

Abstract
Workers exposed to aerosolized dust present in concentrated animal feeding operations (CAFOs) are susceptible to inflammatory lung diseases, such as chronic obstructive pulmonary disease. Extracts of dust collected from hog CAFOs [hog dust extract (HDE)] are potent stimulators of lung inflammatory responses in several model systems. The observation that HDE contains active proteases prompted the present study, which evaluated the role of CAFO dust proteases in lung inflammatory processes and tested whether protease-activated receptors (PARs) are involved in the signaling pathway for these events. We hypothesized that the damaging proinflammatory effect of HDE is due, in part, to the proteolytic activation of PARs, and inhibiting the proteases in HDE or disrupting PAR activation would attenuate HDE-mediated inflammatory indexes in bronchial epithelial cells (BECs), in mouse lung slices in vitro, and in a murine in vivo exposure model. Human BECs and mouse lung slice cultures stimulated with 5% HDE released significantly more of each of the cytokines measured (IL-6, IL-8, TNF-α, keratinocyte-derived chemokine/CXC chemokine ligand 1, and macrophage inflammatory protein-2/CXC chemokine ligand 2) than controls, and these effects were markedly diminished by protease inhibition. Inhibition of PARs also blunted the HDE-induced cytokine release from BECs. In addition, protease depletion inhibited HDE-induced BEC intracellular PKCα and PKCε activation. C57BL/6J mice administered 12.5% HDE intranasally, either once or daily for 3 wk, exhibited increased total cellular and neutrophil influx, bronchial alveolar fluid inflammatory cytokines, lung histopathology, and inflammatory scores compared with mice receiving protease-depleted HDE. These data suggest that proteases in dust from CAFOs are important mediators of lung inflammation, and these proteases and their receptors may provide novel targets for therapeutic intervention in CAFO dust-induced airways disease.
Keywords: lung, epithelial cell, proteases, environmental dust, inflammation, protease-activated receptors
exposure to organic dusts from agricultural environments leads to airway inflammation, and workers in agricultural industries are at increased risk for upper and lower airway diseases, including rhinosinusitis, chronic bronchitis, and chronic obstructive pulmonary disease (COPD) (1, 15, 18, 28). In particular, workers in livestock industries such as concentrated animal feeding operations (CAFOs) are susceptible to chronic airway diseases specifically associated with these occupations (22, 28).
In an effort to understand the mechanistic basis of airway inflammation associated with agricultural dusts, previous work from our group has focused on characterizing inflammatory responses induced by aqueous organic dust extracts derived from hog CAFOs [hog dust extract (HDE)] using in vitro cell culture models and in vivo murine models of dust exposure (26, 29, 33, 35, 41, 43). Those studies showed that HDE treatment of bronchial epithelial cells (BECs) results in the sequential activation of protein kinase C (PKC) α and ε that, in turn, leads to inflammatory cytokine/chemokine production (33, 43). In murine studies, a single exposure to HDE resulted in marked neutrophil influx and enhanced inflammatory mediator levels in bronchoalveolar lavage fluid (BALF), and repetitive HDE exposure, while having less dramatic changes in BALF cell/cytokine levels, resulted in dramatic increases in lung histopathology characterized by mononuclear leukocyte cell aggregates and peribronchiolar and perivascular inflammation (29).
To explore the inflammatory components of these complex organic dusts, several studies have investigated the composition of dusts from different agricultural environments, including hog CAFOs. Although fungal and bacterial cell wall components of agricultural dusts contribute to the inflammatory responses, these components do not entirely account for the inflammatory effects observed (27). Many common environmental irritants and aeroallergens, such as extracts from house dust mites, cockroaches, and numerous fungal species, have been reported to exhibit protease activity (2, 23, 30, 31), which is implicated in bronchial inflammation, likely due in part to the release of inflammatory cytokines from BECs (5, 8). Although proteases can exert their effects on cell function in several ways, specific G protein-coupled receptors, termed protease-activated receptors (PARs), have been identified, and these have been shown to mediate many of the physiological responses of cells to proteases (14, 31). Accordingly, we tested whether proteases in HDE contribute to the dust-induced inflammatory response in BECs, in lung tissue slices, and in a murine model of airway disease, and whether this response is mediated through PARs.
Our results demonstrate that the proteases found in dust derived from hog CAFOs play an important role in eliciting the dust-mediated inflammatory responses of BECs, at least partially through activation of PARs, and that these proteases contribute significantly to the airway inflammation associated with acute and repetitive dust exposure in mice. Together, these data reveal new potential targets for the prevention and/or treatment of airway inflammation in individuals exposed to agricultural dust.
MATERIALS AND METHODS
Reagents.
LHC (Laboratory for Human Carcinogenesis) basal medium, growth factor supplements, and Lipofectamine-2000 were purchased from Life Technologies (Carlsbad, CA). Monoclonal anti-human tumor necrosis factor-α (TNF-α) neutralizing antibody, anti-human interleukin (IL)-6 and IL-8 ELISA capture antibodies, biotin-conjugated anti-TNF-α, streptavidin-conjugated horseradish peroxidase, and ELISA substrate kit were all obtained from R&D Systems (Minneapolis, MN). ELISA bridge antibodies were purchased from Biolegend (San Diego, CA), and detection antibodies were from Rockland (Gilbertsville, PA). The PAR-1 antagonist SCH-79797 was obtained from Tocris (Bristol, UK), and PAR-2 inhibitor FSLLRY-NH2 was from BACHEM (Torrence, CA). Small interfering RNAs (siRNAs) for PAR-1 and PAR-2, and nontargeting control siRNA were obtained from Santa Cruz Biotechnology (Dallas, TX). All other reagents not specified were purchased from Sigma-Aldrich (St. Louis, MO).
Aqueous organic dust HDE.
Settled dust was obtained from hog CAFOs housing between 500 and 1,000 animals. Dust samples collected from surfaces >1 m from the floor were sifted through a coarse 0.25-mm sieve before being stored at −20°C in a dessicator. HDE was prepared as previously described (33). Saturated dust extracts containing 100 mg/ml in Hank's balanced salts solution (pH 7.4) (100% HDE) were filter-sterilized (0.22 μm) and stored at −20°C until use. For cultured epithelial cells, a final concentration of 5% HDE was used. For in vivo studies, mice were instilled with a 12.5% HDE solution in sterile saline, consistent with prior studies (29). These respective concentrations have been determined empirically to elicit maximal proinflammatory activation while having no observable cytotoxic effects (by LDH release). HDE diluted to 5% in culture medium contains 2.2 mg/ml total protein (Bradford method, Bio-Rad) and 40 endotoxin units/ml endotoxin (18 ng/ml) as lipopolysaccharide (LAL assay, Sigma). Protease activity was depleted from undiluted HDE used for cell culture and animal experiments by the addition of one of two serine protease inhibitors [phenylmethylsulfonyl fluoride (PMSF), 2 mM; or the more soluble 4-(2-aminoethyl) benzenesulfonyl fluoride-HCl (AEBSF), 4 mM]. Both protease inhibitors become hydrolyzed and inactivated within 4 h in aqueous solution. Mixtures were incubated at 37°C for 2 h with agitation and maintained at 4°C until use (<24 h). Complete (native) HDE was handled identically. While HDE from the same facility was used within each series of experiments, experiments have been performed with dusts from at least two other facilities with similar results.
Measurement of proteolytic activity.
The presence of proteolytic activity was analyzed by 1% gelatin zymography using a modification of the method of Sweetman and Ornstein (37) as summarized by Lantz and Ciborowski (19). Undiluted HDE (100%) was treated with various protease inhibitors: 2 mM PMSF, 4 mM AEBSF, protease inhibitor cocktail (PIC), or soybean trypsin inhibitor (2 mg/ml) for 2 h with agitation at 37°C. Each sample (35 μl) was loaded onto an 8% SDS polyacrylamide gel copolymerized with 1% gelatin. Following electrophoresis, proteins were renaturated in a 2.5% Triton X-100 solution for 30 min, developed in Tris buffer overnight, and visualized by staining with 0.5% Coomassie Brilliant Blue R-250. After destaining (50% methanol, 40% acetic acid), lytic activity was visible as clear bands against the field of undigested substrate. As an additional method for detection of protease activity, the same samples were assayed by a modification of the colorimetric azocoll conversion assay described by Chavira et al. (7). The technique uses collagen coupled to an azo dye as a proteolytic substrate that is converted to a soluble chromogen in the presence of proteases (azocoll, EMD/Millipore, Billerica, MA). Protease activity was calculated by comparing optical density values (absorbance at 520 nm) to a standard curve using trypsin as the reference standard. The exogenous proteases porcine pancreatic elastase (PPE; 1 mg/ml), and trypsin (1 mM) were also assayed for comparison. As AEBSF showed slightly greater efficacy in initial experiments, this inhibitor was preferentially used in the subsequent experiments.
Cell culture.
For most in vitro experiments, the SV40-immortalized human BEC line BEAS-2B (American Tissue Culture Collection, Manassas, VA) was used. Cells were plated on 12-well plates coated with type I collagen and maintained in culture in serum-free medium at 37°C in 5% CO2/95% air. Epithelial cell growth medium consisted of growth factor-supplemented LHC basal medium combined with RPMI containing 1% penicillin/streptomycin and amphotericin B (LHC-9/RPMI, 50:50), as previously described (33). For cytokine release, subconfluent cell monolayers were exposed to complete 5% HDE or to protease-depleted 5% HDE in growth medium for 24 h. No detectable cytotoxic effects were observed under these conditions (TOX-7 LDH release assay, Sigma-Aldrich). Some experiments were also performed using primary normal human BECs (NHBE) isolated and cultured as previously described (3). Briefly, de-identified normal human lungs that were rejected for transplantation were obtained from the International Institute for the Advancement of Medicine. Cells were isolated from the lumen of sectioned bronchi by gentle protease digestion, plated on collagen-coated dishes, and maintained in serum-free medium (BEGM; Lonza, Basel, Switzerland). Cells used for these experiments were passaged less than four times and were derived from two donors. For the sake of conformity, “BEC” is used to denote any epithelial cell (BEAS-2B or NHBE) in the text.
Transfection of cells with PAR siRNA.
Transfection was carried out using a modification of the method described previously (16). Briefly, cells were grown to 75% confluency on 12-well collagen-coated plates in serum-free LHC9-RPMI medium. Lipofectamine-2000 (6 μl) was mixed with 200 μl of serum- and antibiotic-free LHC basal medium for each well transfected, followed by gentle agitation for 10 min. Simultaneously, 12 μl of each siRNA were mixed with 200 μl of LHC basal medium for each well and incubated similarly. The two suspensions were then combined and incubated for an additional 30 min at room temperature. After cell layers were washed gently with PBS, 200 μl of the mixture were added to each well, with the addition of 300 μl of LHC basal medium, and incubated overnight (18 h) at 37°C. Cells were then fed with normal growth medium and maintained for at least 18 h before being used in experiments. For some experiments, cells were identically transfected with nontargeting control siRNA before treatment, with or without 5% HDE. After transfection, cultures were exposed to complete 5% HDE, protease-depleted HDE, or specific inhibitors for PAR-1 (SCH-79797, 0.75 nM) or PAR-2 (FSLLRY-NH2, 45 nM) for 24 h. Culture supernates were collected for cytokine measurements, and cell lysates were prepared for total protein normalization. To assess the efficiency of the siRNA suppression, cell lysates were electrophoresed, and Western blots were probed for PAR-1 and PAR-2 expression. The prominent protein bands observed in control and HDE-stimulated cells (43 kDa for PAR-1; ∼47 kDa for PAR-2) were markedly diminished in siRNA-transfected cell lysates (see Fig. 9F).
Fig. 9.

RNA interference of PAR-1 and PAR-2 inhibit HDE-mediated BEC cytokine release. BEAS-2B cells were transfected with PAR-1 or PAR-2 small interfering RNA (siRNA) 18 h before being challenged with complete 5% HDE, protease depleted HDE, or either the PAR-1 (0.75 nM) or PAR-2 (45 nM) inhibitor in combination with complete HDE. Cytokines were measured in culture supernates 24 h later. Challenge with HDE alone resulted in a robust elevation in cytokine release. This effect was significantly reduced in cells transfected with either PAR-1 (A and B) or PAR-2 (C and D) siRNA. RNA interference had no further effect on the modest cytokine response elicited by either protease-depleted HDE or either PAR inhibitor. E: control nontargeting siRNA did not affect baseline or HDE-stimulated cytokines. Values are means ± SE for 3 experiments and 2 technical replicates per condition. #P < 0.05 vs. control. *P < 0.05 vs. no siRNA. §P < 0.05 vs. HDE alone. F: Western blot demonstrates effect of siRNA transfection on BEAS-2B PAR-1 and PAR-2 protein expression. Separation of blot sections with white spaces in the PAR-2 blot and corresponding β-actin loading control blot indicate noncontinuous lanes from the same blots.
Precision mouse lung slice cultures.
As an integrated model of ex vivo whole tissue responses, a precision-cut mouse lung slice technique was employed. The procedure was adapted from the method reported by Sanderson (34), as previously described (40). In brief, normal healthy mice (C57BL/6J, Jackson Laboratory, Bar Harbor, ME) were euthanized, and their lungs perfused with a warmed 2.5% low melting point agarose solution. After polymerization by cooling, the lungs were removed en bloc, and major lobes were sectioned with an oscillating tissue slicer (OTS-4500 Electron Microscopy Sciences, Hatfield, PA) into 150-μm slices. Five slices were placed into each well of a 12-well culture dish and maintained in serum-free LHC9/RPMI at 37°C with daily replacement of medium to remove contaminating blood cells and agarose. After being allowed to stabilize for 5–8 days, lung slice cultures were challenged with complete 5% HDE, protease-depleted 5% HDE, or with protease inhibitors alone for 24 h. Supernatant medium was then collected, and the tissue was homogenized for total protein determination. Four independent experiments were conducted, and triplicate cytokine measurements were made for each sample by ELISA.
PKC activity assays.
BEAS-2B cells grown to 80–90% confluency on triplicate 60-mm dishes were exposed to complete 5% HDE, protease-depleted HDE, 4 mM AEBSF, or medium alone for either 1 or 6 h. Supernatant media were removed from treated cells, and the cell monolayers were immediately flash frozen in cell lysis buffer, as previously described (42). The cells were collected with a cell lifter, ultrasonically disrupted, and centrifuged at 10,000 g for 30 min at 4°C. The cytosolic fraction was collected, and the pellet was resuspended in cell lysis buffer containing 0.01% Triton X-100 and sonicated again (particulate fraction). PKCα and PKCε isoform activities were determined in crude whole cell cytosolic and particulate fractions by incubating for 15 min at 30°C in a magnesium acetate buffer containing isoform-specific substrate peptides and 10 μCi/ml [γ-32P]ATP. The resulting mixture was then spotted onto P-81 phosphocellulose papers (Whatman, Clinton, NJ) and radioactivity quantified in nonaqueous scintillation cocktail (National Diagnostics, Atlanta, GA). PKC activity was corrected for total protein in the original cell cultures and is expressed as fold change (compared to control) in picomoles of phosphate incorporated per minute per milligram of protein.
Murine model of HDE exposure.
Eight-week-old male C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME) and maintained in a dedicated pathogen-free Association for Assessment and Accreditation of Laboratory Animal Care-accredited facility located on the University of Nebraska Medical Center campus. Mice had unrestricted access to food and water, and all experiments were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee (IACUC protocol no. 10-062-08-EP). Mice were given 50 μl of complete 12.5% HDE, protease-depleted HDE, saline, or the protease inhibitor alone (500 μM AEBSF) by intranasal instillation once (acute, single exposure model) or daily for 3 wk (repeated exposure model) following a previously published protocol (29). Five hours after the final exposure, mice (6 animals per group) were euthanized, and tracheas cannulated for bronchoalveolar lavage (3 fractions × 1 ml each). Cells recovered from the pooled BALF were counted, and slides were made for total cell counts and inflammatory cell differential analysis (Cytopro, ELITech/Wescor, Logan, UT; and DiffQuick, Dade-Behring, Newark, DE). Cell-free BALF supernates were assayed for cytokine content by ELISA.
For analysis of mouse lung histopathology, lungs were removed en bloc after lavage and infused with a 10% formalin/PBS solution at a pressure of 10 cmH2O for 24 h. Fixed lungs were routinely processed, embedded in paraffin, and 4- to 5-μm thick microtomy sections were mounted, deparaffinized, and then stained with hematoxylin/eosin by the University of Nebraska Medical Center Tissue Sciences Facility. Slides were analyzed for inflammatory indicators by a pathologist (W.W.) blinded to treatment conditions, as previously reported (29). Briefly, the pathologist analyzes typical markers of inflammation associated with the response to HDE, including number, size, and location of lymphoid aggregates, inflammation surrounding bronchioles and vasculature, and the presence of diffuse inflammatory features across the lung section in alveolar and bronchiolar compartments. Each microscopic field is then given a numerical score between 0 and 3, correlating with no (0) to severe (3) inflammation. Scores were based on review of a slide for each mouse in each condition with all lung tissue on each slide included in the assessment.
Cytokine measurement.
Cell-free supernatant medium harvested from cell culture, murine lung slice culture, and mouse BALF was assessed using commercially available ELISA development antibody sets (Duoset, R&D Systems, Minneapolis, MN) or with lab-designed immunoassays, as published previously (33). All samples were assayed in duplicate or triplicate within an experiment, and experiments were repeated three or more times each. The limits of detectability for human cytokine assays were as follows: IL-8, 125 pg/ml; IL-6, 60 pg/ml; TNF-α, 15 pg/ml. For murine cytokines, they were as follows: IL-6, 35 pg/ml; keratinocyte-derived chemokine (KC)/CXC chemokine ligand (CXCL) 1, 15 pg/ml; macrophage inflammatory protein-2 (MIP-2)/CXCL2, 52 pg/ml; TNF-α, 25 pg/ml.
Statistical analysis.
Data are presented as the means ± SE for replicate values pooled from three or more parallel experiments. The number of values used for statistical analysis is indicated in each figure legend. All pairwise comparisons were tested using a one-way ANOVA with Tukey's post hoc analysis (GraphPad Prism software, San Diego, CA). Statistical significance was accepted when P values were ≤ 0.05.
RESULTS
Protease activity is present in swine confinement dust extract.
Analysis of proteolytic activity by gelatin zymography and azocoll assay confirmed the presence of several distinct protease activities in HDE (Fig. 1). To further characterize these protease activities, the protease inhibitors PMSF, AEBSF, PIC, and soybean-derived trypsin inhibitor were combined with HDE before analysis by zymography (Fig. 1A) and the quantitative azocoll colorimetric assay (Fig. 1B). All inhibitory preparations effectively blocked the majority of the proteolytic activity in HDE, although none completely eradicated it. In addition, preparations of purified PPE and bovine trypsin were analyzed to estimate the molecular size and proteolytic strength of the HDE proteases. At least five major bands of activity were detected in HDE with a range of molecular sizes, and they were variably sensitive to inactivation by treatment with the protease inhibitors (Fig. 1A). While PIC significantly reduced protease activities in cell-free experiments, subsequent protease activity depletion experiments in living systems utilized the serine protease-specific inhibitors PMSF or AEBSF due to their efficacy and lack of cytotoxicity.
Fig. 1.
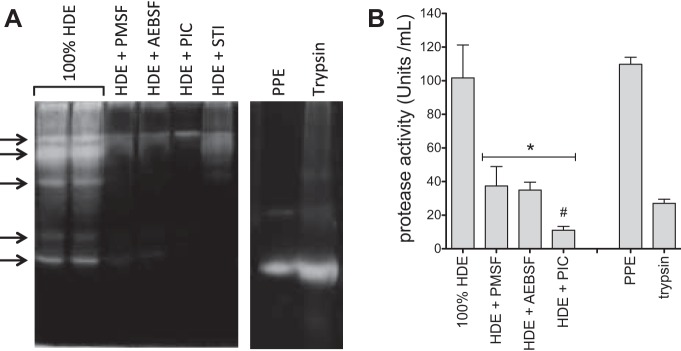
Hog dust extract (HDE) contains proteases that are effectively depleted by protease inhibitor treatment. Undiluted HDE (100% HDE) was treated with various protease inhibitors [2 mM phenylmethylsulfonyl fluoride (PMSF), 4 mM 4-(2-aminoethyl) benzenesulfonyl fluoride-HCl (AEBSF), protease inhibitor cocktail (PIC), or 2 mg/ml porcine trypsin]. Each sample was analyzed by 1% gelatin zymography for the presence of proteolytic activity (indicated at arrows by white clearing/banding; A) or assayed for protease activity by colorimetric azocoll conversion assay (B). Exogenous proteases, porcine pancreatic elastase (PPE, 1 mg/ml), and trypsin (1 mM) were also assayed for comparison. A is representative of 5 or more similar experiments. B depicts means ± SE for 3 independent experiments, with 3 technical replicates per condition. *P < 0.05 vs. 100% HDE. #P < 0.05 vs. HDE + PMSF and HDE + AEBSF. STI, soybean trypsin inhibitor.
HDE-induced human epithelial cell and murine lung tissue cytokine release is attenuated by protease depletion.
Our laboratory previously demonstrated that HDE treatment augments cytokine release from BECs (33). We tested whether and to what degree the protease activity in HDE contributed to cytokine release. As in our laboratory's previous studies, treatment of subconfluent monolayers BECs (BEAS-2B and primary NHBEs) with 5% HDE resulted in a rapid and sustained increase of IL-6 and IL-8/CXCL8 release (43). When HDE was depleted of protease activity with either PMSF or AEBSF, HDE-induced IL-6 and IL-8 were significantly diminished for both cell types at 24 h posttreatment [for BEAS-2B, IL-6 levels were decreased from 2,456 ± 279 to 785 ± 16 pg/ml after AEBSF treatment; AEBSF lowered IL-8 levels from 6,595 ± 348 to 3,313 ± 149 pg/ml (Fig. 2, A and B); for NHBE, AEBSF reduced IL-6 from 992 ± 44 to 593 ± 48 pg/ml, and IL-8 from 7,704 ± 853 to 3,554 ± 493 pg/ml (Fig. 2, C and D)]. To test for nonspecific effects of the protease inhibitors, a known protease-independent cytokine activator present in HDE, peptidoglycan (PGN; 10 μg/ml), was treated with AEBSF (which had the most potent effect on cytokine release in our HDE treatment studies) in an identical manner before challenge of BEAS-2B cells in culture. Protease activity depletion had no effect on the PGN-stimulated cytokine release (Fig. 2).
Fig. 2.

Depletion of proteases dampens the proinflammatory effect of HDE on bronchial epithelial cells (BECs) in culture. Undiluted HDE was treated with one of two broad-spectrum protease inhibitors (PMSF, 2 mM, or AEBSF, 4 mM). SV40-immortalized human BEC line (BEAS-2B; A and B) and normal human BECs (NHBE; C and D) cultures were then exposed to untreated (complete) 5% HDE, protease-depleted 5% HDE, control medium, or the inhibitors alone for 24 h. Supernates were assayed for interleukin (IL)-6 (A and C) and IL-8 (B and D) by ELISA. As a control experiment, the gram-positive bacterial component peptidoglycan (PGN; 200 μg/ml) was likewise treated with 4 mM AEBSF and was then diluted to 10 μg/ml before being used to stimulate cells (shaded bars). Values are means ± SE from 3 (A and B) or 4 (C and D) parallel experiments with 2 technical replicates per condition. *P < 0.05 vs. complete HDE. NSD, no significant difference.
To confirm these results in a more physiologically complex tissue-based model, precision-cut murine lung slice cultures were prepared and challenged with complete and protease-depleted HDE, as described above, and supernatant conditioned medium was collected after 24 h for cytokine analysis by ELISA. Depletion of proteases by either PMSF or AEBSF significantly inhibited the HDE-induced production of IL-6, TNF-α, CXCL1, and CXCL2 (Fig. 3) (e.g., AEBSF treatment reduced murine IL-6 from 841 ± 49 to 238 ± 68 pg/ml; KC from 1,783 ± 514 to 809 ± 117 pg/ml; TNF-α from 795 ± 96 to 271 ± 79 pg/ml; and MIP-2 from 1,386 ± 99 to 413 ± 85 pg/ml; P < 0.05 for all). Neither PMSF nor AEBSF alone had an effect on cytokine/chemokine release. These results demonstrate that, in both BEC cultures and murine lung tissue cultures, proteases in HDE mediate a significant fraction of its inflammatory cytokine/chemokine responses.
Fig. 3.

HDE-induced cytokine production is inhibited by protease depletion in mouse lung slice cultures. Precision-cut 150-μm-thick lung slices from naive mice were cultured in growth medium for 3–8 days before being challenged with complete 5% HDE, protease-depleted HDE, medium alone, or protease inhibitors alone for 24 h. Supernates from the ex vivo cultures were assayed for mouse cytokines and normalized for total protein in the homogenized tissue. Values are mean cytokine release ± SE for data from 4 independent experiments and 3 technical replicates for each condition. KC, keratinocyte-derived chemokine; CXCL, CXC chemokine ligand; TNF-α, tumor necrosis factor-α; MIP-2, macrophage inflammatory protein-2. *P < 0.05 vs. complete HDE. #P < 0.05 vs. all other conditions.
HDE-induced intrapulmonary inflammatory cell influx, neutrophilia, and cytokine release are diminished by protease depletion in an acute exposure mouse model.
Mice (6 mice per group) were intranasally challenged with a 12.5% solution of complete HDE, protease-depleted HDE, AEBSF alone, or with saline vehicle alone following an established instillation technique (29). Consistent with prior studies, 5 h following intranasal challenge with HDE, elevated inflammatory cell influx with increased neutrophils was evident compared with saline controls (29). Both the HDE-induced total cell influx and neutrophilia were significantly reduced by protease depletion (2.3-fold decrease in total cells; 3.2-fold decrease in neutrophils) (Fig. 4A). Neither HDE exposure nor protease depletion of HDE had an effect on macrophage infiltration. Furthermore, HDE treatment elicited a prominent accumulation of cytokines in BALF compared with saline instillation, whereas depletion of HDE proteases resulted in a marked inhibition of all four cytokines measured compared with complete HDE exposure (protease depletion reduced murine IL-6 from 1,068 ± 189 to 291 ± 64 pg/ml; TNF-α from 1,018 ± 199 to 272 ± 71 pg/ml; KC from 855 ± 129 to 361 ± 58 pg/ml; and MIP-2 from 205 ± 44 to 90 ± 14 pg/ml, P < 0.05 for all) (Fig. 4B). Protease inhibitor treatment alone did not alter cytokine production. Taken together, these results suggest that protease activity is an important component of HDE that mediates a significant fraction of the acute inflammatory response in vivo.
Fig. 4.

HDE-stimulated mouse lung inflammation is modulated by removal of proteases in a single exposure model. Mice challenged with a single intranasal instillation of complete 12.5% HDE, protease-depleted HDE, protease inhibitor alone (AEBSF), or with saline were killed 5 h later, and their lungs lavaged for recovery of inflammatory cell infiltrates (A) and measurement of proinflammatory cytokines (B). Total cell counts, neutrophil fraction, and cytokines levels in complete HDE-treated mouse bronchoalveolar lavage fluid (BALF) were markedly increased compared with control mice, whereas these inflammatory markers were significantly reduced in mice given protease-depleted HDE. Each symbol represents the mean of 5 fields counted for each mouse (A) and the mean of 2 technical replicates per mouse for cytokines (B). Values are means ± SE for 6 mice per condition. *P < 0.05 vs. complete 12.5% HDE.
Protease depletion attenuates HDE-stimulated inflammation in a repeated exposure mouse model.
Previous investigations indicate that repetitive exposure to HDE leads to a BALF inflammatory response that is significantly higher than saline treatment, but dampened compared with single exposure. However, mice develop dramatic lung pathology with repetitive HDE exposure, characterized by lymphoid aggregate accumulation and bronchiolar/alveolar inflammation (29). To determine whether HDE proteases are similarly important in a repetitive exposure model, mice were treated with complete 12.5% HDE or protease-depleted HDE daily for 3 wk. The total cellularity and neutrophil counts in BALF of mice repeatedly challenged with HDE was significantly elevated compared with saline-treated mice. Protease depletion of HDE significantly reduced HDE-induced total cell and neutrophil counts in BALF recovered from repeatedly challenged mice (2.4-fold decrease in total cells, 3-fold decrease in neutrophils, Fig. 5A). Similar to single-exposure studies, HDE and protease-depleted HDE did not significantly alter macrophage accumulation in the lungs. In addition, protease depletion significantly reduced HDE-induced IL-6, CXCL1, and CXCL2 levels (for IL-6, from 130 ± 11 to 45 ± 7 pg/ml; for CXCL1 from 113 ± 15 to 58 ± 5 pg/ml; and for CXCL2 from 188 ± 13 to 67 ± 8 pg/ml), although no significant reduction in TNF-α levels was observed (Fig. 5B). Consistent with prior studies demonstrating a chronic inflammatory adaptation response (29), the magnitude of the HDE-induced cytokine/chemokine production was attenuated following 3-wk exposure compared with the single acute exposure.
Fig. 5.

Inflammatory effects of repetitive exposure of mice to HDE persist for up to 3 wk. Lavage fluid collected from mice challenged repeatedly (3 wk) by intranasal instillations of complete 12.5% HDE shows heightened inflammatory markers compared with control conditions [inflammatory cell infiltrates (A) and cytokine release (B)], but the response was somewhat blunted compared with single instillation (Fig. 4). Depletion of proteases in HDE diminished the proinflammatory response. Each symbol represents the mean of 5 fields counted for each mouse (A) and the mean of 2 technical replicates per mouse for cytokines (B). Values are means ± SE for 6 mice per condition. *P < 0.05 vs. complete 12.5% HDE.
To examine the histopathological effects of HDE protease depletion on repeatedly challenged mice, lungs were microscopically examined for inflammatory changes, and inflammatory scores were assigned by a pathologist blinded to treatment conditions, as previously described (29). Lung tissue from the saline and the inhibitor-alone-exposed mice demonstrated very few inflammatory cells (Fig. 6A). In contrast, lung tissue from mice challenged daily for 3 wk with complete HDE demonstrated a consistent pattern of peribronchiolar and alveolar inflammation, characterized by a mixture of mononuclear and neutrophilic cellular aggregates, as well as diffuse inflammatory cell infiltration in the alveolar parenchyma. By microscopic review and blinded inflammatory scoring, mice exposed to protease-depleted HDE exhibited a significant scored reduction in leukocytic aggregates, as well as in the scored degree of diffuse alveolar inflammation, compared with complete HDE-challenged mice (Fig. 6). Collectively, these results indicate that the protease activity in HDE is important in mediating the inflammatory response in the repetitive HDE exposure animal model.
Fig. 6.

Chronic exposure to HDE induces accumulation of inflammatory cells in mouse lungs; protease-depletion attenuates the effect. A: histological assessment of lung inflammation in mice challenged with complete HDE daily for 3 wk demonstrates markedly heightened perivascular and peribronchiolar inflammatory cell accumulation compared with mice treated with saline or protease inhibitor alone. Depletion of proteases in HDE results in reduction of the inflammatory effect. B: this observation is supported by semiquantitative inflammatory scores of lymphoid aggregates and inflammatory markers in the alveolar and bronchiolar compartments. Values are means ± SE for one entire lung cross section examined for each mouse. *P < 0.05 vs. AEBSF and saline. #P < 0.05 vs. complete HDE.
Sequential HDE-induced PKCα and PKCε activation is protease dependent.
Previously, our laboratory demonstrated the importance of the protein kinase isoforms PKCα and PKCε in the release of HDE-induced proinflammatory cytokines IL-6, IL-8, and TNF-α in BECs (33, 43). In the present study, we tested whether the proteases in HDE influence the stimulation of PKC activity, by depleting protease activity in HDE before stimulating BEAS-2B cells in vitro. Both PKCα (1 h) and PKCε (6 h) activities in BEAS-2B lysates were significantly increased after exposure to complete HDE, as previously reported (43), whereas protease-depleted HDE treatment did not activate either kinase (Fig. 7). These results provide a potential mechanism by which proteases affect the BEC inflammatory response to HDE by contributing to the PKC activation that mediates subsequent cytokine release.
Fig. 7.
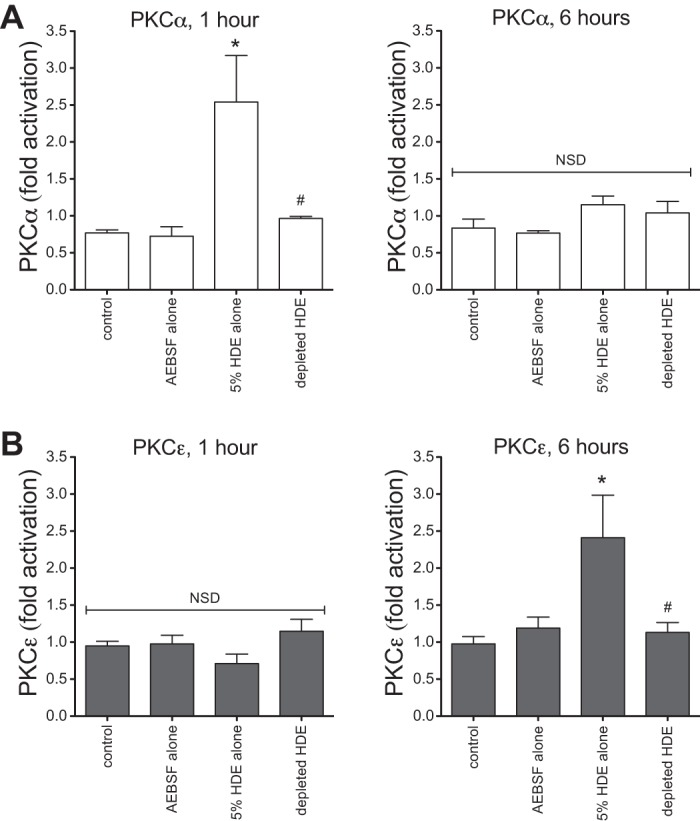
Proteases are involved in HDE-induced epithelial protein kinase C (PKC) isoform activation. HDE challenge rapidly increases BEAS-2B PKCα activation within 1 h, but, by 6 h, PKCα activity returns to baseline levels. A: depletion of proteases from HDE inhibits its stimulatory effect at both 1 and 6 h. HDE treatment fails to induce PKCε activity at 1 h after exposure, but significantly promotes PKCε activity by 6 h. B: as with PKCα, protease-depleted HDE has no effect on PKCε activity at either time point. Three independent experiments were performed, and 4 technical replicates were recorded for each condition. Values are means ± SE. *P < 0.05 vs. control. #P < 0.05 vs. complete 5% HDE.
PAR-1 and PAR-2 are involved in the protease-induced proinflammatory cytokine response.
To determine whether HDE proteases act by activating PARs, selective inhibitors of PAR-1 and PAR-2 were tested for their effects on cytokine/chemokine release in both BEAS-2B and NHBE. Cultured BEC were pretreated with various concentrations of the PAR-1 small molecule inhibitor SCH-79797 (0.01–10 nM) or the PAR-2 peptide inhibitor FSLLRY-NH2 (0.5–50 nM) for 1 h before being challenged with complete 5% HDE for 24 h, after which culture supernates were collected for cytokine measurements. Inhibition of either PAR-1 or PAR-2 dose-dependently reduced the HDE-stimulated release of IL-6 and IL-8 [for BEAS-2B, the maximum dose of SCH-79797 reduced IL-6 from 2,139 ± 216 to 1,493 ± 137 pg/ml and IL-8 from 6,658 ± 492 to 3,932 ± 387 pg/ml (Fig. 8, A and B); the highest dose of FSLLRY reduced IL-6 from 1,667 ± 90 to 813 ± 39 pg/ml, and IL-8 from 7,871 ± 360 to 3,172 ± 376 pg/ml (Fig. 8, C and D); P < 0.05 vs. HDE alone, for all comparisons]. Similar inhibition of HDE stimulated cytokines was observed for primary NHBE treated in an identical manner in separate experiments (Fig. 8, E–H). Suppression of PAR-1 or PAR-2 in BEAS-2B cells by transfection with siRNAs that specifically target the two receptors also significantly blunted the cytokine-enhancing effects of HDE treatment compared with control cells (Fig. 9, A–D). Neither the PAR inhibitors nor inactivation of proteases in HDE reduced cytokine stimulation in cells pretreated with PAR siRNAs, indicating that PAR-1 and PAR-2 are significant contributors to the proinflammatory signaling associated with the HDE proteases. Transfection with nontargeting control siRNA had no effect on control or HDE-stimulated cytokines (Fig. 9E), and the efficacy of the siRNA transfection was confirmed by Western blot (Fig. 9F).
Fig. 8.

Pharmacological inhibitors of protease-activated receptor (PAR)-1 (A, B, E, F) and PAR-2 (C, D, G, H) dose-responsively blunt dust extract-enhanced BEC cytokine release. BEAS-2B and NHBE were exposed to 5% HDE in the presence or absence of various concentrations of the inhibitor for PAR-1 (SCH-79797; A, B, E, F) or PAR-2 (FSLLRY-NH2; C, D, G, H) for 24 h. Supernatant medium was recovered and assayed for cytokine release by ELISA. In BEAS-2B, nanomolar concentrations of both PAR-1 (A and B) and PAR-2 (C and D) inhibitors dose-dependently attenuated the HDE-induced cytokines. Similar results were observed for NHBE cultures treated identically (E–H). Values are means ± SE for 3 independent experiments, with 2 technical replicates recorded per condition. *P < 0.05 vs. HDE alone. HBEC, human BEC.
DISCUSSION
Individuals working in hog CAFOs are at risk for developing chronic airway diseases as a result of their occupational exposure to organic dust. Dusts from these environments are complex and composed of a mixture of components. The gram-positive bacterial cell wall component PGN and the gram-negative component lipopolysaccharide have been shown to be important in mediating the inflammatory responses associated with HDE (27). However, these components alone do not account for all of the inflammatory effects resulting from agricultural exposures. Thus we examined other components of HDE that could be responsible for mediating the inflammatory airway responses in both in vitro BEC models and in vivo murine models of dust extract exposure.
Both endogenous and environmental proteases are known to be involved in inflammatory airway responses, including the promotion or exacerbation of cystic fibrosis, COPD, and allergic asthma, among other airway inflammatory disease states (6, 12, 23, 30, 38). In light of the identification of protease activity in a variety of environmental allergens and irritants, it is not surprising that CAFO dust extract was found to contain potentially harmful proteolytic activity as well.
One of the first lines of defense against the inhalation of dust and other exogenous agents into the lungs occurs at the level of the bronchial epithelium. In addition to providing a mechanical barrier within the airways, BECs also play an important role in the early inflammatory response to inhaled irritants. Indeed, BECs are directly activated by protease activity in a variety of different exposure models (12). Proteases found in allergens from fungi have been shown to induce the expression of proinflammatory cytokines IL-6 and IL-8 in airway and nasal epithelial cells (13), and proteases found in cockroach and house dust mite allergen extracts have also been shown to increase the TNF-α-induced release of IL-8 from BECs (24). Instillation of the protease PPE into mouse lungs is a commonly used animal model of emphysema, further establishing the likely relevance of proteases to airways diseases.
To determine the role of proteases in eliciting the proinflammatory responses to HDE in BECs, we depleted the protease activity of HDE using the serine protease inhibitors PMSF or AEBSF. Both inhibitors exhibit broad specificity and lack cytotoxic effects. Also critical is their irreversible inhibition of proteases and their rapid hydrolytic degradation in aqueous solution; for these experiments, it is essential that there be no remaining protease inhibitor activity in the protease-depleted dust that could inhibit epithelial cell proteases that might also contribute to the inflammatory response. Both PMSF and AEBSF inhibited much of the protease activity of HDE (Fig. 1), which corresponded to reductions in HDE-stimulated cytokine release (Fig. 2) and activation of PKCα and PKCε (Fig. 7), both of which are known features of HDE-induced proinflammatory responses in BECs (33, 43).
Many of the actions of extracellular proteases on lung epithelial cells are mediated by their cleavage and activation of PAR-2, including the effects of fungal, cockroach, and dust mite allergen proteases; in contrast, PAR-1 plays a minor, if any, role for these agents (2, 17, 24, 30, 36). PARs are involved in bronchial fibroblast proliferation, epithelial cell wound healing, and mucus hypersecretion (10, 20, 21). Our data showing that PAR-1/2 antagonism (Fig. 8) or knockdown (Fig. 9) significantly reduce BEC inflammatory responses to HDE suggest that the proteases in HDE act through both PAR-2 and PAR-1, and that a significant portion of HDE protease effects appear to be mediated via activation of PAR-1 and PAR-2. The involvement of both PAR-1 and PAR-2 in responses to HDE may be explained by the fact that HDE is a complex mixture and contains multiple proteases, whereas other studies (such as those using dust mite or cockroach allergens) focused on a single or a limited number of well-defined proteases that may elicit more specific PAR activation patterns. Another potential explanation could be due to the commercially available inhibitors used in these investigations potentially having unintended off-target effects on non-PAR G protein-coupled receptors (25). Studies to further investigate potential combined effects of dual antagonism are warranted. In addition, an important goal for future studies will be to establish whether different proteases mediate the specific activation of PAR-1 vs. PAR-2; these two PARs are known to have distinct protease activation specificities, with PAR-1 being activated strongly by thrombin but weakly by trypsin, and PAR-2 being activated selectively by trypsin but not by thrombin (31). Interestingly, these PARs are also inhibited by some proteases, including elastase, so a complex combination of stimulatory and inhibitory effects of proteases on PAR activity may occur in response to HDE. Thus deciphering the specific actions of PAR-1 and PAR-2 in HDE-stimulated BECs will provide greater understanding of how these receptors function singly or cooperatively to induce a proinflammatory response to HDE.
Mouse lung slice ex vivo cultures retain the tissue and cellular components of the intact organ and thus represent a transition model, allowing closer approximation to the complexities of an in vivo system. When exposed to protease-depleted HDE, lung slices exhibited a blunted proinflammatory response compared with complete HDE-stimulated cultures (Fig. 3). The similarity in magnitude of protease-dependent inhibition of HDE-stimulated cytokine release between epithelial cell monocultures and the lung tissue model suggest that the effects of HDE protease activity on the lung may be independent of recruited inflammatory cells. In the murine in vivo exposure model, depletion of protease activity from HDE significantly reduced the airway inflammatory markers that are characteristic of single and repetitive exposures to HDE. These data suggest that the proteases in HDE may be promoting and/or potentiating the airway inflammation that is associated with both acute and repetitive exposure to agricultural dusts.
Ongoing studies are aimed at identifying the specific proteases present in HDE and which proteases mediate the pro-inflammatory effects of HDE on BECs. Although zymography is an effective tool for the detection and separation of protease activities, interactions between the enzymes and the gel during electrophoresis prevent precise determination of the molecular weights and identification of the specific proteases that are present. It is likely that relevant proteases in HDE include serine proteases, because PMSF and AEBSF specifically inhibit serine proteases. Identifying the specific HDE proteases that mediate airway inflammation could point to novel therapeutic approaches for ameliorating or treating swine CAFO dust-induced lung disease. Alternatively, monitoring for these proteases in the environments surrounding CAFOs could prove effective in reducing or eliminating disease in those exposed in neighboring communities but outside of the CAFO environment per se.
The studies reported here focus on the contribution of proteases to lung inflammation, but a variety of other dust components and pathways have been shown to contribute to the lung inflammatory response to dust exposure. Our laboratory has previously shown that endotoxin is not a major contributor to the inflammatory response of BECs or mouse lungs to HDE, whereas PGN does play a major role in the dust-induced inflammatory response (27). Prior investigations have also shown that epidermal growth factor receptor (EGFR) signaling plays a role in the proinflammatory response of BECs to HDE (9). EGFR activation often occurs via transactivation mechanisms involving matrix metalloprotease activation (4, 11, 32). Although protease-mediated transactivation of EGFR signaling pathways is an attractive explanation for the effects of both protease and PAR inhibitors, data to date suggest that the protease activity present in HDE is in fact not responsible for EGFR activation (data not shown).
The potential for therapeutic intervention with PAR antagonists or other protease-inhibiting strategies for treating individuals with airway diseases resulting from these exposures is compelling. Our laboratory has recently shown that cAMP-dependent protein kinase activation can inhibit the effects of HDE on BECs (41). One study has shown that the long-acting β2-adrenergic receptor agonist salmeterol, a standard therapy for COPD, can prevent some of the deleterious effects of PAR-2 activation in airway epithelial cells (39). Some of the effects of β2-adrenergic receptor agonists may be due to inhibition of protease- and PAR-mediated signaling, although this needs to be established more directly.
Collectively, these data reveal a role for the protease components of HDE (and the hog CAFO dust from which it was derived) in promoting and/or perpetuating airway inflammation. The results of these studies support the concept that targeting the protease activity of agricultural and potentially other environmental dusts could provide a novel method for preventing or treating airway inflammation in individuals who work within or are exposed to dusty agricultural environments.
GRANTS
The work reported here was supported by a grant from the Centers for Disease Control National Institute for Occupational Safety and Health (2R01-OH-008539, D. J. Romberger); National Institute of Environmental Health Sciences (R01-ES-019325, J. A. Poole; F32-ES-022913, T. M. Nordgren); and Department of Veteran's Affairs (I01BX000728, T. A. Wyatt). This work was supported in part by the Central States Center for Agricultural Safety and Health (CS-CASH; U54OH010162). This work was also supported by the use of facilities and resources at the Veterans Affairs Nebraska-Western Iowa Healthcare System, Omaha, NE.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s). The authors have no conflicts to disclose.
AUTHOR CONTRIBUTIONS
D.J.R., T.M.N., X.-d.L., and J.A.P. conception and design of research; D.J.R., T.M.N., J.A.P., M.L.T., and T.A.W. interpreted results of experiments; D.J.R. and T.M.N. drafted manuscript; D.J.R., A.J.H., T.M.N., C.P.S., W.W.W., X.-d.L., J.A.P., M.L.T., and T.A.W. edited and revised manuscript; D.J.R., A.J.H., T.M.N., C.P.S., W.W.W., X.-d.L., J.A.P., M.L.T., and T.A.W. approved final version of manuscript; A.J.H., T.M.N., C.P.S., and T.A.W. performed experiments; A.J.H. prepared figures; T.M.N., W.W.W., and T.A.W. analyzed data.
ACKNOWLEDGMENTS
The authors thank Angela Gleason and Christopher Bauer for assistance with the mouse studies, Kristina Bailey for providing primary bronchial epithelial cells, and Lisa Chudomelka for invaluable assistance in the preparation of the manuscript.
REFERENCES
- 1.Respiratory health hazards in agriculture. Am J Respir Crit Care Med 158: S1–S76, 1998. [DOI] [PubMed] [Google Scholar]
- 2.Asokananthan N, Graham PT, Stewart DJ, Bakker AJ, Eidne KA, Thompson PJ, Stewart GA. House dust mite allergens induce proinflammatory cytokines from respiratory epithelial cells: the cysteine protease allergen, Derp 1, activates protease-activated receptor (PAR)-2 and inactivates PAR-1. J Immunol 169: 4572–4578, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Bailey KL, Robinson JE, Sisson JH, Wyatt TA. Alcohol decreases RhoA activity through a nitric oxide (NO)/cyclic GMP (cGMP)/protein kinase G (PKG)-dependent pathway in the airway epithelium. Alcohol Clin Exp Res 35: 1277–1281, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergin DA, Greene CM, Sterchi EE, Kenna C, Geraghty P, Belaaouaj A, Taggart CC, O'Neill SJ, McElvaney NG. Activation of the epidermal growth factor receptor (EGFR) by a novel metalloprotease pathway. J Biol Chem 283: 31736–31744, 2008. [DOI] [PubMed] [Google Scholar]
- 5.Boitano S, Flynn AN, Sherwood CL, Schulz SM, Hoffman J, Gruzinova I, Daines MO. Alternaria alternata serine proteases induce lung inflammation and airway epithelial cell activation via PAR2. Am J Physiol Lung Cell Mol Physiol 300: L605–L614, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brehm A, Geraghty P, Campos M, Garcia-Arcos I, Dabo AJ, Gaffney A, Eden E, Jiang XC, D'Armiento J, Foronjy R. Cathepsin G degradation of phospholipid transfer protein (PLTP) augments pulmonary inflammation. FASEB J 28: 2318–2331, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chavira R, Jr Burnett TJ, Hageman JH. Assaying proteinases with azocoll. Anal Biochem 136: 446–450, 1984. [DOI] [PubMed] [Google Scholar]
- 8.Chiu LL, Perng DW, Yu CH, Su SN, Chow LP. Mold allergen, pen C 13, induces IL-8 expression in human airway epithelial cells by activating protease-activated receptor 1 and 2. J Immunol 178: 5237–5244, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Dodmane PR, Schulte NA, Heires AJ, Band H, Romberger DJ, Toews ML. Airway epithelial epidermal growth factor receptor mediates hogbarn dust-induced cytokine release but not Ca2+ response. Am J Respir Cell Mol Biol 45: 882–888, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ewen D, Clarke SL, Smith JR, Berger C, Salmon G, Trevethick M, Shute JK. The role of protease-activated receptors PAR-1 and PAR-2 in the repair of 16HBE 14o(-) epithelial cell monolayers in vitro. Clin Exp Allergy 40: 435–449, 2010. [DOI] [PubMed] [Google Scholar]
- 11.Hamilton LM, Torres-Lozano C, Puddicombe SM, Richter A, Kimber I, Dearman RJ, Vrugt B, Aalbers R, Holgate ST, Djukanovic R, Wilson SJ, Davies DE. The role of the epidermal growth factor receptor in sustaining neutrophil inflammation in severe asthma. Clin Exp Allergy 33: 233–240, 2003. [DOI] [PubMed] [Google Scholar]
- 12.Jacquet A. Interactions of airway epithelium with protease allergens in the allergic response. Clin Exp Allergy 41: 305–311, 2011. [DOI] [PubMed] [Google Scholar]
- 13.Kauffman HF, Tomee JF, van de Riet MA, Timmerman AJ, Borger P. Protease-dependent activation of epithelial cells by fungal allergens leads to morphologic changes and cytokine production. J Allergy Clin Immunol 105: 1185–1193, 2000. [DOI] [PubMed] [Google Scholar]
- 14.Kawabata A, Kawao N. Physiology and pathophysiology of proteinase-activated receptors (PARs): PARs in the respiratory system: cellular signaling and physiological/pathological roles. J Pharm Sci 97: 20–24, 2005. [DOI] [PubMed] [Google Scholar]
- 15.Kirkhorn SR, Garry VF. Agricultural lung diseases. Environ Health Perspect 108, Suppl 4: 705–712, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobayashi T, Liu X, Wen FQ, Fang Q, Abe S, Wang XQ, Hashimoto M, Shen L, Kawasaki S, Kim HJ, Kohyama T, Rennard SI. Smad3 mediates TGF-beta1 induction of VEGF production in lung fibroblasts. Biochem Biophys Res Commun 327: 393–398, 2005. [DOI] [PubMed] [Google Scholar]
- 17.Kouzaki H, O'Grady SM, Lawrence CB, Kita H. Proteases induce production of thymic stromal lymphopoietin by airway epithelial cells through protease-activated receptor-2. J Immunol 183: 1427–1434, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langley RL. Consequences of respiratory exposures in the farm environment. N C Med J 72: 477–480, 2011. [PubMed] [Google Scholar]
- 19.Lantz MS, Ciborowski P. Zymographic techniques for detection and characterization of microbial proteases. Methods Enzymol 235: 563–594, 1994. [DOI] [PubMed] [Google Scholar]
- 20.Liu C, Li Q, Zhou X, Kolosov VP, Perelman JM. Human airway trypsin-like protease induces mucin5AC hypersecretion via a protease-activated receptor 2-mediated pathway in human airway epithelial cells. Arch Biochem Biophys 535: 234–240, 2013. [DOI] [PubMed] [Google Scholar]
- 21.Matsushima R, Takahashi A, Nakaya Y, Maezawa H, Miki M, Nakamura Y, Ohgushi F, Yasuoka S. Human airway trypsin-like protease stimulates human bronchial fibroblast proliferation in a protease-activated receptor-2-dependent pathway. Am J Physiol Lung Cell Mol Physiol 290: L385–L395, 2006. [DOI] [PubMed] [Google Scholar]
- 22.May S, Romberger DJ, Poole JA. Respiratory health effects of large animal farming environments. J Toxicol Environ Health B Crit Rev 15: 524–541, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Page K. Role of cockroach proteases in allergic disease. Curr Allergy Asthma Rep 12: 448–455, 2012. [DOI] [PubMed] [Google Scholar]
- 24.Page K, Strunk VS, Hershenson MB. Cockroach proteases increase IL-8 expression in human bronchial epithelial cells via activation of protease-activated receptor (PAR)-2 and extracellular-signal-regulated kinase. J Allergy Clin Immunol 112: 1112–1118, 2003. [DOI] [PubMed] [Google Scholar]
- 25.Perez M, Lamothe M, Maraval C, Mirabel E, Loubat C, Planty B, Horn C, Michaux J, Marrot S, Letienne R, Pignier C, Bocquet A, Nadal-Wollbold F, Cussac D, de Vries L, Le Grand B. Discovery of novel protease activated receptors 1 antagonists with potent antithrombotic activity in vivo. J Med Chem 52: 5826–5836, 2009. [DOI] [PubMed] [Google Scholar]
- 26.Poole JA, Alexis NE, Parks C, MacInnes AK, Gentry-Nielsen MJ, Fey PD, Larsson L, Allen-Gipson D, Von Essen SG, Romberger DJ. Repetitive organic dust exposure in vitro impairs macrophage differentiation and function. J Allergy Clin Immunol 122: 375–382, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poole JA, Dooley GP, Saito R, Burrell AM, Bailey KL, Romberger DJ, Mehaffy J, Reynolds SJ. Muramic acid, endotoxin, 3-hydroxy fatty acids, and ergosterol content explain monocyte and epithelial cell inflammatory responses to agricultural dusts. J Toxicol Environ Health A 73: 684–700, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poole JA, Romberger DJ. Immunological and inflammatory responses to organic dust in agriculture. Curr Opin Allergy Clin Immunol 12: 126–132, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poole JA, Wyatt TA, Oldenburg PJ, Elliott MK, West WW, Sisson JH, Von Essen SG, Romberger DJ. Intranasal organic dust exposure-induced airway adaptation response marked by persistent lung inflammation and pathology in mice. Am J Physiol Lung Cell Mol Physiol 296: L1085–L1095, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Post S, Heijink IH, Petersen AH, de Bruin HG, van Oosterhout AJ, Nawijn MC. Protease-activated receptor-2 activation contributes to house dust mite-induced IgE responses in mice. PLoS One 9: e91206, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramachandran R, Hollenberg MD. Proteinases and signalling: pathophysiological and therapeutic implications via PARs and more. Br J Pharmacol 153, Suppl 1: S263–S282, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richter A, O'Donnell RA, Powell RM, Sanders MW, Holgate ST, Djukanovic R, Davies DE. Autocrine ligands for the epidermal growth factor receptor mediate interleukin-8 release from bronchial epithelial cells in response to cigarette smoke. Am J Respir Cell Mol Biol 27: 85–90, 2002. [DOI] [PubMed] [Google Scholar]
- 33.Romberger DJ, Bodlak V, Von Essen SG, Mathisen T, Wyatt TA. Hog barn dust extract stimulates IL-8 and IL-6 release in human bronchial epithelial cells via PKC activation. J Appl Physiol 93: 289–296, 2002. [DOI] [PubMed] [Google Scholar]
- 34.Sanderson MJ. Exploring lung physiology in health and disease with lung slices. Pulm Pharmacol Ther 24: 452–465, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slager RE, Allen-Gipson DS, Sammut A, Heires A, DeVasure J, Von Essen S, Romberger DJ, Wyatt TA. Hog barn dust slows airway epithelial cell migration in vitro through a PKCalpha-dependent mechanism. Am J Physiol Lung Cell Mol Physiol 293: L1469–L1474, 2007. [DOI] [PubMed] [Google Scholar]
- 36.Sun G, Stacey MA, Schmidt M, Mori L, Mattoli S. Interaction of mite allergens Der p3 and Der p9 with protease-activated receptor-2 expressed by lung epithelial cells. J Immunol 167: 1014–1021, 2001. [DOI] [PubMed] [Google Scholar]
- 37.Sweetman F, Ornstein L. Electrophoresis of elastase-like esterases from human neutrophils. J Histochem Cytochem 22: 327–339, 1974. [DOI] [PubMed] [Google Scholar]
- 38.Thibodeau PH, Butterworth MB. Proteases, cystic fibrosis and the epithelial sodium channel (ENaC). Cell Tissue Res 351: 309–323, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winter MC, Shasby SS, Ries DR, Shasby DM. PAR2 activation interrupts E-cadherin adhesion and compromises the airway epithelial barrier: protective effect of beta-agonists. Am J Physiol Lung Cell Mol Physiol 291: L628–L635, 2006. [DOI] [PubMed] [Google Scholar]
- 40.Wyatt TA, Kharbanda KK, McCaskill ML, Tuma DJ, Yanov D, DeVasure J, Sisson JH. Malondialdehyde-acetaldehyde-adducted protein inhalation causes lung injury. Alcohol 46: 51–59, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wyatt TA, Poole JA, Nordgren TM, DeVasure JM, Heires AJ, Bailey KL, Romberger DJ. cAMP-dependent protein kinase activation decreases cytokine release in bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 307: L643–L651, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wyatt TA, Schmidt SC, Rennard SI, Tuma DJ, Sisson JH. Acetaldehyde-stimulated PKC activity in airway epithelial cells treated with smoke extract from normal and smokeless cigarettes. Proc Soc Exp Biol Med 225: 91–97, 2000. [DOI] [PubMed] [Google Scholar]
- 43.Wyatt TA, Slager RE, Heires AJ, Devasure JM, Vonessen SG, Poole JA, Romberger DJ. Sequential activation of protein kinase C isoforms by organic dust is mediated by tumor necrosis factor. Am J Respir Cell Mol Biol 42: 706–715, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]