

Abstract
The reasons for non-resolving thrombosis in chronic thromboembolic pulmonary hypertension (CTEPH) have not been fully elucidated. Despite platelets being implied in its pathogenesis, they have been poorly studied. We hypothesized that platelets would be altered in CTEPH. The aim of our study was to compare selected platelet parameters in CTEPH patients with healthy subjects. The study included healthy subjects (n = 50) and patients with CTEPH (n = 47). We investigated platelet count, mean platelet volume (MPV), and platelet aggregation-spontaneous (SPA) and induced by various concentrations of five agonists. In addition, some other hemostatic parameters were also investigated to provide a comprehensive view on hemostasis. We found a decreased platelet count [212 (171-251) versus 248 (205-408) 109 L-1, P<0.01], higher MPV [11.3 (10.5-11.7) versus 10.1 fL (9.4-10.4), P<0.001] and higher SPA [9.5 (7.1-12.4) versus 5 (1.3-9) %, P<0.001], but a decrease of induced platelet aggregation (only by maximal agonist concentrations) in CTEPH patients compared to controls. These changes were accompanied by a significant increase of plasma fibrinogen, factor VIII, von Willebrand factor (antigen and activity), and plasminogen activator inhibitor. Thus, we can conclude that CTEPH is accompanied by a prothrombotic state, including platelet abnormalities. They reflect a higher platelet turnover/reactivity and specific platelet behavior (impaired aggregation) in these patients. Our findings imply that platelet disorders can contribute to the pathogenesis of CTEPH. However, further research would be desirable to better understand the reason for this finding.
Keywords: Platelets, mean platelet volume, platelet aggregation, thromboembolism
Introduction
Chronic thromboembolic pulmonary hypertension (CTEPH) is a progressive potentially fatal disease, in which it is believed that thromboembolic occlusion of pulmonary vessels due to non-resolving but organizing thrombi gradually leads to significant elevation of pulmonary blood pressure, resulting in pressure overload and failure of the right heart [1-6]. Pulmonary embolism is thought to be the initiating event that despite adequate treatment results only in incomplete resolution. Furthermore, organization of the thromboemboli is associated with progressive vascular remodeling, relevantly contributing to the severity of pulmonary hypertension.
Until now, many aspects of the pathogenesis of CTEPH have been poorly understood. The reasons for incomplete resolution of pulmonary emboli have not been identified. However, several clinical conditions associated with non-resolving thrombosis are accepted [1-6]. Current hypotheses also include a prothrombotic state due to abnormal platelet activation and interactions with the pulmonary vasculature [1]. An alternative hypothesis of the pathogenesis of CTEPH suggests a primary arteriopathy of pulmonary vessels and secondary in situ thrombosis as causes of pulmonary vascular occlusion. It is widely believed that some patients are genetically susceptible to developing this complication, but genetic variants associated with a heightened risk of CTEPH have yet to be determined [1].
Structural changes in the pulmonary vasculature in CTEPH may correlate with its clinical severity [1]. The mechanisms underlying the remodeling of the pulmonary vascular bed have not been fully elucidated but are likely to involve vasoconstriction, inflammation, thrombosis, cell proliferation, and fibrosis [1]. It has been proposed that vascular endothelial damage may result in adherence and activation of platelets favoring thrombosis as well as activation of coagulation pathways.
Platelets may be related to all three basic mechanisms of CTEPH (thrombosis, vasoconstriction, vascular remodeling) through different pathways [7]. They produce, store, and release mediators that may contribute to the initiation or aggravation of CTEPH [7-9].
Despite platelet defects being implied in the pathogenesis of CTEPH, original research supporting this hypothesis is limited. Platelet function has been poorly studied and has not been critically evaluated.
We hypothesized that platelets would be altered in CTEPH and might contribute to its development. Thus, the aim of our prospective study was to investigate platelet count, platelet size and platelet function (platelet aggregation) in patients with severe CTEPH, and compare them with a group of sex- and age-matched healthy subjects. In addition, some related endothelial, coagulation and inflammatory parameters were also investigated, in order to provide a more comprehensive view on hemostasis. These could help to better understand some of the platelet findings.
Materials and methods
The study comprised a control group of healthy subjects (n=50) and CTEPH patients (n=47) matched on sex and age (Table 1). Healthy subjects were recruited from blood donors and staff volunteers. They admitted neither antiplatelet agents nor other drug therapies. The patients were referred to the Department of Cardiology and Angiology, Medical Faculty of Slovak Medical University and National Institute of Cardiovascular Diseases in Bratislava for evaluation and treatment of CTEPH. The study was conducted according to the principles in the Declaration of Helsinki.
Table 1.
General characteristic in healthy subjects and patients with chronic thromboembolic pulmonary hypertension (CTEPH)
| Healthy subjects | CTEPH | |
|---|---|---|
| Number of subjects | 50 | 47 |
| Age median, years | 57 (45-68) | 63 (55-69) |
| Race (%) white | 100 | 100 |
| Female gender, n (%) | 16 (32) | 15 (31.9) |
| Medications | ||
| Warfarin use currently, n (%) | 20 (42.5) | |
| Digoxin, n (%) | 6 (12.8) | |
| ACE inhibitors/ARB, n (%) | 14/3 (29.8/6.4) | |
| Diuretics, n (%) | 29 (61.7) | |
| Beta-blocker, n (%) | 18 (38.3) | |
| CCB, n (%) | 12 (25.5) | |
| OAD, n (%) | 5 (10,6) | |
| Statin/fibrate, n (%) | 8/1 (17.0/2.1) | |
| Amiodaron, n (%) | 2 (4.3) | |
| INR median | 1.07 (1.04-1.14) | 1.26 (1.08-2.28) |
| PEA, n (%) | - | 17 (36.2) |
| NYHA, n (%) | ||
| I | 7 (14.9) | |
| II | 20 (42.5) | |
| III | 18 (38.3) | |
| IV | 2 (4.3) | |
| Arterial hypertension, n (%) | 0 | 27 (57.4) |
| Diabetes mellitus, n (%) | 0 | 7 (14.9) |
| Bleeding history, n (%) | 0 | 2 (4.3) |
| Atrial arrhythmia, n (%) | 0 | 11 (14.9) |
ACE: angiotensin converting enzyme, ARB: angiotensin II receptor blocker, CCB: calcium channel blocker, OAD: oral antidiabetic drug, INR: international normalized ratio, PEA: pulmonary endarterectomy, NYHA: New York Heart Association functional class.
All patients underwent a thorough initial workup. Data obtained included anatomic diagnosis, clinical functional class, drug use, right heart failure, arrhythmia, hemoptysis, other bleeding diathesis, or thromboembolism. Laboratory variables included complete blood count, capillary oxygen saturation, and serum biochemistry. All patients underwent standard non-invasive and invasive cardiologic examination: electrocardiography, pulmonary function tests, echocardiography, ventilation-perfusion scanning, pulmonary angiography, computed tomography (CT) angiography, and right heart catheterization. All patients had an invasively confirmed severe pulmonary hypertension (by right heart catheterization, mean pulmonary artery pressure > 45 mmHg; pulmonary capillary wedge pressure < 15 mmHg).
The diagnosis of CTEPH was made according to the European Society of Cardiology (ESC) guidelines [3,6]. It was based on clinical presentation (history of pulmonary embolism, progressive dyspnea and signs of right heart failure), echocardiography, ventilation-perfusion scanning, pulmonary angiography, CT angiography, and right heart catheterization. All patients were placed on anticoagulant therapy, but only 20 patients (42.5%) were currently on warfarin (desired International normalized ratio [INR] 2-3 was found in 13 of them, i.e. 65.0%). The rest of them (57.5%) at the time of blood collection were on a bridging therapy by low molecular weight heparin due to planned invasive procedures. Overall, the INR median in CTEPH patients was 1.26 (1.08-2.28).
Treatment other than anticoagulants included oxygen, diuretics, renin-angiotensin-aldosterone system inhibitors, beta-blockers, calcium channel blockers, digoxin, antiarrhythmics, and other drugs. Subjects were excluded if they were taking other medications known to affect platelet function. The crucial clinical data for patients with CTEPH are depicted in Table 1.
Blood samples
Blood samples were obtained for complete blood count, platelet function tests, endothelial markers and selected coagulation variables. Blood was collected after resting comfortably for at least 10 minutes into a Vacuette® 3 mL K3E K3EDTA tube (for complete blood count), Vacuette® 5 mL 9NC Coagulation sodium citrate 3, 2% tubes (for platelet aggregation, endothelial and coagulation parameters) and Vacuette® 5 mL Serum tube with micronized silica particles (for antiphospholipid antibodies), using a Vacuette® 21Gx1 ½ needle (Vacuette® system, Greiner Bio-One, Krems-münster, Austria). All blood collections were carried out between 08:00 and 09:00 AM after overnight fasting with minimal trauma from an antecubital vein. The samples were treated immediately, at least up to 2 h after blood collection.
The complete blood count (including mean platelet volume, MPV) was measured immediately after sampling (within 5-10 min) by Sysmex XT-2000i automated analyzer (Sysmex Europe GmbH, Germany). Platelet function was investigated by light transmission aggregometry, using Helena AggRAM™ platelet aggregometer (Helena, Biosciences Europe). Epinephrine 3 mM, adenosine diphosphate 200 µM, collagen 100 µg mL-1, ristocetin 10 mg mL-1, and arachidonic acid 5 mg mL-1 (Helena Bioscences Europe, Gateshead, Tyne and Wear, United Kingdom) were used as aggregation agonists in platelet aggregation studies.
Platelet-rich plasma (PRP) was obtained by centrifugation at 100 g for 10 min. Platelet count in PRP was adjusted to 200-250. 109 L-1 with autologous platelet-poor plasma obtained by centrifuging PRP at 2,500 g for 10 min. Aggregation agonists and saline were added separately into these plasma samples. We used 50 μL of 0.9% NaCl solution to measure the spontaneous platelet aggregation (SPA). The same amount of aggregation agonists were used to evaluate the inducible aggregation. The aggregation agonists were tested before the examination, using plasma pooled from six blood donors, treated the same way as the examined sample. Detecting both decreased and increased platelet aggregation, we measured aggregation response to agonists at various concentrations: no agonist (spontaneous), epinephrine (30.0, 10.0, 1.0, 0.5 µM), adenosine diphosphate (ADP; 20.0, 2.0, 1.0, 0.5 µM), collagen (1.0 µM), ristocetin (0.1 µM), and arachidonic acid (0.5 mg mL-1). The results were recorded as maximal platelet aggregation (%).
For endothelial markers and coagulation studies, citrated platelet-poor plasma was used, after the centrifugation of the blood tube for 15 minutes at ≥ 1,500 g. All these studies were performed on an automated coagulation analyzer (BCS® XP System, Siemens Healthcare Diagnostics, Marburg, Germany), using test kits by Siemens Healthcare Diagnostics, Marburg, Germany: vWF Ag test kit for the determination of von Willebrand factor antigen (vWF: Ag), INNOVANCE VWF Ac test kit for von Willebrand factor activity (vWF: Ac), coagulation factor VIII and XII deficient plasmas for the coagulation factors VIII and XII activity, Berichrom PAI for plasminogen activator inhibitor type 1 (PAI-1) activity, Multifibren U for fibrinogen, Berichrom Antithrombin III (A) for the functional activity of antithrombin III (AT III) and INNOVANCE D-Dimer for the cross-linked fibrin degradation products (D-dimers) in plasma, according to the instruction manual from the instrument manufacturer.
Serum antibodies against cardiolipin and ß2-glycoprotein were determined by immunoassay, using AESKULISA Cardiolipin-Check and AESKULISA ß2-Glyko-Check (AESKU.DIAGNOSTICS, Wendelsheim, Germany), respectively. Sera with particles were cleared by low speed centrifugation (<1,000 g). After separation, the freshly collected serum samples were used immediately or frozen at -20°C for no longer than 2 weeks. The tests were performed on an automated system BEP 2000 Advance (Siemens Healthcare Diagnostics, Marburg, Germany).
Statistical analysis
GraphPad Prism, version 6.05 (GraphPad Software, Inc., San Diego, USA), was used for all data analyses. The results were presented as median and interquartile range (for non-normally distributed data). Normality testing was performed by D’Agostino-Pearson test. The non-parametric Mann-Whitney U-test was used to compare variables between groups. This test was used including percentage values of some variables, which are equivalent to units (100%=100 units, e.g. for vWF 100%=100 IU dL-1). P values less than 0.05 were considered significant.
Results
Subjects in both control and CTEPH groups were of similar sex and age, with no statistically significant differences. The results of platelet, endothelial and other coagulation parameters in controls and CTEPH patients are shown in Tables 2 and 3.
Table 2.
Platelet characteristics in healthy subjects and patients with chronic thromboembolic pulmonary hypertension (CTEPH)
| Parameter | Healthy subjects | CTEPH | P value |
|---|---|---|---|
| Platelet count (109 L-1) | 248 (205-408) | 212 (171-251) | 0.0012 |
| MPV (fL) | 10.1 (9.4-10.4) | 11.3 (10.5-11.7) | <0.0001 |
| SPA (%) | 5 (1.3-9.0) | 9.5 (7.1-12.4) | <0.0001 |
| MPA (%) EPI 30 µM | 82.9 (79.1-85.9) | 80.1 (79.5-84.2) | 0.0231 |
| MPA (%) EPI 10 µM | 80.9 (74.6-85.5) | 79.1 (69.4-82.2) | NS |
| MPA (%) EPI 1 µM | 79.7 (76.5-84.6) | 77.9 (55.7-81.8) | 0.009 |
| MPA (%) EPI 0.5 µM | 77.7 (20.5-82.3) | 67.5 (30.5-78.6) | NS |
| MPA (%) ADP 20 µM | 86 (82.9-87.9) | 82.7 (74.7-86) | 0.0004 |
| MPA (%) ADP 2 µM | 40.4 (13.6-91.2) | 39.9 (22.6-86.2) | NS |
| MPA (%) ADP 1 µM | 14.4 (6.0-86.8) | 20.7 (9.5-31.9) | NS |
| MPA (%) ADP 0.5 µM | 7.4 (1.1-13) | 12.9 (3.7-20.2) | 0.0497 |
| MPA (%) COL 1.0 µM | 85.5 (81.8-87.9) | 80.7 (70.9-84.4) | <0.0001 |
| MPA (%) RIS 0.1 µM | 88.6 (85.3-91.6) | 85.1 (75.5-89.5) | 0.0045 |
| MPA (%) ARA 0.5 mg mL-1 | 85.7 (82.4-89.3) | 82.7 (74.7-86) | <0.0001 |
MPV: mean platelet volume, SPA: spontaneous platelet aggregation, MPA: maximal platelet aggregation, ADP: adenosine diphosphate, EPI: epinephrine, COL: collagen, RIS: ristocetin, ARA: arachidonic acid. Data are shown as median and interquartile range. Comparisons between healthy subjects and CTEPH patients are analysed by the Mann-Whitney U-test. NS-non-significant values.
Table 3.
Selected endothelial and coagulation variables in healthy subjects and patients with chronic thromboembolic pulmonary hypertension (CTEPH)
| Parameter | Healthy subjects | CTEPH | P value |
|---|---|---|---|
| vWF:Ag (IU dL-1) | 116.5 (94.1-168.5) | 199.5 (181.1-200) | <0.0001 |
| vWF:Ac (%) | 115.5 (87.0-135.9) | 199.8 (173.8-199.8) | <0.0001 |
| Factor VIII (%) | 123.3 (100.4-199.2) | 171.6 (141.7-199.2) | <0.0001 |
| Factor XII (%) | 115.7 (95.7-126.3) | 102.2 (87.0-121.8) | NS |
| PAI-1 (U mL-1) | 2.22 (1.01-2.2) | 2.44 (1.43-3.64) | 0.0327 |
| Fibrinogen (g L-1) | 3.3 (2.6-4.9) | 3.7 (3.2-5.3) | <0.0001 |
| AT III (%) | 99.6 (92.1-107.3) | 97.8 (83.3-108) | NS |
| D-dimers (mg L-1) | 0.24 (0.19-0.38) | 0.29 (0.18-0.7) | NS |
| ACLA (U mL-1) | 0.88 (0.49-1.56) | 0.699 (0.34-1.67) | NS |
| Anti-ß2-GP (U mL-1) | 0.47 (0.18-0.99) | 0.73 (0.25-1.84) | NS |
vWF: Ag-von Willebrand factor antigen, vWF: Ac-von Willebrand factor activity, PAI-1: plasminogen activator inhibitor type 1, AT III: antithrombin III, ACLA: anti-cardiolipin antibodies, anti-ß2-GP: anti-ß2-glycoprotein. Data are shown as median and interquartile range. Comparisons between healthy subjects and CTEPH patients are analysed by the Mann-Whitney U-test. NS-non-significant values.
In CTEPH patients, we found a decreased platelet count (P=0.0012), higher MPV (P<0.0001), higher SPA (P<0.0001), but a decrease of platelet aggregation induced by all standard concentrations of various agonists-epinephrine 30.0 µM (P=0.0231), ADP 20 µM (P=0.0004), collagen 1.0 µM (P<0.0001), ristocetin 0.1 µM (P=0.0045), and arachidonic acid 0.5 mg mL-1 (P<0.0001). Besides a borderline increase of platelet aggregation induced by 0.05 µM ADP (P=0.0497) in CTEPH patients, we did not observe any changes in platelet aggregation induced by low (“submaximal”) concentrations of epinephrine (10.0, 1.0, and 0.5 µM) and ADP (2.0, 1.0 µM) compared to controls (Figure 1). In CTEPH patients, we found an increase of plasma fibrinogen, factor VIII, vWF: Ag, vWF: Ac (P<0.0001, for all 4 of these parameters), and PAI-1 (P=0.0327) compared to controls.
Figure 1.
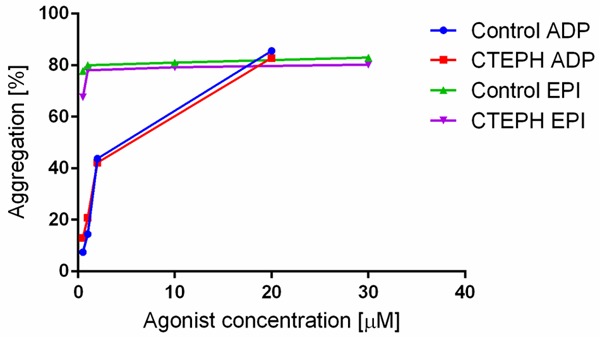
Platelet aggregation induced by various concentrations of adenosine diphosphate (ADP) and epinephrine (EPI) agonists in a control group of healthy subjects and in patients with chronic thromboembolic pulmonary hypertension (CTEPH).
Discussion
The inter-individual variation in platelet reactivity may contribute to the risk of thrombosis in one individual, but protection in another [10]. It is known that markers of platelet activation are increased in patients with systemic arterial hypertension [11-13], but only a few reports on platelet function in pulmonary arterial hypertension (PAH)/CTEPH are available [7,9,14-18]. Therefore, we investigated selected platelet parameters in patients with CTEPH, as a specific type of pulmonary hypertension.
Along with the platelet count, MPV can be an indicator of platelet turnover. A high MPV indicates increased production of platelets. If combined with a low platelet count, this indicates a condition that results in destruction of platelets. In our CTEPH patients, lower platelet count was associated with higher MPV. This may reflect a chronic process of platelet activation, consumption or destruction, in which the production of new (younger, bigger and more potent) platelets is not sufficient and may lead to thrombocytopenia. Our results were in agreement with findings of higher MPV in PAH patients in two other studies currently available [14,15]. Unlike them, we investigated platelet parameters according to the etiology because some differences between various PAH types can be hypothesized. In a large population study, increased MPV was reported as a predictor for venous thromboembolism, supporting the concept that platelets are important in its pathogenesis [19].
Potential limitations of our study included previous findings that MPV increased slightly during the time from blood sampling to MPV determination due to platelet swelling in vitro using ethylenediaminetetraacetic acid (EDTA) as an anticoagulant [20]. Nevertheless, we were able to adjust for this, because time from blood sampling until analysis (in one laboratory, using a single type of analyzer) was carefully controlled for in all participants. Thus, we do not believe that this limitation significantly altered our results or the conclusions that should be drawn from them.
It is not clear whether thrombocytopenia seen in various PAH patients is an incidental finding, caused by platelet consumption in pulmonary vasculature, or as a result of platelet shearing due to pulmonary microangiopathy [7]. On the other hand, it was demonstrated that circulating platelets from idiopathic PAH patients with thrombocytopenia are mature, suggesting that the low platelet levels in these patients are not explained by increased platelet consumption [16].
In the present study, platelet aggregation measurements were used to establish platelet phenotypes. Platelet aggregometry is a conventional method for the assessment of platelet function and remains the ‘gold standard’ [21-23]. We evaluated maximal platelet aggregation (MPA) without stimulation (SPA) and MPA-induced by several agonists. Measurement of SPA may be a suitable index of platelet reactivity [24]. It is a predictive risk marker of arterial occlusions, coronary events and mortality [21,24,25]. In our patients with CTEPH, marked SPA increase was found. This was consistent with the results of some previous studies indicating an increase of another markers, which are implicated for platelet activation such as ratios of the P-selectin positive platelets, activated GPIIb/IIIa-positive platelets, or platelet hyper-responsiveness to ex vivo thrombin stimulation when evaluated for surface markers in CTEPH patients [17].
We used standard (“maximal”) agonist concentrations to identify individuals whose platelets demonstrated a low response to stimulation. On the other hand, we also used lower threshold (“submaximal”) concentrations to identify those with robust response to stimulation, who may represent a high-risk group for thrombotic events [10]. Despite expectations, we did not find a typical phenotype of agonist-induced platelet hyperactivity in patients with CTEPH. However, in CTEPH patients compared to healthy subjects, a significant platelet dysfunction after standard concentrations (but not after most of the low concentrations) of agonists was observed. This “paradoxical” platelet behavior in our CTEPH patients is similar to the phenotype of the platelets observed by other authors in idiopathic PAH [16]. They showed a defect in platelet ability to be activated in vitro by thrombin receptor-activating protein (TRAP). In some of these patients, even 10 times the concentration of TRAP could not elicit the same response as the control.
Marked abnormalities in platelet reactivity in CTEPH raise important questions about their determinants. One hypothesis is that some genetic factors underlie the observed differences in platelet reactivity. Partly, a decrease of platelet aggregation may be due to the drug treatment used, which is supposed to affect the platelet function [12,13]. It is accepted that CTEPH/PAH are associated with a specific platelet phenotype. Defective platelet aggregation in patients with idiopathic PAH may, at least in part, be due to abnormalities in nitric oxide (NO) levels and endothelial NO synthase (eNOS) expression, which could impact the ability of the platelets to regulate their own function appropriately [16]. This was in agreement with our results in CTEPH patients, in which already threshold concentrations of agonists led to sufficient platelet aggregation (even higher with ADP 0.05 µM), while standard concentrations revealed an impaired platelet aggregation. However, further research would be desirable to better understand the reason for this finding.
Furthermore, other coagulation and endothelial parameters were significantly increased in the plasma of our CTEPH patients: fibrinogen, factor VIII, vWF: Ag, vWF: Ac, and PAI-1. All of these can be increased as a result of damages to the vascular endothelium and/or in response to inflammation. vWF is secreted into plasma from endothelial cells and plays a crucial role in platelet adhesion and aggregation. It can also be increased in patients with idiopathic PAH and may be a predictor of long-term prognosis [16]. Elevated plasma factor VIII was the first prothrombotic factor identified in a large proportion of CTEPH patients [26]. Since elevated D-dimer levels are observed in all conditions with increased coagulation activation, normal values in our CTEPH patients may reflect effective anticoagulation (by warfarin and/or heparin). Similarly, D-dimer was not increased in patients with CTEPH in another study [17].
It is not clear whether all these abnormalities are primary and contributory to CTEPH development, or secondary to this disease. In our study, we did not find any patients with antiphospholipid syndrome and/or elevation of antiphospholipid antibodies.
It still remains unknown whether antiplatelet therapy, combined with anticoagulant therapy, is warranted in patients with CTEPH. The specific antiplatelet drugs have not been studied sufficiently in these patients [7]. Demonstration of an increased platelet turnover by other methods could offer a rationale for this approach. Meanwhile, these therapeutic considerations have to be evaluated very carefully on an individual basis [6,7,9]. On the basis of our results, at least in some selected high-risk CTEPH patients with low risk of bleeding, aspirin therapy in combination with warfarin could be considered.
Lifelong anticoagulant therapy is the gold standard in the management of CTEPH. The purpose is to prevent both recurrent venous thromboembolism and in situ pulmonary artery thrombosis. Prolonged therapy of venous thromboembolism is associated with fewer recurrent emboli, and this effect appears to be larger than the increased risk of bleeding [1]. However, no randomized clinical trial has studied the role of anticoagulants in CTEPH [7].
Although pulmonary endarterectomy is the preferred treatment for patients with CTEPH, new options for pharmacological treatment (like bosentan or riociguat) have shown clinical benefits [27,28]. Their effect on platelet function in CTEPH could be of interest.
On the basis of this study we can conclude that severe CTEPH is accompanied by platelet abnormalities. They can be related to a prothrombotic state, which occurs very frequently in these patients. Further studies could be proposed to better clarify the pathophysiological role of platelets in CTEPH and to address the question of suitable antithrombotic/antiplatelet therapy.
Disclosure of conflict of interest
None.
References
- 1.Hoeper MM, Mayer E, Simonneau G, Rubin LJ. Chronic thromboembolic pulmonary hypertension. Circulation. 2006;113:2011–2020. doi: 10.1161/CIRCULATIONAHA.105.602565. [DOI] [PubMed] [Google Scholar]
- 2.Bonderman D, Wilkens H, Wakounig S, Schäfers HJ, Jansa P, Lindner J, Simkova I, Martischnig AM, Dudczak J, Sadushi R, Skoro-Sajer N, Klepetko W, Lang IM. Risk factors for chronic thromboembolic pulmonary hypertension. Eur Respir J. 2009;33:325–331. doi: 10.1183/09031936.00087608. [DOI] [PubMed] [Google Scholar]
- 3.Galiè N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, Beghetti M, Corris P, Gaine S, Gibbs JS, Gomez-Sanchez MA, Jondeau G, Klepetko W, Opitz C, Peacock A, Rubin L, Zellweger M, Simonneau G, Vahanian A, Auricchio A, Bax J, Ceconi C, Dean V, Filippatos G, Funck-Brentano C, Hobbs R, Kearney P, McDonagh T, McGregor K, Popescu BA, Reiner Z, Sechtem U, Sirnes PA, Tendera M, Vardas P, Widimsky P, Sechtem U, Al Attar N, Andreotti F, Aschermann M, Asteggiano R, Benza R, Berger R, Bonnet D, Delcroix M, Howard L, Kitsiou AN, Lang I, Maggioni A, Nielsen-Kudsk JE, Park M, Perrone-Filardi P, Price S, Domenech MTS, Vonk-Noordegraaf A, Zamorano JL. ESC Committee for Practice Guidelines. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2009;30:2493–2537. doi: 10.1093/eurheartj/ehp297. [DOI] [PubMed] [Google Scholar]
- 4.Lang IM, Pesavento R, Bonderman D, Yuan JX. Risk factors and basic mechanisms of chronic thromboembolic pulmonary hypertension: a current understanding. Eur Respir J. 2013;41:462–468. doi: 10.1183/09031936.00049312. [DOI] [PubMed] [Google Scholar]
- 5.Hoeper M, Madani MM, Nakanishi N, Meyer B, Cebotari S, Rubin LJ. Chronic thromboembolic pulmonary hypertension. Lancet Respir Med. 2014;2:573–582. doi: 10.1016/S2213-2600(14)70089-X. [DOI] [PubMed] [Google Scholar]
- 6.Konstantinides S, Torbicki A, Agnelli G, Danchin N, Fitzmaurice D, Galie N, Gibbs JSR, Huisman M, Humbert M, Kucher N, Lang I, Lankeit M, Lekakis J, Maack C, Mayer E, Meneveau N, Perrier A, Pruszczyk P, Rasmussen LH, Schindler TH, Svitil P, Vonk-Noordegraaf A, Zamorano JL, Zompatori M. 2014 ESC Guidelines on the diagnosis and management of acute pulmonary embolism. Eur Heart J. 2014;35:3033–3073. doi: 10.1093/eurheartj/ehu283. [DOI] [PubMed] [Google Scholar]
- 7.Zanjani KS. Platelets in Pulmonary Hypertension: a Causative Role or a Simple Association? Iran J Pediatr. 2012;22:145–157. [PMC free article] [PubMed] [Google Scholar]
- 8.Herve P, Humbert M, Sitbon O, Parent F, Nunes H, Legal C, Garcia G, Simonneau G. Pathobiology of pulmonary hypertension. The role of platelets and thrombosis. Clin Chest Med. 2001;22:451–458. doi: 10.1016/s0272-5231(05)70283-5. [DOI] [PubMed] [Google Scholar]
- 9.Margey RJ, Gaine S, Peace A, Tedesco T, Kenny D. Heightened platelet reactivity defines chronic thromboembolic pulmonary hypertension from other subtypes of pulmonary hypertension. J Amer Coll Cardiol. 2010;55 Suppl A171.E1605. [Google Scholar]
- 10.Bray PF. Platelet hyperreactivity: predictive and intrinsic properties. Hematol Oncol Clin North Am. 2007;21:633–45. v–vi. doi: 10.1016/j.hoc.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Remková A, Kováčová E, Príkazská M, Kratochvíľová H. Thrombomodulin as a marker of endothelium damage in some clinical conditions. Eur J Intern Med. 2000;2:79–84. doi: 10.1016/s0953-6205(00)00066-2. [DOI] [PubMed] [Google Scholar]
- 12.Remková A, Kratochvíľová H, Ďurina J. Impact of the therapy by renin-angiotensin system targeting antihypertensive agents perindopril versus telmisartan on prothrombotic state in essential hypertension. J Hum Hypertens. 2008;22:338–345. doi: 10.1038/sj.jhh.1002328. [DOI] [PubMed] [Google Scholar]
- 13.Remková A, Remko M. The role of renin-angiotensin system in prothrombotic state in essential hypertension. Physiol Res. 2010;59:13–23. doi: 10.33549/physiolres.931525. [DOI] [PubMed] [Google Scholar]
- 14.Can MM, Tanboga IH, Demircan HC, Ozkan A, Koca F, Keles N, Sonmez K, Kaymaz C, Serebruany V. Enhanced hemostatic indices in patients with pulmonary arterial hypertension: an observational study. Thromb Res. 2010;126:280–282. doi: 10.1016/j.thromres.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 15.Varol E, Uysal BA, Ozaydin M. Platelet indices in patients with pulmonary arterial hypertension. Clin Appl Thromb Hemost. 2011;17:E171–174. doi: 10.1177/1076029610394438. [DOI] [PubMed] [Google Scholar]
- 16.Aytekin M, Aulak KS, Haserodt S, Chakravarti R, Cody J, Minai OA, Dweik RA. Abnormal platelet aggregation in idiopathic pulmonary arterial hypertension: role of nitric oxide. Am J Physiol Lung Cell Mol Physiol. 2012;302:L512–L520. doi: 10.1152/ajplung.00289.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yaoita N, Shirakawa R, Fukumoto Y, Sugimura K, Miyata S, Miura Y, Nochioka K, Miura M, Tatebe S, Aoki T, Yamamoto S, Satoh K, Kimura T, Shimokawa H, Horiuchi H. Platelets are highly activated in patients of chronic thromboembolic pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2014;34:2486–2494. doi: 10.1161/ATVBAHA.114.304404. [DOI] [PubMed] [Google Scholar]
- 18.Mogi K, Nakajima N, Masuda M, Hayashida N, Pearce Y, Nakaya M. A Study on the role of platelet function in patients with chronic pulmonary thromboembolism. Ann Thorac Cardiovasc Surg. 2001;7:133–137. [PubMed] [Google Scholar]
- 19.Brækkan SK, Mathiesen EB, Njølstad I, Wilsgaard T, Størmer J, Hansen JB. Mean platelet volume is a risk factor for venous thromboembolism: the Tromsø Study. J Thromb Haemost. 2010;8:157–162. doi: 10.1111/j.1538-7836.2009.03498.x. [DOI] [PubMed] [Google Scholar]
- 20.Machin SJ, Briggs C. Mean platelet volume: a quick, easy determinant of thrombotic risk? J Thromb Haemost. 2010;8:146–147. doi: 10.1111/j.1538-7836.2009.03673.x. [DOI] [PubMed] [Google Scholar]
- 21.Yee DL, Sun CW, Bergeron AL, Dong J, Bray PF. Aggregometry detects platelet hyperreactivity in healthy individuals. Blood. 2005;106:2723–2729. doi: 10.1182/blood-2005-03-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yee DL, Bergeron AL, Sun CW, Dong JF, Bray PF. Platelet hyperreactivity generalizes to multiple forms of stimulation. J Thromb Haemost. 2006;4:2043–2050. doi: 10.1111/j.1538-7836.2006.02089.x. [DOI] [PubMed] [Google Scholar]
- 23.Remkova A, Janusicova A, Remko M. Is antiplatelet therapy always effective? Vnitř Lék. 2012;58:904–914. [PubMed] [Google Scholar]
- 24.Bampalis VG, Brantl SA, Siess W. Why and how to eliminate spontaneous platelet aggregation in blood measured by multiple electrode aggregometry. J Thromb Haemost. 2012;10:1710–1714. doi: 10.1111/j.1538-7836.2012.04819.x. [DOI] [PubMed] [Google Scholar]
- 25.Trip MD, Cats VM, van Capelle FJ, Vreeken J. Platelet hyperactivity and prognosis of survivors of myocardial infarction. N Engl J Med. 1990;322:1549–1554. doi: 10.1056/NEJM199005313222201. [DOI] [PubMed] [Google Scholar]
- 26.Bonderman D, Turecek PL, Jakowitsch J, Weltermann A, Adlbrecht C, Schneider B, Kneussl M, Rubin LJ, Kyrle PA, Klepetko W, Maurer G, Lang IM. High prevalence of elevated clotting factor VIII in chronic thromboembolic pulmonary hypertension. Thromb Haemost. 2003;90:372–376. doi: 10.1160/TH03-02-0067. [DOI] [PubMed] [Google Scholar]
- 27.Ghofrani HA. Treatment of pulmonary arterial hypertension (PAH): updated recommendations of the Cologne Consensus Conference 2011. Int J Cardiol. 2011;154(Suppl 1):S20–S33. doi: 10.1016/S0167-5273(11)70490-9. [DOI] [PubMed] [Google Scholar]
- 28.Ghofrani HA, D’Armini AM, Grimminger F, Hoeper MM, Jansa P, Kim NH, Mayer E, Simonneau G, Wilkins MR, Fritsch A, Neuser D, Weimann G, Wang C CHEST-Study Group. Riociguat for the treatment of CTEPH. N Engl J Med. 2013;369:319–329. doi: 10.1056/NEJMoa1209657. [DOI] [PubMed] [Google Scholar]