

Abstract

Hit-to-lead efforts resulted in the discovery of compound 19, a potent CYP11B2 inhibitor that displays high selectivity vs related CYPs, good pharmacokinetic properties in rat and rhesus, and lead-like physical properties. In a rhesus pharmacodynamic model, compound 19 displays robust, dose-dependent aldosterone lowering efficacy, with no apparent effect on cortisol levels.
Keywords: Aldosterone synthase, CYP11B2, hit-to-lead, hypertension
Aldosterone is a steroid hormone produced in the adrenal zona glomerulosa, and is one of several endogenous ligands that bind the mineralocorticoid receptor (MR).1,2 Binding of aldosterone to MR yields a complex that can activate gene transcription, ultimately leading to the production of proteins such as ENaC, the epithelial sodium channel responsible for reabsorption of sodium in the kidney. Classically, this sodium reabsorption and its attendant water retention and blood volume increase were thought to drive aldosterone’s known ability to increase blood pressure. However, more recent studies indicate that aldosterone raises blood pressure primarily by promoting vasoconstriction, and by acting in the CNS to increase central sympathetic drive.3 In addition to its effects on blood pressure (BP), aldosterone is also known to produce BP-independent organ damage via inflammatory and pro-fibrotic pathways, and to play a role in insulin resistance.4
Aldosterone synthase (CYP11B2) is a mitochondrial cytochrome P450 (CYP) enzyme that catalyzes the final three steps of aldosterone biosynthesis. Compounds that inhibit CYP11B2 should thus inhibit the formation of aldosterone, and may be useful as treatments for hypertension, heart failure, and diabetes. Small molecule CYP11B2 inhibitors have been reported in the literature, and one, LCI-699, has recently been shown to lower blood pressure in the clinic, thus providing validation for the use of CYP11B2 inhibitors as treatments for hypertension.5−10
Steroid-11β-hydroxylase (CYP11B1) is a related enzyme that catalyzes the formation of glucocorticoids such as cortisol, an important regulator of glucose metabolism. Human CYP11B2 and CYP11B1 are >93% homologous, and given the physiological importance of cortisol, selectivity for inhibition of CYP11B2 vs 11B1 is required. LCI-699 displays only ∼4-fold selectivity for inhibition of human CYP11B2 vs 11B1 in cell-based in vitro assays.7 In the clinic, doses of LCI-699 higher than 0.5 mg elicited suppression of cortisol levels, presumably as a result of CYP11B1 inhibition. Selectivity greater than that exhibited by LCI-699 would thus be desired to derisk the potential for adverse effects resulting from cortisol suppression.
Related CYPs 17 and 19, though less homologous to CYP11B2, play important roles in the conversion of steroidal precursors such as pregnenelone and progesterone to end products such as estrone and testosterone. Selectivity with regard to these CYPs as well as the major hepatic CYPs involved in drug metabolism is also required.
Our goal is to discover selective CYP11B2 inhibitors as treatments for hypertension. Previous studies by part of our team identified a range of attractive starting points for hit-to-lead studies.8 Of these, the pyridyl lactams first reported by the Hartmann group appeared particularly promising.10 Subsequent work in a related triazole series, described herein, has led to the discovery of potent CYP11B2 inhibitors that display selectivity vs related CYPs, good pharmacokinetic profiles, and robust aldosterone lowering in a rhesus pharmacodynamic model.
Triazole compounds were synthesized as described in the Supporting Information section, and were tested for inhibition of human CYP11B2 and CYP11B1. The preparation of these compounds has also been reported previously.11
The pyridyl lactams originally reported by Hartmann and co-workers displayed potent and selective inhibition of CYP11B2, but modest PK profiles. In an effort to improve both PK properties and structural novelty, we converted the bicyclic lactam to a tricyclic triazole. Compounds in this triazole series were optimized with respect to CYP11B2 potency, selectivity vs CYP11B1 and other CYPs, lipophilic ligand efficiency (LLE), and PK properties.12 We began by synthesizing a variety of triazoles bearing an attached pyridine. As shown in Table 1, the unsubstituted triazole 1 displayed moderately potent CYP11B2 inhibition. Triazoles substituted with small- or medium-sized alkyl groups such as 2–4 displayed similar potencies, with LLE decreasing as the lipophilicity of the triazole R group increased. Incorporation of larger alkyl groups such as cyclohexyl (compound 5) or tert-butyl (not shown) afforded less potent analogs. When compared to cyclohexyl derivative 5, the sterically similar phenyl analogue 6 proved to be considerably more potent, suggesting a beneficial pi-type interaction between 6 and the target. Unfortunately, while 6 displayed the most potent CYP11B2 inhibition in this series, it was an even more potent inhibitor of CYP17 and CYP19 (see Table 6 for details). Additional analogs were prepared that contained substituted phenyl or heteroaromatic R groups (not shown), but like 6, these also displayed potent CYP17 and CYP19 inhibition. On the basis of these data, the unsubstituted and methyl-substituted triazoles appeared to provide the best combination of CYP11B2 inhibition, B2/B1 selectivity, and lipophilic ligand efficiency, and thus were featured in subsequent studies.
Table 1. Effect of Triazole Substitution on CYP11B2 and B1 Inhibition and LLE.
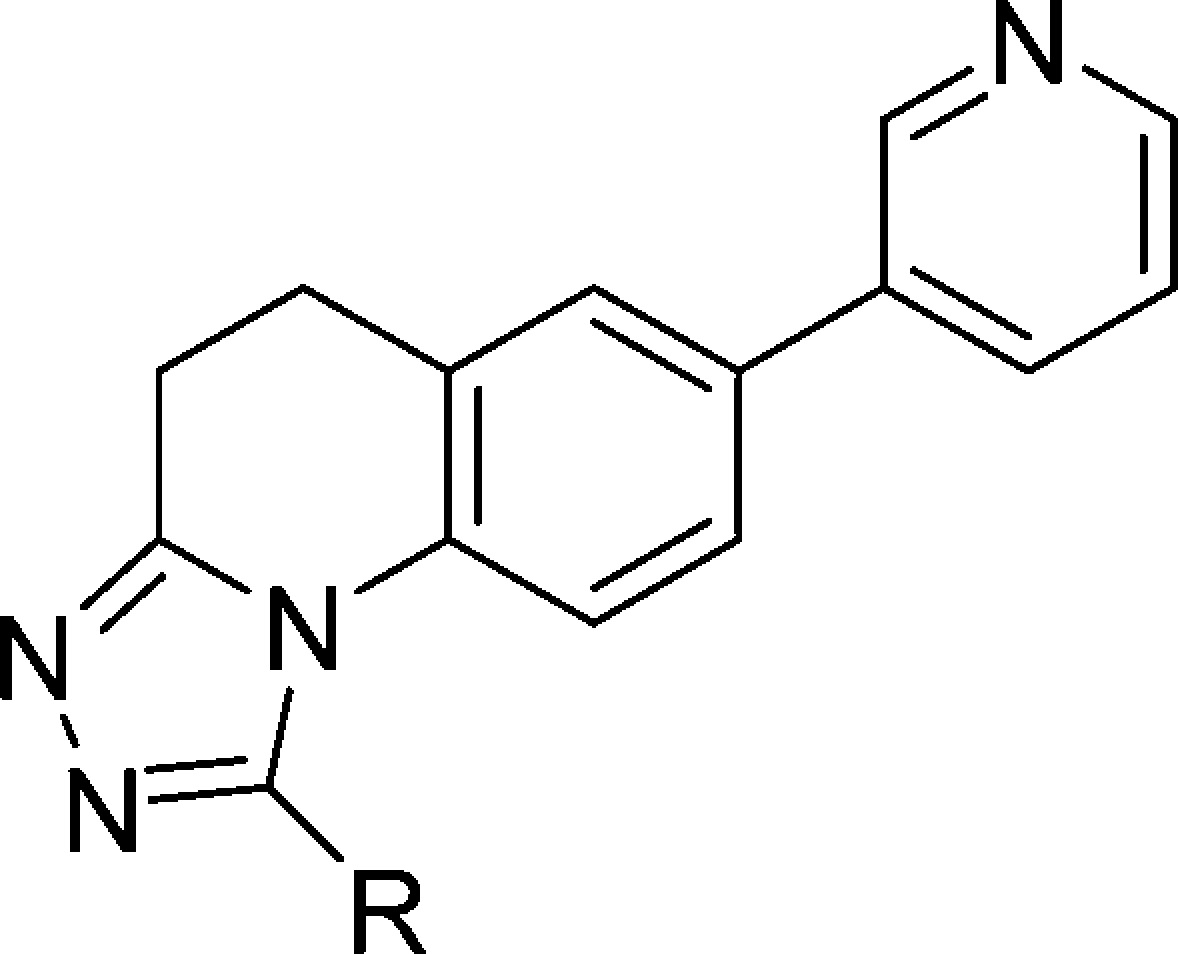
| Cpd | R | CYP11B2a (IC50, nM) | CYP11B1a (IC50, nM) | B1/B2b | LLEc |
|---|---|---|---|---|---|
| 1 | H | 300 | >10000 | >33 | 4.59 |
| 2 | Me | 423 | >10000 | >24 | 4.18 |
| 3 | c-Pr | 487 | >10000 | >21 | 3.36 |
| 4 | CF3 | 506 | >8333 | >16 | 3.02 |
| 5 | Cy | 2400 | >8333 | >3 | 1.30 |
| 6 | Ph | 103 | 1357 | 13 | 3.13 |
IC50s calculated from n ≥ 2, see Supporting Information for details.
Ratio of hCYP11B1 IC50/hCYP11B2 IC50.
Ligand lipophilic efficiency; LLE = pIC50 – aLogP98.
Table 6. Activity of Selected Compounds at Related Human CYP Enzyme Targets.
| Cpd | hCYP11B2a (IC50, nM) | hCYP17a (IC50, nM) | hCYP19a (IC50, nM) | hCYP3A4a (IC50, nM) |
|---|---|---|---|---|
| 6 | 103 | 84 | 21 | 36510 |
| 9 | 58 | >10000 | >10000 | >50000 |
| 15 | 19 | >10000 | 11160 | >50000 |
| 19 | 64 | >10000 | >30000 | >50000 |
IC50s calculated from n ≥ 2, see Supporting Information for details.
With optimized triazole substituents in hand, we next examined the effects of substitution on the pyridine ring, the presumed site of heme binding. Prior work had established that substitution of the 2- or 6-positions adjacent to the pyridine nitrogen was not tolerated. Indeed, substitution at either of those sites with even a methyl group typically abolished all activity. We thus focused our efforts at the 4- and 5-positions, with an emphasis on substituents that had previously proven optimal. As shown in Table 2, substitution of the 5-position with lipophilic electron-withdrawing groups such as fluoro (compound 7) or trifluoromethyl (compound 8) afforded no improvement in potency or B2/B1 selectivity relative to unsubstituted parent compound 2. In contrast, substitution with a methyl ester as in 9 yielded improvements in both potency and LLE. Unfortunately, ester replacements that might be more stable in vivo such as amides 10 and 11 or methyl sulfone 12 were inactive. Phenyl sulfone 13 displayed potent CYP11B2 inhibition, but its lower LLE relative to compounds like 9 reflects an undesired increase in lipophilicity. Substitution of the 5-position with a tertiary alcohol as in 14 gave a compound with reduced potency, but an improved PK profile (see Table 3). Conversion of alcohol 14 to the corresponding methyl ether 15 afforded a 10-fold improvement in potency and an attendant increase in LLE. Tertiary methyl ester 16 matched the potency of 15, but as with 9–12 above, more metabolically stable ester replacements, including a variety of amides and heterocycles, typically proved less potent (data not shown). Finally, isoquinoline 17 delivered the best combination of CYP11B2 inhibition, B2/B1 selectivity, and LLE observed in this series. Taken together, these results provided several promising starting points for subsequent lead optimization, and suggested that further exploration of substitution at the 4- and 5-positions was warranted.
Table 2. Effect of Pyridine Substitution on CYP11B2 and B1 Inhibition and LLE.
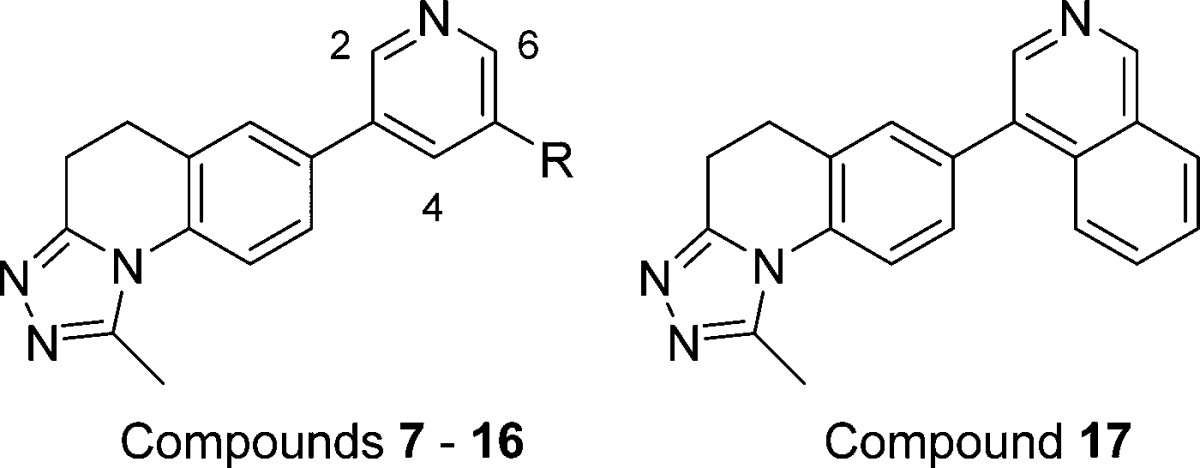
| Cpd | R | hCYP11B2a (IC50, nM) | hCYP11B1a (IC50, nM) | LLEb |
|---|---|---|---|---|
| 7 | F | 558 | >10000 | 3.86 |
| 8 | CF3 | 486 | >10000 | 3.18 |
| 9 | CO2CH3 | 58 | 2198 | 5.19 |
| 10 | C(O)NHCH3 | >8333 | >8333 | <3.68 |
| 11 | C(O)N(CH3)2 | >8333 | >8333 | <3.47 |
| 12 | SO2CH3 | >8333 | >8333 | <3.36 |
| 13 | SO2Ph | 40 | 1501 | 4.10 |
| 14 | C(CH3)2OH | 283 | >8333 | 4.38 |
| 15 | C(CH3)2OCH3 | 19 | 219 | 5.14 |
| 16 | C(CH3)2 CO2CH3 | 19 | 317 | 4.76 |
| 17 | 5 | 407 | 5.20 |
IC50s calculated from n ≥ 2, see Supporting Information for details.
Ligand lipophilic efficiency; LLE = pIC50 – aLogP98.
Table 3. Rat Pharmacokinetic Profiles of Selected Compoundsa.
| Cpd | Mic Clintb | F (%)c | AUCN (po)d | Clpe | t1/2f |
|---|---|---|---|---|---|
| 2 | 55 | 6 | 0.10 | 38 | 0.2 |
| 14 | <20 | 19 | 0.19 | 50 | 1.5 |
| 15 | 107 | 10 | 0.03 | 177 | 0.3 |
| 17 | 33 | 75 | 0.27 | 150 | 0.4 |
See Supporting Information for experimental details.
Intrinsic clearance in rat liver microsomes (mL/min/kg).
Bioavailability.
Area under the curve, normalized for dose (μM·h·kg/mg).
Plasma clearance (mL/min/kg).
Half-life (h).
Selected triazoles were profiled in a rat pharmacokinetic assay, and in assays that determined rat plasma protein binding and intrinsic clearance in rat liver microsomes.13 The parent methyl triazole 2 displayed a weak profile, with moderate intrinsic and plasma clearance, and low oral bioavailability and exposure (Table 3). A metabolite identification study, wherein compound 2 was incubated with rat liver microsomes and the products of oxidative metabolism identified by mass spectrometry, implicated conversion of the pyridine to a pyridine N-oxide as the primary form of oxidative metabolism. With that in mind, we next profiled triazole 14, which contained a bulky tertiary alcohol group that could potentially block enzymatic access to the pyridine. Compound 14 did in fact display lower intrinsic clearance than 2 in rat liver microsomes, as well as improved oral bioavailability and exposure. Somewhat surprisingly, it also displayed higher plasma clearance, indicating the involvement of non-CYP-mediated clearance pathways. These might involve, among other things, aldehyde oxidase (AO)-mediated oxidation of the pyridine or phase II conjugation of the tertiary alcohol. Alternatively, as 14 exhibits a low logD of 0.6 and good aqueous solubility, it might be cleared renally as intact parent. Compound 15, the methyl ether derivative of 14, was submitted for PK determination, but it displayed significantly higher intrinsic and plasma clearance, suggesting the ether methyl as a site of metabolism. Finally, potent triazole 17 was also profiled, but it too suffered high plasma clearance and, like 14, exhibited a disconnect between its intrinsic and plasma clearance rates. In this case, as isoquinolines are known substrates for AO, the higher plasma clearance of 17 might be explained by AO-mediated oxidation of its isoquinoline domain.14
Compound 14 displayed an initial profile that was promising, but suffered from non-CYP-mediated clearance that limited its potential. We speculated that its low lipophilicity (logD = 0.6) might underlie undesired renal clearance of water-soluble parent compound, and/or suboptimal absorption through cell membranes. If that were true, we would be in the somewhat unusual (yet easily remedied) position of needing to increase the lipophilicity of our compounds. To test this hypothesis, we prepared analogs of 14 that replaced a methyl group at the R,1 R2, or R3 position with a more lipophilic trifluoromethyl group. As shown in Table 4, replacement of the triazole R1 methyl with a trifluoromethyl gave 18, a compound with a logD nearly a full unit higher than that of 14. Unfortunately, compound 18 displayed only modest CYP11B2 inhibition and low B2/B1 selectivity. Replacement of the R2 or R3 methyl groups with trifluoromethyl was more productive, affording a pair of enantiomers, 19 and 20, that were more potent and selective than either 18 or 14.15 In fact, since the R3 trifluoromethyl of 19 confers added potency, compound 19 is able to maintain an LLE equal to 14’s despite its higher lipophilicity. Finally, replacement of both the R1 and R3 methyls or the R1 and R2 methyls with trifluoromethyl yielded enantiomers 21 and 22. While the lipophilicities of these compounds fall well within what is often considered to be the ideal range (logD = 1–3), their modest potencies and B2/B1 selectivities prevented them from receiving further consideration.
Table 4. Effects of Trifluoromethylation at R1, R2, and R3.
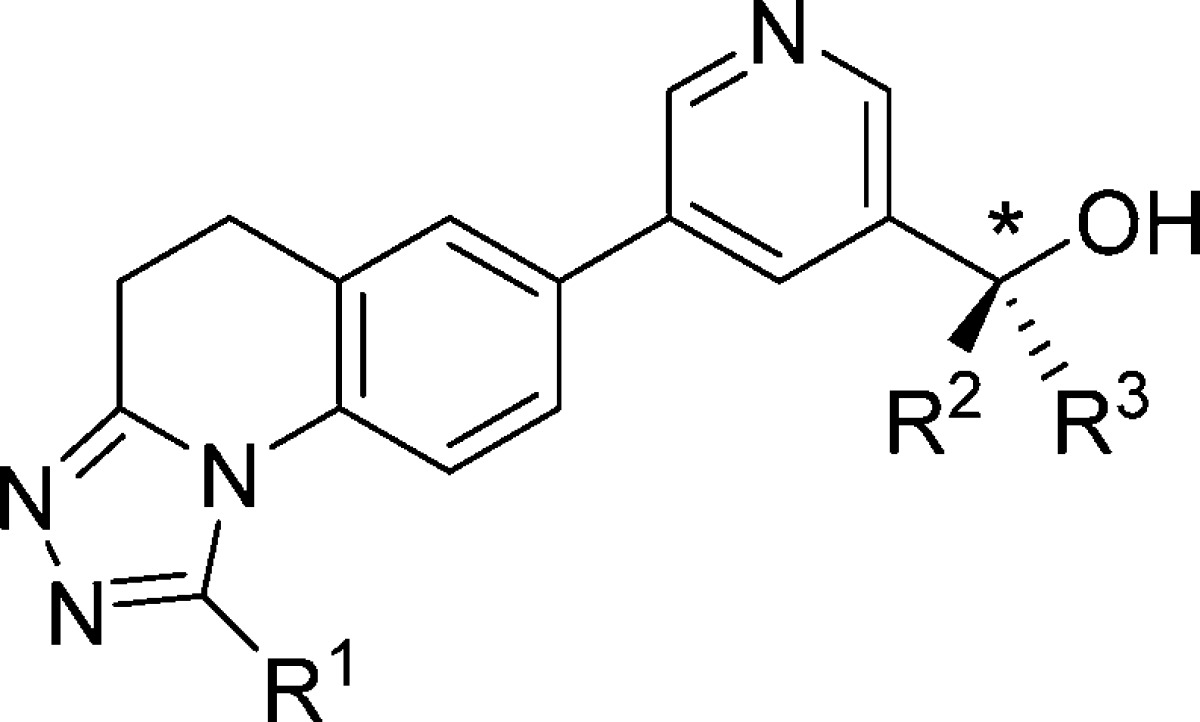
| Cpd | * | R1 | R2 | R3 | hCYP11B2a (IC50, nM) | hCYP11B1a (IC50, nM) | B1/B2b | LLEc | logDd |
|---|---|---|---|---|---|---|---|---|---|
| 14 | CH3 | CH3 | CH3 | 283 | >8333 | 29 | 4.38 | 0.6 | |
| 18 | CF3 | CH3 | CH3 | 526 | 1473 | 3 | 3.02 | 1.5 | |
| 19 | (S) | CH3 | CH3 | CF3 | 64 | 4402 | 69 | 4.36 | 1.2 |
| 20 | (R) | CH3 | CF3 | CH3 | 109 | >8333 | 76 | 4.13 | 1.2 |
| 21 | (S) | CF3 | CH3 | CF3 | 461 | 887 | 2 | 2.42 | 2.2 |
| 22 | (R) | CF3 | CF3 | CH3 | 476 | >8333 | >18 | 2.41 | 2.2 |
IC50s calculated from n ≥ 2, see Supporting Information for details.
Ratio of hCYP11B1 IC50/hCYP11B2 IC50.
Ligand lipophilic efficiency; LLE = pIC50 – aLogP98.
Determined experimentally by HPLC.
By increasing lipophilicity, we were able to significantly improve pharmacokinetic properties in this series. When tested in rats, trifluoromethylated compounds 18 and 19 showed reduced plasma clearance and significantly higher oral bioavailability and exposure relative to compound 14 (Table 5). In the case of compound 19, the improvement in plasma clearance may be attributable to the reduction in free fraction, but in the case of compound 18, additional factors may contribute. The improvements in oral exposure observed with 18 and 19 exceed what would be expected based on the reductions in plasma clearance, and indicate improved absorption for these compounds as well. Notably, key compound 19 displays a strong PK profile in both rat and rhesus, with low intrinsic and plasma clearance, a significant free fraction, high oral exposure, and in rhesus, a half-life of 7.8 h.
Table 5. Rat and Rhesus Pharmacokinetic Profiles of Selected Compoundsa.
| Cpd | logDb | species | PPBc | Mic Clintd | F (%)e | AUCN (po)f | Clpg | t1/2h |
|---|---|---|---|---|---|---|---|---|
| 14 | 0.6 | rat | 67 | <20 | 19 | 0.19 | 50 | 1.5 |
| 19 | 1.2 | rat | 25 | <20 | 87 | 1.88 | 21 | 1.2 |
| 18 | 1.5 | rat | 30 | <20 | 83 | 3.87 | 10 | 2.9 |
| 19 | 1.2 | rhesus | 31 | <20 | 100 | 16.26 | 3 | 7.8 |
Assay details reported in Supporting Information.
Determined experimentally by HPLC.
Plasma protein binding, reported as % free compound.
Intrinsic clearance in liver microsomes (mL/min/kg).
Bioavailability.
Area under the curve, normalized for dose (μM·h·kg/mg).
Plasma clearance (mL/min/kg).
Half-life (h).
Compounds from this series exhibited high selectivity versus related CYP targets. As exemplified by the representatives in Table 6, triazoles typically displayed high selectivity for inhibition of CYP11B2 vs mitochondrial CYPs 17 and 19, and hepatic CYPs 3A4, 2C9, and 2D6 (data for 2C9 and 2D6 not shown). Aryl substituted triazoles such as compound 6 were the exception, as these showed potent and undesirable inhibition of CYPs 17 and 19.
A benchmark compound from this series also displayed high selectivity versus a broad range of additional targets. Compound 19 was tested against a panel of 37 enzymes, receptors and ion channels, and at a concentration of 10 μM, displayed <40% activity against all targets in the screen. Compound 19 was also tested in assays that measure block of ion channels important for cardiac functioning. Specifically, it was tested in a binding assay that measures displacement of 35S-labeled MK-0499, a known hERG blocker, and in functional assays that measure block of Nav1.5 and Cav1.2.16 In each of these assays, compound 20 exhibited an IC50 > 30 μM, indicating minimal risk for cardiovascular adverse effects.
Compounds from this series were potent inhibitors of human and rhesus CYP11B2 (Table 7), but were typically much less potent inhibitors of rat CYP11B2 (data not shown). Given the low identity/homology between human and rat CYP11B2 (∼80%), and the higher homology between human and rhesus CYP11B2 (∼95%), this is perhaps not surprising. As a result, key compounds could not be evaluated for aldosterone lowering efficacy in previously described rat models, and were instead evaluated in a rhesus pharmacodynamic model.
Table 7. Activity of Selected Compounds at Human and Rhesus CYP11B2.
IC50s calculated from n ≥ 2, see Supporting Information for details.
Compound 19 displayed robust, dose-dependent aldosterone lowering efficacy when dosed i.v. in a rhesus pharmacodynamic model. Details of the experimental protocol employed in this model have been recently described.17 In this model, monkeys are anesthetized to maintain stable baseline levels of aldosterone. Thus, even though compound 19 displays a PK profile suitable for oral dosing, the use of anesthetized animals necessitated i.v. dosing. Compound 19 was administered to anesthetized rhesus monkeys at i.v. doses of 0.003, 0.01, 0.03, 0.1, 0.3, and 1.0 mg/kg. Plasma levels of aldosterone and 11-deoxycortisol (11-DOC), a biosynthetic precursor of cortisol, were quantified by LC–MS at various time points up to 180 min postdose, allowing the determination of AUCs for aldosterone and 11-DOC over that time course. Plasma concentrations of compound 19 were measured at 90 min postdose, and free (unbound) concentrations were calculated based on 19’s 31% free fraction in rhesus plasma.
When dosed i.v. at 0.1, 0.3, and 1.0 mg/kg, compound 19 produced statistically significant reductions in aldosterone AUC of 48%, 71%, and 98%, respectively, when compared to baseline (Figure 1). Compound 19 achieved these effects at free plasma concentrations 90 min postdose of 19, 73, and 279 nM, respectively. The reduction in aldosterone AUC effected by a 1.0 mg/kg dose of compound 19 is comparable to that produced by a 1.0 mg/kg i.v. dose of Fadrozole, a potent but poorly selective CYP19/CYP11B2/CYP11B1 inhibitor that has been shown to reduce blood pressure in rodent preclinical models of hypertension.18−20 Additionally, at doses up to 1.0 mg/kg, compound 19 does not produce an increase in 11-DOC AUC, suggesting that CYP11B1, the enzyme that converts 11-DOC to cortisol, has not been significantly inhibited. In contrast, a 1.0 mg/kg dose of Fadrozole, which displays potent inhibition of both CYP11B2 and CYP11B1, elicits a significant increase in 11-DOC AUC. These data demonstrate that, in an acute setting, compound 19 is capable of producing significant reductions in rhesus plasma aldosterone levels with no apparent effect on cortisol levels.
Figure 1.
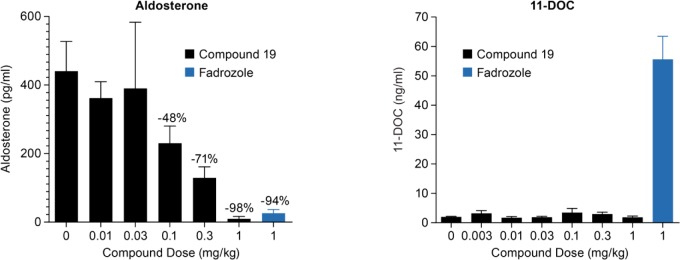
Effects of compound 19 on plasma AUCs of aldosterone and 11-deoxycortisol (11-DOC) in rhesus monkeys.
In these initial experiments, compound 19 displays good agreement between in vitro and in vivo assay results. At a free plasma concentration of 19 nM, compound 19 produces a 48% reduction in aldosterone AUC, which is consistent with its potent inhibition of rhesus CYP11B2 (IC50 = 12 nM). Additionally, at free plasma concentrations up to 279 nM, no reduction in 11-DOC AUC and no apparent inhibition of CYP11B1 are observed, which would be expected given compound 19’s weaker inhibition of rhesus CYP11B1 (IC50 = 3483 nM).
Aldosterone is a clinically validated biomarker for blood pressure, which itself is a strongly validated biomarker for cardiovascular morbidity and mortality. In a large (n = 524) Phase II clinical trial, 0.5 mg of LCI-699 dosed twice daily produced a mean 34% reduction in plasma aldosterone, along with significant reductions in mean systolic (−9.7 mmHg) and diastolic (−4.7 mmHg) blood pressure at 8 weeks.5 Compound 19’s profound ability to reduce plasma aldosterone levels (>95%) in rhesus thus suggests that a compound from this series with a suitable profile would be efficacious at reducing blood pressure in a clinical setting.
In summary, we have reported the discovery and hit-to-lead optimization of a novel series of triazole CYP11B2 inhibitors. A benchmark compound from this series, compound 19, displays potent inhibition of human and rhesus CYP11B2, high selectivity versus related CYP targets, and a good pharmacokinetic profile in rat and rhesus. In a rhesus pharmacodynamic model, compound 19 exhibits robust, dose-dependent aldosterone lowering efficacy. At doses and plasma levels that produce a 98% reduction in plasma aldosterone AUC, compound 19 elicits no increase in the AUC of 11-deoxycortisol, a biosynthetic precursor of cortisol. Additional lead optimization in this series has been completed, and will be reported in due course.
Glossary
ABBREVIATIONS
- 11-DOC
11-deoxycortisol
- AO
aldehyde oxidase
- AUC
area under curve
- BP
blood pressure
- CNS
central nervous system
- ENaC
epithelial sodium channel
- LLE
lipophilic ligand efficiency
- MR
mineralocorticoid receptor
- PK
pharmacokinetics
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00048.
Assay protocols, synthesis schemes, representative procedures for the synthesis of compounds 1, 16, 17, and 19 and related intermediates, and X-ray crystallographic data for intermediate 48 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Carey R. M. Aldosterone and cardiovascular disease. Curr. Opin. Endocrinol., Diabetes Obes. 2010, 17, 194–198. 10.1097/MED.0b013e3283390fa4. [DOI] [PubMed] [Google Scholar]
- Shavit L.; Lifschitz M. D.; Epstein M. Aldosterone blockade and the mineralocorticoid receptor in the management of chronic kidney disease: current concepts and emerging treatment paradigms. Kidney Int. 2012, 81, 955–968. 10.1038/ki.2011.505. [DOI] [PubMed] [Google Scholar]
- Tomaschitz A.; Pilz S.; Ritz E.; Obermayer-Pietsch B.; Pieber T. R. Aldosterone and arterial hypertension. Nat. Rev. Endocrinol. 2010, 6, 83–93. 10.1038/nrendo.2009.263. [DOI] [PubMed] [Google Scholar]
- Gilbert K. C.; Brown N. J. Aldosterone and inflammation. Curr. Opin. Endocrinol., Diabetes Obes. 2010, 17, 199–204. 10.1097/MED.0b013e3283391989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun D. C.; White W. B.; Krum H.; Guo W.; Bermann G.; Trapani A.; Lefkowitz M. P.; Menard J. Effects of a novel aldosterone synthase inhibitor for treatment of primary hypertension. Circulation 2011, 124, 1945–1955. 10.1161/CIRCULATIONAHA.111.029892. [DOI] [PubMed] [Google Scholar]
- Azizi M.; Amar L.; Menard J. Aldosterone synthase inhibition in humans. Nephrol., Dial., Transplant. 2013, 28, 36–43. 10.1093/ndt/gfs388. [DOI] [PubMed] [Google Scholar]
- Meredith E. L.; Ksander G.; Monovich L. G.; Papillon J. P. N.; Liu Q.; Miranda K.; Morris P.; Rao C.; Burgis R.; Capparelli M.; Hu Q.-Y.; Singh A.; Rigel D. F.; Jeng A. Y.; Beil M.; Fu F.; Hu C.-W.; LaSala D. Discovery and in vivo evaluation of potent dual CYP11B2 (Aldosterone Synthase) and CYP11B1 inhibitors. ACS Med. Chem. Lett. 2013, 4, 1203–1207. 10.1021/ml400324c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a recent review of aldosterone synthase inhibitors, see:Hu Q.; Yin L.; Hartmann R. W. Aldosterone synthase inhibitors as promising treatments for mineralocorticoid dependent cardiovascular and renal diseases. J. Med. Chem. 2014, 57, 5011–5022. 10.1021/jm401430e. [DOI] [PubMed] [Google Scholar]
- Cerny M. A. Progress toward clinically useful aldosterone synthase inhbitors. Curr. Top. Med. Chem. 2013, 13, 1385–1401. 10.2174/1568026611313120003. [DOI] [PubMed] [Google Scholar]
- Lucas S.; Heim R.; Ries C.; Schewe K. E.; Birk B.; Hartmann R. W. In vivo active aldosterone synthase inhibitors with improved selectivity: lead optimization providing a series of pyridine substituted 3,4-dihydro-1H-quinolin-2-one derivatives. J. Med. Chem. 2008, 51, 8077–8087. 10.1021/jm800888q. [DOI] [PubMed] [Google Scholar]
- Hoyt S. B.; Petrilli W. L.; London C.; Xiong Y.; Taylor J. A.; Ali A.; Lo M.; Henderson T. J.; Hu Q.; Hartmann R.; Yin L.; Heim R.; Bey E.; Saxena R.; Samanta S. K.; Kulkarni B. A.. Aldosterone Synthase Inhibitors. WO 2012148808, 11/01/2012.
- Lipophilic ligand efficieny (LLE) was calculated using the equation LLE = pIC50 – aLogP98, as first proposed by:Leeson P. D.; Springthorpe B. Nat. Rev. Drug Discovery 2007, 6, 881–890. 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- For detailed experimental protocols, see Supporting Information section.
- Pryde D. C.; Dalvie D.; Hu Q.; Jones P.; Obach R. S.; Tran T.-D. Aldehyde oxidase: an enzyme of emerging importance in drug discovery. J. Med. Chem. 2010, 53, 8441–8460. 10.1021/jm100888d. [DOI] [PubMed] [Google Scholar]
- The absolute configurations of compounds 19–22 were established by X-ray crystallographic analysis; see Supporting Information for details.
- Wang J.; Della Penna K.; Wang H.; Karczewski J.; Connolly T. M.; Koblan K. S.; Bennett P. B.; Salata J. J. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H256. 10.1152/ajpheart.00220.2002. [DOI] [PubMed] [Google Scholar]
- Cai T.-Q.; Stribling S.; Tong X.; Xu L.; Wisniewski T.; Fontenot J. A.; Struthers M.; Akinsanya K. O. Rhesus monkey model for concurrent analyses of pharmacodynamics, pharmacokinetics and in vivo selectivity of aldosterone synthase inhibitors. J. Pharmacol. Toxicol. Methods 2015, 71, 137–146. 10.1016/j.vascn.2014.09.011. [DOI] [PubMed] [Google Scholar]
- Rigel D. F.; Fu F.; Beil M.; Hu C.-W.; Liang G.; Jeng A. Y. Pharmacodynamic and pharmacokinetic characterization of the aldosterone synthase inhibitor FAD286 in two rodent models of hyperaldosteronism: comparison with the 11beta-hydroxylase inhibitor metapyrone. J. Pharmacol. Exp. Ther. 2010, 334, 232–243. 10.1124/jpet.110.167148. [DOI] [PubMed] [Google Scholar]
- LaSala D.; Shibanaka Y.; Jeng A. Y. Coexpression of CYP11B2 or CYP11B1 with adrenodoxin and adrenodoxin reductase for assessing the potency and selectivity of aldosterone synthase inhibitors. Anal. Biochem. 2009, 394, 56–61. 10.1016/j.ab.2009.07.025. [DOI] [PubMed] [Google Scholar]
- Huang B. S.; White R. A.; Jeng A. Y.; Leenen F. H. H. Role of central nervous system aldosterone synthase and mineralocorticoid receptors in salt-induced hypertension in Dahl salt-sensitive rats. Am. J. Physiol. Reg. Integr. Comp. Physiol. 2009, 296, R994–R1000. 10.1152/ajpregu.90903.2008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.