho-AKT (Ser473, Cell Signaling), and phospho-S6 (Ser235/236 Cell Signaling), immunohistochemistry was performed on FFPE sections, as previously described.32 Briefly, sections were de-waxed and rehydrated before endogenous peroxidases and nonspecific epitopes were blocked. Incubation with the respective primary antibody was performed overnight, followed by counterstaining and visualization, as previously described.32 Images were acquired with an Olympus BX51 microscope and Image-Pro+ (Version 6.0).
Immunofluorescent staining and TUNEL
Freshly cut FFPE sections were evaluated by immunofluorescent staining, as previously described.6 The following primary antibodies were used: anti-GRP78 (C-20; Santa Cruz Biotechnology; 1:50), anti-phospho-AKT (Ser473) (D9E XP; Cell Signaling Technology; 1:50), anti-phospho-S6 (Ser235/236, Cell Signaling; 1:100), and anti-S6 (Cell Signaling; 1:100). DAPI was used for nuclear staining. Immunofluorescence was analyzed by Zeiss LSM 510 confocal microscope with LSM 510 Version 4.2 SP1 acquisition software. Confocal images were acquired with 40X and 100X oil lens, and processed with LSM Image Browser R4.2 and Adobe Photoshop CS5.
Apoptosis was measured using the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) fluorescent kit (Promega, Madison, WI) on FFPE sections, according to manufacturer’s instructions, as previously described.44 The percentage of TUNEL-positive cells was determined using ImageJ.
Western blot analysis
Snap frozen murine uteri collected at necropsy were homogenized (OMNI tissue master) in cold M-PER (mammalian protein extraction reagent) supplemented with cComplete, Mini and phosStop phosphatase inhibitor (Roche, Mannheim, Germany) on ice. The protein was extracted after the lysate was centrifuged (4°C, 14,000 rpm, 10 min), and quantified using the Bradford assay kit (Bio-Rad Laboratories, Hercules, CA). As previously described,19 20 μg of lysate was run on a 10% SDS-PAGE gel then transferred to PVDF membranes. Nonspecific epitopes were blocked with Odyssey blocking buffer (Li-COR) and membranes were incubated with the appropriate primary antibodies – anti-GRP78 (1:2000, Abcam), anti-pAKT (Ser473) (1:1000, Cell Signaling), anti-AKT (1:1000), and anti-PTEN (1:1000, Cell Signaling) overnight at 4°C. Vinculin (1:5000, Sigma) served as a loading control. Odyssey secondary antibodies (goat anti-rabbit IRDye680 and goat anti-mouse IRDye800) were used as appropriate, and proteins were visualized and quantified using the Li-COR Odyssey CLx Infrared Imaging System (LI-COR, Lincoln, NE).
In vivo monoclonal antibody treatment
Three-week-old cPtenf/fGrp78+/+ mice were randomized into two treatment groups: IgG-vehicle and MAb159, a highly specific anti-GRP78 antibody. The mice were treated with either IgG-vehicle or MAb159 (10 mg/kg) by intraperitoneal injection twice weekly for 4 weeks after which time mice were euthanized. Uteri were collected at necropsy for analysis.
Statistical analysis
Data regarding mouse and uterine weights are expressed as mean ± SD. The Mann-Whitney rank-sum test was used to analyze nonparametric and non-normally distributed data. Log-rank analyses were used to compare survival curves. A 2-tailed p-value <0.05 was considered statistically significant. Analyses were performed using GraphPad (Prism Version 5, La Jolla, CA).
Supplementary Material
Acknowledgments
Sources of Support: This work was supported by the USC Department of Obstetrics/Gynecology Seed Grant (YGL), Stop Cancer Career Development Award (YGL), NIH Grant R01 CA027607 (ASL), NIH Grant K08 CA175161 (YGL), and NCI Cancer Center Support Grant (P30 CA014089, ASL/YGL). Microscopy at the Cell and Tissue Imaging Core of the USC Research Center for Liver Diseases was supported by NIH Grant P30 DK048522.
The authors thank the USC Norris Comprehensive Cancer Center Translational Pathology Core for tissue processing and the USC Research Center for Liver Diseases Cell and Tissue Imaging Core. The authors would also like to thank the members of the Lee lab, particularly Wan-Ting Chen, Genyuan Zhu, and Michelle Pong for their input and assistance throughout this project.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iglesias DA, Bodurka DC. Personalized care in uterine cancer. Clin Adv Hematol Oncol. 2012;10:797–805. [PMC free article] [PubMed] [Google Scholar]
- 4.Leslie KK, Thiel KW, Goodheart MJ, De Geest K, Jia Y, Yang S. Endometrial cancer. Obstet Gynecol Clin North Am. 2012;39:255–268. doi: 10.1016/j.ogc.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daikoku T, Hirota Y, Tranguch S, Joshi AR, DeMayo FJ, Lydon JP, et al. Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer Res. 2008;68:5619–5627. doi: 10.1158/0008-5472.CAN-08-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu Y, Wey S, Wang M, Ye R, Liao CP, Roy-Burman P, et al. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc Natl Acad Sci USA. 2008;105:19444–19449. doi: 10.1073/pnas.0807691105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wey S, Luo B, Tseng CC, Ni M, Zhou H, Fu Y, et al. Inducible knockout of GRP78/BiP in the hematopoietic system suppresses Pten-null leukemogenesis and AKT oncogenic signaling. Blood. 2012;119:817–825. doi: 10.1182/blood-2011-06-357384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conley-LaComb MK, Saliganan A, Kandagatla P, Chen YQ, Cher ML, Chinni SR. PTEN loss mediated Akt activation promotes prostate tumor growth and metastasis via CXCL12/CXCR4 signaling. Mol Cancer. 2013;12:85. doi: 10.1186/1476-4598-12-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cordero-Espinoza L, Hagen T. Increased concentrations of fructose 2,6-bisphosphate contribute to the Warburg effect in phosphatase and tensin homolog (PTEN)-deficient cells. J Biol Chem. 2013;288:36020–36028. doi: 10.1074/jbc.M113.510289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 11.He L, Hou X, Kanel G, Zeng N, Galicia V, Wang Y, et al. The critical role of AKT2 in hepatic steatosis induced by PTEN loss. Am J Pathol. 2010;176:2302–2308. doi: 10.2353/ajpath.2010.090931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen WT, Tseng CC, Pfaffenbach K, Kanel G, Luo B, Stiles BL, et al. Liver-specific knockout of GRP94 in mice disrupts cell adhesion, activates liver progenitor cells, and accelerates liver tumorigenesis. Hepatology. 2014;59:947–957. doi: 10.1002/hep.26711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581:3641–3651. doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014;14:263–276. doi: 10.1038/nrc3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2013;32:805–818. doi: 10.1038/onc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, Zanetti M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc Natl Acad Sci USA. 2011;108:6561–6566. doi: 10.1073/pnas.1008942108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bifulco G, Miele C, Di Jeso B, Beguinot F, Nappi C, Di Carlo C, et al. Endoplasmic reticulum stress is activated in endometrial adenocarcinoma. Gynecol Oncol. 2012;125:220–225. doi: 10.1016/j.ygyno.2011.11.045. [DOI] [PubMed] [Google Scholar]
- 18.Lee E, Nichols P, Spicer D, Groshen S, Yu MC, Lee AS. GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res. 2006;66:7849–7853. doi: 10.1158/0008-5472.CAN-06-1660. [DOI] [PubMed] [Google Scholar]
- 19.Gray MJ, Mhawech-Fauceglia P, Yoo E, Yang W, Wu E, Lee AS, et al. AKT inhibition mitigates GRP78 (glucose-regulated protein) expression and contribution to chemoresistance in endometrial cancers. Int J Cancer. 2013;133:21–30. doi: 10.1002/ijc.27994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmandt RE, Iglesias DA, Co NN, Lu KH. Understanding obesity and endometrial cancer risk: opportunities for prevention. Am J Obstet Gynecol. 2011;205:518–525. doi: 10.1016/j.ajog.2011.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuo K, Gray MJ, Yang DY, Srivastava SA, Tripathi PB, Sonoda LA, et al. The endoplasmic reticulum stress marker, glucose-regulated protein-78 (GRP78) in visceral adipocytes predicts endometrial cancer progression and patient survival. Gynecol Oncol. 2013;128:552–559. doi: 10.1016/j.ygyno.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer. 2004;4:966–977. doi: 10.1038/nrc1505. [DOI] [PubMed] [Google Scholar]
- 24.Wu J, Kaufman RJ. From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 2006;13:374–384. doi: 10.1038/sj.cdd.4401840. [DOI] [PubMed] [Google Scholar]
- 25.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 26.Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J. 2011;434:181–188. doi: 10.1042/BJ20101569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Liu R, Ni M, Gill P, Lee AS. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J Biol Chem. 2010;285:15065–15075. doi: 10.1074/jbc.M109.087445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Tseng CC, Tsai YL, Fu X, Schiff R, Lee A. Cancer cells resistant to therapy promote cell surface relocalization of GRP78 which complexes with PI3K and enhances PI(3,4,5)P3 production. PLoS One. 2013;8:e80071. doi: 10.1371/journal.pone.0080071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gonzalez-Gronow M, Selim MA, Papalas J, Pizzo SV. GRP78: a multifunctional receptor on the cell surface. Antioxid Redox Signal. 2009;11:2299–2306. doi: 10.1089/ARS.2009.2568. [DOI] [PubMed] [Google Scholar]
- 30.Sato M, Yao VJ, Arap W, Pasqualini R. GRP78 signaling hub a receptor for targeted tumor therapy. Adv Genet. 2010;69:97–114. doi: 10.1016/S0065-2660(10)69006-2. [DOI] [PubMed] [Google Scholar]
- 31.Arap MA, Lahdenranta J, Mintz PJ, Hajitou A, Sarkis AS, Arap W, et al. Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell. 2004;6:275–284. doi: 10.1016/j.ccr.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 32.Liu R, Li X, Gao W, Zhou Y, Wey S, Mitra SK, et al. Monoclonal antibody against cell surface GRP78 as a novel agent in suppressing PI3K/AKT signaling, tumor growth and metastasis. Clin Cancer Res. 2013;19:6802–6811. doi: 10.1158/1078-0432.CCR-13-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wey S, Luo B, Lee AS. Acute inducible ablation of GRP78 reveals its role in hematopoietic stem cell survival, lymphogenesis and regulation of stress signaling. PLoS One. 2012;7:e39047. doi: 10.1371/journal.pone.0039047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cali G, Insabato L, Conza D, Bifulco G, Parrillo L, Mirra P, et al. GRP78 mediates cell growth and invasiveness in endometrial cancer. J Cell Physiol. 2014;229:1417–1426. doi: 10.1002/jcp.24578. [DOI] [PubMed] [Google Scholar]
- 35.Pootrakul L, Datar RH, Shi SR, Cai J, Hawes D, Groshen SG, et al. Expression of stress response protein Grp78 is associated with the development of castration-resistant prostate cancer. Clin Cancer Res. 2006;12:5987–5993. doi: 10.1158/1078-0432.CCR-06-0133. [DOI] [PubMed] [Google Scholar]
- 36.Li J, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med. 2006;6:45–54. doi: 10.2174/156652406775574523. [DOI] [PubMed] [Google Scholar]
- 37.Lee E, Nichols P, Groshen S, Spicer D, Lee AS. GRP78 as potential predictor for breast cancer response to adjuvant taxane therapy. Int J Cancer. 2011;128:726–731. doi: 10.1002/ijc.25370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhuang L, Scolyer RA, Lee CS, McCarthy SW, Cooper WA, Zhang XD, et al. Expression of glucose-regulated stress protein GRP78 is related to progression of melanoma. Histopathology. 2009;54:462–470. doi: 10.1111/j.1365-2559.2009.03242.x. [DOI] [PubMed] [Google Scholar]
- 39.Zhang LH, Yang XL, Zhang X, Cheng JX, Zhang W. Association of elevated GRP78 expression with increased astrocytoma malignancy via Akt and ERK pathways. Brain Res. 2011;1371:23–31. doi: 10.1016/j.brainres.2010.11.063. [DOI] [PubMed] [Google Scholar]
- 40.Misra UK, Deedwania R, Pizzo SV. Activation and cross-talk between Akt, NF-{kappa}B, and unfolded protein response signaling in 1-LN prostate cancer cells consequent to ligation of cell surface-associated GRP78. J Biol Chem. 2006;281:13694–13707. doi: 10.1074/jbc.M511694200. [DOI] [PubMed] [Google Scholar]
- 41.Shin BK, Wang H, Yim AM, Le Naour F, Brichory F, Jang JH, et al. Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J Biol Chem. 2003;278:7607–7616. doi: 10.1074/jbc.M210455200. [DOI] [PubMed] [Google Scholar]
- 42.Hill R, Li Y, Tran LM, Dry S, Calvopina JH, Garcia A, et al. Cell intrinsic role of COX-2 in pancreatic cancer development. Mol Cancer Ther. 2012;11:2127–2137. doi: 10.1158/1535-7163.MCT-12-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bokhman JV, Chepick OF, Volkova AT, Vishnevsky AS. Can primary endometrial carcinoma stage I be cured without surgery and radiation therapy? Gynecol Oncol. 1985;20:139–155. doi: 10.1016/0090-8258(85)90135-0. [DOI] [PubMed] [Google Scholar]
- 44.Liu R, Ferguson BD, Zhou Y, Naga K, Salgia R, Gill PS, et al. EphB4 as a therapeutic target in mesothelioma. BMC Cancer. 2013;13:269. doi: 10.1186/1471-2407-13-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.