

Abstract
Clopidogrel nonresponsiveness increases the recurrence of cardiovascular events in patients undergoing percutaneous coronary intervention (PCI). Previous studies found that genetic variants such as single nucleotide polymorphisms (SNPs) of CYP2C19 and PON1 may influence clopidogrel response, cause high platelet reactivity (HPR) and increase cardiovascular events. However, these studies were inconsistent and inconclusive, especially in the Eastern Asian population. In this study, we investigated the effects of genetic variants on clopidogrel response and clinical outcomes in Chinese patients undergoing PCI. A total of 336 acute coronary syndrome patients undergoing PCI were included, 53 (15.77%) of whom were categorized as HPR. Among the 10 SNPs studied (ABCB1, CYP2C19*2, CYP2C19*3, CYP2C19*4, CYP2C19*17, CYP3A4, CYP3A5, ITGB3, P2Y12 and PON1 Q192R), the CYP2C19*4 and P2Y12 variants were not found in our population. The PON1 192Q and CYP2C19*2 alleles were significantly higher in the HPR group compared with the normal platelet reactivity (NPR) group (P=0.033 and 0.038, respectively), while the other SNPs were not significantly different between the two groups. Platelet aggregation of the PON1 192Q allele carriers was significantly higher than that of non-carriers both at baseline and 1 month after PCI (P=0.010 and 0.024, respectively); this was the case for CYP2C19*2 allele carriers, as well (P=0.005 and 0.003, respectively). The risk of major adverse cardiovascular event (MACE) increased with the presence of the PON1 192Q and CYP2C19*2 alleles during 6-month follow-up (P=0.012 and 0.003, respectively). In conclusion, both the PON1 Q192R and CYP2C19*2 variants are associated with HPR and an increased risk of ischemic events in Chinese patients undergoing PCI.
Keywords: Clopidogrel, platelet reactivity, single nucleotide polymorphisms, percutaneous coronary intervention
Introduction
Currently, percutaneous coronary intervention (PCI) is used as a primary treatment for coronary heart disease (CHD). Clopidogrel- and aspirin-based dual antiplatelet therapy (DAT) is widely used in the treatment of CHD patients undergoing PCI and has been proven to reduce the risk of thrombotic events after PCI [1,2]. However, despite the routine use of DAT, some patients still experienced recurring cardiovascular events after stent implantation; this is considered to be highly associated with clopidogrel nonresponsiveness or resistance [3-5].
Genetic variance is thought to be a major factor influencing platelet response to clopidogrel [6,7]. Some single nucleotide polymorphisms (SNPs), including CYP2C19, ABCA1, and PON1, have been investigated regarding their roles in clopidogrel response. However, powerful evidence only exists with regard to the effects of CYP2C19*2 variant on clopidogrel reactivity [8,9]. The results regarding other SNPs, such as ABCA1 and PON1, were inconsistent or controversial [7,10,11].
Gene polymorphisms vary among different ethnicities and races. Some SNPs, such as CYP2C19*2 and *3, had a higher prevalence in the Eastern Asian population than in the Western European population [12,13]. The effects of SNPs on clopidogrel responsiveness and clinical outcomes have not been fully elucidated in the Eastern Asian population. In this study, we investigated the effects of genetic variants on platelet reactivity to clopidogrel and clinical outcomes in Chinese patients undergoing PCI.
Methods
Ethics statement
The study protocol was approved by the medical ethics committee of Xinhua Hospital, Shanghai Jiaotong University School of Medicine. Written informed consent was obtained from all participants. The data were recorded anonymously. If the participants asked to withdraw, their data were destroyed.
Study population
Acute coronary syndrome (ACS) patients presenting to Xinhua Hospital, Shanghai Jiaotong University School of Medicine between January 2012 and March 2013 were considered for enrollment in our prospective observational study. The inclusion criteria were as follows: age above eighteen years, had undergone PCI and stent implantation, received a loading dose of 300 mg clopidogrel and aspirin followed by 75 mg clopidogrel and 100 mg aspirin daily, and could be followed up for more than six months after PCI. The major exclusion criteria were as follows: no regular medication, concomitant administration of oral anticoagulation agents or other antiplatelet agents, contraindications to clopidogrel or aspirin, active bleeding and bleeding diatheses, total platelet count less than 100×109/L, hematologic disorder, severe hepatic or renal insufficiency, pregnancy or malignancies.
Study design
All patients received a 300 mg loading dose of clopidogrel and aspirin before PCI followed by 75 mg clopidogrel per day for 12 months and 100 mg aspirin per day for life. All interventions were performed according to the current standard guidelines [14,15]. Stent types were chosen by the operator. If a glycoprotein IIb/IIIa inhibitor was required, tirofiban was administered. Low-molecular-weight heparin was used before angiography in all patients.
Platelet function assay
Blood samples were collected in vacutainer tubes containing 3.2% lithium heparin and trisodium citrate at baseline (just before PCI) and 1 month after PCI. The tubes were inverted three times to ensure the blood was completely mixed with the anticoagulant. Then, platelet reactivity was detected by the Thrombelastograph (TEG) Hemostasis Analyzer (Haemoscope Corp, Niles, IL, USA). A detailed description of this method has been outlined previously [14]. Platelet aggregation in response to adenosine diphosphate (ADP) was calculated with computerized software using the following formula: %Aggregation = [(MAADP - MAFibrin)/(MAThrombin - MAFibrin)] × 100. MAADP is the ADP-induced clot strength, reflecting the contribution of platelets not inhibited by clopidogrel. MAFibrin is the fibrin-induced clot strength, representing the contribution of fibrin alone to clot strength. MAThrombin is the thrombin-induced clot strength, representing the maximal potential platelet reactivity of the patient.
Genotyping
Genomic DNA was extracted from peripheral blood leukocytes and stored at -80°C until analysis. The following 10 SNPs within seven genes were selected: ABCB1 (rs1045642), CYP2C19*2 (rs4244285), CYP2C19*3 (rs4986893), CYP2C19*4 (rs28399504), CYP2C19*17 (rs12248560), CYP3A4 (rs4646437), CYP3A5 (rs776746), ITGB3 (rs5918), P2Y12 (rs2046934) and PON1 Q192R (rs662). The Sequenom MassARRAY (Sequenom Inc., San Diego, California, USA) platform was used for SNP genotyping. The whole process was conducted according to the manufacturer’s instructions. Briefly, the primers for polymerase chain reaction (PCR) amplification and single base extension were designed with Sequenom Assay Design 3.1 software. PCR amplification, shrimp alkaline phosphatase treatment, and primer extension reactions were performed with the iPLEX Gold assay. Extension reaction products were automatically transferred to the 384-SpectroCHIP. Mass signals of alleles were detected by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Finally, the genotyping results were analyzed using Sequenom MassARRAY TYPER software. For quality control, repeat genotyping was performed on random duplicate samples (n=50); the concordance rate was 100%, indicating that the genotyping was correct.
Outcome measures
The pharmacodynamic endpoint was high platelet reactivity (HPR), as assessed by TEG platelet-mapping assays. HPR was defined as ≥70% ADP-induced aggregation with 2 μmol/L ADP, as measured by TEG [15]. The clinical endpoint was the incidence of a major adverse cardiovascular event (MACE), which was defined as a composite event including cardiovascular death, myocardial infarction, target vessel revascularization, stent thrombosis or stroke.
Statistical analysis
Statistical analyses were performed with SPSS software, version 17 (SPSS Inc., Chicago, IL, USA). Continuous variables were expressed as the mean ± standard deviation, and categorical variables were expressed as frequencies and percentages. Study population characteristics were compared between HPR and normal platelet reactivity (NPR) patients by means of the chi-square test or Student’s t-test. The Hardy-Weinberg equilibrium was assessed by the chi-square test. Allelic frequency was estimated by gene counting. Kaplan-Meier analysis was used to assess the cumulative event-free survival for MACE. Multivari-ate Cox regression analysis was utilized to identify independent associations between underling polymorphisms and clinical outcomes after adjustment for potential confounders. All analyses used two-sided P values of <0.05.
Results
Study population characteristics
The study flowchart is shown in Figure 1. A total 336 patients were included in the study, all of whom were from the Chinese Han population. Among them, 53 (15.77%) were categorized as HPR patients. The clinical data of the HPR and NPR groups are shown in Table 1. There were no significant differences among gender, age, risk factors and other general information between the two groups (P>0.05).
Figure 1.
Study flow diagram.
Table 1.
Study population characteristics according to HPR
| Overall (n=336) | HPR (n=53) | NPR (n=283) | P | |
|---|---|---|---|---|
| Age | 66.5±10.5 | 69.2±11.1 | 66.6±10.4 | 0.119 |
| Male | 223 (66.4%) | 35 (66.0%) | 198 (70.0%) | 0.569 |
| LVEF (%) | 63±7.4 | 62±5.3 | 63±7.7 | 0.366 |
| BMI | 25.3±3.3 | 25.8±2.4 | 25.2±3.5 | 0.263 |
| Platelet reactivity | ||||
| ADP-induced aggregation (%) | 39.2±28.0 | 84.1±10.7 | 30.8±21.4 | <0.001 |
| Cardiovascular risk factors | ||||
| Smoking | 148 (44.0%) | 18 (40%) | 130 (44.7%) | 0.630 |
| Hypertension | 232 (69.0%) | 28 (62.2%) | 204 (70.1%) | 0.300 |
| DM | 81 (24.1%) | 13 (28.9%) | 68 (23.4%) | 0.455 |
| Dyslipidemia | 161 (47.9%) | 26 (49.1%) | 135 (47.7%) | 0.856 |
| Medical history | ||||
| Previous MI | 55 (16.4%) | 8 (15.1%) | 47 (16.6%) | 0.785 |
| Previous PCI | 49 (14.6%) | 7 (13.2%) | 43 (14.8%) | 0.709 |
| PAD | 25 (7.4%) | 4 (7.5%) | 21 (7.4%) | 0.974 |
| Stroke | 32 (9.5%) | 4 (8.9%) | 28 (9.6%) | 1.000 |
| Clinical presentation | ||||
| UA | 96 (28.6%) | 12 (22.6%) | 84 (29.7%) | 0.325 |
| AMI | 240 (71.4%) | 41 (77.4%) | 199 (70.3%) | 0.325 |
| Concomitant medication | ||||
| ACEI/ARB | 283 (84.2%) | 42 (79.2%) | 241 (85.2%) | 0.278 |
| β-blockers | 297 (88.4%) | 47 (88.7%) | 250 (88.3%) | 0.943 |
| Statin | 100 (29.8%) | 13 (24.5%) | 87 (30.7%) | 0.364 |
| PPI | 147 (43.8%) | 22 (41.5%) | 125 (44.2%) | 0.720 |
| GPI | 65 (19.3%) | 12 (22.6%) | 53 (18.7%) | 0.508 |
| Coronary intervention procedure | ||||
| LAD-related | 211 (62.8%) | 35 (66.0%) | 176 (62.2%) | 0.595 |
| Amount of stents | 1.6±0.5 | 1.7±0.3 | 1.6±0.5 | 0.160 |
| Length of stents (mm) | 44.0±22.6 | 46.4±18.1 | 43.6±23.3 | 0.408 |
HPR: high platelet reactivity; NPR: normal platelet reactivity; LVEF: left ventricular ejection fraction; BMI: body mass index; ADP: adenosine diphosphate; DM: diabetes mellitus; MI: myocardial infarction; PCI: percutaneous coronary intervention; PAD: peripheral arterial disease; UA: unstable angina; AMI: acute myocardial infarction; ACEI: angiotensin-converting enzyme inhibitors; ARB: angiotensin II receptor blockers; PPI: proton pump inhibitors; GPI: glycoprotein IIb/IIIa inhibitors; LAD: left anterior descending artery.
Genotype and high platelet reactivity
Among the ten SNPs, the CYP2C19*4 (rs28399504) and P2Y12 (rs2046934) variants were not found in the study population. The PON1 192Q and CYP2C19*2 alleles were significantly more frequent in the HPR group than in the NPR group (P=0.033 and 0.038, respectively). The other SNPs were not significantly different between the two groups (Table 2). All SNPs were in Hardy-Weinberg equilibrium.
Table 2.
Distribution of genetic variant alleles according to HPR of clopidogrel
| SNP | Overall (n=336) | HPR (n=53) | NPR (n=283) | P | OR | 95% CI |
|---|---|---|---|---|---|---|
| ABCB1 (rs1045642) | 0.678 | 0.879 | 0.477-1.618 | |||
| CC | 129 (38.4%) | 19 (35.8%) | 110 (38.9%) | |||
| CT or TT | 207 (61.6%) | 34 (64.2%) | 173 (61.1%) | |||
| CYP2C19*2 (rs4244285) | 0.038 | 0.516 | 0.275-0.971 | |||
| GG | 145 (43.2%) | 16 (30.2%) | 129 (45.6%) | |||
| GA or AA | 191 (56.8%) | 37 (69.8%) | 154 (54.4%) | |||
| CYP2C19*3 (rs4986893) | 0.071 | 0.474 | 0.207-1.082 | |||
| GG | 302 (89.5%) | 44 (83.0%) | 258 (91.2%) | |||
| GA | 34 (10.1%) | 9 (17.0%) | 25 (8.8%) | |||
| CYP2C19*17 (rs12248560) | 0.595 | NA | NA | |||
| CC | 330 (98.2%) | 53 (100%) | 277 (97.9%) | |||
| CT | 6 (1.8%) | 0 (0%) | 6 (2.1%) | |||
| CY3A4 (rs4646437) | 0.828 | 1.081 | 0.537-2.173 | |||
| CC | 256 (76.2%) | 41 (77.4%) | 215 (76.0%) | |||
| CT or TT | 80 (23.8%) | 12 (22.6%) | 68 (24.0%) | |||
| CYP3A5 (rs776746) | 0.512 | 0.822 | 0.457-1.478 | |||
| GG | 185 (55.1%) | 27 (50.9%) | 158 (55.8%) | |||
| GA or AA | 151 (44.9%) | 26 (49.1%) | 125 (44.2%) | |||
| ITGB3 (rs5918) | 1.000 | NA | NA | |||
| TT | 334 (99.4%) | 53 (100%) | 281 (99.3%) | |||
| CT | 2 (0.6%) | 0 (0%) | 2 (0.7%) | |||
| PON1 Q192R (rs662) | 0.033 | 0.495 | 0.257-0.952 | |||
| GG | 133 (39.6%) | 14 (26.4%) | 119 (42.0%) | |||
| GA or AA | 203 (60.4%) | 39 (73.6%) | 164 (58.0%) |
SNP: single nucleotide polymorphism; HPR: high platelet reactivity; NPR: normal platelet reactivity; NA: not applicable.
Effects of the PON1 Q192R and CYP2C19*2 variants on platelet reactivity
Platelet aggregation at baseline and 1 month after PCI are shown in Table 3. Platelet aggregation in patients with the PON1 192Q allele was significantly higher than that in non-carriers at both time points (P=0.010 and 0.024, respectively). However, there was no significant difference between PON1 192QQ and PON1 192QR (P=0.624 and 0.454, respectively). Similarly, platelet aggregation was significantly higher in patients with CYP2C19*2 allele than in non-carriers at these time points (P=0.005 and 0.003, respectively). However, there were no significant differences between CYP2C19*2/*2 and CYP2C19*1/*2 (P=0.486 and 0.441, respectively). We further divided all of the patients into the following four groups: PON1 192Q (-) and CYP2C19*2 (-), PON1 192Q (+) only, CYP2C19*2 (+) only, PON1 192Q (+) and CYP2C19*2 (+). Compared with the first group, platelet aggregation was significantly increased in the other groups (P=0.029, 0.014 and <0.001 at baseline, P=0.036, 0.005 and 0.004 at 1 month, respectively; Figure 2). Among the latter three groups, platelet aggregation did not differ significantly.
Table 3.
Platelet reactivity of PON1 Q192R and CYP2C19*2 (%)
| PON1 192RR n=133 | PON1 192QR n=154 | PON1 192QQ n=49 | PON1 192Q allele n=203 | CYP2C19*1/*1 n=145 | CYP2C19*1/*2 n=144 | CYP2C19*2/*2 n=47 | CYP2C19*2 allele n=191 | |
|---|---|---|---|---|---|---|---|---|
| Baseline | 34.6±6.9 | 41.6±8.2 | 44.0±7.2 | 42.2±5.8 | 34.4±4.6 | 42.1±7.8 | 45.5±10.7 | 42.9±9.0 |
| 1 month after PCI | 25.2±3.4 | 29.7±4.0 | 32.4±4.8 | 30.4±3.9 | 25.5±2.8 | 31.9±5.0 | 34.8±5.4 | 32.6±6.3 |
Figure 2.
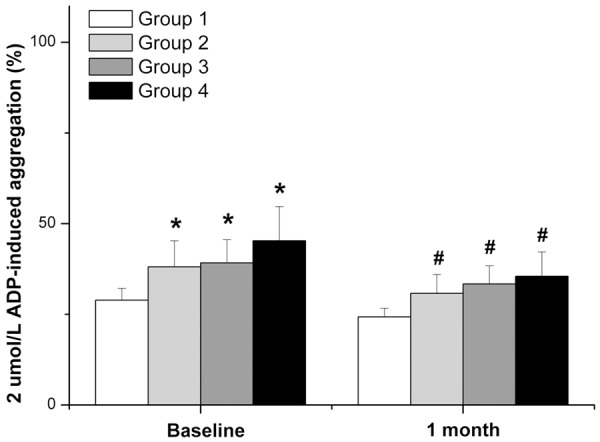
Platelet reactivity of PON1 Q192R and CYP2C19*2 allele carriers. Group 1: PON1 192Q (-) and CYP2C19*2 (-); Group 2: PON1 192Q (+) only; Group 3: CYP2C19*2 (+) only; Group 4: PON1 192Q (+) and CYP2C19*2 (+). Compared with Group 1, *P or #P<0.05.
Effects of the PON1 Q192R and CYP2C19*2 variants on clinical outcome
During 6-month follow-up, 49 patients experienced MACE. Among them, 36 were PON1 192Q allele carriers, who experienced more MACE than non-carriers (P=0.043). 39 patients with MACE were CYP2C19*2 allele carriers, who also experienced more MACE than non-carriers (P=0.001). After all patients were divided into the four groups mentioned above, only one MACE occurred in the PON1 192Q (-) and CYP2C19*2 (-) group, eight in the PON1 192Q (+) only group, 12 in the CYP2C19*2 (+) only group, and 27 in the PON1 192Q (+) and CYP2C19*2 (+) group. Compared with the first group, MACE increased significantly in the other groups (P=0.027, 0.003 and <0.001, respectively). Among the latter three groups, platelet aggregation did not significantly differ.
Kaplan-Meier analysis showed that the risks of MACE were increased in PON1 192Q and CYP2C19*2 allele carriers during follow-up (P=0.012 and 0.003, respectively; Figure 3). Multivariable Cox regression analysis showed that the adjusted risk of MACE for patients with the PON1 192Q allele was 1.928 times that among non-carriers (95% CI: 1.022-3.636, P=0.043), while for of patients with CYP2C19*2 allele was 3.183 times that among non-carriers (95% CI: 1.589-6.378, P=0.001).
Figure 3.
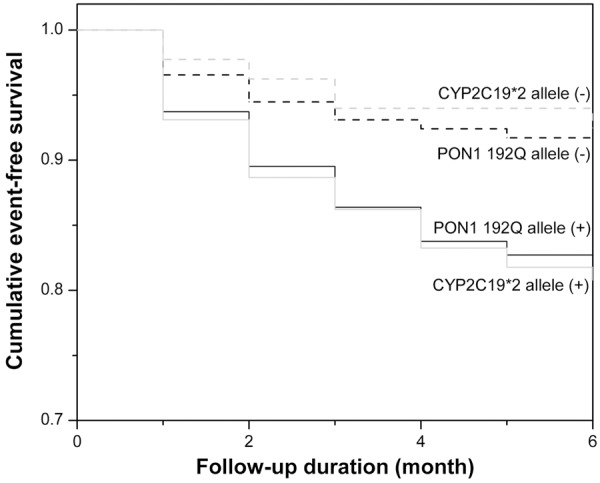
Cumulative Kaplan-Meier estimates of the rates of MACE according to PON1 Q192R and CYP2C19*2 allele carriers during follow-up.
Discussion
The study included 10 SNPs among seven genes. ABCB1 is related to the active transport of the prodrug clopidogrel by the intestinal membrane [16]. CYP2C19, CYP3A4 and CYP3A5 are associated with the formation of the active product of clopidogrel in the liver [8]. The P2Y12 gene encodes the platelet membrane protein P2Y12 receptor, which is specifically antagonized by clopidogrel [17]. ITGB3 encodes the platelet glycoprotein IIb/IIIa receptor, which plays a critical role in platelet aggregation and is coupled with the P2Y12 receptor [18]. Studies suggested that some SNPs of these genes could influence the intestinal absorption, activation and platelet reactivity of clopidogrel, but their conclusions were inconsistent [6,19]. In Chinese populations in particular, the effects of these SNPs on clopidogrel responsiveness have not been fully elucidated. In our study, only the CYP2C19*2 and PON1 Q192R among the ten SNPs were significantly different between the HPR and NPR groups.
CYP2C19, an important member of the second subfamily of Cytochrome P450 enzymes, plays an important role in converting clopidogrel into a functional active metabolite in the liver. Studies have shown that CYP2C19*2 can reduce platelet reactivity to clopidogrel in the Caucasian population, while CYP2C19*17 can increase the platelet response to clopidogrel [20]. Our study found that the CYP2C19*2 allele had a significantly higher prevalence in the HPR group compared with the NPR group in the Chinese population, while CYP2C19*17 was not significantly different between the two groups. This phenomenon may be related to ethnic factors. In the Chinese population, the prevalence of CYP2C19*17 (1.8%) was much lower than in the Caucasian population. In addition, we found that MACE increased significantly in CYP2C19*2 allele carriers. CYP2C19*2 may cause increased cardiovascular ischemic events by weakening clopidogrel bioactivation.
As a type of hepatic phospholipid hydrolase, PON1 has been demonstrated to inhibit lipoprotein peroxidation. Several studies showed that PON1 polymorphisms were associated with the occurrence of coronary and intracranial atherosclerosis [21,22]. Bhattacharyya T et al. demonstrated that PON1 has cardioprotective effects by reducing systemic oxidative stress and prospective cardiovascular risk [25]. In 2011, Bouman et al. first found that PON1 is the key enzyme of clopidogrel bioactivation; PON1 catalyzes 2-oxo-clopidogrel (clopidogrel intermediate) into the pharmacologically active thiol metabolite [23]. The PON1 192QQ allozyme showed lower catalytic efficiency for 2-oxo-clopidogrel, which might explain why the PON1 192QQ genotype could significantly reduce the antiplatelet effect of clopidogrel and induce HPR, thereby increasing in-stent thrombosis in the Caucasian population. However, some subsequent studies by other researchers did not support this view [24-26]. Several recent studies evaluated the relationship between PON1 variants and clopidogrel response in Chinese patients. Wu H et al. found that among ACS patients, the PON1 192QQ/QR genotype could lead to HPR [27]. However, another study performed by Zhang L et al. did not find a significant association between the PON1 192QQ/QR genotype and HPR [28]. In Zhang’s study, HPR was defined as >50% ADP-induced aggregation, while a more strict criterion (≥70% ADP-induced aggregation) has now been widely adopted. The loose definition of HPR may be the main cause of their negative results. In our study, we demonstrated that the PON1 192Q allele had a higher prevalence in the HPR group and that platelet reactivity in PON1 192Q allele carriers was higher than in non-carriers, supporting Wu’s view on the positive relationship between the PON1 192Q allele and HPR. Furthermore, we found that the PON1 192Q allele was associated with an increased incidence of MACE in a 6-month follow-up period, which might be due to the comprehensive effects of the PON1 192 Q allele on lipids and the response to clopidogrel.
In addition, we investigated the change in platelet reactivity over time. We found that compared with acute cases of ACS, the platelet reactivity of the entire population declined in the chronic phase, 1 month after PCI. This may be because platelet function was activated by stress, the intervention and other factors in the acute phase of ACS but was stabilized in the chronic phase as these internal and external influences reduced or disappeared. Notably, platelet aggregation in patients with the PON1 192Q or CYP2C19*2 allele was still higher than in non-carriers, suggesting that the effects of the PON1 192Q or CYP2C19*2 allele on platelet aggregation were persistent over time.
In our study, both the PON1 192Q and CYP2C19*2 alleles influenced platelet reactivity and increased the risk of MACE. However, by comparing platelet reactivity and MACE between the PON1 192Q (+) only and CYP2C19*2 (+) only groups, we found that CYP2C19*2 seemed to have greater effects than PON1 192Q. This observation requires further investigation in a larger population with a longer follow-up duration.
There are several limitations in our study. First, this is a single-center study performed in East China. Multi-center studies conducted across China will help to improve the research power and representation of the Chinese population. Second, despite the positive findings in our study, the sample size is relatively small. Future studies with larger sample sizes are warranted. Third, due to the limited conditions, platelet function was detected by only one method (TEG) in the study. Whether different platelet function testing methods could influence the research results requires further investigation.
In conclusion, both the PON1 Q192R and CYP2C19*2 variants are associated with HPR and an increased risk of ischemic events during 6-month follow-up in Chinese patients undergoing PCI. Their effects on platelet aggregation are persistent over time. The CYP2C19*2 allele seems to have a greater effect on platelet aggregation and MACE than the PON1 192Q allele. Future studies with a larger population and a long-term follow-up duration are warranted.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81270209), the Shanghai Committee of Science and Technology of China (No. 12JC1406400) and the Doctor Innovation Fund of Shanghai Jiaotong University School of Medicine (No. BXJ201224). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure of conflict of interest
None.
References
- 1.Sabatine MS, Cannon CP, Gibson CM, Lopez-Sendon JL, Montalescot G, Theroux P, Claeys MJ, Cools F, Hill KA, Skene AM, McCabe CH, Braunwald E. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation. N Engl J Med. 2005;352:1179–1189. doi: 10.1056/NEJMoa050522. [DOI] [PubMed] [Google Scholar]
- 2.Jneid H, Anderson JL, Wright RS, Adams CD, Bridges CR, Casey DE Jr, Ettinger SM, Fesmire FM, Ganiats TG, Lincoff AM, Peterson ED, Philippides GJ, Theroux P, Wenger NK, Zidar JP. 2012 ACCF/AHA Focused Update of the Guideline for the Management of Patients With Unstable Angina/Non-ST-Elevation Myo- cardial Infarction (Updating the 2007 Guideline and Replacing the 2011 Focused Update): A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2012;60:645–681. doi: 10.1016/j.jacc.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Gurbel PA, Tantry US. Clopidogrel response variability and the advent of personalized antiplatelet therapy: A bench to bedside journey. Thromb Haemost. 2011;106:265–271. doi: 10.1160/TH11-03-0167. [DOI] [PubMed] [Google Scholar]
- 4.Muller I, Besta F, Schulz C, Massberg S, Schonig A, Gawaz M. Prevalence of clopidogrel non-responders among patients with stable angina pectoris scheduled for elective coronary stent placement. Thromb Haemost. 2003;89:783–787. [PubMed] [Google Scholar]
- 5.Gurbel PA, Tantry US. Clopidogrel resistance? Thromb Res. 2007;120:311–321. doi: 10.1016/j.thromres.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 6.Campo G, Miccoli M, Tebaldi M, Marchesini J, Fileti L, Monti M, Valgimigli M, Ferrari R. Genetic determinants of on-clopidogrel high platelet reactivity. Platelets. 2011;22:399–407. doi: 10.3109/09537104.2011.579648. [DOI] [PubMed] [Google Scholar]
- 7.Fefer P, Matetzky S. The genetic basis of platelet responsiveness to clopidogrel. A critical review of the literature. Thromb Haemost. 2011;106:203–210. doi: 10.1160/TH11-04-0228. [DOI] [PubMed] [Google Scholar]
- 8.Mega JL, Close SL, Wiviott SD, Shen L, Hockett RD, Brandt JT, Walker JR, Antman EM, Macias W, Braunwald E, Sabatine MS. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med. 2009;360:354–362. doi: 10.1056/NEJMoa0809171. [DOI] [PubMed] [Google Scholar]
- 9.Yin T, Miyata T. Pharmacogenomics of clopidogrel: evidence and perspectives. Thromb Res. 2011;128:307–316. doi: 10.1016/j.thromres.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 10.Price MJ, Murray SS, Angiolillo DJ, Lillie E, Smith EN, Tisch RL, Schork NJ, Teirstein PS, Topol EJ. Influence of genetic polymorphisms on the effect of high- and standard-dose clopidogrel after percutaneous coronary intervention: the GIFT (Genotype Information and Functional Testing) study. J Am Coll Cardiol. 2012;59:1928–1937. doi: 10.1016/j.jacc.2011.11.068. [DOI] [PubMed] [Google Scholar]
- 11.Lewis JP, Shuldiner AR. Paraoxonase 1 Q192R variant and clopidogrel efficacy: fact or fiction? Circ Cardiovasc Genet. 2012;5:153–155. doi: 10.1161/CIRCGENETICS.112.962910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeong YH, Tantry US, Kim IS, Koh JS, Kwon TJ, Park Y, Hwang SJ, Bliden KP, Kwak CH, Hwang JY, Gurbel PA. Effect of CYP2C19*2 and *3 loss-of-function alleles on platelet reactivity and adverse clinical events in East Asian acute myocardial infarction survivors treated with clopidogrel and aspirin. Circ Cardiovasc Interv. 2011;4:585–594. doi: 10.1161/CIRCINTERVENTIONS.111.962555. [DOI] [PubMed] [Google Scholar]
- 13.Man M, Farmen M, Dumaual C, Teng CH, Moser B, Irie S, Noh GJ, Njau R, Close S, Wise S, Hockett R. Genetic variation in metabolizing enzyme and transporter genes: comprehensive assessment in 3 major East Asian subpopulations with comparison to Caucasians and Africans. J Clin Pharmacol. 2010;50:929–940. doi: 10.1177/0091270009355161. [DOI] [PubMed] [Google Scholar]
- 14.Rivard GE, Brummel-Ziedins KE, Mann KG, Fan L, Hofer A, Cohen E. Evaluation of the profile of thrombin generation during the process of whole blood clotting as assessed by thrombelastography. J Thromb Haemost. 2005;3:2039–2043. doi: 10.1111/j.1538-7836.2005.01513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bliden KP, DiChiara J, Tantry US, Bassi AK, Chaganti SK, Gurbel PA. Increased risk in patients with high platelet aggregation receiving chronic clopidogrel therapy undergoing percutaneous coronary intervention: is the current antiplatelet therapy adequate? J Am Coll Cardiol. 2007;49:657–666. doi: 10.1016/j.jacc.2006.10.050. [DOI] [PubMed] [Google Scholar]
- 16.Silverton L, Dean M, Moitra K. Variation and evolution of the ABC transporter genes ABCB1, ABCC1, ABCG2, ABCG5 and ABCG8: implication for pharmacogenetics and disease. Drug Metabol Drug Interact. 2011;26:169–179. doi: 10.1515/DMDI.2011.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bouman HJ, van Werkum JW, Rudez G, Hackeng CM, Leebeek FW, ten Cate H, ten Berg JM, de Maat MP. The relevance of P2Y(12)-receptor gene variation for the outcome of clopidogrel-treated patients undergoing elective coronary stent implantation: a clinical follow-up. Thromb Haemost. 2012;107:189–191. doi: 10.1160/TH11-05-0306. [DOI] [PubMed] [Google Scholar]
- 18.Di Castelnuovo A, de Gaetano G, Benedetta Donati M, Iacoviello L. Platelet glycoprotein IIb/IIIa polymorphism and coronary artery disease: implications for clinical practice. Am J Pharmacogenomics. 2005;5:93–99. doi: 10.2165/00129785-200505020-00002. [DOI] [PubMed] [Google Scholar]
- 19.Momary KM, Dorsch MP, Bates ER. Genetic causes of clopidogrel nonresponsiveness: which ones really count? Pharmacotherapy. 2010;30:265–274. doi: 10.1592/phco.30.3.265. [DOI] [PubMed] [Google Scholar]
- 20.Holmes MV, Perel P, Shah T, Hingorani AD, Casas JP. CYP2C19 genotype, clopidogrel metabolism, platelet function, and cardiovascular events: a systematic review and meta-analysis. Jama. 2011;306:2704–2714. doi: 10.1001/jama.2011.1880. [DOI] [PubMed] [Google Scholar]
- 21.Menini T, Gugliucci A. Paraoxonase 1 in neurological disorders. Redox Rep. 2014;19:49–58. doi: 10.1179/1351000213Y.0000000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuremoto K, Watanabe Y, Ohmura H, Shimada K, Mokuno H, Daida H. R/R genotype of human paraoxonase (PON1) is more protective against lipoprotein oxidation and coronary artery disease in Japanese subjects. J Atheroscler Thromb. 2003;10:85–92. doi: 10.5551/jat.10.85. [DOI] [PubMed] [Google Scholar]
- 23.Bouman HJ, Schomig E, van Werkum JW, Velder J, Hackeng CM, Hirschhauser C, Waldmann C, Schmalz HG, ten Berg JM, Taubert D. Paraoxonase-1 is a major determinant of clopidogrel efficacy. Nat Med. 2011;17:110–116. doi: 10.1038/nm.2281. [DOI] [PubMed] [Google Scholar]
- 24.Lewis JP, Fisch AS, Ryan K, O’Connell JR, Gibson Q, Mitchell BD, Shen H, Tanner K, Horenstein RB, Pakzy R, Tantry US, Bliden KP, Gurbel PA, Shuldiner AR. Paraoxonase 1 (PON1) gene variants are not associated with clopidogrel response. Clin Pharmacol Ther. 2011;90:568–574. doi: 10.1038/clpt.2011.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen DY, Wang CY, Wen MS, Lee TH, Chu Y, Hsieh MJ, Chang SH, Lee CH, Wang JL, Chen CC, Lu LS, Lee MT, Yeh SJ, Lin FC, Hsieh IC. Paraoxonase-1 is not a major determinant of stent thrombosis in a Taiwanese population. PLoS One. 2012;7:e39178. doi: 10.1371/journal.pone.0039178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sibbing D, Koch W, Massberg S, Byrne RA, Mehilli J, Schulz S, Mayer K, Bernlochner I, Schömig A, Kastrati A. No association of paraoxonase-1 Q192R genotypes with platelet response to clopidogrel and risk of stent thrombosis after coronary stenting. Eur Heart J. 2011;32:1605–1613. doi: 10.1093/eurheartj/ehr155. [DOI] [PubMed] [Google Scholar]
- 27.Wu H, Qian J, Xu J, Sun A, Sun W, Wang Q, Ge J. Besides CYP2C19, PON1 genetic variant influences post-clopidogrel platelet reactivity in Chinese patients. Int J Cardiol. 2013;165:204–206. doi: 10.1016/j.ijcard.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 28.Zhang L, Chen Y, Jin Y, Qu F, Li J, Ma C, Yang J, Xu B, Wang H, Li X, Li Y, Zhang Y, Lu C, Yin T. Genetic determinants of high on-treatment platelet reactivity in clopidogrel treated Chinese patients. Thromb Res. 2013;132:81–87. doi: 10.1016/j.thromres.2013.05.006. [DOI] [PubMed] [Google Scholar]