

Ca2+-activated large-conductance K+ (BKCa) channel activation may be an important strategy to alleviate myocardial stunning, a significant clinical problem. We demonstrate that BKCa activation with the small molecule rottlerin is potently cardioprotective for ischemic insults associated with hypothermic cardioplegic arrest and reperfusion. Activation of BKCa promotes reduced reactive oxygen species and preserves mitochondrial membrane potential.
Keywords: ischemia, mitochondria, potassium channel, reperfusion, reactive oxygen species
Abstract
Mitochondrial Ca2+-activated large-conductance K+ (BKCa) channels are thought to provide protection during ischemic insults in the heart. Rottlerin (mallotoxin) has been implicated as a potent BKCa activator. The purpose of this study was twofold: 1) to investigate the efficacy of BKCa channel activation as a cardioprotective strategy during ischemic cardioplegic arrest and reperfusion (CP/R) and 2) to assess the specificity of rottlerin for BKCa channels. Wild-type (WT) and BKCa knockout (KO) mice were subjected to an isolated heart model of ischemic CP/R. A mechanism of rottlerin-induced cardioprotection was also investigated using H9c2 cells subjected to in vitro CP/reoxygenation and assessed for mitochondrial membrane potential and reactive oxygen species (ROS) production. CP/R decreased left ventricular developed pressure, positive and negative first derivatives of left ventricular pressure, and coronary flow (CF) in WT mice. Rottlerin dose dependently increased the recovery of left ventricular function and CF to near baseline levels. BKCa KO hearts treated with or without 500 nM rottlerin were similar to WT CP hearts. H9c2 cells subjected to in vitro CP/R displayed reduced mitochondrial membrane potential and increased ROS generation, both of which were significantly normalized by rottlerin. We conclude that activation of BKCa channels rescues ischemic damage associated with CP/R, likely via effects on improved mitochondrial membrane potential and reduced ROS generation.
NEW & NOTEWORTHY
Ca2+-activated large-conductance K+ (BKCa) channel activation may be an important strategy to alleviate myocardial stunning, a significant clinical problem. We demonstrate that BKCa activation with the small molecule rottlerin is potently cardioprotective for ischemic insults associated with hypothermic cardioplegic arrest and reperfusion. Activation of BKCa promotes reduced reactive oxygen species and preserves mitochondrial membrane potential.
mitochondrial k+ channels are potential therapeutic targets for improved myocardial protection. Mitochondrial K+ channels reside in the inner mitochondrial membrane and regulate K+ flux to the mitochondrial matrix. Agents that activate the mitochondrial ATP-sensitive K+ channel (mitoKATP) have long been investigated as cardioprotective agents and are known to improve cardiomyocyte and whole heart contractile function, survival, and mitochondrial function following ischemic insults (18, 21, 23, 27, 28, 35). More recently, the Ca2+-activated large-conductance K+ (BKCa) channel has also been recognized as having potent cardioprotective properties. BKCa activation reduces myocardial infarction, promotes survival, and improves cardiac and vascular function following severe ischemic insults (3, 7, 9, 12, 31, 32, 34, 36). In addition, BKCa channels may be more potently cardioprotective than mitoKATP channels (17).
Activation of BKCa channels may also provide effective cardioprotection in the case of reversible ischemic insults associated with cardiac surgery for coronary artery bypass grafting or valve repair. Cardiac surgeries with cardioplegia and cardiopulmonary bypass provide protection to the heart and tissue from damage that could otherwise prove lethal or result in massive myocardial infarction (8, 19, 25). However, surgical cardioprotection is not perfect, and ischemic insults associated with cardiac surgery include reduced contractile function after surgery (myocardial stunning) and propensity for vasoconstriction and vasospasm in the coronary circulation (30).
We recently demonstrated that the small molecule rottlerin (mallotoxin) enhances cardioprotection following ischemic cardioplegic arrest and reperfusion (CP/R) in a rat model of mild ischemic injury and myocardial stunning (10). Early evidence indicated that the cardioprotective effect of rottlerin was mediated by BKCa channels as demonstrated by pharmacological inhibition of the channel with paxilline. In addition, rottlerin can directly activate BKCa channels as demonstrated by stimulation of in vitro and smooth muscle BKCa currents and rottlerin-induced smooth muscle relaxation (13, 15, 38, 41). In addition to displaying superior potency, rottlerin lacks many of the detrimental side effects seen with the most commonly used BKCa activators, NS11021 and NS1619 (6, 36, 41). The purpose of this study was to establish 1) the specificity of rottlerin for BKCa channels and 2) the efficacy and potential mechanism of BKCa activation for improved cardioprotection during ischemic CP/R.
METHODS
Animal studies.
All mouse studies were approved by the Rhode Island Hospital Institutional Animal Care and Use Committee (IACUC). BKCa−/− mice on an FVB background were kindly provided by Andrea L. Meredith at the University of Maryland School of Medicine (26). Wild-type (WT) littermates were used as controls for the majority of experiments. For dose-response studies, FVB mice were purchased from Charles River Laboratories (Wilmington, MA).
Isolated Langendorff perfused model of CP.
Isolated heart model of CP/R was performed similar to previous publications (10, 11). FVB WT and kcnma1−/− mice were anesthetized intraperitoneally (ip) with 80 mg/kg ketamine and 0.5 mg/kg dexmedatomidine and anticoagulated with heparin (2,000 U/kg iv), and the heart was rapidly exposed. The aorta was immediately cannulated and retrograde perfused in Langendorff mode in a water-jacketed organ chamber and perfusion system (IH-SR; Harvard Apparatus, Boston, MA). Following cannulation, the heart was cleaned of excess tissue and vessels, the left atrium was removed, and a balloon was placed in the left ventricle (LV). LV end diastolic pressure was set to ∼8 mmHg at the beginning of the experiment. A temperature probe was placed to monitor coronary effluent temperature. The hearts were perfused in constant pressure mode (∼70 mmHg) with a modified Krebs-Henseleit buffer (KHB) (in mM) (118.00 NaCl, 4.70 KCl, 1.40 CaCl2, 1.70 MgSO4, 24.88 NaHCO3, 6.00 glucose, 1.20 KH2PO4, and 2.00 Na pyruvate) for 30 min to stabilize and record baseline measurements. During baseline measurements, myocardial temperature was maintained at 37°C. Groups subjected to cold crystalloid cardioplegia solution were perfused with St. Thomas Hospital Solution II (in mM) (110.0 NaCl, 16.0 KCl, 16.0 MgCl2, 1.5 CaCl2, and 10.0 NaHCO3). Myocardial cooling during CP was initiated at the onset of cardioplegia infusion via rapidly switching the Langendorff organ chamber and perfusate to a refrigerated circulator. Myocardial temperature was maintained at ∼10°C for the duration of CP. Cardioplegia groups were perfused initially for 2 min, followed by a 1-min infusion at 30, 60, and 90 min of arrest. Following 120 min, the organ chamber and perfusate were switched back to a heating circulator, and the heart was perfused at 70 mmHg with modified KHB. Myocardial temperature was subsequently maintained at 37°C. Indices of ventricular function, perfusion pressure, and myocardial temperature were measured continuously throughout the experiment using an LDS-Ponemah data-acquisition system. Coronary flow measurements were assessed via effluent collection at indicated times.
In vitro cell culture, ROS, and mitochondrial membrane potential (ΔΨm).
H9c2 cells (ATCC) were grown to confluence in 10% FBS DMEM and then switched to 1% FBS DMEM for 1 wk to promote increased cardiomyocyte protein expression (cardiac Troponin I and T, cTnI and cTnT). For reactive oxygen species (ROS) and tetramethylrhodamine ethyl ester (TMRE) assays, cells were seeded in black, clear-bottom, 96-well tissue culture plates. Cells were switched to crystalloid cardioplegia solution bubbled with 5% CO2-95% N2 under anoxic conditions supplemented with either vehicle (DMSO), rottlerin, or the BKCa channel inhibitor paxilline at the indicated doses. Cells were then placed in a hypoxia chamber evacuated with 5% CO2-95% N2 for 3 h at 20°C. For simulated reperfusion, cardioplegia-treated cells were removed from the hypoxic chamber and returned to the cell culture incubator in normoxic 1% FBS DMEM for 30 min. Control cells were maintained in a standard cell culture incubator for 3 h in KHB buffer with or without indicated drugs (DMSO, rottlerin) and switched to drug-free medium for 30 min to simulate reperfusion.
For ROS studies, cells were washed in Tyrodes buffer and then loaded with 20 μM 2,7-dichlorofluorescein diacetate (DCFDA) for 12 min before the end of reperfusion. Cells were again washed in Tyrodes buffer, and the rate of fluorescence increase was assessed (excitation 495, emission 530) with 10 sequential readings every 2 min. Following 20 min, maximum fluorescence was assessed by addition of 10 mM H2O2 and immediately measured. All DCFDA measurements were normalized to maximal H2O2 fluorescence.
For TMRE studies, cells were loaded with 1 μM TMRE, 20 min before the end of reperfusion in media. Loading media was then washed twice with PBS, and TMRE fluorescence was measured (excitation 540, emission 575). Following measurement, wells were treated with 10 μM FCCP for 10 min, and background fluorescence was measured. FCCP fluorescence was subtracted from all readings for each well. All measurements were performed using a Biotek Synergy MX plate reader maintained at 37°C.
Mouse myocyte isolation and ΔΨm measurement.
Mouse myocytes were isolated similarly to previously published rat protocols (10, 11). Mouse hearts were rapidly excised as detailed above, and Langendorff was perfused with perfusion media (MEM) (GIBCO, Carlsbad, CA) (Joklik modification supplemented with 20 μM CaCl2, creatine, taurine, and carnitine) for 5 min. Hearts were the perfused for 40–50 min at 4 ml/min at 37°C with digestion buffer consisting of perfusion buffer supplemented with 400 U/ml collagenase II (Worthington Bioproducts, Lakewood, NJ) and 0.2% BSA. When hearts were noticeably soft, they were removed and immersed in perfusion buffer without collagenase or Ca2+ but with 0.5% BSA (incubation buffer). Myocardial chunks were then mechanically disrupted using transfer pipettes to obtain an even suspension. Following disruption, myocytes were washed once in incubation buffer by centrifugation at 700 g. Myocytes were then resuspended in 10–15 ml of incubation buffer and plated on laminin-coated glass coverslips in a 24-well plate. Cells were used in experiments ∼0.5–1.0 h after being plated to allow attachment. Control and CP cells were incubated with DMSO or 500 nM rottlerin for 3 h in incubation buffer at 37°C or cold hypoxic CP at 20°C. Media or CP was then removed and incubated with 100 nM tetramethylrhodamine methyl ester (TMRM) without permeabilization for 10 min at 37°C. After TMRM loading, myocytes were incubated in Tyrodes buffer for 20 min at 37°C followed by imaging. Myocytes were imaged in XY mode of a laser-scanning Leica SPII confocal microscope. TMRM was excited using a 543-nm HeNe laser at a scanning speed of 200 Hz, and emitted signal was collected at 560–660 nm. Baseline fluorescence was measured over 1–2 min following treatment with 20 μM FCCP to dissipate mitochondrial membrane potential and determine Fmin. ΔΨm was determined as baseline fluorescence (F) normalized to Fmin. ΔΨm = (F − Fmin)/Fmin.
LDH release.
Lactate dehydrogenase (LDH) was measured in coronary effluent and media from H9c2 cells using a cytotox96 nonradioactive cytotoxicity assay kit purchased from Promega (Madison, WI). Briefly, coronary effluent was collected for 1 min at the indicated time points and immediately frozen for later measurement. For H9c2 cells, 1 ml of buffer (Tyrodes buffer) was collected after 30 min of reoxygenation. A sample (50 μl) of effluent or buffer was used in the LDH reaction in 96-well plates according to the manufacturer's instructions. Absorbance was measured at 490 nm.
Statistical analysis.
All tests were performed with Sigma Plot software (SyStat, San Jose, CA) or Excel (t-tests). Analysis of whole-heart ischemia/reperfusion studies were performed with two-way repeated-measure ANOVA and Student-Newman-Keuls post hoc test, *P < 0.05 vs. WT control. For H9c2 cell studies on ROS, two-way RM ANOVA and Student-Newman-Keuls test were used to compare the time course of DCFDA fluorescence, and one-way ANOVA was used on Rank's, with Tukey's test for pairwise comparisons for normalized rates of increase; one-way ANOVA and Dunnett's test were used to compare differences in TMRE fluorescence vs. control. For TMRM in cardiomyocytes, significant differences were determined by one-way ANOVA and Student-Newman-Keuls test. For LDH release, two-way ANOVA was used to determine significance in H9c2 cells, and two-way repeated-measures ANOVA was used for control vs. isolated hearts.
RESULTS
Rottlerin dose dependently increases cardiac function following ischemic CP in mouse hearts.
Supplementation of cardioplegia solution with 500 nM rottlerin significantly improved LV developed pressure (LVDP) and positive and negative first derivatives of LV pressure (+/−dP/dt) in mouse hearts (Fig. 1, A and B). Rottlerin (100 nM) resulted in a nonsignificant trend for improved cardiac function.
Fig. 1.
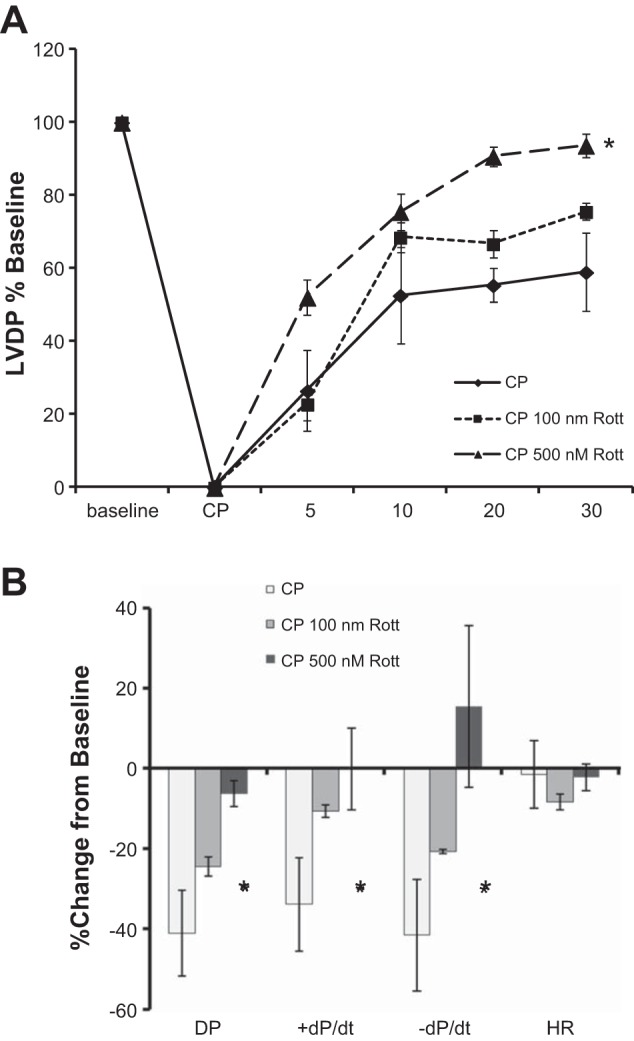
Rottlerin dose dependently increases cardiac contractile function after cardioplegic arrest and reperfusion (CP/R) in isolated mouse hearts. 2-h intermittent cold crystalloid cardioplegia (CP) depresses isolated heart contractile function. Inclusion of rottlerin enhances cardiac function following reperfusion. A: developed pressure (DP), % baseline ± SE, n = 3 (P < 0.05 2-way repeated-measures ANOVA vs. CP). B: summary of hemodynamic data as % change from baseline of the 30-min time point. *P < 0.05 1-Way ANOVA, Student-Newman-Keuls. HR, heart rate; +/−dP/dt, positive and negative first derivatives of left ventricular (LV) pressure.
Rottlerin improves cardiac function via a BKCa channel-dependent mechanism.
Rottlerin significantly improved indices of cardiac function in isolated mouse hearts including LVDP and +/−dP/dt (Fig. 2, A–C). The effects of rottlerin were wholly dependent on BKCa channels, as BKCa knockout (KO) mice treated with and without rottlerin displayed no cardioprotection. Rottlerin had no effect on heart rate (Fig. 2D); however, deletion of the BKCa channel caused a slight depression of heart rate upon reperfusion with or without rottlerin. Figure 1E displays the percentage of change of the 30-min time points compared with baseline. There were no differences in baseline hemodynamic values in any groups as compared with WT controls (Table 1).
Fig. 2.

Rottlerin rescues cold cardioplegia-induced depressed contractile function through activation of Ca2+-activated large-conductance K+ (BKCa) channels. Inclusion of rottlerin (CP + Rott, n = 6) enhances cardiac function following reperfusion but not in BKCa knockout (KO) mice (CP + Rott BKCa−/−). A: DP. B: +dP/dt. C: -dP/dt. D: HR. x-axis, time: baseline − pre-CP function, CP − cardioplegia, 5–30-min reperfusion. *P < 0.05, 2-way repeated-measures (RM) ANOVA, Student-Newman-Keuls vs. wild-type (wt) CP. E: summary of hemodynamic data as % change from baseline of the 30-min time point; n = 8–9/group, *P < 0.05 2-way ANOVA, Student-Newman-Keuls. #Statistically significant effect (P < 0.05) of BKCa KO vs. wt.
Table 1.
Baseline hemodynamic parameters
| Systolic Pressure, mmHg | SE | P Value | Developed Pressure, mmHg | SE | P Value | +dP/dt, mmHg/s | SE | P Value | −dP/dt, mmHg/s | SE | P Value | Coronary Flow, ml/min | SE | P Value | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Wild-type | 84.99 | 2.41 | NA | 76.21 | 2.42 | NA | 3226 | 132 | NA | 2482 | 114 | NA | 2.49 | 0.17 | NA |
| Wild-type + 500 nM rottlerin | 84.68 | 4.60 | 0.95 | 76.02 | 4.55 | 0.97 | 3025 | 181 | 0.39 | 2162 | 140 | 0.10 | 2.54 | 0.38 | 0.91 |
| BKCa−/− | 81.30 | 5.03 | 0.54 | 72.62 | 4.99 | 0.54 | 2974 | 244 | 0.40 | 2311 | 237 | 0.54 | 2.54 | 0.28 | 0.87 |
| BKCa−/− +500 nM Rottlerin | 84.19 | 4.06 | 0.87 | 75.30 | 4.13 | 0.85 | 3325 | 249 | 0.73 | 2513 | 277 | 0.92 | 2.51 | 0.30 | 0.94 |
BKCa, Ca2+-activated large-conductance K+ channel; +/−dP/dt, positive and negative first derivatives of left ventricular pressure.
Rottlerin improves coronary flow via BKCa-mediated vasodilation.
Rottlerin caused a significant increase in coronary flow in WT mice that was absent in BKCa KO mice (Fig. 3). There were no significant differences in CF of BKCa KO mice compared with WT at baseline (Table 1). These results are consistent with rottlerin-dependent activation of vascular BKCa channels and subsequent vasodilation.
Fig. 3.
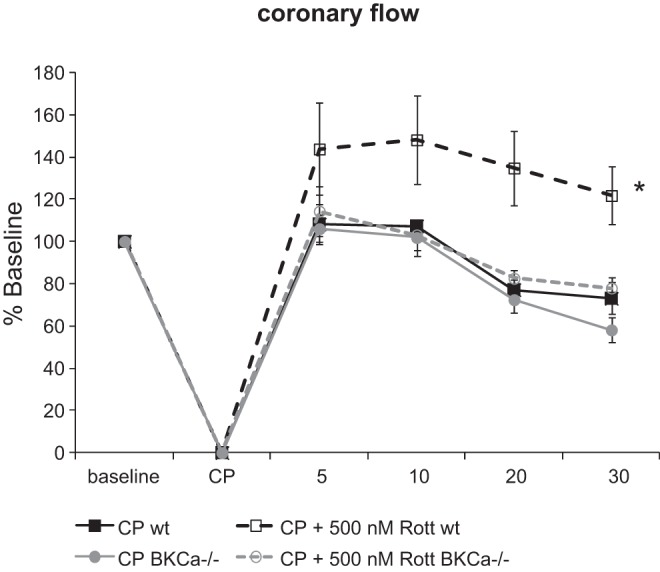
Rottlerin increases coronary flow above baseline in wt hearts following recovery from CP. Coronary flow as % change from baseline to 30 min postreperfusion; n = 5–7/group, *P < 0.05 using 2-way RM ANOVA, Student-Newman-Keuls.
Rottlerin reduces ROS generation in vitro.
Ischemic injury is accompanied by an increased rate of production of ROS in cardiomyocytes, which is detrimental for cell function. To test possible effects of rottlerin on redox balance and mitochondrial membrane potential, we used H9c2 cardiomyoblasts cultured as described in methods. Under these conditions (1 wk, 1% FBS DMEM after confluence), H9c2 cells expressed cardiac markers cTnI (Fig. 4, A and B, red) as well as cTnT (Fig. 4C, red). Cultures were heterogeneous with the majority of cells adopting long multi-nucleate tubes expressing cardiac troponins (red) and actin (green, blue, Fig. 4, B and C). Cultures also contained interspersed single cells, which were absent of cardiac markers but positive for actin (green), which was present in both phenotypes (Fig. 4, B and C). These culturing conditions provided significantly greater expression of cardiac markers than cells supplemented with retinoic acid (data not shown), in contrast to previous reports (24, 29). Cells subjected to in vitro CP/R significantly increased ROS generation over control H9c2 cardiomyoblasts (Fig. 4, D and E). Inclusion of rottlerin in the hypoxic CP solution caused a significant decrease in CP-induced ROS generation, as measured by DCFDA (Fig. 4, D and E).
Fig. 4.

Rottlerin normalizes reactive oxygen species (ROS) following ischemic insults associated with in vitro CP (3 h)/reoxygenation (30 min) in H9c2 embryonic cardiomyocytes. H9c2 cells express cardiac Troponin I (cTnI) (A and B, red), cTnT (C, red), and actin (green, B and C only), in multinucleate tube-like cells (nuclei, blue, B and C only). D and E: rottlerin decreases ROS production measured 30 min postreoxygenation. D: time course of ROS production normalized to Fmax [2,7-dichlorofluorescein (DCF) diacetate intensity/Fmax (H2O2 treatment)]. E: normalized rate as measured in D; n = 12 from 3 independent experiments; *P < 0.05 vs. control, #P < 0.05 vs. control and CP. Pax, paxilline.
Rottlerin normalizes mitochondrial membrane potential after CP/R.
CP/R caused a significant decrease in H9c2 mitochondrial membrane potential (ΔΨm), as measured by TMRE fluorescence. Rottlerin dose dependently reversed depolarization of ΔΨm in CP/R-treated cells, as measured 30 min after reperfusion. In addition, rottlerin hyperpolarized ΔΨm in control myocytes in a dose-dependent manner (Fig. 5). Rottlerin-induced hyperpolarization was also attenuated by the BKCa channel blocker paxilline.
Fig. 5.

Rottlerin normalizes mitochondrial membrane potential (Δψm) following ischemic insults associated with in vitro CP (3 h)/reoxygenation (30 min). A: rottlerin causes increases in ΔΨm 30 min postreoxygenation, as measured by FCCP-normalized tetramethylrhodamine ethyl ester (TMRE) fluorescence; n ≥ 8 from 3 independent experiments, *P < 0.05, 1-way ANOVA Dunnett's test vs. control. All other groups were different from CP alone except CP + 100 nM rottlerin, 1-way ANOVA Dunnett's test. B: representative mouse cardiomyocyte loaded with tetramethylrhodamine methyl ester (TMRM). C: representative TMRM trace of myocyte picture in A. Arrow indicates addition of FCCP. D: relative ΔΨm in control, CP, and CP + rottlerin-treated myocytes. Results similar to H9c2 cells. Minimum n = 6 myocytes/group from at least 2 independent isolations. P values determined via 1-way ANOVA, Student-Newman-Keuls.
To confirm the finding of pronounced hyperpolarization with BKCa treatment in H9c2 cells, we performed similar experiments in isolated mouse cardiomyocytes loaded with the alternative mitochondrial membrane potential indicator TMRM. TMRM fluorescence was measured using a scanning confocal microscope and normalized to 20 μM FCCP treatment (Fig. 5, B and C). Similar to H9c2 cells, 3-h simulated hypoxic cardioplegia followed by 30-min reoxygenation caused a significant decrease in ΔΨm. Inclusion of 500 nM rottlerin caused a significant hyperpolarization following CP/R (Fig. 5D).
CP/R in isolated hearts and cells results in minimal cell damage.
Isolated hearts had minimal LDH release during reperfusion compared with baseline, and there were no significant differences between groups (Fig. 6A). Supporting these observations of limited cardiac injury in CP isolated hearts, there is no detectable necrosis in CP/R-treated hearts, as determined with triphenyltetrazolium chloride staining (data not shown). In vitro simulated CP/R also caused minimal cell damage. There was a main effect for decreased LDH release in CP/R-treated cells compared with control cells, likely attributable to reduced temperature during the hypoxic period. There were no significant differences with rottlerin-treated groups or statistically significant interactions (Fig. 6B).
Fig. 6.
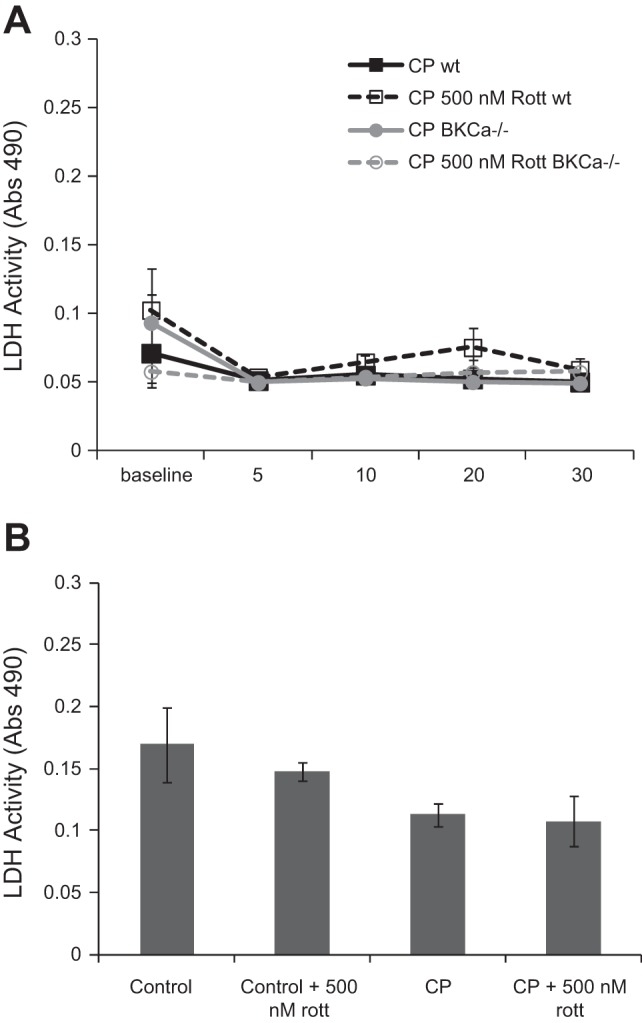
CP/R in isolated hearts and cells results in minimal cell damage. There were no increases in lactate dehydrogenase (LDH) release in perfused mouse hearts or cells following CP/R. A: 2-h CP/R in mouse hearts does not increase LDH release in the coronary effluent above baseline. There were no differences between groups (2-way RM ANOVA vs. wt). B: in vitro hypoxic CP/R does not increase LDH release in cell media. There was a main effect of CP to reduce LDH release. There were no other effects or interactions (2-way ANOVA).
DISCUSSION
The principle findings of this study were the following: 1) rottlerin improves cardiac functional recovery in a mouse model of ischemic CP/R; 2) the beneficial effects of rottlerin are solely dependent on BKCa channels, as there is no improved cardioprotection in BKCa KO mice; and 3) rottlerin likely exerts its effects via decreased ROS and improved mitochondrial membrane repolarization.
Mitochondrial K+ channels, including mitoKATP and BKCa, are implicated in cardioprotection following ischemic insults (23, 28, 35). Both are considered potential end effectors of numerous cardioprotective signaling pathways, including pharmacological and pre- and postconditioning stimuli. Activation of mitoKATP or BKCa channels results in K+ influx into the mitochondrial matrix. The exact mechanism of mitochondrial K+ flux for cardioprotection remains unclear, but possibilities include 1) electrochemical reduction of mitochondrial matrix Ca2+ overload, 2) inhibition of mitochondrial permeability transition pore opening (decreased apoptosis/necrosis), 3) regulation of mitochondrial matrix volume (with subsequent regulation of respiration and/or signaling), and 4) beneficial changes in respiration and/or ROS production (12, 28, 35).
Activation of BKCa channels may mediate enhanced cardioprotection in response to severe ischemia relative to mitoKATP channel activation (17, 32, 36). Expression of BKCa in cardiomyocytes has been controversial. However, numerous studies using mitoplasts and mitochondria have detected BKCa-like effects and currents sensitive to BKCa-modulating compounds (4, 5, 34, 39). Singh et al. (32) recently demonstrated that a COOH-terminal splice variant of the BKCa channel is present in cardiomyocytes and is sufficient to localize the channel to cardiomyocytes. In addition, Wojtovich et al. (36) recently showed that BKCa channel activators limit necrosis in a mouse ischemia model; however, this effect may include protective BKCa activation in additional noncardiomyocyte cell types (i.e., intrinsic neurons and smooth muscle). Demonstrated cardioprotective effects associated with the BKCa channel openers NS1619 and NS11021 following severe ischemia reperfusion injury include reductions in mitochondrial Ca2+ overload, mitochondrial membrane depolarization, increased cell viability, and improved function in whole hearts (7, 31, 36). However, both of these drugs are not without significant drawbacks; at doses consistent with BKCa activation, NS1619 (10–100 μM) and NS11021 can have detrimental side effects such as directly uncoupling mitochondria, promoting nonspecific ion leak, and yielding other off-target effects (1, 3, 35–37).
Rottlerin (mallotoxin) is an extremely potent BKCa channel activator, as shown through patch-clamp studies and induction of BKCa-mediated smooth muscle relaxation (2, 38, 41). Rottlerin activates BKCa channels in a manner distinct from NS1619 and NS11021 (2, 41). It may activate the channel in a manner similar to interaction of the regulatory leucine-rich repeat-containing protein 26 gene (LRRC26) γ-subunit with the BKCa pore-forming α-subunit, as the presence of LRRC26 blocks rottlerin-induced channel activation. The rottlerin-induced significant leftward shift (∼70 mV) of the voltage-dependent activation of the channel (2, 40) may be especially important for activation in the inner mitochondria membrane, where ΔΨm is >150 mV. Originally, rottlerin was reported as a PKC-δ inhibitor. However, rottlerin as a true inhibitor of PKC-δ generated considerable controversy (33). In these studies, we did not detect any increase in PKC-δ phosphorylation (associated with increased PKC activity) in rottlerin-treated mouse myocardium (data not shown). The potential inhibitory dose of rottlerin for PKC-δ is much higher than those reported for BKCa activation (10–100-fold). We also demonstrate that 500 nM rottlerin greatly improves cardiac function following ischemic insults associated with CP/R and that these effects are completely dependent on BKCa channels, as there was no effect in the KO animals (Figs. 1 and 2). The beneficial effects of rottlerin were also consistent with modulation of mitochondrial K+ channels, as rottlerin treatment of ischemic H9c2 cells caused significant reductions in ROS generation and attenuation of CP/R-induced depolarization of mitochondrial membrane potential (Figs. 4 and 5). The unequivocal effect of rottlerin on BKCa channels was also recently demonstrated to alleviate asthma and improve bronchodilation (an important role of BKCa in airway smooth muscle) in WT mice but not BKCa KO animals (15). Collectively, these results indicate that the cardioprotective effects of rottlerin are mediated by BKCa channel activation and that rottlerin may be one of the most promising small molecules available to activate myocardial BKCa channels in vivo.
Interestingly, in our study, rottlerin caused a significant increase in mitochondrial hyperpolarization both in control cells and in cells following ischemic CP/R. Acute activation of BKCa channels has been proposed to cause moderate mitochondrial depolarization attributable to matrix K+ influx. However, some recent studies demonstrated that activation of BKCa channels does not significantly reduce ΔΨm and that large reductions in membrane potential of common BKCa activators may be due to nonspecific drug effects (1, 3, 14, 16). Mitochondrial K+ flux is hypothesized to modulate enhanced respiration possibly attributable to changes in mitochondrial structure and volume (3, 14, 25). In addition, it was recently demonstrated that there is a direct association between electron transport chain (ETC) and BKCa components and that respiration can modulate BKCa activity (5). Our study demonstrates that activation of BKCa channels during ischemia significantly hyperpolarizes ΔΨm. However, unlike many previous studies, ΔΨm was not measured after acute BKCa activation but 30 min after ischemia and subsequent removal of rottlerin-containing solutions. The rottlerin-dependent increase in mitochondrial hyperpolarization may be due to either or both 1) the prevention of mitochondrial damage manifested in permeability-transition pore openings and collapse of ΔΨm during ischemia, thus resulting in enhanced metabolic and functional recovery, and 2) BKCa channel activation directly promoting enhanced respiration. The latter would be supported by rottlerin-dependent increases in ΔΨm in control cells (Fig. 5A). The findings in H9c2 cells were confirmed in mouse isolated cardiomyocytes, which also displayed significant improvements in mitochondrial repolarization at 30 min of reperfusion compared with CP-treated myocytes (Fig. 5, B–D).
We also observed a significant decrease in CP-induced ROS generation upon treatment with rottlerin. Reduced ROS generation is a known consequence of other BKCa-activating compounds. Interestingly, reduced ROS in this model was associated with increased mitochondrial membrane potential following reperfusion. Reduced ROS is most often associated with reduced membrane potential. It is presently unclear how BKCa activation causes reduced ROS and enhanced ΔΨm during reperfusion. One possibility may be that, by limiting ROS during ischemia and early reperfusion, there is less mitochondrial ETC protein damage, allowing more efficient respiration and enhanced ΔΨm during reperfusion. In addition, it should be noted that rottlerin has been identified as a direct antioxidant (20, 22). The ability of paxilline to attenuate rottlerin-induced decreases in ROS and that channel deletion in mice completely blocks rottlerin-induced improved cardiac function argue against a general antioxidant mechanism and support these effects as specific consequences of BKCa activation.
Finally, rottlerin also caused robust increases in coronary flow after CP/R. These effects are consistent with vascular smooth muscle BKCa channel activation, which promotes vasodilation. We have previously demonstrated direct BKCa-dependent vasodilatory properties of rottlerin in pig coronary vessels and mouse aortas. Expression of BKCa channels in cardiomyocyte mitochondria and the plasma membrane of smooth muscle cells allows alleviation of two major consequences of ischemic injury, including impaired energetics and contraction of cardiomyocytes and propensity for spasm and contraction of the coronary vasculature. BKCa-induced enhanced coronary flow will also support improved cardiac function.
There are notable limitations to our study. The rat embryonic cardiomyoblast H9c2 cell line was used in the majority of experiments to determine mechanistic effects of rottlerin. This cell line has a number of limitations, including a lack of contractile activity, embryonic phenotype, and reduced cardiac marker expression. Freshly isolated adult cardiomyocytes are a more physiologically appropriate model. However, their use also has significant drawbacks, which are related to variation in cell quality attributable to enzymatic digestion and limited time for experimental use in culture (hours) compared with the highly reproducible H9c2 cell line. Importantly, we found that rottlerin-induced increases in mitochondrial membrane repolarization were similar in both cell types, indicating that metabolic alterations and signaling in H9c2 cells are relevant for cardiomyocyte pathways. Another limitation of these studies is that these ex vivo and in vitro models are designed to mimic mild ischemic insults associated with CP and cardiopulmonary bypass during surgery. The majority of cardiac surgeries utilize cold blood cardioplegia (4:1 blood to concentrated crystalloid cardioplegia). Potential effects of blood, inflammatory signals associated with cardiopulmonary bypass, and numerous other differences present in the clinic are not accounted for in these mouse and cell models.
In conclusion, we demonstrate that activation of BKCa channels during hyperkalemic CP/R greatly improves myocardial functional recovery, alleviates myocardial stunning, and improves coronary flow, all major consequence of cardiac surgery using CP/R. In addition, the small molecule rottlerin is a potent activator of BKCa channels that promotes robust cardioprotection during ischemic insults.
GRANTS
This work was supported by National Heart, Lung, Blood Institute Grants R00-HL093352 (to R. Clements) and R01-HL121796 (to D. Terentyev) and American Heart Association Grant-in-Aid GRNT20460376 (to R. Clements).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: B.C., D.T., and R.T.C. performed experiments; B.C. and R.T.C. analyzed data; B.C., D.T., and R.T.C. interpreted results of experiments; B.C. and R.T.C. prepared figures; B.C., D.T., and R.T.C. edited and revised manuscript; B.C., D.T., and R.T.C. approved final version of manuscript; R.T.C. conception and design of research; R.T.C. drafted manuscript.
ACKNOWLEDGMENTS
The authors thank Prof. Andrea L. Meredith of the University Of Maryland School of Medicine for generously providing the BKCa KO mice.
REFERENCES
- 1.Aldakkak M, Stowe DF, Cheng Q, Kwok WM, Camara AK. Mitochondrial matrix K+ flux independent of large-conductance Ca2+-activated K+ channel opening. Am J Physiol Cell Physiol 298: C530–C541, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Almassy J, Begenisich T. The LRRC26 protein selectively alters the efficacy of BK channel activators. Mol Pharmacol 81: 21–30, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aon MA, Cortassa S, Wei AC, Grunnet M, O'Rourke B. Energetic performance is improved by specific activation of K+ fluxes through KCa channels in heart mitochondria. Biochim Biophys Acta 1797: 71–80, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bednarczyk P, Koziel A, Jarmuszkiewicz W, Szewczyk A. Large-conductance Ca2+-activated potassium channel in mitochondria of endothelial EA.hy926 cells. Am J Physiol Heart Circ Physiol 304: H1415–H1427, 2013. [DOI] [PubMed] [Google Scholar]
- 5.Bednarczyk P, Wieckowski MR, Broszkiewicz M, Skowronek K, Siemen D, Szewczyk A. Putative structural and functional coupling of the mitochondrial BK channel to the respiratory chain. PLoS One 8: e68125, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bentzen BH, Nardi A, Calloe K, Madsen LS, Olesen SP, Grunnet M. The small molecule NS11021 is a potent and specific activator of Ca2+-activated big-conductance K+ channels. Mol Pharmacol 72: 1033–1044, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Bentzen BH, Osadchii O, Jespersen T, Hansen RS, Olesen SP, Grunnet M. Activation of big conductance Ca2+-activated K+ channels (BK) protects the heart against ischemia-reperfusion injury. Pflügers Arch 457: 979–988, 2009. [DOI] [PubMed] [Google Scholar]
- 8.Bolli R, Marban E. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev 79: 609–634, 1999. [DOI] [PubMed] [Google Scholar]
- 9.Cheng Y, Gu XQ, Bednarczyk P, Wiedemann FR, Haddad GG, Siemen D. Hypoxia increases activity of the BK-channel in the inner mitochondrial membrane and reduces activity of the permeability transition pore. Cell Physiol Biochem 22: 127–136, 2008. [DOI] [PubMed] [Google Scholar]
- 10.Clements RT, Cordeiro B, Feng J, Bianchi C, Sellke FW. Rottlerin increases cardiac contractile performance and coronary perfusion through BKCa++ channel activation after cold cardioplegic arrest in isolated hearts. Circulation 124: S55–S61, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clements RT, Feng J, Cordeiro B, Bianchi C, Sellke FW. p38-MAPK-dependent heat shock protein 27 (HSP27) and αB-crystallin (cryAB) phosphorylation in regulation of myocardial function following cardioplegic arrest. Am J Physiol Heart Circ Physiol 300: H1669–H1677, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clements RT, Terentyev D, Sellke FW. Ca2+-activated K+ channels as therapeutic targets for myocardial and vascular protection. Circ J 79: 455–462, 2015. [DOI] [PubMed] [Google Scholar]
- 13.Cordeiro B, Shinn C, Sellke FW, Clements RT. Rottlerin-induced BKCa channel activation impairs specific contractile responses and promotes vasodilation. Ann Thorac Surg 99: 626–634, 2015. [DOI] [PubMed] [Google Scholar]
- 14.Costa AD, Quinlan CL, Andrukhiv A, West IC, Jaburek M, Garlid KD. The direct physiological effects of mitoKATP opening on heart mitochondria. Am J Physiol Heart Circ Physiol 290: H406–H415, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Goldklang MP, Perez-Zoghbi JF, Trischler J, Nkyimbeng T, Zakharov SI, Shiomi T, Zelonina T, Marks AR, D'Armiento JM, Marx SO. Treatment of experimental asthma using a single small molecule with anti-inflammatory and BK channel-activating properties. FASEB J 27: 4975–4986, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heinen A, Camara AK, Aldakkak M, Rhodes SS, Riess ML, Stowe DF. Mitochondrial Ca2+-induced K+ influx increases respiration and enhances ROS production while maintaining membrane potential. Am J Physiol Cell Physiol 292: C148–C156, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Kang SH, Park WS, Kim N, Youm JB, Warda M, Ko JH, Ko EA, Han J. Mitochondrial Ca2+-activated K+ channels more efficiently reduce mitochondrial Ca2+ overload in rat ventricular myocytes. Am J Physiol Heart Circ Physiol 293: H307–H313, 2007. [DOI] [PubMed] [Google Scholar]
- 18.Lawton JS, Hsia PW, Damiano RJ Jr. The adenosine-triphosphate-sensitive potassium-channel opener pinacidil is effective in blood cardioplegia. Ann Thorac Surg 66: 768–773, 1998. [DOI] [PubMed] [Google Scholar]
- 19.Levitsky S. Protecting the myocardial cell during coronary revascularization. The William W. L. Glenn Lecture. Circulation 114: I339–I343, 2006. [DOI] [PubMed] [Google Scholar]
- 20.Longpre JM, Loo G. Protection of human colon epithelial cells against deoxycholate by rottlerin. Apoptosis 13: 1162–1171, 2008. [DOI] [PubMed] [Google Scholar]
- 21.Maffit SK, Sellitto AD, Al-Dadah AS, Schuessler RB, Damiano RJ Jr, Lawton JS. Diazoxide maintains human myocyte volume homeostasis during stress. J Am Heart Assoc 1: 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maioli E, Torricelli C, Valacchi G. Rottlerin and curcumin: a comparative analysis. Ann NY Acad Sci 1259: 65–76, 2012. [DOI] [PubMed] [Google Scholar]
- 23.McCully JD, Levitsky S. Mitochondrial ATP-sensitive potassium channels in surgical cardioprotection. Arch Biochem Biophys 420: 237–245, 2003. [DOI] [PubMed] [Google Scholar]
- 24.Menard C, Pupier S, Mornet D, Kitzmann M, Nargeot J, Lory P. Modulation of L-type calcium channel expression during retinoic acid-induced differentiation of H9C2 cardiac cells. J Biol Chem 274: 29063–29070, 1999. [DOI] [PubMed] [Google Scholar]
- 25.Mentzer RM., Jr Myocardial protection in heart surgery. J Cardiovasc Pharmacol Ther 16: 290–297, 2011. [DOI] [PubMed] [Google Scholar]
- 26.Meredith AL, Thorneloe KS, Werner ME, Nelson MT, Aldrich RW. Overactive bladder and incontinence in the absence of the BK large conductance Ca2+-activated K+ channel. J Biol Chem 279: 36746–36752, 2004. [DOI] [PubMed] [Google Scholar]
- 27.O'Rourke B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ Res 94: 420–432, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Rourke B, Cortassa S, Aon MA. Mitochondrial ion channels: gatekeepers of life and death. Physiology (Bethesda) 20: 303–315, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pereira SL, Ramalho-Santos J, Branco AF, Sardao VA, Oliveira PJ, Carvalho RA. Metabolic remodeling during H9c2 myoblast differentiation: relevance for in vitro toxicity studies. Cardiovasc Toxicol 11: 180–190, 2011. [DOI] [PubMed] [Google Scholar]
- 30.Ruel M, Khan TA, Voisine P, Bianchi C, Sellke FW. Vasomotor dysfunction after cardiac surgery. Eur J Cardiothorac Surg 26: 1002–1014, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Sakamoto K, Ohya S, Muraki K, Imaizumi Y. A novel opener of large-conductance Ca2+-activated K+ (BK) channel reduces ischemic injury in rat cardiac myocytes by activating mitochondrial KCa channel. J Pharm Sci 108: 135–139, 2008. [DOI] [PubMed] [Google Scholar]
- 32.Singh H, Lu R, Bopassa JC, Meredith AL, Stefani E, Toro L. mitoBKCa is encoded by the Kcnma1 gene, and a splicing sequence defines its mitochondrial location. Proc Natl Acad Sci USA 110: 10836–10841, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soltoff SP. Rottlerin: an inappropriate and ineffective inhibitor of PKCdelta. Trends Pharmacol Sci 28: 453–458, 2007. [DOI] [PubMed] [Google Scholar]
- 34.Soltysinska E, Bentzen BH, Barthmes M, Hattel H, Thrush AB, Harper ME, Qvortrup K, Larsen FJ, Schiffer TA, Losa-Reyna J, Straubinger J, Kniess A, Thomsen MB, Bruggemann A, Fenske S, Biel M, Ruth P, Wahl-Schott C, Boushel RC, Olesen SP, Lukowski R. KCNMA1 encoded cardiac BK channels afford protection against ischemia-reperfusion injury. PLoS One 9: e103402, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walters AM, Porter GA, Brookes PS. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ Res 111: 1222–1236, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wojtovich AP, Nadtochiy SM, Urciuoli WR, Smith CO, Grunnet M, Nehrke K, Brookes PS. A non-cardiomyocyte autonomous mechanism of cardioprotection involving the SLO1 BK channel. PeerJ 1: e48, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wrzosek A. The potassium channel opener NS1619 modulates calcium homeostasis in muscle cells by inhibiting SERCA. Cell Calcium 56: 14–24, 2014. [DOI] [PubMed] [Google Scholar]
- 38.Wu SN, Wang YJ, Lin MW. Potent stimulation of large-conductance Ca2+-activated K+ channels by rottlerin, an inhibitor of protein kinase C-delta, in pituitary tumor (GH3) cells and in cortical neuronal (HCN-1A) cells. J Cell Physiol 210: 655–666, 2007. [DOI] [PubMed] [Google Scholar]
- 39.Xu W, Liu Y, Wang S, McDonald T, Van Eyk JE, Sidor A, O'Rourke B. Cytoprotective role of Ca2+-activated K+ channels in the cardiac inner mitochondrial membrane. Science 298: 1029–1033, 2002. [DOI] [PubMed] [Google Scholar]
- 40.Yan J, Aldrich RW. BK potassium channel modulation by leucine-rich repeat-containing proteins. Proc Natl Acad Sci USA 109: 7917–7922, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zakharov SI, Morrow JP, Liu G, Yang L, Marx SO. Activation of the BK (SLO1) potassium channel by mallotoxin. J Biol Chem 280: 30882–30887, 2005. [DOI] [PubMed] [Google Scholar]