

Abstract
Previous studies have suggested that diabetes significantly impairs the cognitive function. Tangzhining (TZN), as a kind of Traditional Chinese Medicine (TCM), has been widely used to treat diabetes in China. However, the effect of TZN on treatment of diabetes-induced learning and memory deficits has not been well documented. The present study was to investigate the effect of TZN on diabetes-induced learning and memory deficits and delineate the underlying molecular mechanism. Diabetic rats were randomly grouped and treated with various doses of TZN (0.47, 0.94 and 1.4 g/kg) by intraperitoneal injection. Using the Morris water maze, TZN treatment (0.94 g/kg and 1.4 g/kg) reduced markedly the escape latency and path length of diabetic rats. The morphological changes of pyramidal cells in hippocampus of diabetic rats were apparently reversed and improved by TZN treatment, in comparison with that in diabetic rats without TZN treatment. Moreover, the results of Western blot analysis showed that TZN treatment significantly increased the protein expression of glutamic acid decarboxylase (GAD) and excitatory amino acid carrier 1 (EAAC1) in hippocampus of diabetic rats. Furthermore, TZN treatment increased the protein expression of N-methyl-D-aspartic acid (NMDA) receptor subunits including NR1 and NR2B. Taken together, our data suggest that TZN sustains the balance between glutamate (Glu) and GABA by regulating GAD and EAAC1, and maintains the NMDA receptors activity for learning and memory function through regulating the subunits NR1 and NR2B.
Keywords: Diabetes, TZN, learning and memory impairment, N-methyl-D-aspartic acid receptors, glutamic acid decarboxylase, excitatory amino acid carrier 1
Introduction
Diabetes is a chronic disease caused by the body’s inability to produce insulin or by the ineffective use of the insulin produced, seriously affecting the quality of the patients’ life [1,2]. However, the impairment of diabetes on central nervous system has not drawn much attention in the past decades. Diabetic encephalopathy belongs to “Senile Dementia” category of traditional Chinese medicine (TCM) and the patients present clinically mental deterioration, forgetfulness, dullness, less words, and fatigue [3]. In recent years, dementia caused by diabetes has drawn increasing attention [4-6]. Population-based studies have demonstrated that patients with diabetes have higher incidences of dementia [5]. Nowadays, the incidences of diabetes and dementia in elderly people are comparatively higher and expected to increase in future [7,8]. Therefore, better understanding the association between dementia and diabetes is of great importance.
Increasing evidences have suggested that diabetes impairs neurological functions not only on the peripheral nervous system [9-11] but also on the central nervous system [2,12]. It has reported that diabetes significantly impairs physiology parameter such as inhibition of nerve conduction speed [13,14]. The impact of diabetes on cognitive function was firstly reported in the early 20th century [15]. The function of learning and memory was impaired by diabetes [16,17]. A concept of diabetic encephalopathy has been proposed by Dejgaard based on a broad spectrum analysis of brain electrical activity, physiological metabolism, brain morphological and behavior in diabetic patients [18]. Diabetic encephalopathy is characterized by cognitive dysfunctions and neurochemical and structural abnormalities [19]. More recently, a new term of diabetes-associated cognitive decline has been proposed which defines the cognitive impairments such as reduced mental flexibility and slowed psychomotor induced by diabetes [20].
The hippocampus of rodent and human are involved in the long-term potentiation (LTP) which plays an important role in learning and memory [21]. The hippocampal networks consist of excitatory neuronsusing glutamate (Glu) as a neurotransmitter and inhibitory interneurons using gamma-amino butyric acid (GABA) as transmitter molecules [22,23]. Previous studies have reported that dynamic changes between Glu and GABA play an important role in regulating LTP function [24]. Glu exerts a leading role in the formation of long-term regulation of synaptic plasticity and transmission of brain excitability [25]. It has been reported that Glu regulates learning and memory through activation of N-methyl-D-aspartic acid (NMDA) receptors [26,27].
TCM has been used to treat various diseases including psychological and physiological diseases in China for thousands of years [28]. Tangzhining (TZN), a TCM formula composed of Coptidis Rhizoma, Panacis Majoris Rhizoma, Puerariae Lobatae Radix, is capable of improving cognitive function and diabetic animal memory functions in our preliminary test. Herein, we investigated the effect of TZN on cognitive dysfunction in diabetic rats and aimed to elucidate the underlying molecular mechanism.
Materials and methods
Animals and drugs
Eight-week-old male Sprague-Dawley (SD) rats (weighting 220-250 g) were purchased from the Experimental Animal Centre of the Fourth Military Medical University (Xi’an, China). Five rats were kept per cage with water and food ad libitum in a room under temperature (22±1°C), humidity (60%±10%), and a 12 h light/12 h dark cycle. The experimental protocol was approved by the Institutional Animal Care and Use Committee of the Shaanxi University of Chinese Medicine, and was in accordance with the guidelines of the National Research Council Guide for the care and use of laboratory animals. TZN capsule was kindly provided by the Chemical Laboratory of the Shaanxi University of Chinese Medicine.
Experimental design
Diabetic rats were established by injection of Streptozotocin (STZ) in rats. STZ was dissolved in citrate buffer (6 mg/mL) and injected intraperitoneally into rats. The serum was collected from caudal vein and fasting blood glucose was examined. The rats with fasting blood glucose level above 11.1 mmol/L were considered to be diabetic and were used in the experiment. The SD rats were randomly divided into five groups: control, normal rats without treatment; model, diabetic rats without TZN treatment; TZN (0.47 g/kg), diabetic rats treated with TZN (0.47 g/kg); TZN (0.94 g/kg), diabetic rates treated with TZN (0.94 g/kg); TZN (1.40 g/kg), diabetic rats treated with TZN (1.40 g/kg). The rats were fasted (without water deprivation) for 12 h prior to TZN administration.
Morris water maze task
Learning and memory performance was assessed by the Morris water maze task test, which consisted of 5-day training (visible and invisible platform training sessions) and a probe trial on day 6. On day 6, the platform was removed and the probe trial was started. All animals received Morris water maze test after the drug intervention. The Morris water maze consisted of a circular pool (150 cm diameter, wall depth 60 cm) and a platform. Rats were trained to escape from water by swimming to the hidden platform (1.5 cm beneath water surface) whose location could only be identified using distal extra-maze cues attached to the room walls. Water temperature was maintained at 24±2°C. The pool was divided into four quadrants by a computerized tracking/image analyzer system (video camcorder coupled to the computational tracking system). The platform was placed in the middle of the quadrant and remained unmoved during the training experiment. One hundred and twenty seconds (s) was set as the longest escape latency (more than 120 s was recorded as 120 s). If the rats were unable to find the platform within 120 s, they would have a rest for 15 s on the platform (each test interval was 15 s). After the trainings, all the rats were tested in Morris water maze task, and escape latency in finding the platform, movement distance and number of crossing the platform target were recorded. All rats were euthanized after the test, and the brains were rapidly dissected, isolated and fixed in 4% paraformaldehyde for hematoxylin and eosin staining (HE) staining. The remains hippocampus tissues were taken out and stored at -80°C for biochemical analysis.
Histopathological analysis
Hippocampus tissues were fixed in 4% of paraformaldehyde solution for 24 hours, and then made into coronal paraffin sections for HE staining. The morphology changes of hippocampus were observed under a fluorescence microscope (Leica, Germany).
Western blot analysis
Brain tissues were homogenated in lysis buffer (Beyotime Institute of Biotechnology, Haimen, Jiangsu, China). Protein concentration in each sample was measured by using a BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Equal amounts of proteins (30 μg) were separated by 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by transferred to nitrocellulose membranes (Pall Corporation, Port Washington, NY, USA). The membranes were then blocked with 5% nonfat milk in Tris-buffered saline (TBS) buffer containing 0.05% Tween-20 (TBST) for 2 h at 37°C. Primary antibody was then added and incubated overnight at 4°C. Subsequently, the membranes were washed three times with TBST (each for 5 min) before incubation with anti-rabbit secondary antibodies (1:6,000) for 70 min at room temperature. Additional washes were carried out as described above, and immune-reactive proteins on the membrane were detected using Immobilon Western HRP Substrate (Millipore, Boston, MA, USA). The signal intensity of the respective bands was quantified and normalized to β-actin expression. The experiment was repeated independently three times. Primary antibodies used in the experiment were as follows: rabbit anti-EAAC1 and NR2A antibodies (EPITOMICS, Burlingame, CA, USA); rabbit anti-GAD, NR1, and β-actin antibodies (Bioworld, St. Louis Park, MN, USA); rabbit anti-NR2B antibodies (Abcam, Cambridge, UK). Secondary antibody was purchased from Thermo Corporation (Waltham, MA, USA).
Statistical analysis
Data were expressed as mean ± SD. Statistical analysis was performed using SPSS version 11.5 (SPSS Inc., Chicago, IL, USA). Statistical difference was analyzed by one-way analysis of variance followed by Bonferroni post hoc. A P value less than 0.05 was considered statistically significant.
Results
Effects of TZN on diabetes-induced learning and memory impairment
In the present study, we first assessed the effect of TZN on cognitive function in diabetic rats by Morris water maze. The data showed that the escape latency (Table 1) and path length (Table 2) in diabetic rats was significantly longer than normal controls during the four days of learning. Interestingly, TZN treatment (0.94 g/kg and 1.4 g/kg) markedly reduced the escape latency and path length of diabetic rats following the four days of training. However, TZN treatment (0.47 g/kg) had no apparent effect on escape latency and path length of diabetic rats.
Table 1.
Effect of TZN on latency time in the Morris water maze test in rats (n=10)
| Day | Control | Model | TZN | ||
|---|---|---|---|---|---|
|
| |||||
| 0.47 g/kg | 0.94 g/kg | 1.40 g/kg | |||
| 1d | 60.84±3.39 | 116.56±11.27** | 104.50±10.61 | 95.02±7.80# | 86.15±8.98## |
| 2d | 56.39±5.76 | 114.55±11.80** | 106.95±13.37 | 84.33±9.48## | 78.79±7.06## |
| 3d | 45.87±5.42 | 101.37±10.33** | 102.86±12.47 | 81.89±7.98# | 65.41±9.47## |
| 4d | 31.89±8.65 | 90.39±8.95** | 85.30±8.59 | 75.56±7.04# | 46.41±5.86## |
| Σx | 48.75±5.81 | 105.72±10.5** | 99.90±11.26 | 84.20±8.07# | 69.19±8.62## |
P<0.01 vs. Control;
P<0.05 vs. Model;
P<0.01 vs. Model.
Table 2.
Effect of TZN on path length in the Morris water maze test in rats (n=10)
| Day | Control | Model | TZN | ||
|---|---|---|---|---|---|
|
| |||||
| 0.47 g/kg | 0.94 g/kg | 1.40 g/kg | |||
| 1d | 967.29±28.32 | 1779.48±89.88** | 1636.26±65.63 | 1354.77±122.29## | 1119.35±100.59## |
| 2d | 842.49±59.39 | 1686.26±119.28** | 1596.61±19.58 | 1172.36±98.38## | 960.90±52.59## |
| 3d | 606.62±16.29 | 1332.69±230.34** | 1401.9±118.95 | 1036.38±76.69# | 842.66±87.75## |
| 4d | 425.71±34.57 | 1141.64±125.82** | 1173.0±121.57 | 765.71±115.29## | 622.99±77.12## |
| Σx | 710.53±34.64 | 1485.02±141.33** | 1451.94±81.43 | 1082.31±103.16## | 1016.14±91.69## |
P<0.01 vs. Control;
P<0.05 vs. Model;
P<0.01 vs. Model.
Effects of TZN on pathological changes of hippocampus in diabetic rats
To validate whether TZN had an effect on pathological changes of hippocampus, we further analyzed the pathomorphology of hippocampus in diabetic rats treated with TZN. The results exhibited that pyramidal cells were structurally integrated and arranged in neat rows in the CA1 region of control rats (Figure 1A). In contrast, pyramidal cells were degranulated, and arranged irregularly in diabetic rats (Figure 1B). Furthermore, pyramidal cells were in triangular or irregular shape; blurred nucleolus and nuclear membrane, and condensed and aggregated chromatin could be also observed in the hippocampus of diabetic rats (Figure 1B). As expected, TZN treatments (1.40 g/kg, Figure 1C; 0.94 g/kg, Figure 1C and 0.49 g/kg, Figure 1C) apparently reversed these morphological changes of pyramidal cells in hippocampus of diabetic rats.
Figure 1.
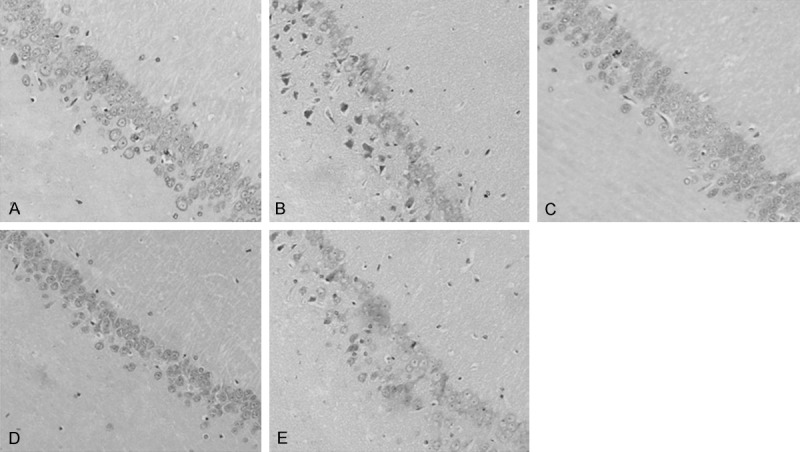
Effect of TZN on the pathologic morphology of hippocampus (CA1) in diabetic rats. A. Control group. B. Diabetic group without TZN treatment. C. Diabetic rats treated with 1.40 g/kg TZN. D. Diabetic rats treated with 0.94 g/kg TZN. E. Diabetic rats treated with 0.47 g/kg TZN. Conventional formalin-paraffin sections of hippocampus (CA1) were stained with HE and observed at ×200 magnification.
Likewise, similar results were observed in CA3 region of hippocampus. In the CA3 region of control group, pyramidal cells were loosely arranged in multi-levels (Figure 2A), whereas pyramidal cells were arranged irregularly and the cell shapes were in triangular or irregular shapes in diabetic rats (Figure 2B). Furthermore, nucleolus and nuclear membrane were blurred; chromatin were condensed and aggregated in CA3 region in diabetic rats (Figure 2B). With TZN (1.40 g/kg, Figure 2C; 0.94 g/kg, Figure 2D and 0.49 g/kg, Figure 2E) treatments, these morphological changes of pyramidal cells were reversed. Taken together, these results suggested that TZN treatments improved the hippocampus pathological changes in diabetic rats.
Figure 2.
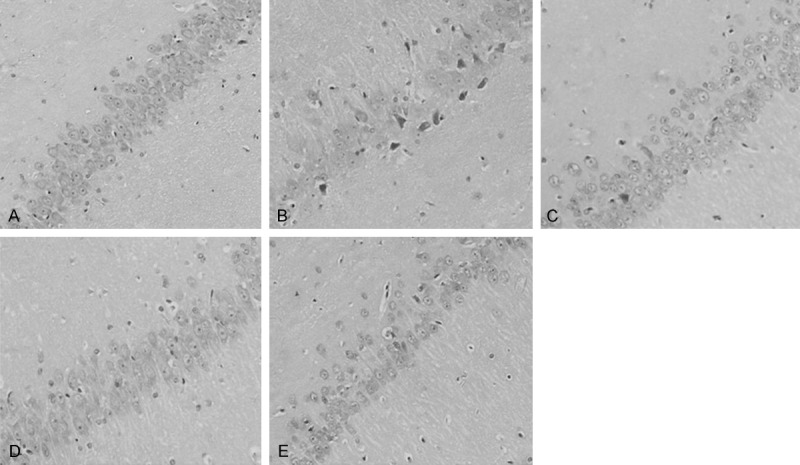
Effect of TZN on pathologic morphology of hippocampus (CA3) in diabetic rats. A. Control group. B. Diabetic group without TZN treatment. C. Diabetic rats treated with 1.40 g/kg TZN. D. Diabetic rats treated with 0.94 g/kg TZN. E. Diabetic rats treated with 0.47 g/kg TZN. Conventional formalin-paraffin sections of hippocampus (CA3) were stained with HE and observed at ×200 magnification.
Effects of TZN on GAD and EAAC1 protein expression
Glutamic acid decarboxylase (GAD) has been suggested to promote Glu to form inhibitory neurotransmitter GABA which improves cognitive function [29,30]. Excitatory amino acid carrier 1 (EAAC1) transporter plays an important role in preventing Glu-induced toxicity [31]. Considering the critical roles of GAD and EAAC1 in regulation of neuronal cellular function, we further investigated the effect of TZN on the protein expression of GAD and EAAC1 in hippocampus by Western blot analysis. The results showed that both of GAD and EAAC1 protein expression levels were significantly reduced in diabetic rats as compared with control rats, whereas diabetic rats treated with TZN (0.94 g/kg and 1.4 g/kg) showed a significant increase in the protein expression of GAD and EAAC1 in comparison with model group (Figure 3A and 3B). However, TZN (0.47 g/kg) had no apparently effect on the protein expression of GAD and EAAC1. The results suggested that TZN (0.94 g/kg and 1.4 g/kg) increased GAD and EAAC1 expression in hippocampus of diabetic rats.
Figure 3.
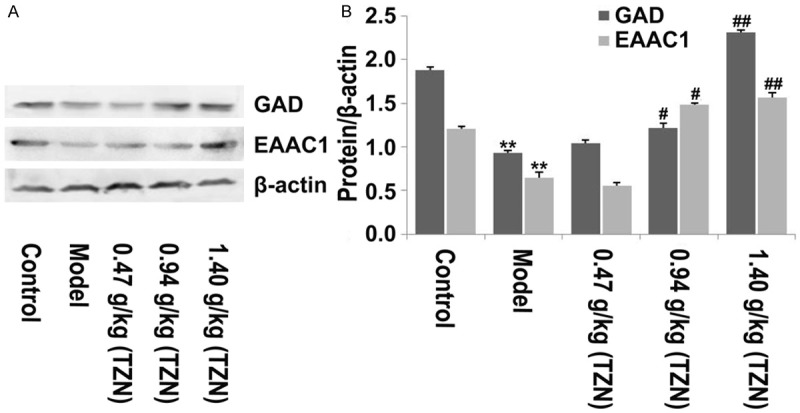
The effect of TZN on GAD and EAAC1 expression in hippocampus of rats. A. Western blot analysis of GAD and EACC1 protein expression in hippocampus of rats from different groups with antibodies indicated. β-actin was used as the internal control. B. Relative protein expression was quantified using Image-Pro Plus 6.0 software and normalized to β-actin. **P<0.01 vs. control group; #P<0.05 and ##P<0.01 vs. model group.
Effects of TZN on NMDA receptor subunit protein expression
NMDA receptor has been reported to play a critical role in regulation of learning and memory [32]. We next detected the effect of TZN on the subunits of NMDA receptor. The results of Western blot analysis showed that the protein expression of NR1 and NR2B in the hippocampus of diabetic rats was significantly decreased as compared with control rats. Additionally, no significant difference of NR2A expression was observed among control, model and TAN treatment groups. TZN treatment (0.94 g/kg and 1.40 g/kg) significantly increased NR1 and NR2B protein expression in the hippocampus of diabetic rats (Figure 4A, 4B). However, low dose TZN treatment (0.47 g/kg) had no significant effect on the protein expression of NR1 and NR2B as compared with model group. Taken together, our data implied that TZN mainly regulated the expression of NMDA receptor subunits NR1 and NR2B in hippocampus of diabetic rats.
Figure 4.
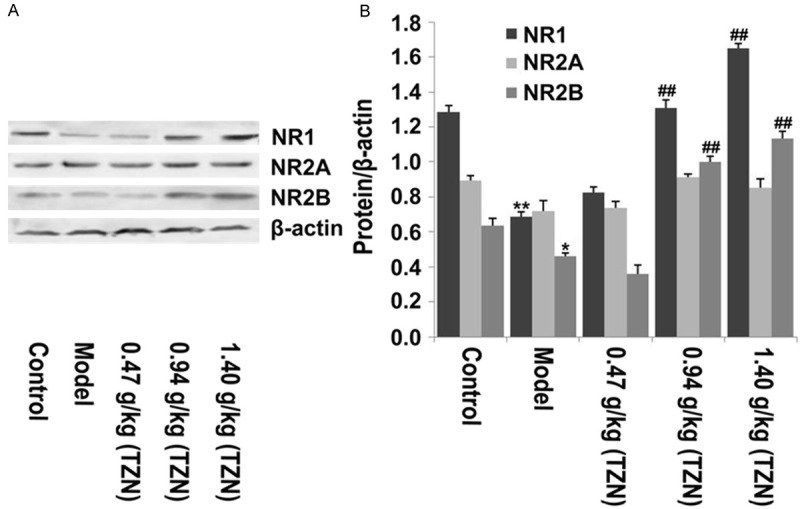
The effect of TZN on NMDA receptor expression. A. Western bolt analysis was performed to detect the protein expression of NR1, NR2A and NR2B in hippocampus of rats from different groups with antibodies indicated. β-actin was used as internal control. B. Relative protein expression was quantified using Image-Pro Plus 6.0 software and normalized to β-actin. *P<0.05 and **P<0.01 vs. control group; ##P<0.01 vs. model group.
Discussion
TZN has been widely used to treat diseases in China due to its ability of deceasing blood glucose and lipid, and improving intelligence and body’s functions. However, the role of TZN in treatment of the cognitive dysfunction in diabetes is unknown. Here, our study for the first time provided evidences that TZN improved cognitive dysfunction in diabetic rats and delineated the potential molecular basis of TZN in treatment of cognitive function.
In the present study, we found that high dose of TZN (0.94 g/kg and 1.4 g/kg) effectively increased the learning and memory function which was impaired in diabetic rats. Pyramidal cells has been suggested to regulate excitability and play an important role in control of cognitive function [33]. Here, our data demonstrated the morphology of pyramidal cells in hippocampus were apparently impaired in diabetic rats. Intriguingly, TZN treatments significantly improved these abnormal pathological changes in hippocampus of diabetic rats. Glu has been reported to be overproduced under pathologic conditions leading excessive excitability and neuronal death [25]. Therefore, removal and inactivation of excessive Glu is beneficial to decreased neurotoxicity. The inhibitory transmitter molecule GABA plays a negative feedback role in regulation of learning and memory [34]. Studies have demonstrated that GABA is significantly decreased in hippocampus of aged rats, and forced expression of GABA improves learning and memory functions [29]. It has been reported that GAD catalyzes Glu to synthesize GABA by its decarboxylase ability, and thereby protects hippocampal neuron against neurotoxicity [35]. In the present study, we demonstrated that high dose of TZN significantly increased GAD protein expression in hippocampus of diabetic rats. Herein, we also revealed that TZN markedly increased EAAC1 in hippocampus of diabetic rats. EAAC1 as a glutamate transporter has also been reported to play a critical role in Glu reuptake and neurotoxicity elimination [31]. Taken together, our data suggested that TZN maintained homeostasis between Glu and GABA through regulation of GAD and EACC1, which might be contributed to increasing cognitive function in diabetic rats.
NMDA receptor-dependent LTP has been proposed to be critically associated with learning and memory [21,36,37]. NMDA receptors consists of two essential NR1 subunits and two or three NR2 subunits [38-40]. NR2 subunits including NR2A and NR2B are the regulatory subunit of NMDA receptors and different subunits of NR2 imparts different characteristics on NMDA receptors function [41]. It has been found that NR2A-containing NMDA receptors play a more important role in rapid kinetics [42]. In regulation of NMDA receptor-dependent synaptic function, NR2A-containing NMDA receptors were suggested to be preferentially targeted to synaptic sites and NR2B-containing NMDA receptors tended to be targeted to extrasynaptic sites [43,44]. Therefore, the alterations between NR2A and NR2B ratio may significantly impair NMDA receptors-mediated synaptic responses and cortical development [45,46]. It has demonstrated that aberrant expression of NR1 resulted in a loss of NMDA receptor activities [32] and knockdown of NR1 exhibits significant impairment on learning and memory of mouse [47]. Overexpression of NR2B resulted in an enhancement of LTP in hippocampus and the mice exhibited better performance in behavioral experiment [48]. Knockdown of NR2B led to a decrease in the memory of mice, while re-expression of NR2B resulted in a significant increase in the memory of mice [41]. Increasing studies have suggested that NR2B plays important roles in the development of neuronal plasticity and the formation of learning and memory formation [49]. In consistent with these findings, we demonstrated that the subunits of NMDA receptor including NR1 and NR2B were significantly decreased in diabetic rats. However, the subunit NR2A was not apparently impaired in diabetic rats. Notably, high dose of TZN was found to be capable of increasing the protein expression of NR1 and NR2B in hippocampus of diabetic rats. These data implied that TZN might improve the learning and memory functions of diabetic rats through regulation of the NMDA receptor subunits NR1 and NR2B.
In summary, our data demonstrated that TZN treatment could improve and increase the learning and memory functions of diabetic rats. The possible molecular mechanism may be that TZN maintains the balance between Glu and GABA through regulating GAD and EAAC1, and sustains the NMDA receptors activity for learning and memory function through regulating the NMDA receptor subunits NR1 and NR2B. Therefore, TZN possesses the functions of improving cognitive dysfunction in diabetes. However, further studies are warranted to validate the effects and delineate the precise molecular mechanism of TZN in treatment of cognitive dysfunction in diabetes.
Acknowledgements
Financial support from the National Natural Science Foundation of China (NSFC; Grant No. 81102805) and the Innovation Program on Science and Technology Project of Shaanxi Province (2011KTCQ03-02) is acknowledged. The Project of Science and Technology Research and Development of Shannxi Province (Grant No. 2011K16-04-04).
Disclosure of conflict of interest
None.
References
- 1.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87:4–14. doi: 10.1016/j.diabres.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Gispen WH, Biessels GJ. Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci. 2000;23:542–549. doi: 10.1016/s0166-2236(00)01656-8. [DOI] [PubMed] [Google Scholar]
- 3.Yan JL, Chen YQ. Traditional Chinese medical understanding on pathogenesis of senile dementia. CJTCMP. 2008;7:030. [Google Scholar]
- 4.Gudala K, Bansal D, Schifano F, Bhansali A. Diabetes mellitus and risk of dementia: A meta-analysis of prospective observational studies. J Diabetes Investig. 2013;4:640–650. doi: 10.1111/jdi.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 2006;5:64–74. doi: 10.1016/S1474-4422(05)70284-2. [DOI] [PubMed] [Google Scholar]
- 6.Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O'Brien PC, Palumbo PJ. The risk of dementia among persons with diabetes mellitus: a population-based cohort study. Ann N Y Acad Sci. 1997;826:422–427. doi: 10.1111/j.1749-6632.1997.tb48496.x. [DOI] [PubMed] [Google Scholar]
- 7.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–787. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 8.Hallschmid M, Higgs S, Thienel M, Ott V, Lehnert H. Postprandial administration of intranasal insulin intensifies satiety and reduces intake of palatable snacks in women. Diabetes. 2012;61:782–789. doi: 10.2337/db11-1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bloomgarden ZT. Diabetic retinopathy and diabetic neuropathy. Diabetes Care. 2007;30:760–765. doi: 10.2337/dc07-zb03. [DOI] [PubMed] [Google Scholar]
- 10.Bloomgarden ZT. Diabetic neuropathy. Diabetes Care. 2007;30:1027–1032. doi: 10.2337/dc07-zb04. [DOI] [PubMed] [Google Scholar]
- 11.Dobretsov M, Romanovsky D, Stimers JR. Early diabetic neuropathy: triggers and mechanisms. World J Gastroenterol. 2007;13:175–191. doi: 10.3748/wjg.v13.i2.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biessels GJ, Gispen WH. The impact of diabetes on cognition: what can be learned from rodent models? Neurobiol Aging. 2005;26(Suppl 1):36–41. doi: 10.1016/j.neurobiolaging.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 13.Whiteley SJ, Tomlinson DR. Motor nerve conduction velocity and nerve polyols in mice with short-term genetic or streptozotocin-induced diabetes. Exp Neurol. 1985;89:314–321. doi: 10.1016/0014-4886(85)90092-5. [DOI] [PubMed] [Google Scholar]
- 14.Mayer JH, Tomlinson DR. Prevention of defects of axonal transport and nerve conduction velocity by oral administration of myo-inositol or an aldose reductase inhibitor in streptozotocin-diabetic rats. Diabetologia. 1983;25:433–438. doi: 10.1007/BF00282524. [DOI] [PubMed] [Google Scholar]
- 15.Miles W, Root H. Psychologic tests applied to diabetic patients. Arch Int Med. 1922;30:767–777. [Google Scholar]
- 16.Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, Kurinami H, Shinohara M, Rakugi H, Morishita R. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A. 2010;107:7036–7041. doi: 10.1073/pnas.1000645107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryan CM, Williams TM. Effects of insulin-dependent diabetes on learning and memory efficiency in adults. J Clin Exp Neuropsychol. 1993;15:685–700. doi: 10.1080/01688639308402589. [DOI] [PubMed] [Google Scholar]
- 18.Dejgaard A, Gade A, Larsson H, Balle V, Parving A, Parving HH. Evidence for diabetic encephalopathy. Diabet Med. 1991;8:162–167. doi: 10.1111/j.1464-5491.1991.tb01564.x. [DOI] [PubMed] [Google Scholar]
- 19.Sima AA, Kamiya H, Li ZG. Insulin, C-peptide, hyperglycemia, and central nervous system complications in diabetes. Eur J Pharmacol. 2004;490:187–197. doi: 10.1016/j.ejphar.2004.02.056. [DOI] [PubMed] [Google Scholar]
- 20.Mijnhout GS, Scheltens P, Diamant M, Biessels GJ, Wessels AM, Simsek S, Snoek FJ, Heine RJ. Diabetic encephalopathy: A concept in need of a definition. Diabetologia. 2006;49:1447–1448. doi: 10.1007/s00125-006-0221-8. [DOI] [PubMed] [Google Scholar]
- 21.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 22.Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 23.Somogyi P, Klausberger T. Defined types of cortical interneurone structure space and spike timing in the hippocampus. J Physiol. 2005;562:9–26. doi: 10.1113/jphysiol.2004.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taneera J, Jin Z, Jin Y, Muhammed SJ, Zhang E, Lang S, Salehi A, Korsgren O, Renstrom E, Groop L, Birnir B. gamma-Aminobutyric acid (GABA) signalling in human pancreatic islets is altered in type 2 diabetes. Diabetologia. 2012;55:1985–1994. doi: 10.1007/s00125-012-2548-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sickmann HM, Waagepetersen HS, Schousboe A, Benie AJ, Bouman SD. Brain glycogen and its role in supporting glutamate and GABA homeostasis in a type 2 diabetes rat model. Neurochem Int. 2012;60:267–275. doi: 10.1016/j.neuint.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 26.Panza F, D’Introno A, Colacicco AM, Capurso C, Del Parigi A, Caselli RJ, Pilotto A, Argentieri G, Scapicchio PL, Scafato E, Capurso A, Solfrizzi V. Current epidemiology of mild cognitive impairment and other predementia syndromes. Am J Geriatr Psychiatry. 2005;13:633–644. doi: 10.1176/appi.ajgp.13.8.633. [DOI] [PubMed] [Google Scholar]
- 27.Peila R, Rodriguez BL, Launer LJ. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes. 2002;51:1256–1262. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- 28.Lao L, Xu L, Xu S. Integrative Pediatric Oncology. Springer; 2012. Traditional chinese medicine; pp. 125–135. [Google Scholar]
- 29.Abdulla FA, Abu-Bakra MA, Calaminici MR, Stephenson JD, Sinden JD. Importance of forebrain cholinergic and GABAergic systems to the age-related deficits in water maze performance of rats. Neurobiol Aging. 1995;16:41–52. doi: 10.1016/0197-4580(95)80006-d. [DOI] [PubMed] [Google Scholar]
- 30.Ali F, Rowley M, Jayakrishnan B, Teuber S, Gershwin ME, Mackay IR. Stiff-person syndrome (SPS) and anti-GAD-related CNS degenerations: protean additions to the autoimmune central neuropathies. J Autoimmun. 2011;37:79–87. doi: 10.1016/j.jaut.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 31.Lane MC, Jackson JG, Krizman EN, Rothstein JD, Porter BE, Robinson MB. Genetic deletion of the neuronal glutamate transporter, EAAC1, results in decreased neuronal death after pilocarpine-induced status epilepticus. Neurochem Int. 2014;73:152–158. doi: 10.1016/j.neuint.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krugel U, Koles L, Illes P. Integration of neuronal and glial signalling by pyramidal cells of the rat prefrontal cortex; control of cognitive functions and addictive behaviour by purinergic mechanisms. Neuropsychopharmacol Hung. 2013;15:206–213. [PubMed] [Google Scholar]
- 34.Obrietan K, van den Pol AN. GABAB receptor-mediated regulation of glutamate-activated calcium transients in hypothalamic and cortical neuron development. J Neurophysiol. 1999;82:94–102. doi: 10.1152/jn.1999.82.1.94. [DOI] [PubMed] [Google Scholar]
- 35.Xu L, Anwyl R, Rowan MJ. Spatial exploration induces a persistent reversal of long-term potentiation in rat hippocampus. Nature. 1998;394:891–894. doi: 10.1038/29783. [DOI] [PubMed] [Google Scholar]
- 36.Bear MF, Abraham WC. Long-term depression in hippocampus. Annu Rev Neurosci. 1996;19:437–462. doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- 37.Kemp N, Bashir ZI. Long-term depression: a cascade of induction and expression mechanisms. Prog Neurobiol. 2001;65:339–365. doi: 10.1016/s0301-0082(01)00013-2. [DOI] [PubMed] [Google Scholar]
- 38.Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11:327–335. doi: 10.1016/s0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- 39.Behe P, Stern P, Wyllie DJ, Nassar M, Schoepfer R, Colquhoun D. Determination of NMDA NR1 subunit copy number in recombinant NMDA receptors. Proc Biol Sci. 1995;262:205–213. doi: 10.1098/rspb.1995.0197. [DOI] [PubMed] [Google Scholar]
- 40.Premkumar LS, Auerbach A. Stoichiometry of recombinant N-methyl-D-aspartate receptor channels inferred from single-channel current patterns. J Gen Physiol. 1997;110:485–502. doi: 10.1085/jgp.110.5.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loftis JM, Janowsky A. The N-methyl-D-aspartate receptor subunit NR2B: localization, functional properties, regulation, and clinical implications. Pharmacol Ther. 2003;97:55–85. doi: 10.1016/s0163-7258(02)00302-9. [DOI] [PubMed] [Google Scholar]
- 42.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 43.Stocca G, Vicini S. Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol. 1998;507:13–24. doi: 10.1111/j.1469-7793.1998.013bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hestrin S. Developmental regulation of NMDA receptor-mediated synaptic currents at a central synapse. Nature. 1992;357:686–689. doi: 10.1038/357686a0. [DOI] [PubMed] [Google Scholar]
- 46.Barth AL, Malenka RC. NMDAR EPSC kinetics do not regulate the critical period for LTP at thalamocortical synapses. Nat Neurosci. 2001;4:235–236. doi: 10.1038/85070. [DOI] [PubMed] [Google Scholar]
- 47.Bi C, Cui Y, Mao Y, Dong S, Zhang J, Sun X. The effect of early auditory deprivation on the age-dependent expression pattern of NR2B mRNA in rat auditory cortex. Brain Res. 2006;1110:30–38. doi: 10.1016/j.brainres.2006.06.056. [DOI] [PubMed] [Google Scholar]
- 48.Lim IA, Merrill MA, Chen Y, Hell JW. Disruption of the NMDA receptor-PSD-95 interaction in hippocampal neurons with no obvious physiological short-term effect. Neuropharmacology. 2003;45:738–754. doi: 10.1016/s0028-3908(03)00276-4. [DOI] [PubMed] [Google Scholar]
- 49.Khan AM, Curras MC, Dao J, Jamal FA, Turkowski CA, Goel RK, Gillard ER, Wolfsohn SD, Stanley BG. Lateral hypothalamic NMDA receptor subunits NR2A and/or NR2B mediate eating: immunochemical/behavioral evidence. Am J Physiol. 1999;276:R880–891. doi: 10.1152/ajpregu.1999.276.3.R880. [DOI] [PubMed] [Google Scholar]