

Abstract
Objective
To evaluate the safety and efficacy of hCDR1 (Edratide) in patients with systemic lupus erythematosus (SLE).
Methods
Patients (n=340) with SLE ≥4 ACR criteria (4–11, mean 7) with active disease (SLEDAI-2K of 6–12). Patients were on average 7.1 years post-diagnosis and their organ involvement was mainly musculoskeletal, mucocutaneous and haematologic. Placebo or Edratide was administered subcutaneously weekly at doses of 0.5, 1.0 or 2.5 mg. The co-primary endpoints were SLEDAI-2K SLE Disease Activity and Adjusted Mean SLEDAI (AMS) reduction in patients compared with controls using a landmark analysis. Secondary outcomes were improvement in British Isles Lupus Assessment Group (BILAG) Responder Index and medicinal flare analysis.
Results
Edratide was safe and well tolerated. The primary endpoints based solely on SLEDAI-2K and AMS were not met. The secondary predefined endpoint, BILAG, was met for the 0.5 mg Edratide arm in the intention to treat (ITT) cohort (N=316) (OR=2.09, p=0.03) with trends in the 1.0 and 2.5 mg doses. There was also a positive trend in the Composite SLE Responder Index of the ITT cohort. Post hoc analysis showed that the BILAG secondary endpoint was also met for the 0.5 mg Edratide for a number of subgroup dose levels, including low or no steroids, seropositivity and patients with 2 grade BILAG improvement.
Conclusions
The favourable safety profile and encouraging clinically significant effects noted in some of the endpoints support the need for additional longer term Edratide studies that incorporate recent advances in the understanding and treatment of SLE, including steroid treatment algorithms, and using a composite primary endpoint which is likely to include BILAG.
Trial registration number
Keywords: Systemic Lupus Erythematosus, Treatment, Autoimmune Diseases
Key messages.
Edratide demonstrated efficacy in one and possibly more clinically meaningful endpoints.
Dose ranging studies demonstrated the 0.5 mg subcutaneous weekly was the most effective dose.
There were no safety signals in this 26 week study.
Introduction
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease characterised by the production of autoantibodies, and by impaired function of T and B cells.1 2 The pathogenesis of SLE has been shown to involve B and T cell apoptosis,3 4 cytokines5 6 and impaired regulatory T cell function.7 8
Due to the complex nature of the disease, progress in developing new SLE treatment modalities has been slow.9 10 Until the approval by the Food and Drug Administration in 2011 of belimumab,11 no new drugs had been approved for the treatment of SLE since 1955. There is therefore a clear need for new therapeutic agents.12
hCDR1 (Edratide) is a novel synthetic peptide of 19 amino acid residues (H-G-Y-Y-W-S-W-I-R-Q-P-P-G-K-G-E-E-W-I) based on the complementarity-determining region 1 (CDR1) of a human anti-DNA mAb that expresses a major idiotype denoted 16/6 Id.13 14 Treatment with hCDR1 leads to a cascade of events that culminate in the downregulation of SLE-associated autoreactive T and B cells and in the clinical amelioration of lupus. hCDR1 is therefore a candidate for treatment of patients with SLE.15
In mouse models of SLE, treatment with hCDR1 significantly reduced immune complex deposits in the kidney, and resulted in improvement in proteinuria and leucopenia. The treatment downregulated anti-dsDNA-specific Abs and had no significant effect on the 16/6 Id-specific antibody response while immunomodulating cytokines and apoptosis and upregulating regulatory T cells.16–18 In addition, hCDR1 was also shown to significantly and specifically downregulate IFN-α gene expression.19
In a large animal (porcine) model, hCDR1 administration resulted in improvement in the clinical SLE-related manifestations. In addition, the treatment reduced the gene expression of the pathogenic cytokines interleukin 10 (IL-10), tumor necrosis factor α (TNFα), IFN-γ and interleukin 1β (IL-1β) and elevated the expression of TGF-β, the antiapoptotic molecule Bcl-xL and the suppressive master gene, Foxp3.20
In vitro, hCDR1 downregulated the proliferation of peripheral blood mononuclear cells (PBMC) of patients with SLE.12 Furthermore, incubation of hCDR1 with PBMC of patients with SLE diminished the gene expression of pathogenic cytokines and of T cell apoptosis and upregulated the expression of TGF-β leading to the expansion of functional regulator T cells.21
A small-scale human study22 examined gene expression in PBMC of nine patients with lupus who were treated for 26 weeks with Edratide. Disease activity was assessed by SLEDAI-2K and the British Isles Lupus Assessment Group (BILAG) scores. Edratide restored the global immune dysregulation of patients with lupus by downregulating in vivo gene expression of apoptosis, BLyS and pathogenic cytokines, and upregulating immunosuppressive molecules associated with a significant decrease in SLEDAI-2K and BILAG scores in the Edratide treated patients.22
This report describes a phase II double-blind, placebo-controlled, dose-ranging study of the safety and efficacy of Edratide in patients with active SLE who were receiving standard SLE treatment.
Materials and methods
Study design
The Study to Evaluate the Tolerability, Safety and Effectiveness of Edratide in the Treatment of Lupus (PRELUDE) was a phase II, multinational (USA, Canada, Mexico, France, Germany, UK, Spain, Holland, Italy, Russia, Hungary and Israel) multicentre, randomised, double–blind, placebo-controlled study to assess the efficacy, tolerability and safety of Edratide in patients with mild-to-moderate SLE.
Eligible subjects were randomised in a 1:1:1:1 ratio into one of the following four treatment groups: 0.5 mg Edratide, 1 mg Edratide, 2.5 mg Edratide, Placebo for Edratide. Edratide was administered weekly as a subcutaneous injection prepared from freeze-dried (lyophilised) powder containing Edratide acetate in doses corresponding to 0.5 mg, 1.0 mg or 2.5 mg of Edratide free base and/or 120 mg sulfobutylether beta-cyclodextrin sodium (SBECD, Captisol).
Inclusion criteria
A SLEDAI-2K score of 6–12 and stable concomitant medications (prednisone <40 mg, non-steroidal anti-inflammatory drugs (NSAIDs) antimalarials) for at least 1 month prior to baseline.
Exclusion criteria
Active central nervous system (CNS) lupus, active lupus nephritis (protein to creatinine ratio limit of ≥0.75 and/or no active urine sediments), treatment with azathioprine (AZA), methotrexate (MTX) within 4 weeks prior to baseline or treatment with any other immunosuppressive drugs within 3 months prior to baseline. Change of any dose of antimalarial agents during the last month prior to randomisation. Any investigational drug or immunomodulating medication taken within 3 months prior to randomisation. Any cytotoxic agents (eg, cyclophosphamide, mycophenolate mofetil, ciclosporin, leflunomide, chlorambucil) or any immunosuppressive agents or immunomodulators not mentioned above (eg, infliximab, etanercept), intravenous Ig or plasmapheresis in the 3 months prior to randomisation.
Steroid dosing
The study comprised two stages. Eight weeks of a combination phase with steroids, in which subjects were treated with Edratide (or placebo) in addition to their ongoing steroid dose to allow the induction of Edratide activity. This was to be followed by 18 weeks of a steroid withdrawal phase where steroid dosage was tapered down. Briefly, the steroid withdrawal algorithm was as follows: during the first 8 weeks, based on the patient’s condition, the investigator could reduce the steroid dose by up to 30% or, alternatively, if the patient was not in stable remission, could increase the steroid dose up to a daily dose of 60 mg. Starting week 9, a 10-week tapering process began based on the patient’s steroid dose. For example, a patient who entered on 30 mg steroids and maintained that for the first 8 weeks would reduce the steroid dose for 2 weeks to 20 mg, and if there was no deterioration in the patient’s condition, the dose was then reduced to 17.5 mg for 2 weeks. This process continued until the patient reached the target level of 7.5 mg.
If the disease worsened during the withdrawal phase, the patient was instructed to contact the site investigator and steroids could be tapered up according to the algorithm. When deemed clinically feasible by the investigator, tapering down was resumed.
Safety assessments
The incidence and frequency of adverse events (AEs) were categorised according to a regulatory accepted dictionary. Data were tabulated by treatment group, gender, maximal severity, maximal outcome, maximal action taken and maximal relationship to the tested drug. At baseline and weeks 4, 8, 12, 16, 20, 24 and 26/termination, urinalysis was performed and blood samples were obtained for haematology and clinical chemistry analyses.
Outcomes
Since this was a phase II study aiming to assess efficacy and appropriate dosing, we were interested in having as much data on drug as possible and therefore used the Last Observed Value (LOV) approach.
Primary
The primary outcome of the study was a significant difference in the reduction in SLEDAI-2K and SLEDAI Adjusted Mean SLEDAI (AMS)23 between drug and placebo groups. The SLEDAI-2K assesses 16 clinical manifestations and eight laboratory measures. SLEDAI-2K was assessed at screening visit, baseline visit, visit weeks 4, 8, 12, 16, 20, 24 and 26/termination and at each disease exacerbation confirmation visit.
Secondary
British Isles Lupus Assessment Group
The first secondary outcome measure was BILAG Index.24 The BILAG assessment consists of 86 questions, some based on the patient’s history, some on examination findings and others on laboratory results. The scoring system used was A=12; B=5; C=1; D/E=0.25
BILAG responders
For this study, several terms were used:
Substantial responder (SR)—all systems at LOV are either C or D/E, provided that at least one system was either A or B at baseline.
Non-responder (NR)—same BILAG score in LOV as in baseline in all systems or new A or B in at least one system compared with baseline (other systems may improve or deteriorate).
Partial responder (PR)—at least one system improved, at least one system did not improve from A or B at baseline to C or D/E at LOV and no other system deteriorates from C or D/E at baseline to A or B at LOV.
This study was concluded at the end of 2007, prior to the development of the systemic lupus responder index-4 (SRI-4) and BICLA indices. As a consequence, a post hoc responder index was implemented, which is very similar to the above measures. This outcome measure, Composite SLE Responder Index was designated cSRI.
Composite SLE Responder Index (cSRI). SLE composite indices are composite measures of disease activity and responders are designated based on criteria of response and no deterioration measures. In the past, three validated indices were used: SELENA-SLE Disease Activity Index (SELENA-SLEDAI); Physician Global Assessment (PGA) and the BILAG 2004.26 In the present study, cSRI was based on two validated indices, BILAG and SLEDAI only, that is, improvement in BILAG but no deterioration in SLEDAI-2 K. It should be noted that at the time this study was performed the present-day systemic lupus responders indices (SRIs) were not available, but the cSRI used in the study was very similar.
Flare analysis was performed according to accepted protocols.27 Mild-to-moderate flare was defined as a change in SLEDAI of ≥3 points or new onset/worsening of skin manifestations, stomatitis, serositis, arthritis, fever (oral temperature), or an increase of the subject's prednisone dosage, but not exceeding 0.5 mg/kg/day, or adding of NSAIDs/Plaquenil, or an increase in PGA ≥1.0 (but not more than 2.5).
Severe flare was defined as an increase of total SLEDAI to >12 points (by at least 3 points) or new onset or worsening of CNS-SLE, vasculitis, nephritis, myositis, platelets <60 000, haemolytic anaemia with Hb <7 g/dL requiring hospitalisation for SLE, or an increase of the subject's prednisone, exceeding 0.5 mg/kg/day, new immunosuppressive drug or increased PGA to >2.5.
Flare definition by BILAG Index was defined as a new A or B score in at least one system according to the BILAG Index. Moderate disease flare was defined as a new B score following a C, D or E. A severe flare was defined as a score of A in any system having previously been a lower activity score (ie, B/C/D/E).
Statistical analysis
The principal statistical analysis of SLEDAI-2K change from baseline to LOV was carried out using analysis of covariance (ANCOVA—SAS general linear models procedure), with treatment group the independent variable and the covariates medical centre, SLEDAI-2K score at baseline, PO steroids dose at baseline and damage score (SLICC—ACR-DI) at baseline.
AMS represents the time-weighted average of disease activity as measured by SLEDAI-2K score according to AMSi=AUCi/ei, where AUCi=SLEDAI-2K area under curve on change from baseline for the ith subject and ei=total exposure (in weeks) of the ith subject.
BILAG responder analyses were performed using multinomial regression (SAS PROC GENMOD procedure), with treatment group serving as an independent variable. Medical centre, per os (PO) steroids dose at baseline, damage score (SLICC—ACR-DI) at baseline as well as BILAG numeric total score at baseline were included as covariates.
Comparisons between groups for medicinal flare analyses were done using logistic regression (SAS GENMOD procedure). Medical centre, PO steroids dose at baseline, damage score (SLICC—ACR-DI) at baseline and indicator whether the subject had A or at least 2B's at baseline were included as covariates. Medicinal flare was defined as new or increased oral prednisolone dose by at least 5 mg/day (or equivalent), from baseline or any intravenous or intramuscular steroids dose; new immunosuppressive drugs or new or increased dose of antimalarial drugs.
Human subject protection
The study was conducted in accordance with the ethical principles of the Declaration of Helsinki. All participating sites received approval from an institutional review board or ethics committee before patient enrolment. All patients provided written informed consent before any study-related procedures were performed.
Results
Study population
Four hundred and fifty-five subjects were screened for this study; 115 were screening failures. The reasons for non-enrolment were 70.4% did not meet inclusion/exclusion criteria, 20% decided to withdraw and for 9.6% the reason was recorded as ‘Other’.
Figure 1 shows the subgroups of subjects in the study. Thirteen subjects on 0.5 mg Edratide, 13 subjects on 1.0 mg Edratide, 11 subjects on 2.5 mg Edratide and seven subjects on placebo terminated the study prematurely.
Figure 1.

Patient distribution over the course of the study.
See online supplementary table B for additional details about the study population. Demographic characteristics are summarised in table 1. The four groups are comparable in the distribution of age, sex, race and BMI.
Table 1.
Edratide: demographic characteristics and disease activity scores at baseline by treatment group
| TV4710/201 (PRELUDE) | 0.5 mg Edratide (N=84) | 1.0 mg Edratide (N=87) | 2.5 mg Edratide (N=82) | Placebo (N=87) | All (N=340) |
|---|---|---|---|---|---|
| Sex | |||||
| Female N (%) | 81 (96.4%) | 82 (94.3%) | 77 (93.9%) | 80 (92.0%) | 320 (94.1%) |
| Male N (%) | 3 (3.6%) | 5 (5.7%) | 5 (6.1%) | 7 (8.0%) | 20 (5.9%) |
| Race | |||||
| Asian/Oriental N (%) | 1 (1.2%) | 2 (2.3%) | 1 (1.2%) | 2 (2.3%) | 6 (1.8%) |
| Black or African American N (%) | 7 (8.3%) | 5 (5.7%) | 5 (6.1%) | 8 (9.2%) | 25 (7.4%) |
| Caucasian N (%) | 64 (76.2%) | 73 (83.9%) | 66 (80.5%) | 67 (77.0%) | 270 (79.4%) |
| Hispanic N (%) | 3 (3.6%) | 2 (2.3%) | 4 (4.9%) | 3 (3.4%) | 12 (3.5%) |
| Other N (%) | 9 (10.7%) | 5 (5.7%) | 6 (7.3%) | 7 (8.0%) | 27 (7.9%) |
| Age (years) Mean±SD |
40.2±11.0 | 40.8±1.8 | 39.0±11.5 | 38.8±11.7 | 39.7±11.5 |
| BMI Mean±SD |
27.3±7.2 | 26.2±6.2 | 27.4±6.4 | 25.6±5.9 | 26.6±6.5 |
| SLEDAI score Mean |
8.5 | 8.8 | 9.0 | 8.7 | 8.8 |
| BILAG score Mean |
8.8 | 8.0 | 8.2 | 8.1 | 8.3 |
| Years from SLE diagnosis Mean±SD | 7±6.5 | 8.5±7.8 | 8.1±7.1 | 7±6.9 | 7.6±7.1 |
At baseline, 84 patients in the 0.5 mg dose, 87 in the 1 mg dose, 82 in the 1.5 mg dose and 87 on placebo had either BILAG A or B.
BILAG, British Isles Lupus Assessment Group; PRELUDE, A Study to Evaluate the Tolerability, Safety and Effectiveness of Edratide in the Treatment of Lupus; SLE, systemic lupus erythematosus.
There was no statistical difference between the treatment groups in terms of steroids at baseline (table 2).
Table 2.
Steroids at baseline (given per Os, mg equivalent doses to prednisolone)
| TV4710/201 (PRELUDE) | 0.5 mg Edratide (N=84) |
1 mg Edratide (N=87) |
2.5 mg Edratide (N=82) |
Placebo (N=87) |
|---|---|---|---|---|
| Mean | 9 | 9.7 | 9.3 | 9.9 |
| SD | 7.7 | 8.2 | 8.1 | 7.7 |
| Minimum | 0 | 0 | 0 | 0 |
| Median | 7.5 | 9.6 | 10 | 10 |
| Maximum | 40 | 30 | 35 | 30 |
| Proportion of pts with steroid dose ≤7.5 | 51.2% (43) | 48.3% (42) | 42.7% (35) | 43.68% (38) |
| Proportion of pts 7.5 < dose <20 | 38.1% (32) | 35.6% (31) | 43.9% (36) | 40.2% (35) |
| Proportion of pts with steroid dose ≥20 | 10.7% (9) | 16.1% (14) | 13.4% (11) | 16.1% (14) |
PRELUDE, A Study to Evaluate the Tolerability, Safety and Effectiveness of Edratide in the Treatment of Lupus.
Safety
Adverse events (AEs)
AE incidence was similar in all groups: 82.1%, 77%, 75.6% and 79.3% on 0.5 mg Edratide, 1 mg Edratide, 2.5 mg Edratide and placebo, respectively (table 3).
Table 3.
Edratide common adverse events (AEs)—incidence presented by preferred/high-level term and dose and sorted by the risk ratio of pooled Edratide doses versus placebo
| Pooled Edratide doses (N=253) | 0.5 mg Edratide (N=84) |
1 mg Edratide (N=87) |
2.5 mg Edratide (N=82) |
Placebo (N=87) | ||
|---|---|---|---|---|---|---|
| Preferred term | Percentage of subjects | Percentage of subjects | Percentage of subjects | Percentage of subjects | Percentage of subjects | Risk ratio of Pooled Edratide versus Placebo |
| Oedema peripheral | 4.3 | 6.0 | 5.7 | 1.2 | 1.1 | 3.8 |
| Liver function analyses | 3.6 | 3.6 | 3.4 | 3.7 | 1.1 | 3.3 |
| Weight increased | 3.2 | 1.2 | 5.7 | 2.4 | 1.1 | 2.8 |
| Hypertension (HTN)/labile HTN/blood pressure (BP) Increased/hypertensive crisis/hypertensive encephalopathy | 7.5 | 9.5 | 2.3 | 12.2 | 3.4 | 2.2 |
| Fungal vaginal infection | 2.4 | 4.8 | 1.1 | 1.2 | 1.1 | 2.2 |
| International normalised ratio (INR)/INR increased | 2.4 | 2.4 | 4.6 | – | 1.1 | 2.2 |
| Hypoaesthesia | 2.4 | 2.4 | 3.4 | 1.2 | 1.1 | 2.1 |
| Urticarias | 2.0 | 3.6 | 2.3 | – | 1.1 | 1.8 |
| Back pain | 7.9 | 7.1 | 5.7 | 11.0 | 4.6 | 1.7 |
| Diarrhoea | 5.9 | 3.6 | 6.9 | 7.3 | 3.4 | 1.7 |
| Dizziness | 4.0 | 2.4 | 5.7 | 3.7 | 2.3 | 1.7 |
| Depression | 2.0 | 1.2 | 1.1 | 3.7 | 1.1 | 1.7 |
| Dyspnoea | 2.0 | 1.2 | 3.4 | 1.2 | 1.1 | 1.7 |
| Palpitations | 2.0 | 2.4 | 2.3 | 1.2 | 1.1 | 1.7 |
| Nausea and vomiting symptoms | 11.9 | 14.3 | 9.2 | 12.2 | 8.0 | 1.5 |
| Injection site reactions | 17.8 | 17.9 | 16.1 | 19.5 | 12.6 | 1.4 |
| Insomnia | 3.2 | 4.8 | 2.3 | 2.4 | 2.3 | 1.4 |
| Seasonal allergy | 1.6 | 1.2 | – | 3.7 | 1.1 | 1.4 |
| Arthralgia/musculoskeletal pain | 5.9 | 8.4 | 4.6 | 4.9 | 4.6 | 1.3 |
| Cough | 4.3 | 3.6 | 6.9 | 2.4 | 4.6 | 0.9 |
| Dermatitis and eczema | 2.0 | – | 1.1 | 4.9 | 2.3 | 0.9 |
| Herpes viral infections | 4.3 | 2.4 | 8.0 | 2.4 | 0 | 0 |
| Vertigo/vertigo positional | 4.0 | 4.8 | 4.6 | 2.4 | 0 | 0 |
| Migraine | 3.2 | 1.2 | 6.9 | 1.2 | 0 | 0 |
| Tachycardia/tachycardia paroxysmal | 3.2 | 3.6 | 4.6 | 1.2 | 0 | 0 |
| Pharyngitis | 2.4 | 3.6 | 1.1 | 2.4 | 0 | 0 |
| Sciatica | 2.0 | 1.2 | 3.4 | 1.2 | −0 | 0 |
| Dysgeusia | 2.0 | 0 | – | 6.1 | 0 | 0 |
| Abdominal pain upper | 2.0 | 0 | 3.4 | 2.4 | 0 | 0 |
Serious adverse events (SAEs)
Serious adverse events (SAEs) were reported in 6 (7.1%) subjects from the 0.5 mg Edratide group, 10 (11.5%) subjects from the 1.0 mg group, 8 (9.8%) subjects from the 2.5 mg group and 9 (10.3%) subjects from the placebo group. SAE incidence for the pooled Edratide group was similar to placebo: 9.5% versus 10.3% with no statistical differences in the risk ratio of pooled Edratide versus placebo. Most common for both Edratide and placebo were infection-related SAEs: 3.2% (N=4 on 0.5 mg, 2 on 1 mg, 2 on 2.5 mg) versus 4.6% in the placebo group (N=2).
Efficacy
SLEDAI-2K and AMS
The PRELUDE study failed to meet its two primary endpoints. There was no statistically significant difference between or within the four arms at baseline and at any of the three visits. Overall, a mean reduction from the baseline SLEDAI-2K score of about 35% was noted in all treatment arms, including placebo.
British Isles Lupus Assessment Group
Predefined BILAG responder analysis of the intention to treat (ITT) cohort showed a statistically significant difference (p=0.03) between the 0.5 mg Edratide arm (n=76) and the placebo arm (n=83) (figure 2A). The 1 mg and 2.5 mg doses produced non-significant responses.
Figure 2.

Disease activity as measured by British Isles Lupus Assessment Group (BILAG) responder analysis in different groups. (A) BILAG responder analysis at Last Office Visit (LOV) versus Baseline predefined analysis on intention to treat (ITT) cohort (0.5 mg arm n=76; Placebo arm n-83). (B) BILAG responder analysis of patients receiving steroids <20 mg/day at baseline (0.5 mg arm n=68; Placebo arm n=69). (C) BILAG responder analysis of seropositive patients (anti-DNA>30 IU at baseline) (0.5 mg arm n=26; Placebo arm n=31).
When the predefined definition of the BILAG SRs was applied to the cohort of 292 subjects (85% of the ITT cohort), with prednisolone dose <20 mg at baseline, 40% (n=27) of the subjects treated with 0.5 mg Edratide had a substantial response, while only 19% (n=13) of the placebo arm were SRs (p=0.007, OR=2.75) (figure 2B).
In a small group of patients who received no steroids at baseline and who were treated with 0.5 mg Edratide, 54% (n=7) of the Edratide group were SRs as opposed to 13% (n=2) of the placebo group (p=0.05).
A clinically significant effect was identified in seropositive patients (anti-DNA >30 IU). Forty-six per cent (n=12) of the SR group were treated with Edratide as compared with 26% (n=8) with placebo. In addition, 68% (n=21) of the non-responder group were treated with placebo, while 42% (n=11) (p=0.05) were treated with 0.5 mg Edratide (figure 2C).
In the ITT group, post hoc analysis of flare scoring showed that while 17% (n=14) of subjects on 0.5 mg Edratide had a medicinal flare, 29% (n=25) of the subjects on placebo flared (p=0.039, OR=0.43) (figure 3).
Figure 3.
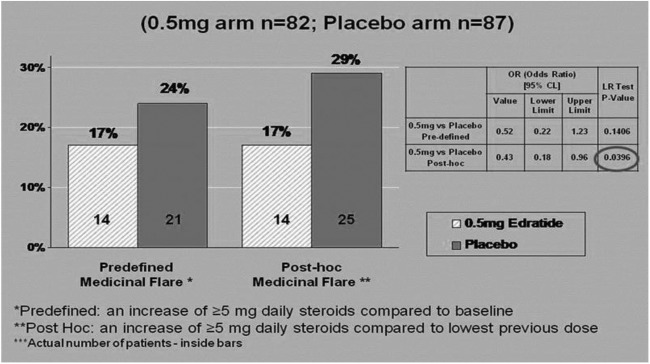
Medicinal flare analysis on intention to treat (ITT) cohort.
In addition, cSRI analysis substantiated the positive effects of Edratide with 34% (n=26) of the 0.5 mg Edratide group being positive responders as compared with 20% (n=17) of the placebo group (p=0.058) (figure 4).
Figure 4.

Composite systemic lupus erythematosus (SLE) Responder Index (cSRI) on intention to treat (ITT) cohort.
Covariate analysis did not find relation between response and body weight. The disease activity clusters that showed at least a trend towards response are summarised in table 4.
Table 4.
Endpoints with at least a trend for the 0.5 mg dose as compared with placebo (intention to treat (ITT) or <20 mg steroid dose cohort)
| End point | Predefined/post hoc | Level of significance |
|---|---|---|
| Disease activity cluster | ||
| BILAG responders (SR, PR,NR) | Predefined-statistical analysis plan (SAP) | p<0.05 (ITT, <20) |
| Total BILAG score (medians) | Predefined-SAP | p<0.05 (<20), trend (ITT) |
| BILAG responders (3 points) | Post hoc | p<0.05 (<20), trend (ITT) |
| BILAG responders (5 points) | Post hoc | p<0.05 (ITT) |
| BILAG MC system | Post hoc | p<0.05 (<20), trend (ITT) |
| SLEDAI decreased to ≤2 | Post hoc | Trend (ITT, <20) |
| Flare cluster | ||
| Medicinal flare (5 mg from baseline) | Predefined-SAP | Trend (ITT, <20) |
| Medicinal flare (5 mg from lowest point from previous dose) | Post hoc | p<0.05 (ITT) |
| Medicinal flare (10 mg from lowest point from previous dose) | Post hoc | Trend (ITT) |
BILAG, British Isles Lupus Assessment Group; NR, no response; PR, partial response; SR, substantial response.
Discussion
This phase II, randomised, controlled trial had a number of goals: (1) to examine the efficacy of Edratide in treating SLE and to select the appropriate outcome measure to detect efficacy; (2) to determine whether there is a subset of patients for which Edratide may be most efficacious; (3) to determine an appropriate dose for future studies; (4) to examine how corticosteroids may influence the effects of Edratide and (5) to monitor for any safety-related issues. As discussed below, while the trial did not meet its primary endpoints, the trial successfully addressed each of the items above.
Edratide demonstrated efficacy in one and possibly more clinically meaningful endpoints. The 0.5 mg dose showed a significant increase in BILAG SRs (and a trend in a combined BILAG and SLEDAI-2K Responder Index) as compared with the placebo group.
While various BILAG analyses and flare definitions supported the possible advantage of Edratide over placebo, neither of the SLEDAI-2K post hoc analyses yielded clinically or statistically meaningful effect. It should be remembered that the SLEDAI-2K captures only complete resolution of a manifestation score, while the BILAG score is more nuanced in that it also captures partial improvement or worsening of SLE manifestations.
In the ITT cohort, there was a significant difference between 0.5 mg Edratide and the placebo group in BILAG substantial response, and similar trends were observed in the 1.0 and 2.5 mg dose groups. In addition, this study demonstrated that Edratide may be more effective in patients receiving lower or no steroids as well as in seropositive patients. As noted above, there was a significant difference between 0.5 mg Edratide and the placebo group in BILAG substantial response for patients with prednisolone dose <20 mg at baseline.
The median total BILAG score also supported the idea of a possible advantage of the 0.5 mg dose versus placebo and the non-parametric test yielded p=0.038 on the prednisolone dose <20 mg at baseline cohort (data not shown).
This study also helped determine the appropriate dose of Edratide for future studies. In this trial, the 0.5 mg dose was the optimal dose in all outcome measures. The fact that the low dose was the most effective is a well-known phenomenon in immunology where, for induction of tolerance, low doses of the tolerogens used are required.28 29
This study also demonstrated that corticosteroids masked the effects of Edratide. Due to their strong immunosuppressive effect on the clinical manifestations, corticosteroids have been shown in animal studies30 to mask, but not interfere with, the beneficial effect of hCDR1.
The same phenomenon was seen in this PRELUDE study. During the first 8 weeks of the study, the protocol stated that the baseline steroid dose be kept stable (no more than 30% reduction), while during the subsequent period the protocol suggested a gradual tapering-down scheme to a minimum of 7.5 mg prednisolone per day. Non-compliance with the tapering-down scheme was not regarded as a protocol violation. During the entire course of the study, an increase of up to 60 mg of the prednisone dose was permitted. Thus, the continued use of steroids in certain patient groups may have masked the effects of Edratide. The fact that this interference by steroids was not identically manifested in SLEDAI and BILAG may simply reflect the fact that SLEDAI did not detect a partial improvement of a manifestation.
Corticosteroids play an important role in the management of active SLE, but are also a major contributor to the damage accumulation over time. More effective management of disease activity by Edratide could lead to the reduction or elimination of steroid use, thus reducing its toxicity.
Finally, this trial demonstrated that the SAEs seen in the Edratide group were not statistically higher than those noted in the placebo group and indicated the favourable safety profile of Edratide at the doses used.
In summary, the results of this study indicate that Edratide appears to be safe and the lowest dose (0.5 mg) appears to be efficacious in suppressing SLE disease activity. At the time this study was performed, it was generally accepted that efficacy of SLE-modifying agents could be shown at 6 months. However, it has subsequently been shown that the minimum time for proving efficacy has been determined to be approximately 1 year.31 Future studies with Edratide will therefore be longer term, will use one of the newly developed responder indices as the endpoint, include seropositive patients, modify the definition of acute lupus nephritis to protein to creatinine ratio limit of ≥0.75 and active urine sediments and also implement a strict steroid withdrawal algorithm.
Acknowledgments
The authors thank Dr Edna Mozes for assistance in analysis and interpretation of the data, Dr Dan Goldstaub for assistance in acquisition, analysis and interpretation of the data and Dr Charles Hurwitz for assistance in drafting the article.
Footnotes
Contributors: All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. MBU had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Funding: TEVA Pharmaceuticals.
Competing interests: All three authors are paid consultants for XTL Biopharmaceuticals Ltd which is involved in possible commercial development of Edratide.
Ethics approval: University Health Network Research Ethics Board.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: All authors were involved in the study conception and design, acquisition of data and analysis and interpretation of data.
Declaration: The study was conducted in accordance with the Declaration of Helsinki.
References
- 1.Hahn BH. An overview of the pathogenesis of systemic lupus erythematosus. In: Wallace DJ, Hahn BH, eds. Dubois’ lupus erythematosus. 5th edition 1993:69–76. [Google Scholar]
- 2.Nagy G, Koncz A, Perl A. T and B cell abnormalities in systemic lupus erythematous. Crit Rev Immunol 2005;25:123–40. doi:10.1615/CritRevImmunol.v25.i2.30 [DOI] [PubMed] [Google Scholar]
- 3.Trebeden-Negre H, Weill B, Fournier C et al. . B cell aptosis accelerates the onset of murine lupus. Eur J Immunol 2003;33:1603–12. doi:10.1002/eji.200323665 [DOI] [PubMed] [Google Scholar]
- 4.Navratil JS, Ahearn JM. Apoptosis, clearance mechanisms, and the development of systemic lupus erythematosus. Curr Rheumatol Rep 2001;3:191–8. doi:10.1007/s11926-001-0018-1 [DOI] [PubMed] [Google Scholar]
- 5.Horowitz DA, Jacob CO. The cytokine network in the pathogenesis of systemic lupus. Semin Immunopathol 1994;16:181–200. [DOI] [PubMed] [Google Scholar]
- 6.Dean GS, Tyrrell-Price J, Crawley E et al. . Cytokines and systemic lupus erythematosus. Ann Rheum Dis 2000;59:243–51. doi:10.1136/ard.59.4.243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paust S, Cantor H. Regulatory T cells and autoimmune disease. Immunol Rev 2005;204:195–207. doi:10.1111/j.0105-2896.2005.00247.x [DOI] [PubMed] [Google Scholar]
- 8.Sakaguchi S, Yamaguchi T, Nomuta T et al. . Regulatory T cells and immune tolerance. Cell 2008;133:775–87. doi:10.1016/j.cell.2008.05.009 [DOI] [PubMed] [Google Scholar]
- 9.Eisenberg R. Why can't we find a new treatment for SLE? 2009;32:223–30. doi:10.1016/j.jaut.2009.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haubitz M. New and emerging treatment approaches to lupus. Biologics 2010;4:263–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Navarra SV, Guzman RM, Gallacher AE et al. . Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled phase 3 trial. Lancet 2011;377:721–31. doi:10.1016/S0140-6736(10)61354-2 [DOI] [PubMed] [Google Scholar]
- 12.Sthoeger Z, Sharabi A, Mozes E. Novel approaches to the development of targeted therapeutic agents. J Autoimmun 2014;54:60–71. doi:10.1016/j.jaut.2014.06.002 [DOI] [PubMed] [Google Scholar]
- 13.Sthoeger ZM, Dayan M, Tcherniack A et al. . Modulation of autoreactive responses of peripheral blood lymphocytes of patients with systemic lupus erythematosus by peptides based on human and murine anti-DNA antibodies. Clin Exp Immunol 2003;131:385–92. doi:10.1046/j.1365-2249.2003.02058.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shoenfeld Y, Isenberg DA, Rauch J et al. . Idiotypic cross-reactions of monoclonal human lupus autoantibodies. J Exp Med 1983;58:718–30. doi:10.1084/jem.158.3.718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mozes A, Sharabi E. A novel tolerogenic peptide, hCDR1, for the specific treatment of systemic lupus erythematosus. Autoimmun Rev 2010;10:22–6. doi:10.1016/j.autrev.2010.07.004 [DOI] [PubMed] [Google Scholar]
- 16.Luger D, Dayan M, Zinger H et al. . A peptide based on the complementarity determining region 1 of a human monoclonal autoantibody ameliorates spontaneous and induced lupus manifestations in correlation with cytokine immunomodulation. J Clin Immunol 2004;24:579–90. doi:10.1007/s10875-004-6245-2 [DOI] [PubMed] [Google Scholar]
- 17.Mauermann N, Sthoeger Z, Zinger H et al. . Amelioration of lupus manifestations by a peptide based on the complementarity determining region 1 of an autoantibody in severe combined immunodeficient (SCID) mice engrafted with peripheral blood lymphocytes of systemic lupus erythematosus (SLE) patient. Clin Exp Immunol 2004;137:513–20. doi:10.1111/j.1365-2249.2004.02559.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharabi A, Zinger H, Zborowsky M et al. . A peptide based on the complementarity-determining region 1 of an autoantibody ameliorates lupus by upregulating CD4+CD25+ cells and TGF-β. Proc Natl Acad Sci 2006;103:8810–5. doi:10.1073/pnas.0603201103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sthoeger Z, Zinger H, Sharabi A et al. . The tolerogenic peptide, hCDR1, down-regulates the expression of interferon-α in murine and human systemic lupus erythematosus. PLoS ONE 2013;8:e60394 http://dx.doi.org/10.1371/journal.pone.0060394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharabi A, Dayan M, Zinger H et al. . A new model of induced experimental systemic lupus erythematosus (SLE) in pigs and its amelioration by treatment with a tolerogenic peptide. J Clin Immuno 2010;30:34–44. doi:10.1007/s10875-009-9326-4 [DOI] [PubMed] [Google Scholar]
- 21.Sthoeger ZM, Sharabi A, Dayan M et al. . The tolerogenic peptide, hCDR1, down-regulates pathogenic cytokines and apoptosis and upregulates immunosuppressive molecules and regulatory T cells in peripheral blood mononuclear cells of lupus patients. Hum Immunol 2009;70:139–45. doi:10.1016/j.humimm.2009.01.018 [DOI] [PubMed] [Google Scholar]
- 22.Sthoeger ZM, Sharabi A, Molad Y et al. . Treatment of lupus patients with a tolerogenic peptide, hCDR1 (Edratide). Immunomodulation of gene expression. J Autoimmunity 2009;33:77–82. doi:10.1016/j.jaut.2009.03.009 [DOI] [PubMed] [Google Scholar]
- 23.Gladman DD, Ibanez D Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol 2002;2:288–91. [PubMed] [Google Scholar]
- 24.Hay EM, Bacon PA, Gordon C et al. . The BILAG index: a reliable and valid instrument for measuring clinical disease activity in systemic lupus erythematosus. Q J Med 1993;7:447–58. [PubMed] [Google Scholar]
- 25.Cresswell L, Yee CS, Farewell V et al. . Numerical scoring for the classical BILAG index. Rheumatology (Oxford) 2009;48:1548–52. doi:10.1093/rheumatology/kep183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luijten KM, Tekstra J, Bijlsma JW et al. . The Systemic Lupus Erythematosus Responder Index (SRI); a new SLE disease activity assessment. Autoimmun Rev 2012;11:326–9. doi:10.1016/j.autrev.2011.06.011 [DOI] [PubMed] [Google Scholar]
- 27.Petri M, Buyon J, Kim Mimi. Classification and definition of major flares in SLE clinical trials. Lupus 1999;8:685–91. doi:10.1191/096120399680411281 [DOI] [PubMed] [Google Scholar]
- 28.Apostolou I, von Boehmer H. In vivo instruction of suppressor commitment in naïve T cells. J Exp Med 2004;199:1401–8. doi:10.1084/jem.20040249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang HK, Michaels MA, Berner BR et al. . Very low-dose tolerance with nucleosomal peptide controls lupus and induces potent regulatory T cell subsets. J Immunol 2005;174:3247–55. doi:10.4049/jimmunol.174.6.3247 [DOI] [PubMed] [Google Scholar]
- 30.Sharabi A, Haviv H, Zinger H, et al. Amelioration of murine lupus by a peptide, based on the complimentarity determining region-1 of an auto antibody as compared to dexamethasone: different effects on cytokines and aptosis. Clin Immunol 2006;119:146–55. doi:10.1016/j.clim.2006.01.007 [DOI] [PubMed] [Google Scholar]
- 31.Anonymous. Guidance for industry-systemic lupus erythematosus-developing medical products for treatment. s.l: : U.S. Department of Health and Human Services, 2010. [Google Scholar]